

Abstract
Calcium-binding proteins containing local circuit neurons are distributed ubiquitously in the human cerebral cortex where they colocalize with a subpopulation of cells that contain GABA. Several reports using a variety of pathological models, including Alzheimer’s disease (AD), have suggested that cells containing calcium-binding proteins are resistant to pathological insults. In this report, we test the hypothesis that AD pathology can differentially affect parvalbumin-containing cells depending on their location in the entorhinal cortex and the state of projection neurons with which they are associated. Using cases with different quantities of AD pathology, we determined the density of immunostaining for parvalbumin in the entorhinal cortex, and we correlated this with the concomitant pathological lesions in the various layers of this cortex.
Our results show a clear decrease in parvalbumin immunostaining in some parts of the entorhinal cortex when AD neuropathological markers are present. As the density of pathological markers in the entorhinal cortex becomes greater and more widespread, there is a decrease of parvalbumin immunostaining in additional layers, although in all cases, some cells persist.
Parvalbumin-containing neurons are clearly vulnerable in AD, but not because of neurofibrillary tangle formation. Instead, they are rendered vulnerable only after substantial loss of projection neurons; only then do they, too, become part of the lesion.
Keywords: parahippocampal gyrus, calcium-binding proteins, GABA, Alzheimer’s disease, entorhinal cortex, parvalbumin immunohistochemistry
Since the elucidation of the chemical/pathological correlates of Parkinson’s disease nearly five decades ago, it has been tempting to believe that other degenerative diseases may be linked to specific neurochemically identifiable systems. An early example of this arose from the pioneering biochemical studies of Davies and Maloney (1976) and Perry et al. (1977), who reported altered levels of cortical acetylcholine in Alzheimer’s disease (AD) and the subsequent demonstration of cholinergic cell loss in some AD patients (Whitehouse et al., 1981; Hansen et al., 1988; Geula and Mesulam, 1989; Giacobini, 1990). However, it is clear that the neurochemistry of degenerative diseases is a complex issue (Morrison et al., 1987; Lowe et al., 1990;Akiyama et al., 1989; Terry et al., 1991; Palmer and DeKosky, 1993;Auchus et al., 1994; Jaarsma et al., 1994). For example, the viability of a chemically specific neuron relates as well to the health of projection neurons with which it is associated and to targets on which their axons end. Thus, cholinergic neurons may be targeted directly by the disease, but their axons also terminate in cortex undergoing atrophy and degeneration. It is unclear which of the two lead to cholinergic neuron vulnerability. Importantly, the same could be said about cortical local circuit neurons because by definition they associate with projection neurons that are the targets for pathology in AD. When it is argued that local circuit neurons (calcium-binding protein and NADPH-d-positive neurons) are resistant to change in AD (Ferrer et al., 1991; Hof et al., 1991, 1993; Heizmann and Braun, 1992;Mufson and Brandabur, 1994), it implies both that they are not vulnerable to the particular pathology of the disorder and also that they are unaltered by to the loss of their own postsynaptic targets and other trophically related interactions that they may have with them.
In our estimation, the fate of local circuit neurons in AD has not been adequately assessed, because investigators have focused on the neocortex, where it is well known that only some layers are affected and, even these, in a subtotal manner. A more stringent test would be to assess vulnerability of local circuit neurons (such as the ones containing the calcium-binding protein parvalbumin) in a part of the cortex where a large number of neurons are diseased and where an entire layer of projection neurons is destroyed. Even though it is known that some parvalbumin-containing neurons have long axons, it is likewise clear that a majority have extensive local connections, contacting postsynaptic targets on projection neurons in their vicinity (Braak et al., 1991; Baimbridge et al., 1992; Beall and Lewis, 1992; Tuñon et al., 1992; Andressen et al., 1993).
Entorhinal cortex changes in AD [neurofibrillary tangles (NFTs) and neuritic plaques (NPs)] have a unique laminar distribution and temporal ordering (Hyman et al., 1984; Braak and Braak, 1992). In recent reports (Van Hoesen and Solodkin, 1993, 1994; Solodkin and Van Hoesen, 1996), we proposed that NFTs target neuronal populations that form modules. On the other hand, parvalbumin-containing neurons do not develop NFTs (Iwamoto and Emson, 1991), but are in close association with the entorhinal modules, which in some cases have a complete investment of tangles (Solodkin and Van Hoesen, 1993). Hence, the objective of the present report was to determine whether the parvalbumin neurons associated with the entorhinal modules are concomitantly reduced when NFTs develop in the projection neurons of the modules or whether, in parallel to isocortical layers, they are resistant to degeneration and somehow survive.
