

Abstract
In contrast to its inhibitory role in mature neurons, GABA can exert excitatory actions in developing neurons, including mediation of increases in cytosolic Ca2+. Modulation of this excitatory activity has not been studied previously. We used Ca2+ digital imaging with Fura-2 to test the hypothesis that neuropeptide Y (NPY) would depress GABA-mediated Ca2+ rises in neurons cultured from the developing suprachiasmatic nucleus (SCN). SCN neurons were chosen as a model system for this study because SCN neurons are primarily GABAergic, they express high levels of NPY and GABA receptors, and functionally, NPY causes profound phase-shifts in SCN-generated circadian rhythms.
Vigorous GABA-mediated Ca2+ activity was found in young SCN neurons that were maintained in vitro for 4–14 d. NPY showed a dose-dependent rapid depression of the amplitude of Ca2+ rises generated by GABA released from presynaptic SCN axons. NPY exerted a long-term depression of cytosolic Ca2+ in the majority of neurons tested, which lasted more than 1 hr after NPY washout. The magnitude of the NPY depression was dose-dependent. NPY did not affect Ca2+ levels when GABAAreceptor activity was blocked by bicuculline; however, when bicuculline and NPY were withdrawn from the perfusion solution, the subsequent Ca2+ rise was either significantly reduced or completely absent, suggesting that the NPY receptor was activated in the absence of elevated intracellular Ca2+ and GABAA receptor activity, and that the latent effect of NPY was revealed only after depolarizing GABA stimulation was renewed. Pretreating neurons with pertussis toxin greatly reduced the ability of NPY to depress GABAergic Ca2+ rises, suggesting that the NPY modulation of the GABA activity was based largely on a mechanism involving pertussis toxin-sensitive Gi/Go proteins.
NPY receptor stimulation depressed (<30%) postsynaptic Ca2+ rises evoked by GABA (20 μm) application in the presence of tetrodotoxin (TTX). The effects of NPY were mimicked by the NPY Y1 receptor agonist [Pro34,Leu31] NPY and the Y2 receptor agonist NPY 13–36 and by peptide YY (PYY). Together, our data suggest that the Y1 and Y2 type NPY receptors act both presynaptically and postsynaptically to depress GABA-mediated Ca2+ rises. If related mechanisms exist in peptide modulation of inhibitory GABA activity in mature neurons, this could underlie long-term changes in the behavior of neurons of the SCN necessary for phase-shifting the circadian clock by NPY. NPY also modulated GABA responses in neuroendocrine neurons from the hypothalamic arcuate nucleus. NPY thus can play an important role in evoking long-term depression of GABA-mediated Ca2+ activity in these developing neurons, allowing NPY-secreting cells to modulate the effects of GABA on neurite outgrowth, gene expression, and physiological stimulation. This is the first example of such a cellular memory: that is, long-term Ca2+ depression based on modulation of depolarizing GABA activity.
Keywords: NPY, GABAA receptor, suprachiasmatic nucleus, arcuate nucleus, calcium, neuroendocrine, modulation
A number of studies have shown that GABA acts as an excitatory transmitter during neuronal development, depolarizing the membrane potential and eliciting a cytosolic Ca2+increase (Yuste and Katz, 1991; Horvath et al., 1993; Yamashita and Fukuda, 1993). The probable mechanism through which GABA elicits cytosolic Ca2+ increases in hypothalamic neurons seems to involve activation of the GABAAreceptor; subsequent Cl− efflux attributable to a developmental depolarized Cl− reversal potential causes membrane depolarization and Ca2+entry via voltage-activated Ca2+ channels (Obrietan and van den Pol, 1995; Chen et al, 1996).
GABA modulation of cytosolic Ca2+ could affect various events during neuronal development. Ca2+alters the rate and direction of neurite growth (Mattson and Kater, 1987) and influences gene expression (Vaccarino et al., 1992; Bading et al., 1993). Similarly, GABA increases neurite outgrowth of brain and retinal neurites (Spoerri, 1988; Michler, 1990; Barbin et al., 1993) and modulates synapse formation in cultured cells (Meier et al., 1984;Hansen et al., 1987). Later in development, GABA reverses roles and assumes its classic function as an inhibitory neurotransmitter, suppressing membrane depolarizing excitatory activity. A previously unstudied question that we address is whether the Ca2+-elevating ability of GABA can be modulated during development by peptides such as neuropeptide Y (NPY).
NPY is a 36 amino acid peptide that is found throughout the central and peripheral nervous system from the time of neurogenesis to adulthood (Allen et al., 1984; Chronwall et al., 1985; Woodhams et al., 1985;Barnea et al., 1991). Its neuromodulatory role is widespread and complex. At the cellular level, NPY has been reported to act either presynaptically (via the NPY Y2 receptor) or postsynaptically (via the NPY Y1 receptor) to regulate neuronal excitability (Khanna et al., 1993; Simonneaux et al., 1994). Modulation of Ca2+ influx through voltage-activated Ca2+ channels may be one mechanism through which NPY regulates neural activity (Walker et al., 1988; Wiley et al., 1990,1993; McQuiston et al., 1996). Many physiological and behavioral effects of NPY also have been identified. For example, the infusion of NPY into the hypothalamus modulates circulating levels of the pituitary hormone corticosterone (Albers et al., 1990), stimulates bouts of feeding that can last several hours (Stanley and Leibowitz, 1985), and alters body temperature (Jolicoeur et al., 1995).
A brain region well suited for the study of NPY modulation of GABA-mediated activity is the suprachiasmatic nucleus (SCN) of the hypothalamus. The SCN has a large number of presynaptic GABAergic axons, and most neurons in the SCN synthesize GABA (Card and Moore, 1984; van den Pol and Tsujimoto, 1985; Decavel and van den Pol, 1990;Morely and Flood, 1990; Moore and Speh, 1993). Frequent synaptic contacts between peptidergic and GABAergic cells are found in the SCN (van den Pol and Gorcs, 1986). The SCN functions as the circadian pacemaker of the brain (for review, see van den Pol and Dudek, 1993). This circadian rhythm is in turn entrained to the environmental light cycle via photic input from the optic nerve. In addition to this direct photic pathway, NPY- and GABA-containing afferents of neurons from the intergeniculate leaflet (IGL) form an indirect pathway by which photic information is transmitted to the SCN (Swanson et al., 1974; Card and Moore, 1989; Botchkina and Morin, 1995). Damage to, or electrical stimulation of, NPY-containing neurons of the IGL results in a phase-shift of the circadian rhythm (Harrington et al., 1985;Harrington and Rusak, 1986; Johnson et al., 1989; Rusak et al., 1989). Behavioral and brain-slice studies have shown that NPY applied to the SCN can phase-shift circadian rhythms by 1 hr or more (Albers and Ferris, 1984; Medanic and Gillette, 1993; Shibata and Moore, 1993;Huhman and Albers, 1994) and that the effects of NPY can be inhibited by the GABAA receptor antagonist bicuculline (Huhman et al., 1995). Because extensive phase shifts in the circadian rhythm can be generated by brief exposure to light or by a single exposure to NPY, a long-duration change in cellular behavior would be expected. The mechanisms used by NPY to exert a long-lasting effect on neuronal activity have not been examined.
The studies presented here suggest that NPY not only can modulate the excitatory actions of GABA in developing neurons but also can cause a long-term depression of GABA-elicited Ca2+activity, a form of cellular learning, mediated through Y1 and Y2 receptors with both pre- and postsynaptic components.
MATERIALS AND METHODS
Tissue culture. Two techniques were used to dissect the SCN from embryonic day 19–21 (E19–21) Sprague–Dawley rats. In the first technique, the SCN was dissected from whole brains using anatomical markers to identify its location. The SCN was identified by its position at the ventral surface of the hypothalamus, extending laterally from the third ventricle and directly above the optic chiasm. Tissue was washed immediately three times in standard tissue culture medium (glutamate- and glutamine-free minimal essential medium, 10% fetal bovine serum, 100 U/ml penicillin/streptomycin, and 6 gm/l glucose) and then incubated in a papain digestion solution (Earle’s balanced salt solution containing 10 units/ml papain, 500 μm EDTA, 1.5 μmCaCl2, 0.2 mg/mll-cysteine) for 30 min. The papain solution was removed by aspiration after the tissue was pelleted by centrifugation. The tissue was then triturated into a single-cell suspension and plated onto poly-d-lysine-coated glass coverslips (22 × 22 mm). The protocol for the coverslip preparation included washing with mild detergent, rinsing three times with distilled water, and then autoclaving. High local neuronal density was ensured by plating the cells within a 7 mm diameter glass ring placed on the coverslip. The glass ring was removed 60 min after plating. Cultures were maintained in standard tissue culture medium at 37°C and 5% CO2 in a Napco 6100 incubator. The arcuate nucleus, situated at the ventral surface of the hypothalamus, caudal to the SCN, and on either side of the third ventricle was cultured in a manner identical to that described above for the SCN. Cytosine arabinofuranoside (1 μm) was added to the tissue culture medium of cultures maintained over 6 d to reduce astrocyte proliferation.
Because the first dissection method likely included cells from areas outside the SCN, we used a second technique in which the SCN was microdissected precisely. Whole neonatal brain was chilled to 4°C and then cut into 400–500 μm coronal slices using a tissue chopper. The slice containing the SCN was identified by morphological markers. A polished 22 gauge needle was used to punch out the SCN. Punched SCN tissue was placed briefly in the papain digestion solution described above to help wash off cellular debris and then was placed on poly-d-lysine-coated glass coverslips. No trituration was performed, thereby allowing SCN neurons to be cultured as an intact organotypic block with general neuroanatomical features and local connections intact. Cultures were maintained as described above.