MATERIALS AND METHODS
Human brain tissue from 10 control (ages ranging from 55 to 81 years) and 22 AD cases (ages ranging from 58 to 92 years), all with autolysis times of <6 hr, was processed for immunohistochemistry.
AD was diagnosed using the CERAD (Consortium to Establish a Registry for Alzheimer’s Disease) (Mirra et al., 1991) and Khachaturian (1985)criteria, which include the diagnosis of clinical dementia plus the presence of NFTs and NPs in the hippocampal–parahippocampal areas as well as in other isocortical areas. The AD cases examined had a duration of clinical dementia ranging from 2 to 20 years.
The immunohistochemical methods used have been described previously (Van Hoesen and Solodkin, 1993). Briefly, 50 μm sections obtained from parahippocampal gyrus blocks, fixed by immersion in 3% paraformaldehyde/15% picric acid, were incubated in one of the following primary antibodies: mouse anti-parvalbumin (monoclonal, 1:80,000, Swant, Switzerland), mouse anti-A68 (Alz-50, monoclonal, 1:1000, Abbott Pharmaceutical, Chicago, IL and also donated by Dr. P. Davies, Albert Einstein University), or mouse anti-amyloid precursor protein (APP; 10D5, 1:100, donated by Dr. B. D. Schenk, Athena Neurosciences). Antibody visualization was achieved using biotin–avidin complex (Jackson ImmunoResearch, West Grove, PA) followed by a reaction with 5% DAB/0.03% H2O2. The primary antibodies were dissolved in normal serum containing 0.4% Triton X-100, whereas the secondary antibody contained 0.1% Triton X-100. For specificity control, one of the incubation steps was omitted, resulting in a complete suppression of the immunostaining. Additionally, selected sections were counterstained with Thioflavine-S or methylene blue.
Data were collected with a light microscope coupled to a computer and other analytical devices. For the description of the distribution of the immunostaining in the human entorhinal cortex, Lorente de Nó’s (1933) nomenclature was used. For assessment of cell numbers, all of the immunolabeled cell bodies located in layer II associated with the cell islands were counted in double-labeled sections. This allowed the identification of this layer, especially in severe AD cases in which atrophy was very pronounced. For layer III, cell counting was based on the stereological optical dissector technique using systematic random samples (Srivastava at al., 1993;West, 1994). Layer IIIb width was estimated at the midpoint of the anterior parahippocampal gyrus between the tentorial notch and the collateral sulcus. Statistical analysis of the data was done using Pearson’s correlation coefficient and one-way ANOVA followed by Dunnett’s multiple comparison test. Data are presented as mean ± SE.
Using the entorhinal cortex subdivisions proposed by Insausti et al. (1995), all measurements were done at the level of the intermediate entorhinal cortex.
RESULTS
Distribution of parvalbumin immunostaining in control cases
Parvalbumin-immunostained elements in the entorhinal cortex of control cases have a distinct distribution in both vertical and horizontal planes. This and the cellular characteristics of the entorhinal cortex are shown in Figure 1, Aand B.
Fig. 1.