Calcium digital imaging. Coverslips initially were incubated in standard HEPES perfusion solution (10 HEPES, 137 mm NaCl, 25 mm glucose, 5 mm KCl, 1 mmMgCl2, 3 mmCaCl2, pH 7.4) containing 5 μm Fura-2 acetoxymethyl ester for 30 min at 37°C. Neurons were then washed three times and allowed to recover for 15 min before the start of the experiment. Coverslips were placed in a laminar flow perfusion chamber (Forscher et al., 1987). Rapid changes in the content of the perfusion solution were accomplished by placing an individual port for each perfusion solution at the laminar glass interface. Solution passed through the 180 μl perfusion chamber as a straight wave, thereby minimizing mixing of different perfusion solutions. Complete washout of the chamber was achieved in ∼5 sec. This rapid and complete washout is one great advantage of using monolayer cultured cells for studying long-term or latent effects of neuroactive compounds.
Cells were imaged on a Nikon Diaphot 300 inverted microscope with an Olympus DApo 40× objective with high UV light transmittance. Ca2+ responses were recorded from the neuronal soma. Neurons were identified by their phase-bright appearance. Occasionally, NMDA also was used to verify the identity of neurons. Only healthy neurons responded to NMDA; astrocytes did not. All experiments were performed at room temperature.
A Sutter filter wheel controlled by a Lambda 10 microprocessor was used to switch the excitation light from a 150 W xenon lamp between 340 and 380 nm. Light emitted from the neurons was passed back through the microscope, a 480 nm filter, and then focused on a Hamamatsu 2400 silicon intensified target video camera. Excitation light was attenuated 90% to inhibit photobleaching and phototoxicity, thereby allow recording sessions of >90 min. Data from up to 64 neurons could be recorded simultaneously by a 486 PC computer with Fluor software (Universal Imaging, West Chester, PA). Sixteen video frames (500 msec) of data were recorded from both wavelengths every 2 sec. A shutter was used to block excitation light between the periods of data acquisition. Data were collected in the form of digitized, background-subtracted, single-cell ratiometric values. As described by Grynkiewicz et al. (1985), ratiometric fluorescent values were converted to free Ca2+ values with the equation [Ca2+]i = Kd(R − Rmin)/(Rmax − R), where R is the ratio of the two fluorescence intensities, Rmin is the ratio in the absence of Ca2+, andRmax is the ratio in a saturating concentration of Ca2+. TheKd for Fura 2 + Ca2+was taken to be 224 nm (Grynkiewicz et al., 1985). Calibrated Ca2+ data from single neurons were transferred to an Apple 840AV computer and analyzed with Igor Pro software (WaveMetrics).
For assays that measured the effect of NPY receptor agonists on endogenous GABA-mediated Ca2+ rises, we initially determined the average Ca2+ level in the presence of bicuculline for the 15 sec immediately preceding the removal of bicuculline from the perfusion solution. The Ca2+increase after bicuculline removal was determined by averaging the Ca2+ rise over the 15 sec period immediately preceding the application of NPY receptor agonist. The mean Ca2+ change in the presence of NPY receptor agonist was determined by averaging the Ca2+levels over a 15 sec period 100 sec after the application of NPY. Data from individual neurons were then pooled as a population mean. Responses are reported as the mean Ca2+ rise from basal Ca2+ level (in the presence of bicuculline) ± SEM. For assays that measured peak exogenously evoked Ca2+ rises, the mean basal Ca2+ level over the 15 sec immediately preceding the evoked Ca2+ rise was averaged. This value was subtracted from the peak-evoked Ca2+ level to determine the maximal evoked Ca2+ rise. Data from individual neurons were then pooled as a population mean. Responses are reported as the mean/peak Ca2+ rise from mean basal Ca2+ level ± SEM. Experiments were performed on neurons after 3–5 d in vitro, unless stated otherwise.
Cytosine arabinofuranoside, GABA, muscimol, NMDA, benextramine, and poly-d-lysine were acquired from Sigma.d,l-2-Amino-5-phosphonopentanoic acid (AP5), CNQX, baclofen, bicuculline, nimodipine, and TTX were acquired from Research Biochemicals International. NPY, NPY 13–36, [Pro34,Leu31] NPY, and PYY were acquired from Peninsula Labs. Papain was acquired from Worthington Biochemicals (Freehold, NJ), DMEM from Gibco (Gaithersburg, MD), C2-NPY from Bachem California (Torrance, CA), and Fura-2 acetoxymethyl ester from Molecular Probes (Eugene, OR).
RESULTS
Depression of neuronal Ca2+ by NPY
As described previously (Obrietan and van den Pol, 1995), GABA secreted from neurons derived from the developing SCN induced sustained Ca2+ elevations in a majority of neurons. In the present study, the ability of NPY to modulate GABA-mediated Ca2+ increases was assayed using neurons derived from E19–E21 SCN after 4 d in vitro (DIV) (Fig.1). To determine a baseline Ca2+level, neurons were perfused initially with the GABAA receptor antagonist bicuculline (20 μm). There was a rapid elevation in Ca2+ after bicuculline was removed from the perfusion solution.
Fig. 1.

A, The removal of the GABAA receptor antagonist bicuculline (BIC) (20 μm) from the perfusion solution caused an immediate Ca2+ rise in neurons from the SCN cultured for 4 d in vitro (DIV). The addition of NPY (100 nm) caused a rapid depression in the GABAergic Ca2+ rise. The ability of neurons to recover to pre-NPY Ca2+levels differed significantly, with some showing a modest recovery (A2, A3) and others not recovering (A1). B, The effects of NPY on endogenous, GABA-mediated, Ca2+ rises persisted with time in culture (11 DIV). C, The effect of NPY on GABA-mediated Ca2+ rises was concentration-dependent. The Ca2+ rise from a bicuculline-defined baseline was determined 15 sec before and 100 sec after the application of NPY. These values were normalized such that the Ca2+ rise just before the application of NPY (white bar) was set equal to 100%. Error barsrepresent the SEM. N refers to the total number of neurons assayed. The Ca2+ rise defined from a bicuculline (20 μm) baseline was determined 15 sec before and 100 sec after the application of NPY receptor agonists (see Materials and Methods for a full description). D, Control experiments show that in the absence of NPY, the Ca2+ rise was maintained for an extended time. Ca2+ levels in the presence of bicuculline were usually between 45 and 80 nm. Vertical bars to the right of each neuron represent the calibrated Ca2+ level. Horizontal bars represent the time scale for a group of neurons. The ionotropic glutamate receptor antagonists AP5 and CNQX were present in all perfusion solutions to eliminate the potentially complicating interactions between NPY and glutamatergic neurons.
Figure 1A shows that the application of NPY (100 nm) to the perfusion solution caused a rapid and dramatic depression in the GABA-mediated Ca2+level. In the presence of bicuculline, the resting Ca2+ level was 67 ± 2 nm. After bicuculline was removed from the perfusion solution, the Ca2+ level increased to 124 ± 3 nm, a 56 nmCa2+ rise. The application of NPY reduced the Ca2+ rise to 16 ± 2 nm, representing a statistically significant (p < 0.001, one-tailed t test) 71% drop in the GABA-mediated Ca2+ level (n = 111). In some neurons, NPY was nearly as effective as bicuculline at depressing Ca2+ levels. The Ca2+ level of 59 of 111 neurons was depressed to within 10 nm of the bicuculline-defined baseline. These data are from five independent experiments. Only one experiment was performed per coverslip. We compared the response of cells from each of five coverslips to determine whether there was a significant difference in means or variances. Using ANOVA tests, we found no difference either in the responses to bicuculline removal or in the NPY-mediated depression of Ca2+ rises between different coverslips.
Figure 1C shows that the magnitude of the NPY-mediated depression of GABAergic Ca2+ levels was dose-dependent, with statistically maximal effects exerted at 100 nm. A much smaller effect was seen with 5 and 15 nm NPY. Neurons not stimulated with NPY exhibited extended and stable Ca2+ rises from baseline on bicuculline removal (Fig. 1D).
A very interesting finding was that NPY exerted a long-term Ca2+ depression that lasted well after NPY was removed from the perfusion solution. The neuron shown in Figure1A1 is an example of this extended effect; Ca2+ was depressed to near baseline levels and remained depressed after the removal of NPY. As a measure of the long-term effectiveness of NPY, we found that >68% of SCN neurons did not recover at least 50% of their pre-NPY Ca2+level by 6 min after a 2 min NPY application (n = 115). In the absence of NPY receptor stimulation, no neuron had a GABA-mediated Ca2+ rise decrease by 50% over a 6 min period (n = 25).
We assayed the efficacy of NPY (100 nm) after 11 DIV to determine whether its effects on GABA-mediated Ca2+ persisted during later periods of neuronal development (Fig. 1B). As with 4 DIV neurons, NPY was a potent inhibitor of GABAergic Ca2+ rises after 11 DIV. Long-term Ca2+ depression was not associated with small pre-NPY Ca2+ rises. NPY induced long-term Ca2+ depression in neurons with a relatively large Ca2+ rise (Fig. 1B1), whereas the NPY-mediated Ca2+ depression in a neuron with a relatively small Ca2+ rise (Fig.1B2) was rapidly reversible, recovering much more rapidly. For all endogenous activity assays, neurons were perfused constantly with the broad spectrum ionotropic glutamate receptor antagonists AP5 (100 μm) and CNQX (10 μm) to remove any complicating effects of glutamate-mediated Ca2+ rises and NPY/glutamate interactions.
NPY-mediated long-term Ca2+ depression
To better assess NPY actions as a long-term depressor of GABA-mediated Ca2+ levels, SCN neurons were imaged for >1 hr after a single 2 min application of NPY. Figure2A shows four neurons that were recorded simultaneously. The removal of bicuculline initiated a large Ca2+ increase in all neurons. The application of NPY (100 nm) to the perfusion solution drastically reduced the Ca2+ levels in all neurons. Neurons showed two general responses to the removal of NPY. The first group (Fig. 2A1–A2) did not recover to pre-NPY Ca2+ levels even after 60 min. Although NPY conferred a long-term reduction in the Ca2+level, these neurons were still synaptically active; Ca2+ levels were elevated relative to levels in the presence of bicuculline, and intermittent periods of increased activity were visible. The second group (Fig. 2A3–A4) showed varied Ca2+ recovery rates after the removal of NPY. Of the 27 neurons recorded simultaneously for 40 min, 12 neurons showed >50% recovery, whereas 15 neurons showed <50% recovery to pre-NPY Ca2+ levels. This experiment was repeated with neurons from three additional coverslips with similar results. Spontaneous Ca2+ depression and recovery of the type seen with the addition and washout of NPY was never observed.
Fig. 2.
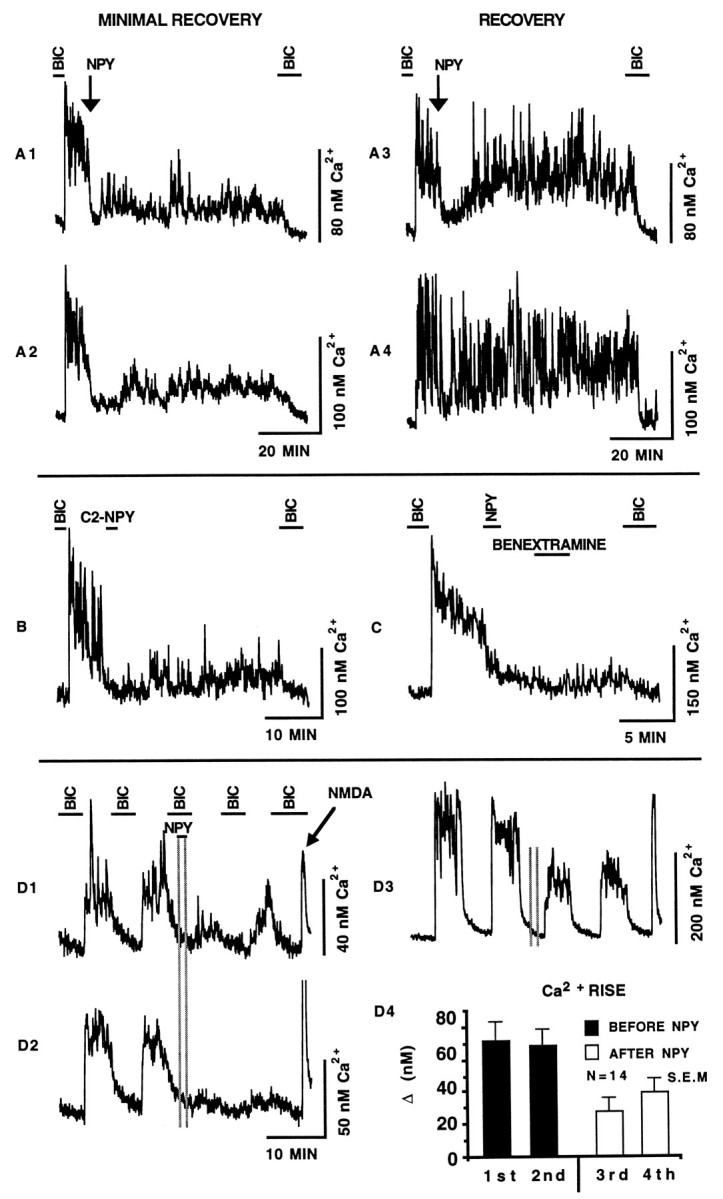
A, The ability of NPY to depress endogenous GABA-mediated Ca2+ rises was assayed for >60 min. The Ca2+ rise initiated by the removal of bicuculline (BIC) (20 μm) was depressed by a 2 min application of NPY (100 nm) (arrow). Representative SCN neurons were divided into two groups: those that showed a MINIMAL RECOVERY (A1–A2) and those that showed a rapidRECOVERY (A3–A4). Note that the reintroduction of bicuculline at the end of the experiment reduced the Ca2+ level in all neurons. B, The addition of the truncated NPY analog C2-NPY (100 nm) caused a rapid and sustained Ca2+ depression. Unlike NPY, C2-NPY has very low nonspecific binding characteristics and therefore tends not to remain associated with cells. C, The NPY receptor antagonist benextramine (10 μm) was added to the perfusion solution 3 min after NPY (100 nm) washout in an attempt to displace NPY that may have remained bound to the NPY receptor. Little effect was detected. D, NPY exhibited a latent regulation of GABAergic Ca2+ rises. The Ca2+ levels of the neurons shown in D1–D3 were elevated reversibly by the removal of bicuculline (20 μm). During the third bicuculline application, neurons were pulsed with NPY (100 nm) (vertical gray lines). Subsequent Ca2+rises induced by the removal of bicuculline were reduced drastically. The arrow points to the 30 sec application of the glutamate receptor agonist NMDA (30 μm).D4 is a graphical representation of the effect of NPY;1st and 2nd refer to the two bicuculline-sensitive Ca2+ rises before the addition of NPY; 3rd and 4th refer to the two bicuculline-sensitive Ca2+ rises after the pulse of NPY. The glutamate receptor antagonists AP5 (100 μm) and CNQX (10 μm) were in all solutions except during NMDA application. Error bars represent SEM.
Additional experiments were performed to explore the mechanism by which NPY triggers long-term Ca2+ depression. Specifically, because NPY may bind nonspecifically to membranes (McLean et al., 1990), NPY may exert long-term effects by remaining cell-associated and rebinding to the NPY receptor, thereby affecting Ca2+ levels for extended periods after presumptive NPY washout. To reduce this possibility, we used the truncated NPY analog C2-NPY that lacks structural elements involved in nonspecific binding (McLean et al., 1990). Therefore, C2-NPY should rapidly wash out of the perfusion solution. Data were collected and pooled from three different coverslips. Figure 2B shows that the addition of C2-NPY (100 μm) to the perfusion solution caused a rapid depression of the GABAergic Ca2+ rise. The Ca2+ rise immediately before C2-NPY application was 57 ± 5 nm; C2-NPY administration decreased the Ca2+ rise to 27 ± 2 nm, representing a 47% drop in Ca2+ levels (n = 37). The Ca2+ level was depressed after the withdrawal of C2-NPY and remained depressed for the duration of the experiment (>30 min). C2-NPY triggered long-term Ca2+ depression in 37% of neurons (n = 37). The general appearance of the extended Ca2+ depression triggered by C2-NPY is virtually identical to the extended Ca2+ depression elicited by NPY (Fig. 2A1–A2). By 20 min after its addition, C2-NPY still depressed Ca2+ by 51%; in comparison, NPY depressed Ca2+ by 43%. These results are not consistent with the idea that the long-term effect is attributable to NPY stickiness. Our data suggest that NPY agonists act rapidly to initiate long-term depression.
To test further the possibility that the long-term effect may have been caused by residual NPY binding repeatedly to NPY receptors after bath washout, the putative NPY receptor antagonist benextramine was used. This antagonist has been reported to block Y1 and Y2 receptors, but it may not be fully effective at all NPY receptors (Li et al., 1991;Doughty et al., 1992; Penner et al., 1993; Palea et al., 1995). If residual NPY was binding repeatedly to NPY receptors, and thereby causing long-term calcium depression, the displacement of NPY with benextramine should cause a rapid Ca2+ elevation. The addition of benextramine (10 μm, 4 min application) 3 min after the withdrawal of NPY (100 nm) had no effect on the level of Ca2+ depression triggered by the addition of NPY (Fig. 2C). The level of depression during benextramine treatment (third min of application) was 77% (the Ca2+ rise before NPY application was 60 ± 6 nm; the NPY-mediated Ca2+decrease during benextramine application was 14 ± 3 nm; n = 20). For neurons not treated with benextramine, the Ca2+ rise before NPY application was 55 ± 7 nm; NPY decreased the Ca2+ rise to 17 ± 4 nm, representing a 69% drop in activity (n = 24). Data were collected over an identical time after NPY application for the two groups. These results provide additional evidence suggesting that the long-term effect of NPY is not attributable to NPY rebinding beyond the time it was perfused onto the neurons.
To understand the physiological events necessary for NPY to depress GABA-mediated Ca2+ increases, we tested the hypothesis that NPY would depress Ca2+ activity even if applied in the absence of a GABA-mediated Ca2+ rise. Figure 2D1–D3 shows that when NPY was applied and removed in the presence of bicuculline, it exerted no detectable effect on cytosolic Ca2+; however, the Ca2+ increase after the removal of bicuculline was significantly lower than the Ca2+increase before NPY application. This effect is shown in the neuron shown in Figure 2D2, where GABA-mediated Ca2+ increases are absent after NPY application. The bar graph in Figure 2D4 quantifies the endogenous GABA-mediated Ca2+ rises before and after the addition of NPY. The second Ca2+ rise after the application of NPY was slightly higher than the first, suggesting that neurons were beginning to recover from NPY. This recovery is seen clearly in the neuron shown in Figure 2D1. These results demonstrate that the efficacy of NPY does not seem to be dependent on elevated cytosolic Ca2+.
Mechanisms of NPY-induced Ca2+ depression
The efficacy of several different NPY agonists was assayed in an attempt to identify the NPY receptor subtype responsible for the depression of GABA-mediated Ca2+ rises. Figure3A,B shows that both the Y2 receptor-specific agonist NPY 13–36 (100 nm) and the Y1 receptor-specific agonist [Pro34,Leu31] NPY (100 nm) were effective at reducing Ca2+. Figure 3C shows that the NPY receptor agonist PYY (100 nm) was also effective. In addition to these immediate effects, long- term Ca2+ depression could be induced by all three agonists.
Fig. 3.