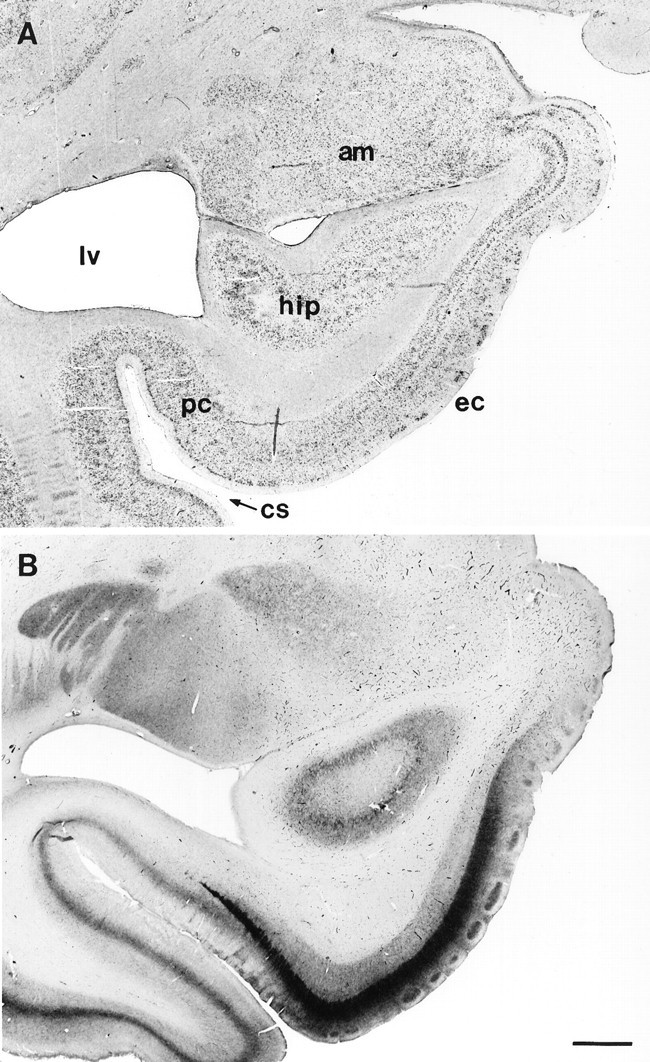
Low-magnification photographs of the human medial temporal lobe and entorhinal cortex (Brodmann’s area 28) in the coronal plane. A, Thionin staining for Nissl substance depicting the location of the entorhinal cortex. am, Amygdala; lv, lateral ventricle; hip, hippocampal formation; pc, perirhinal cortex; ec, entorhinal cortex; cs, collateral sulcus. B, Parvalbumin immunostaining at the same entorhinal level as in A in a control subject. Note the clear, dense staining in the islands of layer II as well as the dense band of immunoreactivity in layer IIIb. Scale bar, 2 mm.
Parvalbumin immunostaining clearly delimits the superficial layers. For example, layer I is devoid of neuronal cell bodies containing this calcium-binding protein, but it has a dense mesh of tangentially oriented axons (some with varicosities). Layer II is notable because the large multipolar and modified pyramidal cells that form the islands in this layer are not immunoreactive for parvalbumin, but deep to them, there are numerous small cell bodies that are (Fig. 2). These cells, for the most part, are oriented in the horizontal plane, and a majority have dendrites and axons directed toward the cell island superficial to them, as well as to neighboring islands. The latter creates the impression that they are interconnecting adjacent clusters of cells. The dendritic and axonal arborizations within the cell islands themselves are so dense that they form an intricate mesh that can be seen even with the unaided eye. The mean number of parvalbumin-positive cells per cell island in layer II was 3.87 ± 0.05 and was not correlated with the age of the subjects (r = −0.42; p = 0.35). In summary, the assessment of parvalbumin immunolabeled elements located in layer II is not subtle because the cell bodies are found close to the lower border of the islands and the neurites form a dense mesh among the neurons of the islands.
Fig. 2.