NPY receptor subtypes. A, The application of NPY (NPY 13–36) (100 nm) reduced GABA-mediated Ca2+ rises. Similar results were seen for [Pro34,Leu31] NPY (NPY PRO-34) (100 nm) (B), and PYY (100 nm) (C). For each pair shown, the top neuron is an example of a long-term effect, and the bottom shows neurons that showed either a smaller effect (A) or a more substantial recovery (B, C). D, Pretreatment of SCN neurons with pertussis toxin (200 ng/ml) for 20 hr before the start of the experiment blocked the NPY-mediated depression of GABAergic Ca2+ rises. E, GABAergic Ca2+ rises are in large part dependent on L-type Ca2+ channel activity. The response of a representative neuron shows that the addition of the L-type Ca2+ channel blocker nimodipine (1 μm) depressed the endogenous Ca2+ rise. NPY (100 nm) had no effect in the presence of nimodipine. F, Bar graph representation of the efficacy of NPY receptor agonists. The Ca2+ rise defined from a bicuculline (20 μm) baseline was determined 15 sec before and 100 sec after the application of NPY receptor agonists. Values were normalized such that the Ca2+ rise just before the application of NPY (white bar) was set equal to 100%. The agonists used are shown along the x-axis. The efficacy of NPY is shown for 11 DIV and 4 DIV. NPY PRO-34 refers to [Pro34,Leu31] NPY. NPY+PTX refers to the efficacy of NPY when the neurons were pretreated with pertussis toxin. The breakdown by group for the total number of neurons assayed (n = 440) was 4 DIV NPY = 111, 11 DIV NPY = 10, NPY 13–36 = 81, NPY PRO-34 = 98, PYY = 75, and NPY+PTX = 65. Only data from neurons with a Ca2+ rise >20 nm are included. Glutamate receptor antagonists AP5 (100 μm) and CNQX (10 μm) were in all solutions. NPY receptor agonist-mediated Ca2+ depression was statistically different from pre-NPY Ca2+ levels for each group (p < 0.001). BIC, Bicuculline. Error bars represent SEM.
We tested the hypothesis that NPY worked through a G-protein-related mechanism to depress Ca2+ levels. Data were combined from three different coverslips. Figure 3D shows that the response of SCN neurons to NPY was reduced drastically after treatment with the Gi/Goinhibitor pertussis toxin (200 ng/ml) for 20 hr before the start of the experiment. After bicuculline removal, the Ca2+level increased to 139 ± 5 nm, a 68 nm rise. The application of NPY reduced the Ca2+ rise to 54 ± 4 nm, representing a 21% Ca2+ decrease (n = 65). On the basis of normalized Ca2+ depressions, this depression is substantially (p < 0.01, one-tailed t test) smaller than NPY-mediated depression in the absence of pertussis toxin treatment described above (71% decrease, n = 111). A bar graph representation comparing the short-term effects of NPY receptor agonists on GABA-mediated Ca2+ rises is shown in Figure3F. The control rise represents the normalized GABA-mediated Ca2+ increase just before the application of NPY receptor agonists.
Next we tested whether NPY had an effect on Ca2+during L-type voltage-activated Ca2+ channel inhibition. We showed previously that Ca2+ rises evoked by the application of GABA can be depressed (86%) by nimodipine (Obrietan and van den Pol, 1995). Figure 3E shows that endogenous GABAergic Ca2+ rises were depressed by the application of nimodipine (1 μm) and that NPY (100 nm) had little effect in the presence of nimodipine (n = 19).
Postsynaptic GABA-evoked Ca2+ rises
Several NPY receptor agonists were used to determine whether the NPY receptor exerted a postsynaptic effect on GABA-evoked Ca2+ rises. Thirty second applications of GABA (20 μm) were added repeatedly to the perfusion solution in either the absence or the presence of NPY receptor agonists (100 nm). All perfusion solutions contained TTX (1 μm) to inhibit neurotransmitter release and thereby eliminate effects that NPY may have on presynaptic GABA release from effects that NPY may have on postsynaptic responses to GABA. Figure 4A shows that GABA-evoked Ca2+ rises were depressed by the coadministration of NPY. The mean GABA-evoked Ca2+ rise in the absence of NPY was 115 ± 7 nm. After NPY was added, the mean Ca2+ rise was 82 ± 5 nm, representing a statistically significant (p < 0.001, one-tailed t test) 29% decrease (n = 147 from five experiments). Of 147 cells, 15% showed a NPY-mediated depression in GABA-evoked Ca2+ rises. Half of the responding cells showed a depression that was characterized by an extended depression (>50%) that persisted through the second application of GABA (10 min post-NPY) (Fig. 4A, top neuron). The other half was characterized by a rapid recovery of GABA potency after NPY withdrawal (Fig. 4A, bottom neuron). The ability of NPY to depress GABA-evoked Ca2+ rises even after NPY withdrawal suggests there may be a postsynaptic component to the long-term Ca2+ depression characterized in experiments focusing on endogenous GABA-release assays.
Fig. 4.

To determine whether NPY exerted a postsynaptic effect, synaptic communication was blocked with TTX (1 μm), and GABA-induced Ca2+ rises were evoked repeatedly in combination with NPY receptor stimulation. A, The application ofGABA (20 μm) (arrows) to the perfusion solution elicited reproducible Ca2+rises. The administration of NPY (100 nm) starting 45 sec before GABA application caused a large reduction in the level of GABA-induced Ca2+ rises. NPY-dependent depression of GABA-induced Ca2+ increases either persisted (top neuron) or diminished (bottom neuron) after NPY was washed out. Cells were pulsed with NMDA (30 μm) to demonstrate that neurons were healthy and would respond to Ca2+-mobilizing transmitters at the end of an experimental series (wide arrow). Similar results were seen for NPY 13–36 (B), [Pro34,Leu31] NPY (NPY PRO-34) (C), and PYY(D). E, The effect of NPY on high K+-induced Ca2+ rises was small. F, The GABAA receptor-specific agonist muscimol (MUSCIMOL) (10 μm) elicited a Ca2+ rise, whereas the GABAB receptor-specific agonist baclofen (BACLOFEN) (10 μm) had no effect.G, GABA-evoked Ca2+ rises could be inhibited by the coadministration of bicuculline (BICUCULLINE) (20 μm).
Similar results were seen with Y2 and Y1 receptor-specific agonists (Fig. 4, B and C, respectively) and PYY (Fig.4D). NPY 13–36 (a Y2 agonist) was the most effective agonist, depressing GABA-evoked Ca2+ rises by >48% (n = 96). PYY induced a Ca2+depression similar to NPY, depressing GABA-evoked Ca2+ by >34% (n = 38). High (25 mm) K+-induced Ca2+ rises were also depressed by the coadministration of NPY (Fig. 4E) (9%, n = 57). In the absence of NPY, the high K+-induced Ca2+ rise was 328 ± 12 nm. After NPY was added, the mean Ca2+ rise decreased to 299 ± 11 nm. The relative efficacy of NPY receptor agonists on evoked Ca2+ rises is shown in Figure 5; the first and second GABA application elevated Ca2+ to nearly equal levels, whereas all NPY receptor agonists significantly reduced the GABA-evoked Ca2+ rise (p < 0.001, one-tailed t test). Separate experiments confirmed that GABA-induced Ca2+ rises were caused exclusively by activation of GABAA receptors; the GABAA receptor-specific agonist muscimol induced a Ca2+ rise, whereas the GABAB agonist baclofen did not alter baseline Ca2+ levels (Fig. 4F). Similarly, GABA-induced Ca2+ increases were blocked completely by the GABAA receptor antagonist bicuculline (20 μm) (Fig. 4G).
Fig. 5.

NPY receptor agonists suppressed GABA-evoked Ca2+ rises. Data compare sequential GABA-evoked Ca2+ rises. The graph shows that the peak response to the first control GABA-evoked Ca2+rise was not different from the second control GABA-evoked Ca2+ rise (white bars). The second control GABA-evoked Ca2+ rise was normalized and set equal to 100% and then compared with the first GABA-evoked Ca2+ rise in the presence of NPY receptor agonist. All NPY receptor agonists caused a statistically significant depression in the peak Ca2+ rise (p < 0.01). Data from high K+-induced Ca2+ increases (25 mm K+NPY) were analyzed in an identical manner to GABA-evoked responses. NPY PRO-34 refers to [Pro34,Leu31] NPY.N refers to the total number of neurons assayed. Error bars represent SEM. Only data from neurons with a Ca2+ rise >20 nm were analyzed.
SCN microslices
To simulate more closely in vivo conditions, the SCN was punched out of slices of neonatal rat brain and maintained as an organotypic mass. This minimizes the disruption of synaptic contacts and leaves the majority of neuronal processes intact. After 4 DIV, the ability of NPY to depress GABAergic Ca2+ rises was assayed (Fig. 6A). In the presence of bicuculline, the resting Ca2+ level was 69 ± 5 nm. After bicuculline was removed, the Ca2+ level increased to 111 ± 3 nm, a 42 nm rise. NPY reduced the Ca2+ rise to 13 ± 4 nm, representing a statistically significant (p < 0.001, one-tailed t test) 69% drop in the GABA-mediated Ca2+ level (n = 14). As described earlier, the Ca2+-depressing-effect of NPY lasted long after NPY was removed from the perfusion solution.
Fig. 6.

NPY (100 nm) depressed GABA-mediated Ca2+ rises in neurons from SCN cultured punches (A, B) and in neurons cultured from the arcuate nucleus (C, D). A,C, Removal of bicuculline (BIC) (20 μm) elicited a Ca2+ rise that was depressed by the addition of NPY. Glutamate receptor antagonists AP5 (100 μm) and CNQX (10 μm) were maintained in the perfusion solution during endogenous activity experiments. B, D, NPY depressed Ca2+ rises evoked by the application of GABA (20 μm) to the perfusion solution. TTX (1 μm) was included in all evoked-response perfusion solutions in B and D.
Figure 6B shows that the application of GABA (20 μm) elicited a reproducible Ca2+ rise and that the peak level of the rise could be depressed by NPY (100 nm). The mean GABA-evoked Ca2+ rise in the absence of NPY was 113 ± 13 nm. After NPY was added, the mean Ca2+ rise was 79 ± 6 nm, representing a statistically significant (p < 0.001, one-tailed t test) Ca2+ rise decrease of 30% (n = 36). The efficacy of NPY at depressing GABA-mediated Ca2+ rises in neurons from cultured microslices was similar to the results described for NPY using standard dissociation and culturing procedures.
Arcuate nucleus
To test the hypothesis that NPY depression of GABA-mediated Ca2+ rises found in SCN neurons are representative of a general mechanism of NPY action in the hypothalamus, we performed parallel experiments with neurons cultured from the hypothalamic arcuate nucleus. The arcuate nucleus is highly enriched in neurons that synthesize NPY and contains many neuroendocrine neurons that regulate pituitary hormone release. NPY exerts many effects on the endocrine system, and some of these are long-lasting (Albers et al., 1990). This, in combination with the documented long-term regulatory role of NPY in pituitary function, makes the study of NPY-mediated modulation of GABAergic Ca2+ rises in the arcuate nucleus of keen interest. After 4 DIV, the efficacy of NPY was assayed. Figure6C shows that the GABA-induced Ca2+rise in arcuate neurons was depressed by NPY (100 nm). The results from the arcuate nuclei are similar to those from the SCN; NPY dramatically depressed GABA-mediated Ca2+ levels, and the effect persisted subsequent to the removal of NPY. The mean Ca2+ rise after the removal of bicuculline and just before the addition of NPY was 40 ± 4 nm. After NPY was added to the perfusion solution, the mean Ca2+ rise was depressed to 14 ± 6 nm, representing a statistically significant (p < 0.001, one-tailed t test) 65% decrease (n = 19).
NPY was also effective in depressing postsynaptic GABA-evoked Ca2+ rises (Fig. 6D). The mean GABA-evoked Ca2+ rise in the absence of NPY was 84 ± 8 nm. The mean Ca2+rise after the application of NPY was 47 ± 5 nm, representing a statistically significant (p < 0.001, one-tailed t test) 44% decrease in the Ca2+ (n = 35).
DISCUSSION
Ours is the first study examining transmitter modulation of GABA-mediated Ca2+ transients during development. Data presented show clearly that NPY receptor activation has a profound modulatory effect on SCN and arcuate neural activity, capable of depressing the endogenous GABA-mediated Ca2+level and maintaining the level of depression long after the removal of NPY. The long-term depression appeared to be mediated through both presynaptic and postsynaptic mechanisms and could be generated with Y1 and Y2 NPY receptor-specific agonists and with PYY.
Mechanisms
The ability of NPY to depress GABA-evoked Ca2+ rises in the presence of TTX suggests that the NPY receptor can regulate neural activity postsynaptically. NPY-mediated postsynaptic modulation of neural activity has been shown under several different conditions. Shibata and Moore (1993) found that circadian firing rates of SCN neurons in slice preparations were modulated by NPY even in the presence of TTX, suggesting a postsynaptic site of action for NPY. A novel finding of our study was that both the Y1 and Y2 receptor-specific agonists depressed postsynaptic GABA-evoked Ca2+ rises. On the basis of the known pharmacological selectivity of the two receptor-specific agonists used (NPY 13–36 and [Pro34,Leu31] NPY) (Fuhlendorff et al., 1990; Krstenansky et al., 1990), it is unlikely that the efficacy of the agonists was the result of agonist/receptor cross-reactivity. Rather, these results suggest that both receptor subtypes are expressed postsynaptically, modulating GABA-evoked Ca2+ rises. PYY was used to discriminate the Y1 and Y2 NPY receptors from the Y3 NPY receptor, which is not stimulated by PYY (Grundemar et al., 1991; Wahlestedt et al., 1992). The efficacy of PYY closely matched the efficacy of NPY, suggesting that the Y3 type receptor did not contribute to NPY receptor agonist-elicited responses.
In other brain regions, NPY modulates Ca2+channel currents (Hirning et al., 1990; Wiley et al., 1990, 1993;Klapstein and Colmers, 1992). We have shown previously that the GABAA receptor elicits Ca2+rises primarily by opening voltage-activated Ca2+channels (Obrietan and van den Pol, 1995). K+-evoked Ca2+ rises were less affected by NPY than were GABA-evoked Ca2+rises. This could be attributable to differences in membrane conductance for these two conditions, the level of depolarization, the overall magnitude of the Ca2+ rise, and the different reversal potentials for K+ and Cl− relative to the resting membrane potential.
We found a relatively large difference when comparing the effectiveness of NPY at depressing endogenous GABA-mediated Ca2+ rises with the effectiveness of NPY at depressing postsynaptic GABA-evoked Ca2+ rises (in the presence of TTX). A higher percentage of cells showed an effect when NPY was introduced into cultures showing endogenous GABA release compared with GABA-evoked responses in the presence of TTX. NPY depressed >70% of the endogenous GABA-mediated Ca2+ rise, whereas NPY depressed only <30% of the evoked Ca2+ response to GABA. A greater effect of NPY presynaptically than postsynaptically in modulating GABA transmission is the most likely explanation for this difference. Several studies have shown that release of neurotransmitters can be regulated by NPY receptor stimulation on presynaptic axons (Westfall et al., 1987; Yokoo et al., 1987; Colmers et al., 1988; Walker et al., 1988; Martire et al., 1993; Greber et al., 1994). Alternatively, NPY effects at the level of the cell body could reduce GABA release from the axon terminals of that neuron.
Y1 and Y2 type NPY receptors have been shown to be G-protein-coupled. Depending on the cell type in which they are expressed, either is capable of decreasing the accumulation of adenosine 3′,5′-cyclic monophosphate or stimulating the synthesis of inositol trisphosphate (Perney and Miller, 1989; Bleakman et al., 1992; Herzog et al., 1992;Larhammar et al., 1992). We found that inhibiting Gi/Go protein activity with pertussis toxin dramatically reduced the NPY depression of spontaneous GABAergic Ca2+ rises. The ability of NPY to depress spontaneous glutamatergic Ca2+ rises in cortical neurons also has been shown to be pertussis-toxin sensitive (Bleakman et al., 1992).
Long-term Ca2+ depression
A novel finding in the present study was that the Ca2+-depressing ability of NPY lasted well after NPY was washed away (>60 min). Long-term Ca2+depression was particularly evident in experiments with neurons that were spontaneously releasing GABA from presynaptic axons, but it was also seen, although much less frequently, in experiments with GABA-evoked Ca2+ rises in SCN neurons. This depression may represent an underlying mechanism responsible for some of the extended phase-shifting effects of NPY if NPY modulates GABA in mature neurons as it does in developing ones, and by virtue of the retinal projection to NPY/GABA-containing cells of the IGL that project to the SCN, it may also represent an underlying mechanism of light-induced phase shifts. Several studies have given hints of long-lasting effects of NPY on neural electrical activity (Shibata and Moore, 1988; Albers et al., 1990); however, data interpretation is difficult, as these experiments were performed in brain slices where the exchange of solutions is very slow. Because of the nonspecific binding characteristics of NPY, pharmacologically active concentrations of NPY could persist in thick slices for prolonged periods (hours) after the drug had been removed from the perfusion solution. This problem was eliminated in the present study by using synaptically coupled neurons in monolayer tissue culture where total washout of drugs should be achieved much more easily. Furthermore, we found long-term Ca2+ depression using the nonsticky NPY agonist C2-NPY, and we were not able to reduce the NPY-mediated long-term Ca2+ depression with the NPY receptor antagonist benextramine, suggesting that NPY receptor stimulation initiates second messenger pathways with effects that far outlast NPY application. Interestingly, although NPY depressed GABA-mediated Ca2+ rises in most neurons, the long-term effects were not found in all neurons examined. This may be attributable to different NPY or GABA receptors expressed in the cells and to different mechanisms of coupling to G-protein-regulated cascades of second messengers. Alternatively, if the SCN cells are undergoing circadian rhythms in vitro that are out of phase with each other (Bos and Mirmiran, 1990; Welsh et al., 1995), NPY may exert a long-term effect only at specific times of the cycle of the cells.
In addition to its long-term effects on circadian rhythms, NPY also exerts a long-lasting effect on stimulation of food intake and alters the timing of hormone secretion (McDonald et al., 1985, 1989; Stanley and Leibowitz, 1985; Albers et al., 1990). In the present study, we show that long-term NPY modulation of GABA activity is not restricted to the SCN, but is also found in neurons from other regions of the hypothalamus such as the neuroendocrine arcuate nucleus, suggesting that a common long-term effect of NPY receptor activation may be to depress GABA activity in hypothalamic neurons regulating various hypothalamic functions.
The effect of NPY on GABA-mediated Ca2+ is difficult to place within any specific model of long-term depression described previously (Linden and Connor, 1995). Both the magnitude and the frequency of the NPY effects were greater in experiments involving presynaptic release of GABA than in experiments with the addition of exogenous GABA, leading us to postulate that the effect of NPY in reducing Ca2+ levels of long periods of time may be attributable to reduction of synaptic efficacy of GABAergic axons. This would represent a form of long-term synaptic depression (LTD), although confirming electrophysiological evidence for this is still needed. NPY exerted a latent depression of GABA-mediated Ca2+ rises. When NPY was applied and removed from the perfusion solution in the presence of bicuculline, subsequent GABA-mediated Ca2+ rises were depressed significantly or eliminated completely, thus suggesting that the long-term Ca2+-depressing ability of NPY was not dependent on postsynaptic depolarization and subsequent elevated Ca2+. This is contrary to some described forms of LTD, where increased Ca2+ levels were required for LTD induction (Wickens and Abraham, 1991; Christie and Abraham, 1992; Mulkey and Malenka, 1992). Activation of the ionotropic NMDA-selective glutamate receptor is thought to be involved in the initiation of LTD in many regions of the brain (Dudek and Bear, 1992; Jaffe et al., 1992). Although we found previously that NPY also can modulate glutamate actions in hypothalamic neurons (van den Pol et al., 1995), ionotropic glutamate receptor activity did not contribute to the extended depression of endogenous GABAergic Ca2+ levels in our experiments; all endogenous activity assays were performed in the presence of glutamate receptor antagonists, thereby eliminating glutamate receptor responses and any effect endogenously released glutamate could have contributed to Ca2+ levels. It is possible that other transmitters were released simultaneously with GABA under conditions of spontaneous release, but because GABA receptor antagonists completely blocked Ca2+ transients, GABA must be a critical transmitter mediating these effects.