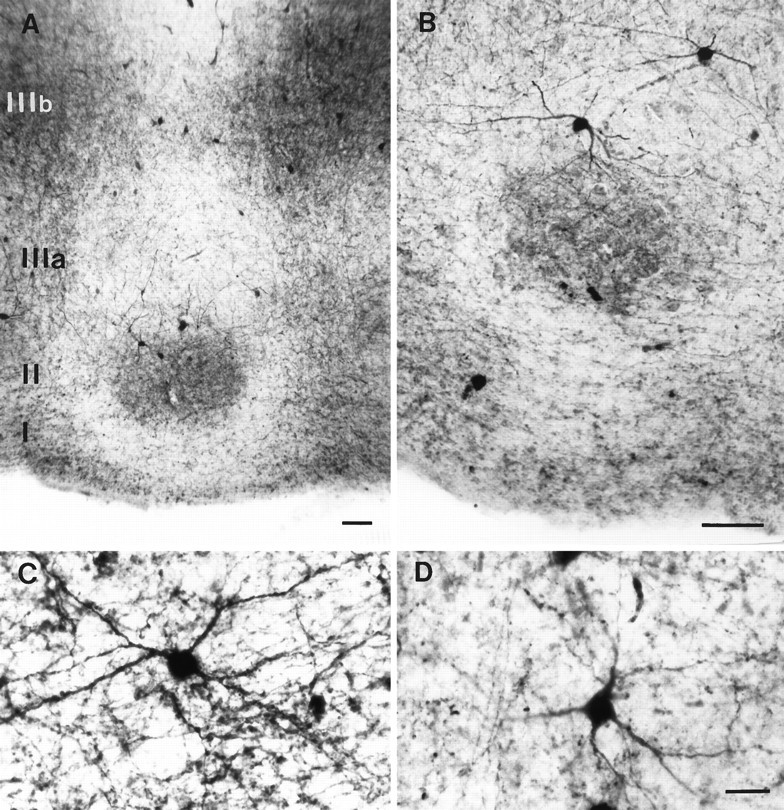
Parvalbumin immunostaining in the entorhinal cortex of control subjects. A, The distribution of the immunolabel in layer II is clearly segregated from that of layer IIIb.B, Higher-magnification parvalbumin-positive neurons. Parvalbumin neurons in layer II form a dense mesh of neurites within the cell islands. The cell bodies, however, are preferentially located along the deeper border of these islands. C, D, Examples of parvalbumin neurons in layers II and IIIb, respectively. Note the dense staining of the cell bodies and the smooth and long appearance of the dendrites. Scale bars: A, B, 100 μm; C, D, 25 μm.
In layer III, the distribution of parvalbumin-immunoreactive cells changes. Layer III, as seen in parvalbumin-stained tissue, can be subdivided in two sublayers (IIIa and IIIb). The more superficial (IIIa) is characterized by the presence of few immunoreactive cell bodies, and those present are located mainly in the interspaces between the cell islands of layer II. These have a vertical orientation with their neurites running toward layer I on the one hand, and deeper layers on the other. This creates the impression of columns between the cell islands (Fig. 2A). Additionally, in some instances, there are parvalbumin cell aggregates deeper and in register to the cell islands of layer II. The distribution of parvalbumin cells in layer IIIb is completely different. The density of neurites increases so dramatically that it forms a dense band of immunoreactivity that stops abruptly at the border with lamina dissecans (Fig.1B). Embedded in this band of immunoreactivity are numerous parvalbumin-positive cell bodies. The width of this band of immunoreactive neurites has a mean of 781 ± 23 μm in control cases. In the same group, the average number of cells per unit area in layer III was 4.34 ± 0.75. Pearson’s correlation coefficient between these values and age was low (r = −0.23, p = 0.58 andr = −0.36, p = 0.34, respectively).
Deeper layers (IV–VI) are characterized by a decrease in the density of cells immunolabeled for parvalbumin. Those present are elongated and larger cells that send dendrites toward more superficial layers. The density of these decreases even more as layer VI is approached.
Distribution of pathological markers in AD cases
In the human entorhinal cortex, the neuropathological markers of AD (NFTs and NPs) have a clear and discrete laminar distribution. For example, the first neuronal elements to develop NFTs (as seen with both Alz-50 and Thioflavine-S staining) are the large multipolar and pyramidal cells that form the cell islands in layers II and IIIa (Figs.3A,B, 4C–G). As the duration of the illness increases, additional neuronal elements are affected with the same type of lesions. These neurons are located mainly in layer IV (Fig. 4D–G), followed in long duration of illness cases by layers IIIb, V, and VI.
Fig. 3.