GABA and NPY during development
GABA may be the principal Ca2+-elevating neurotransmitter during early development in the SCN and in other regions of the hypothalamus. An equimolar concentration of GABA elicits a higher Ca2+ rise than glutamate does in young, but not older, developing neurons (Obrietan and van den Pol, 1995). This is probably the result of the relatively early expression of the GABAA receptor in neural development, preceding glutamate receptor expression in many brain regions, including the hypothalamus (Meinecke and Rakic, 1992; Walton et al., 1993; Chen et al., 1995; van den Pol et al., 1995).
A physiological role for early developmental expression of GABA has been shown in the CNS. Hippocampal cells from 5- to 7-d-old rat pups can be depolarized by GABAA receptor agonists (Fiszman et al., 1990). Depolarizing potentials during the first week of postnatal development appeared to be the result of GABA release (Ben-Ari et al., 1989). Similarly, application of GABA in early embryogenesis elicited action potentials in the chick spinal cord that disappeared later in development (Obata et al., 1978). GABA-induced depolarization and subsequent Ca2+ entry would affect a myriad of developmentally regulated processes. GABAA receptor agonists also initiate chemokinesis in developmentally immature spinal cord neurons (Behar et al., 1994). GABAA receptor antagonists decrease neurite length of cultured hippocampal neurons (Barbin et al., 1993). More recently, LoTurco et al. (1995) found that DNA synthesis in cortical progenitor cells was decreased by GABA.
Several studies have shown that similar to GABA and GABAA receptors, NPY and NPY receptors are present during the early stages of neurogenesis. In rat brain, NPY can be detected before E15 (Allen et al., 1984; Woodhams et al., 1985). High levels of NPY are found in neurons cultured from E18 hypothalamus (Wahle et al., 1993). Furthermore, NPY receptor mRNA has been found during embryogenesis in the developing brain (Larhammar et al., 1992;Janzin et al., 1993). Lateral geniculate nucleus neurons begin to extend NPY-containing processes toward the SCN during embryogenesis in hamster brain (Botchkina and Morin, 1995), a time when the developing SCN first starts to show circadian rhythmicity of 2 deoxyglucose uptake; this occurs at E19 in rats (Reppert, 1992).
The ability of NPY to depress GABA-mediated Ca2+ rises and transients, and to do so for an extended period, would make NPY an agent capable of modulating many of the putative trophic effects GABA has been postulated to exert in the developing brain.
Footnotes
This research was supported by National Institutes of Health Grants NS16296, NS10174, and NS34887, the National Science Foundation, and Air Force Office of Scientific Research. We thank Dr. H. C. Heller for encouragement and L. Haak for suggestions on earlier versions of the manuscript.
Correspondence should be addressed to Anthony N. van den Pol, Section of Neurosurgery, Yale University, School of Medicine, 333 Cedar Street, New Haven, CT 06520.
REFERENCES
- 1.Albers H, Ferris C. Neuropeptide Y: role in light-dark cycle entrainment of hamster circadian rhythms. Neurosci Lett. 1984;50:163–168. doi: 10.1016/0304-3940(84)90480-4. [DOI] [PubMed] [Google Scholar]
- 2.Albers H, Ottenweller J, Liou S, Lumpkin M, Anderson E. Neuropeptide Y in the hypothalamus: effect on corticosterone and single-unit activity. Am J Physiol. 1990;258:R376–R382. doi: 10.1152/ajpregu.1990.258.2.R376. [DOI] [PubMed] [Google Scholar]
- 3.Allen J, McGregor G, Woodhams P, Polak J, Bloom S. Ontogeny of a novel peptide, neuropeptide Y (NPY) in rat brain. Brain Res. 1984;303:197–200. doi: 10.1016/0006-8993(84)90230-0. [DOI] [PubMed] [Google Scholar]
- 4.Bading H, Ginty D, Greenberg M. Regulation of gene expression in hippocampal neurons by distinct calcium signaling pathways. Science. 1993;260:181–186. doi: 10.1126/science.8097060. [DOI] [PubMed] [Google Scholar]
- 5.Barbin G, Pollard H, Gaïarsa J, Ben-Ari Y. Involvement of GABAA receptors in the outgrowth of cultured hippocampal neurons. Neurosci Lett. 1993;152:150–154. doi: 10.1016/0304-3940(93)90505-f. [DOI] [PubMed] [Google Scholar]
- 6.Barnea A, Hajibeigi A, Cho G, Magni P. Regulated production and secretion of immunoreactive neuropeptide Y by aggregating fetal brain cells in cultures. Neuroendocrinology. 1991;54:7–13. doi: 10.1159/000125844. [DOI] [PubMed] [Google Scholar]
- 7.Behar T, Schaffner A, Colton C, Somogyi R, Olah Z, Lehel C, Barker J. GABA-induced chemokinesis and NGF-induced chemotaxis of embryonic spinal cord neurons. J Neurosci. 1994;14:29–38. doi: 10.1523/JNEUROSCI.14-01-00029.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ben-Ari Y, Cherubini E, Corradetti R, Gaïarsa J. Giant synaptic potentials in immature rat CA3 hippocampal neurones. J Physiol (Lond) 1989;416:303–325. doi: 10.1113/jphysiol.1989.sp017762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bleakman D, Harrison N, Colmers W, Miller R. Investigation into neuropeptide Y-mediated presynaptic inhibition in cultured hippocampal neurones of the rat. Br J Pharmacol. 1992;107:334–340. doi: 10.1111/j.1476-5381.1992.tb12747.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bos NPA, Mirmiran M. Circadian rhythms in spontaneous neuronal discharges of the cultured suprachiasmatic nucleus. Brain Res. 1990;511:158–162. doi: 10.1016/0006-8993(90)90235-4. [DOI] [PubMed] [Google Scholar]
- 11.Botchkina G, Morin L. Specialized neuronal and glial contributions to development of the hamster lateral geniculate complex and circadian visual system. J Neurosci. 1995;15:190–201. doi: 10.1523/JNEUROSCI.15-01-00190.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Card J, Moore R. The suprachiasmatic nucleus of the golden hamster: immunohistochemical analysis of cell and fiber distribution. Neuroscience. 1984;13:415–431. doi: 10.1016/0306-4522(84)90240-9. [DOI] [PubMed] [Google Scholar]
- 13.Card J, Moore R (1989) Organization of lateral geniculate-hypothalamic connections in the rat. 284:135–147. [DOI] [PubMed]
- 14.Chen G, Trombley PQ, van den Pol AN. GABA receptors precede glutamate receptors in hypothalamic development; differential regulation by astrocytes. J Neurophysiol. 1995;74:1473–1484. doi: 10.1152/jn.1995.74.4.1473. [DOI] [PubMed] [Google Scholar]
- 15.Chen G, Trombley PQ, van den Pol AN (1996) Excitatory actions of GABA in developing hypothalamus. J Physiol (Lond), in press. [DOI] [PMC free article] [PubMed]
- 16.Christie B, Abraham W. NMDA-dependent heterosynaptic long-term depression in the dentate gyrus of anesthetized rats. Synapse. 1992;10:1–6. doi: 10.1002/syn.890100102. [DOI] [PubMed] [Google Scholar]
- 17.Chronwall B, DiMaggio D, Massari V, Pickel V, Ruggiero D, O’Donohue T. The anatomy of neuropeptide Y containing neurons in the rat brain. Neuroscience. 1985;15:1159–1181. doi: 10.1016/0306-4522(85)90260-x. [DOI] [PubMed] [Google Scholar]
- 18.Colmers W, Lukowiak K, Pittman Q. Neuropeptide Y action in the rat hippocampal slice: site and mechanism of presynaptic inhibition. J Neurosci. 1988;8:3827–3837. doi: 10.1523/JNEUROSCI.08-10-03827.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Decavel C, van den Pol A. GABA: a dominant transmitter in the hypothalamus. J Comp Neurol. 1990;302:1019–1037. doi: 10.1002/cne.903020423. [DOI] [PubMed] [Google Scholar]
- 20.Doughty M, Li K, Hu L, Chu S, Tessel R. Benextramine-neuropeptide Y (NPY) binding site interactions: characterization of tritiated-NPY binding site heterogeneity in rat brain. Neuropeptides. 1992;23:169–180. doi: 10.1016/0143-4179(92)90119-h. [DOI] [PubMed] [Google Scholar]
- 21.Dudek S, Bear M. Homosynaptic long-term depression in area CA1 of hippocampus and effects of N -methyl-d-aspartate receptor blockade. Proc Natl Acad Sci USA. 1992;89:4363–4367. doi: 10.1073/pnas.89.10.4363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fiszman M, Novotny E, Lange G, Barker J. Embryonic and early postnatal hippocampal cells respond to nanomolar concentrations of muscimol. Dev Brain Res. 1990;53:186–193. doi: 10.1016/0165-3806(90)90005-j. [DOI] [PubMed] [Google Scholar]