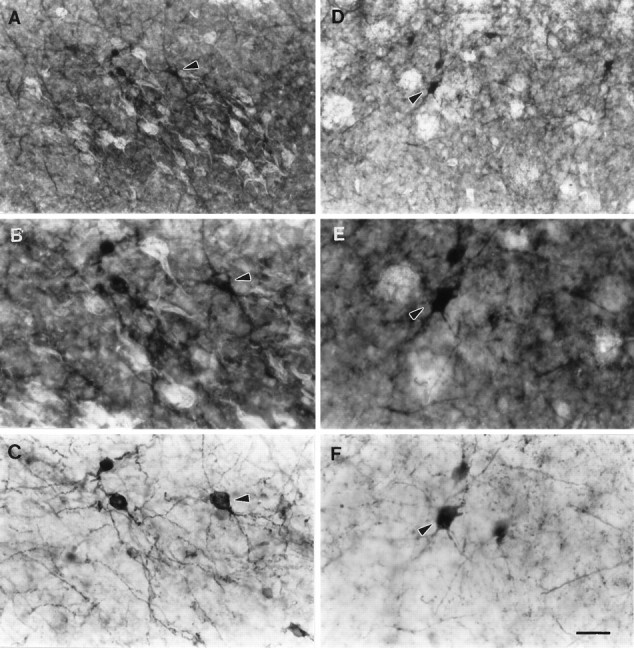
Double staining with Thioflavine-S and parvalbumin immunohistochemistry in AD cases. B and E are higher magnifications of A and D, respectively. Note the proximity of NFTs and parvalbumin cells in layer II (A, B) and NPs and parvalbumin cells in layer IIIb (D, E). Parvalbumin immunostaining in layers II and IIIb taken from B and E, respectively (without epifluorescence illumination). Note especially in C, the irregular density of the cell bodies plus the limited and contorted appearance of their dendrites. Additionally, note the numerous fragmented neurites found in the background. Arrowheadspoint to the same neurons in A–C andD–F, respectively. Scale bar: A,D, 65 μm; B, C, E, F, 25 μm.
Fig. 4.
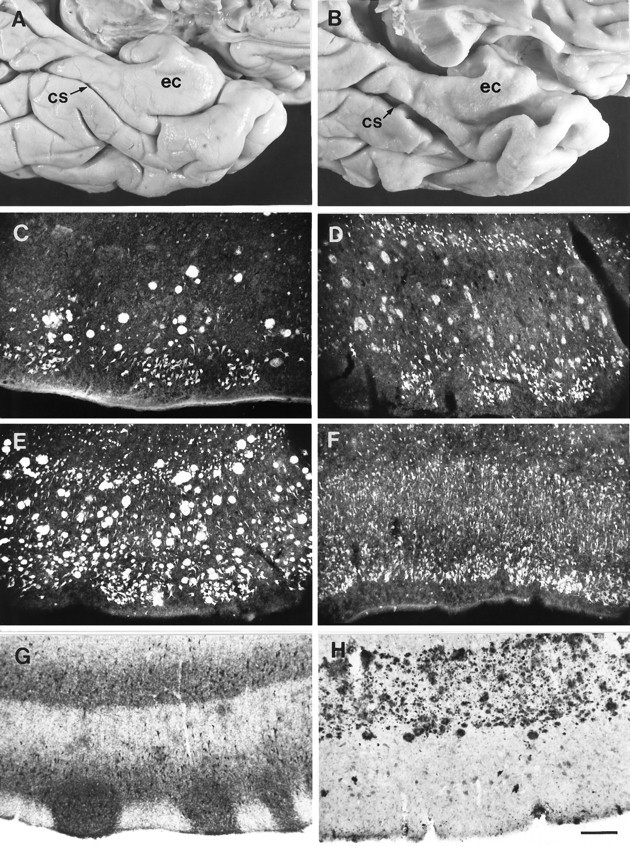
Neuropathological markers on the entorhinal cortex of AD cases. A, Gross view of the ventromedial temporal cortex of a control subject. ec, Entorhinal cortex;cs, collateral sulcus. B, Same view as inA taken from an AD patient. Note the atrophic appearance of the entorhinal cortex. C–F, Thioflavine-S histochemical staining depicting the distribution of NFTs and NPs at pathology grades 1–4, respectively. G, Example of brain Tau isoforms immunostaining (epitope to N-terminal residues 2–10; Goedert et al., 1991) with the Alz-50 antibody showing the location of NFTs.H, Amyloid precursor protein (APP) distribution as seen with antibody 10D5, depicting a dense band of plaques in layer IIIb. Scale bar, 200 μm.
The distribution of amyloid deposits as well as the amyloid precursor protein is for the most part limited to layers IIIb (Figs.3D,E, 4C–E,H), and in some cases layer I. The location of plaques in layer IIIb also shows a preference for medial areas, sparing more lateral parts. In some cases, they form mature plaques but, for the most part, they are of the diffuse type. Layer II, on the other hand, does not have noticeable quantities of either β-amyloid or amyloid precursor protein.
Changes of parvalbumin immunostaining in the entorhinal cortex in AD
Because the main distribution of both parvalbumin immunostaining and the lesions of AD are associated with layers II and IIIb, the results will be described by contrasting them (Fig.3A–F).
In general, the most dramatic change in the density of parvalbumin-containing neurons was in layer II. The assessment of the parvalbumin-containing neurons in layer II was guided by the location of the cell islands in double-labeled sections (Fig.3A,B). In all cases studied, there was a clear decrease in the parvalbumin immunostaining in this layer, evidenced by a reduction, and even disappearance, of the patches formed by dendrites and axons in the cell islands as well as in the number of parvalbumin cell bodies associated with them (Fig.5B–D). The average reduction in the number of parvalbumin cell bodies for all AD cases was 76% (F(4,26)= 13.97, p < 0.00001; Table 1).
Fig. 5.
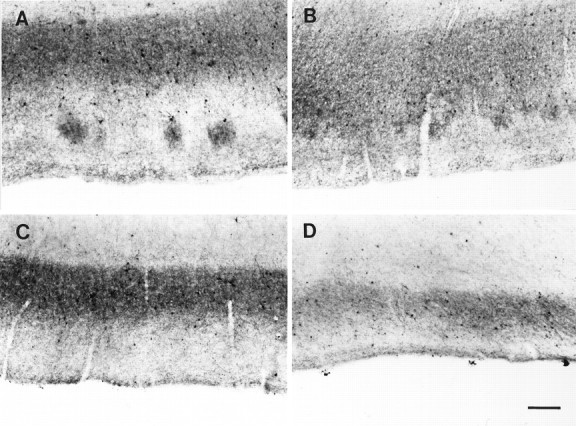
Parvalbumin immunostaining in AD cases.A–D, Distribution of parvalbumin immunostaining at pathological levels 1–4, respectively. Note the eventual complete disappearance of immunolabel in layer II and the consequent reduction on the size of the band of immunoreactivity in layer IIIb. Scale bar, 250 μm.
Table 1.
Mean ± SE of the parvalbumin-immunoreactive cell densities in layers II and III and mean ± SE of the width of layer IIIb
| Control | Grade 1 | Grade 2 | Grade 3 | Grade 4 | AD average | |
|---|---|---|---|---|---|---|
| Layer II cell density | 3.87 ± 0.50 | 1.5 ± 0.45** | 1.31 ± 0.33** | 0.87 ± 0.17** | 0.03 ± 0.03** | 1.07 ± 0.18** |
| Layer III cell density | 4.34 ± 0.75 | 2.73 ± 0.48 | 2.76 ± 0.96 | 3.11 ± 0.70 | 2.5 ± 1 | 2.82 ± 0.17 |
| Layer III width (μm) | 781 ± 23 | 703 ± 81 | 668 ± 39 | 603 ± 29* | 420 ± 20* | 627 ± 28* |
| Years of clinical dementia | 5.5 | 5.6 | 9.3 | 16 | ||
| N | 10 | 4 | 7 | 7 | 4 |
The asterisks denote the values that were statistically different from controls: *p ≤ 0.003, **p < 0.0001.
The initial change observed was a reduction in the density of cell bodies associated with the cell islands of layer II. Concurrently, with a decrease in the density of cells, the quality of immunostaining of parvalbumin neurons was altered and, in many cases, label was only in the proximal dendrites, whereas in control cases, neurons had a Golgi-like appearance with immunoreactive secondary and even tertiary processes (Figs. 3E,F and2C,D, respectively).
Another frequent observation was that parvalbumin cell bodies located in layer II had weak immunostaining and, in some cases, the reaction product was not distributed homogeneously throughout the cell body. Instead, it formed a halo around the periphery of the soma, giving a ghost-like appearance and the impression of cell degeneration.
Parvalbumin immunostaining in layer IIIb of AD cases was more variable. In some cases, it was clearly affected, whereas in others, it was not. The overall changes seen were a decrease in the density of the immunostaining of the band formed by the neurites and a reduction in its width (F(4,26) = 7.61, p = 0.0004; Fig. 5A–D). The density of parvalbumin cells did not differ between the Alzheimer’s cases and controls (F(4,26) = 0.83, p = 0.52; Table1). In contrast to layer II, the reduction of immunostaining was in no case as pronounced as in the former layer, where in most instances, it was completely devoid of parvalbumin immunostaining. In layer IIIb, even in the cases with the largest alterations of immunostaining, there were some parvalbumin-immunolabeled cell bodies and neurites still present. Additionally, in layers II and IIIb of all Alzheimer’s cases, we did not find neurons containing both NFTs and parvalbumin immunoreactivity.