- 23.Forscher P, Kaczmarek L, Buchanan J, Smith S. Cyclic AMP induces changes in distribution and transport of organelles within growth cones of Aplysia bag cell neurons. J Neurosci. 1987;7:3600–3611. doi: 10.1523/JNEUROSCI.07-11-03600.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fuhlendorff J, Gether U, Aakerlund L, Langeland-Johansen N, Thogersen H, Melberg S, Olsen U, Thastrup O, Schwartz T. [Leu31, Pro34]neuropeptide Y: a specific Y1 receptor agonist. Proc Natl Acad Sci USA. 1990;87:182–186. doi: 10.1073/pnas.87.1.182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Greber S, Schwarzer C, Sperk G. Neuropeptide Y inhibits potassium-stimulated glutamate release through Y2 receptors in rat hippocampal slices in vitro. Br J Pharmacol. 1994;113:737–740. doi: 10.1111/j.1476-5381.1994.tb17055.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Grundemar L, Wahlestedt C, Reis D. Neuropeptide Y acts at an atypical receptor to evoke cardiovascular depression and to inhibit glutamate responsiveness in the brainstem. J Pharmacol Exp Ther. 1991;258:633–638. [PubMed] [Google Scholar]
- 27.Grynkiewicz G, Poenie M, Tsien R. A new generation of calcium indicators with greatly improved fluorescence properties. J Biol Chem. 1985;260:3440–3450. [PubMed] [Google Scholar]
- 28.Hansen G, Belhage B, Schousboe A, Meier E. Temporal development of GABA agonist induced alterations in ultrastructure and GABA receptor expression in cultured cerebellar granule cells. Int J Dev Neurosci. 1987;5:263–269. doi: 10.1016/0736-5748(87)90037-2. [DOI] [PubMed] [Google Scholar]
- 29.Harrington ME, Rusak B. Lesions of the thalamic intergeniculate leaflet alter hamster circadian rhythms. J Biol Rhythms. 1986;1:309–325. doi: 10.1177/074873048600100405. [DOI] [PubMed] [Google Scholar]
- 30.Harrington ME, Nance DM, Rusak B. Neuropeptide Y immunoreactivity in the hamster geniculo-suprachiasmatic tract. Brain Res Bull. 1985;15:465–472. doi: 10.1016/0361-9230(85)90037-1. [DOI] [PubMed] [Google Scholar]
- 31.Herzog H, Hort Y, Ball H, Hayes G, Shine J, Selbie L. Cloned human neuropeptide Y receptor couples to two different second messenger systems. Proc Natl Acad Sci USA. 1992;89:5794–5798. doi: 10.1073/pnas.89.13.5794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hirning L, Fox A, Miller R. Inhibition of calcium currents in cultured rat myenteric neurons by neuropeptide Y: evidence for direct receptor/channel coupling. Brain Res. 1990;532:120–130. doi: 10.1016/0006-8993(90)91751-2. [DOI] [PubMed] [Google Scholar]
- 33.Horvath G, Acs Z, Mergl Z, Nagy I, Makara G. γ-Aminobutyric acid-induced elevations of intracellular calcium concentrations in pituitary cells of neonatal rats. Neuroendocrinology. 1993;57:1028–1034. doi: 10.1159/000126467. [DOI] [PubMed] [Google Scholar]
- 34.Huhman K, Albers H. Neuropeptide Y microinjected into the suprachiasmatic region phase shifts circadian rhythms in constant darkness. Peptides. 1994;8:1475–1478. doi: 10.1016/0196-9781(94)90126-0. [DOI] [PubMed] [Google Scholar]
- 35.Huhman K, Babagbemi T, Albers H. Bicuculline blocks neuropeptide Y-induced phase advances when microinjected in the suprachiasmatic nucleus of syrian hamsters. Brain Res. 1995;675:333–336. doi: 10.1016/0006-8993(95)00018-l. [DOI] [PubMed] [Google Scholar]
- 36.Jaffe D, Johnston D, Lasser-Ross N, Lisman J, Miyakawa H, Ross W. The spread of Na spikes determines the pattern of dendritic Ca entry into hippocampal neurons. Nature. 1992;357:244–246. doi: 10.1038/357244a0. [DOI] [PubMed] [Google Scholar]
- 37.Janzin E, Zhang X, Söderstrom S, Williams R, Hökfelt T, Ebendal T, Larhammar D. Expression of peptide YY and mRNA for the NPY/PYY receptor of the Y1 subtype in dorsal root ganglia during rat embyrogenesis. Dev Brain Res. 1993;76:105–113. doi: 10.1016/0165-3806(93)90128-w. [DOI] [PubMed] [Google Scholar]
- 38.Johnson RF, Moore RY, Morin LP. Lateral geniculate lesions alter circadian activity rhythms in the hamster. Brain Res Bull. 1989;22:411–422. doi: 10.1016/0361-9230(89)90068-3. [DOI] [PubMed] [Google Scholar]
- 39.Jolicoeur F, Bouali S, Fournier A, St-Pierre S. Mapping of hypothalamic sites involved in the effects of NPY on body temperature and food intake. Brain Res Bull. 1995;36:125–129. doi: 10.1016/0361-9230(94)00176-2. [DOI] [PubMed] [Google Scholar]
- 40.Khanna S, Sibbald J, Day T. Neuropeptide Y modulation of A1 noradrenergic neuron input to supraoptic vasopressin cells. Neurosci Lett. 1993;161:60–64. doi: 10.1016/0304-3940(93)90140-g. [DOI] [PubMed] [Google Scholar]
- 41.Klapstein G, Colmers F. 4-Aminopyridine and low Ca2+ differentiate presynaptic inhibition mediated by neuropeptide Y, baclofen and 2-chloroadenosine in rat hippocampal CA1 in vitro. Br J Pharmacol. 1992;105:470–474. doi: 10.1111/j.1476-5381.1992.tb14277.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Krstenansky J, Owen T, Payne M, Shatzer S, Buck S. C-terminal modifications of neuropeptide Y and its analogs leading to selectivity for the mouse brain receptor over the porcine spleen receptor. Neuropeptides. 1990;17:117–120. doi: 10.1016/0143-4179(90)90073-8. [DOI] [PubMed] [Google Scholar]
- 43.Larhammar D, Blomqvist A, Yee F, Jazin E, Yoo H, Wahlested C. Cloning and functional expression of a human neuropeptide Y/peptide YY receptor of the Y1 type. J Biol Chem. 1992;267:10935–10938. [PubMed] [Google Scholar]
- 44.Li W, Macdonald R, Hexum T. Benextramine irreversibly inhibits iodine-125 neuropeptide Y affinity labeling of the Y2 binding protein in bovine hippocampus. Eur J Pharmacol. 1991;5:89–91. doi: 10.1016/s0922-4106(05)80042-2. [DOI] [PubMed] [Google Scholar]
- 45.Linden D, Connor J. Long-term synaptic depression. Annu Rev Neurosci. 1995;18:319–357. doi: 10.1146/annurev.ne.18.030195.001535. [DOI] [PubMed] [Google Scholar]
- 46.LoTurco J, Owens D, Heath M, Davis M, Kriegstein A. GABA and glutamate depolarize cortical progenitor cells and inhibit DNA synthesis. Neuron. 1995;15:1287–1298. doi: 10.1016/0896-6273(95)90008-x. [DOI] [PubMed] [Google Scholar]
- 47.Martire M, Pistritto G, Mores N, Agnati L, Fuxe K. Region-specific inhibition of potassium-evoked [3H]noradrenaline release from rat brain synaptosomes by neuropeptide Y-(13–36): involvement of NPY receptors of the Y2 type. Eur J Pharmacol. 1993;230:231–234. doi: 10.1016/0014-2999(93)90807-t. [DOI] [PubMed] [Google Scholar]
- 48.Mattson MP, Kater SB. Calcium regulation of neurite elongation and growth cone motility. J Neurosci. 1987;7:4034–4043. doi: 10.1523/JNEUROSCI.07-12-04034.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.McDonald JK, Lumpkin MD, DePaolo LV. Neuropeptide-Y suppresses pulsatile secretion of luteinizing hormone in ovariectomized rats: possible site of action. Endocrinology. 1989;125:186–191. doi: 10.1210/endo-125-1-186. [DOI] [PubMed] [Google Scholar]
- 50.McDonald JK, Lumpkin MD, Samson WK, McCann SM. Neuropeptide Y affects secretion of luteinizing hormone and growth hormone in ovariectomized rats. Proc Natl Acad Sci USA. 1985;82:561–564. doi: 10.1073/pnas.82.2.561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.McLean R, Buck S, Krstenansky J. Examination of the role of the amphipathic alpha-helix in the interaction of neuropeptide Y and active cyclic analogues with cell membrane receptors and dimyristoylphosphatidylcholine. Biochemistry. 1990;29:2016–2022. doi: 10.1021/bi00460a009. [DOI] [PubMed] [Google Scholar]
- 52.McQuiston A, Petrozzoni J, Conner J, Colmers W. Neuropeptide Y1 receptors inhibit N-type calcium currents and reduce transient calcium increases in rat dentate granule cells. J Neurosci. 1996;16:1422–1429. doi: 10.1523/JNEUROSCI.16-04-01422.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Medanic M, Gillette M. Suprachiasmatic circadian pacemaker of rat shows two windows of sensitivity to neuropeptide Y in vitro. Brain Res. 1993;620:281–286. doi: 10.1016/0006-8993(93)90166-k. [DOI] [PubMed] [Google Scholar]
- 54.Meier E, Drejer J, Schousboe A. GABA induces functionally active low-affinity GABA receptors on cultured cerebellar granule cells. J Neurochem. 1984;43:1737–1744. doi: 10.1111/j.1471-4159.1984.tb06102.x. [DOI] [PubMed] [Google Scholar]
- 55.Meinecke D, Rakic P. Expression of GABA and GABAA receptors by neurons of the subplate zone in developing primate occipital cortex: evidence for transient local circuits. J Comp Neurol. 1992;317:91–101. doi: 10.1002/cne.903170107. [DOI] [PubMed] [Google Scholar]
- 56.Michler A. Involvement of GABA receptors in the regulation of neurite growth in cultured embryonic chick tectum. Int J Dev Neurosci. 1990;8:463–472. doi: 10.1016/0736-5748(90)90078-g. [DOI] [PubMed] [Google Scholar]
- 57.Moore R, Speh J. GABA is the principal neurotransmitter of the circadian system. Neurosci Lett. 1993;150:112–116. doi: 10.1016/0304-3940(93)90120-a. [DOI] [PubMed] [Google Scholar]
- 58.Morely J, Flood J. Neuropeptide Y and memory processing. Ann NY Acad Sci. 1990;611:226–231. doi: 10.1111/j.1749-6632.1990.tb48934.x. [DOI] [PubMed] [Google Scholar]
- 59.Mulkey R, Malenka R. Mechanisms underlying induction of homosynaptic long-term depression in area CA1 of the hippocampus. Neuron. 1992;9:967–975. doi: 10.1016/0896-6273(92)90248-c. [DOI] [PubMed] [Google Scholar]
- 60.Obata K, Oide M, Tanaka H. Excitatory and inhibitory actions of GABA and glycine on embryonic chick spinal neurons in culture. Brain Res. 1978;144:179–184. doi: 10.1016/0006-8993(78)90447-x. [DOI] [PubMed] [Google Scholar]
- 61.Obrietan K, van den Pol A. GABA neurotransmission in the hypothalamus: developmental reversal from Ca2+ elevating to depressing. J Neurosci. 1995;15:5065–5077. doi: 10.1523/JNEUROSCI.15-07-05065.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Palea S, Corsi M, Rimland J, Trist D. Discrimination by benextramine between the NPY-Y-1 receptor subtypes present in rabbit isolated vas deferens and saphenous vein. Br J Pharmacol. 1995;115:3–10. doi: 10.1111/j.1476-5381.1995.tb16311.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Penner S, Smyth D, Glavin G. Effects of neuropeptide Y and [Leu31, Pro34] neuropeptide Y on experimental gastric lesion formation and gastric secretion in the rat. J Pharmacol Exp Ther. 1993;266:339–343. [PubMed] [Google Scholar]
- 64.Perney T, Miller R. Two different G-proteins mediate neuropeptide Y and bradykinin-stimulated phospholipid breakdown in cultured rat sensory neurons. J Biol Chem. 1989;264:7317–7327. [PubMed] [Google Scholar]
- 65.Reppert S. Pre-natal development of a hypothalamic biological clock. Prog Brain Res. 1992;93:119–131. doi: 10.1016/s0079-6123(08)64568-9. [DOI] [PubMed] [Google Scholar]
- 66.Rusak B, Meijer JH, Harrington ME. Hamster circadian rhythms are phase-shifted by electrical stimulation of the geniculo-hypothalamic tract. Brain Res. 1989;498:283–291. doi: 10.1016/0006-8993(89)91163-3. [DOI] [PubMed] [Google Scholar]
- 67.Shibata S, Moore R. Neuropeptide Y and vasopressin effects on rat suprachiasmatic nucleus neurons in vitro. J Biol Rhythms. 1988;3:265–276. [Google Scholar]
- 68.Shibata S, Moore R. Neuropeptide Y and optic chiasm stimulation affect suprachiasmatic nucleus circadian function in vitro. Brain Res. 1993;615:95–100. doi: 10.1016/0006-8993(93)91118-c. [DOI] [PubMed] [Google Scholar]
- 69.Simonneaux V, Ouichou A, Craft C, Pevet P. Presynaptic and postsynaptic effects of neuropeptide Y in the rat pineal gland. J Neurochem. 1994;62:2464–2471. doi: 10.1046/j.1471-4159.1994.62062464.x. [DOI] [PubMed] [Google Scholar]
- 70.Spoerri P. Neurotrophic effects of GABA in cultures of embyronic chick brain and retina. Synapse. 1988;2:11–22. doi: 10.1002/syn.890020104. [DOI] [PubMed] [Google Scholar]
- 71.Stanley B, Leibowitz S. Neuropeptide Y injected in the paraventricular hypothalamus: a powerful stimulant of feeding behavior. Proc Natl Acad Sci USA. 1985;82:3940–3943. doi: 10.1073/pnas.82.11.3940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Swanson L, Cowan W, Jones E. An autoradiographic study of the efferent connections of the ventral lateral geniculate nucleus in the albino rat and cat. J Comp Neurol. 1974;156:143–163. doi: 10.1002/cne.901560203. [DOI] [PubMed] [Google Scholar]
- 73.Vaccarino F, Haywark M, Nestler E, Duman R, Tallman J. Differential induction of immediate early genes by excitatory amino acid receptor types in primary cultures of cortical and striatal neurons. Mol Brain Res. 1992;12:233–241. doi: 10.1016/0169-328x(92)90089-t. [DOI] [PubMed] [Google Scholar]
- 74.van den Pol AN, Dudek FE. Cellular communication in the circadian clock, the suprachiasmatic nucleus. Neuroscience. 1993;56:793–811. doi: 10.1016/0306-4522(93)90128-3. [DOI] [PubMed] [Google Scholar]
- 75.van den Pol AN, Gorcs T. Synaptic relationships between neurons containing vasopressin, GRP, VIP, and glutamate decarboxylase immunoreactivity in the the suprachiasmatic nucleus: dual ultrastructural immunocytochemistry with gold substituted silver peroxidase. J Comp Neurol. 1986;252:507–521. doi: 10.1002/cne.902520407. [DOI] [PubMed] [Google Scholar]
- 76.van den Pol AN, Tsujimoto K. Neurotransmitters of the hypothalamic suprachiasmatic nucleus: immunocytochemical analysis of 25 neuronal antigens. Neuroscience. 1985;15:1049–1086. doi: 10.1016/0306-4522(85)90254-4. [DOI] [PubMed] [Google Scholar]
- 77.van den Pol AN, Obrietan K, Cao V, Trombley PQ. Embryonic hypothalamic expression of functional glutamate receptors. Neuroscience. 1995;67:419–439. doi: 10.1016/0306-4522(95)96912-w. [DOI] [PubMed] [Google Scholar]
- 78.van den Pol AN, Obrietan K, Belousov A. NPY modulation of hypothalamic glutamate transmission. Soc Neurosci Abstr. 1995;22:430. [Google Scholar]
- 79.Wahle P, Müller T, Swandulla D. Characterization of neurochemical phenotypes in cultured hypothalamic neurons with immunohistochemistry and in situ hybridization. Brain Res. 1993;611:37–45. doi: 10.1016/0006-8993(93)91774-m. [DOI] [PubMed] [Google Scholar]
- 80.Wahlestedt C, Regunathan S, Reis D (1992) Identification of cultured cells selectively expressing Y1-, Y2-, or Y3-type receptors for neuropeptide Y/peptide YY. Life Sci 50:PL7–12. [DOI] [PubMed]
- 81.Walker M, Ewald D, Perney T, Miller R. Neuropeptide Y modulates neurotransmitter release and Ca2+currents in rat sensory neurons. J Neurosci. 1988;8:2438–2446. doi: 10.1523/JNEUROSCI.08-07-02438.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Walton M, Schaffner A, Barker J. Sodium channels, GABAA receptors, and glutamate receptors develop sequentially on embryonic rat spinal cord cells. J Neurosci. 1993;13:2068–2084. doi: 10.1523/JNEUROSCI.13-05-02068.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Welsh DK, Logothetis DE, Meister M, Reppert SM. Individual neurons dissociated from rat suprachiasmatic nucleus express independently phased circadian firing rhythms. Neuron. 1995;14:697–706. doi: 10.1016/0896-6273(95)90214-7. [DOI] [PubMed] [Google Scholar]
- 84.Westfall T, Carpentier S, Chen X, Beinfeld M, Naes L, Meldrum M. Prejunctional and postjunctional effects of neuropeptide Y at the noradrenergic neuroeffector junction of the perfused mesenteric arterial bed of the rat. J Cardiovasc Pharmacol. 1987;10:716–722. doi: 10.1097/00005344-198712000-00016. [DOI] [PubMed] [Google Scholar]
- 85.Wickens J, Abraham W. The involvement of L-type calcium channels in heterosynaptic long-term depression in the hippocampus. Neurosci Lett. 1991;130:128–132. doi: 10.1016/0304-3940(91)90244-n. [DOI] [PubMed] [Google Scholar]
- 86.Wiley J, Gross R, Lu Y, Macdonald R. Neuropeptide Y reduces calcium current and inhibits acetylcholine release in nodose neurons via a pertussis toxin-sensitive mechanism. J Neurophysiol. 1990;63:1499–1507. doi: 10.1152/jn.1990.63.6.1499. [DOI] [PubMed] [Google Scholar]
- 87.Wiley J, Gross R, Macdonald R. Agonists for neuropeptide Y receptor subtypes NPY-1 and NPY-2 have opposite actions on rat nodose neuron calcium currents. J Neurophysiol. 1993;70:324–330. doi: 10.1152/jn.1993.70.1.324. [DOI] [PubMed] [Google Scholar]
- 88.Woodhams P, Allen Y, McGovern J, Allen J, Bloom S, Balazs R, Polak J. Immunohistochemical analysis of the early ontogeny of the neuropeptide Y system in rat brain. Neuroscience. 1985;15:173–202. doi: 10.1016/0306-4522(85)90131-9. [DOI] [PubMed] [Google Scholar]
- 89.Yamashita M, Fukuda Y. Calcium channels and GABA receptors in the early embryonic chick retina. J Neurobiol. 1993;24:1600–1614. doi: 10.1002/neu.480241205. [DOI] [PubMed] [Google Scholar]
- 90.Yokoo H, Schlesinger D, Goldstein M. The effect of neuropeptide Y (NPY) on stimulation-evoked release of [3H]norepinephrine (NE) from rat hypothalamic and cerebral cortical slices. Eur J Pharmacol. 1987;143:283–286. doi: 10.1016/0014-2999(87)90545-0. [DOI] [PubMed] [Google Scholar]
- 91.Yuste R, Katz L. Control of postsynaptic Ca2+ influx in developing neocortex by excitatory and inhibitory neurotransmitters. Neuron. 1991;6:333–344. doi: 10.1016/0896-6273(91)90243-s. [DOI] [PubMed] [Google Scholar]