Because of the wide range of parvalbumin immunoreactivity in AD cases, we analyzed the data in terms of the quantity of Alzheimer’s pathological changes found in entorhinal layers II and IIIb, focusing on both NFTs and NPs. Four grades of pathological changes were formulated. The rationale behind this grading was not to provide clues for a correlation between clinical symptoms and pathological lesions or even progression of dementia, but only to describe the density of pathology within the entorhinal cortex (Fig. 4C–F).
Grade 1: NFTs observed only in entorhinal layers II and IIIa and NPs as well as β-amyloid deposits in layer IIIb.
Grade 2: NFTs observed in layers II (forming islands of NFTs), IIIa, and IV. NPs in layer IIIb.
Grade 3: NFTs frequency increases and now present in layer IIIb. NPs in layers I, III, V, and VI.
Grade 4: NFTs density high with all layers affected. The location and density of plaques are similar to those of grade 3.
These grades of entorhinal pathology have a good correlation with the number of years with clinical dementia (r = 0.72, p = 0.001) but not with the age of the patient (r = 0.18, p = 0.39). There was also no correlation between age and years with clinical dementia (r = 0.1, p = 0.58).
Parvalbumin immunostaining changes based on grades of entorhinal pathology
Grade 1
The cases in this grade (n = 4) had an average of 5.5 ± 1.8 years with clinical dementia. Parvalbumin immunostaining showed changes in layer II consisting of a decreased density of parvalbumin cell bodies, although the patches of immunoreactive neurites were still present, preserving the patchy appearance seen in control cases (Fig.5A). The average number of parvalbumin cells per island was 1.5 ± 0.9, which is a decrease of 61% compared with controls (Dunnett’s p = 0.001; Fig. 6). However, the cells that were left, for the most part, were darkly stained and indistinguishable from controls. The density of the immunoreactive band in layer III was not different from controls, although its width was reduced slightly from 781 ± 23 μm in controls to an average of 703 ± 81 μm (Table 1, Fig. 6). In summary, grade 1 was characterized by the reduction of immunoreactive cell bodies in layer II and a slight decrease in the width of layer IIIb immunoreactivity.
Fig. 6.
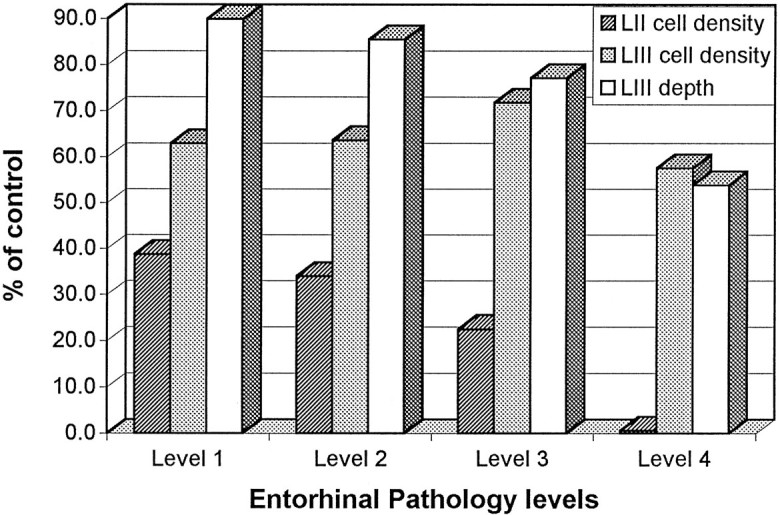
Graphic representation of the relative changes in parvalbumin immunostaining in layers II and III of AD cases. They-axis represents the percent of reduction of the label compared with controls. The x-axis shows the four different pathology levels. The bars depict the density of parvalbumin cells in layers II and IIIb as well as the width of the band of immunoreactivity in layer IIIb. Note that the density of cells in layer II and the width of layer IIIb changed with the progression of the illness, but the density of parvalbumin cells in layer IIIb did not differ from control cases.
Grade 2
The average duration of the illness for this grade was 5.6 ± 3 years (n = 7). Layer II-immunoreactive cell bodies showed a further decrease (66% reduction; Dunnett’s p = 0.00008; Fig. 6, Table 1). However, the most conspicuous change was the large reduction of the patches of immunoreactivity within the cell islands (Fig. 5B). Moreover, the quality of the immunostaining of the remaining neurons was degraded because it was lighter than in the previous grade and, in some cases, was limited to the periphery of the cell bodies (Fig. 3A–C). In layer IIIb, the density of the cells immunoreactive for parvalbumin did not differ from the grade 1 (mean = 2.76 ± 0.96), but the width of the immunoreactivity band was decreased further to an average of 668 ± 39 μm, a reduction of 15% compared with controls (p = 0.09; Fig. 6).
Grade 3
The duration of clinical dementia for these cases (n = 7) was 9.3 ± 3.8 years. Layer II was devoid of immunoreactivity in neurites, and no patches were observed (Fig. 5C). Additionally, the average number of parvalbumin cells per island was decreased further to 0.87 ± 0.44, representing a 77% reduction from controls (Dunnett’s p = 0.00001; Fig. 6, Table 1). Survivors had abnormal staining and were pale with little dendritic staining. Immunostaining within the cell bodies was limited to a peripheral band. The immunoreactivity in layer IIIb showed a further decrease of 23% in its width compared with controls (Dunnett’sp = 0.003; Fig. 6, Table 1), whereas the density of cells within this band remained roughly similar to the two previous grades and controls (mean = 3.11 ± 0.70).
Grade 4
At this stage (n = 4), the average duration of clinical dementia was 16 ± 5 years. Immunostaining in layer II was absent with no patches of neurites, and the reduction in the density of the parvalbumin cells per island reached 99% (Dunnett’s p = 0.00008; Fig. 6, Table 1). Layer IIIb did not show profound changes. The band of immunoreactivity was still present, and there were scattered cell bodies immunolabeled for parvalbumin (mean = 2.5 ± 1.0). The main difference was the width of the band, which on average was 420 ± 20 μm, a 46% reduction compared with controls (Dunnett’sp = 0.0002; Fig. 5D,E). It is interesting to mention that the density of neurites within the band was reduced also, and the remainder had an abnormal appearance with thick and tortuous processes.
DISCUSSION
Our findings reveal a decrease in parvalbumin neurons and neuropil staining in the entorhinal cortex of AD cases that parallels the development of NFTs in the projection neurons of that cortex. These results are interesting because parvalbumin-containing neurons form a subpopulation of GABAergic neurons (Celio, 1986; Kosaka et al., 1987;DeFelipe et al., 1989; Hendry et al., 1989; Lewis and Lund, 1990;VanBrederode et al., 1990; Demeulemeester et al., 1991), and also because previous reports focusing on the isocortex indicate that in AD there is no change in the density of these cells (Ferrer et al., 1991;Fonseca et al., 1993; Hof et al., 1993) but, rather, changes in their morphology (Arai et al., 1987; Satoh et al., 1991; Fonseca et al., 1993). A single biochemical report describes a decrease of >50% in the quantity of parvalbumin in the parahippocampal gyrus (Inaguma et al., 1992). With the exception of this latter report, our findings appear at odds with the negative findings of several investigators. In fact, they are more in line with reports describing reduced parvalbumin immunostaining on Creutzfeldt–Jakob’s disease, stroke, cerebral ischemia, and epilepsy (Johansen et al., 1990; DeFelipe et al., 1993;De Jong et al., 1993; Ferrer et al., 1993; Tortosa and Ferrer, 1993). Many variables determine the vulnerability of neurons within cortical layers and even entire cytoarchitectural areas in AD (Hyman and Gomez-Isla, 1994), but it seems obvious that the presence or absence of calcium-binding proteins in a neuron is not an indicator of whether it survives. Differences in the type of cortex where parvalbumin containing neurons occur is certainly a variable as well as the degree to which projection neurons are compromised.
The entorhinal cortex is a good model to test the vulnerability of parvalbumin neurons, not only because it is one of the areas where the pathological changes of AD occur first and most heavily (Hyman et al., 1984; Braak and Braak, 1991), but also because the distribution of both Alzheimer’s pathological markers and parvalbumin neurons is segregated to some degree. This has enabled us to look objectively at pathological changes and, in the same specimen, assess alterations in immunostaining.
The grades of pathology used to judge our results have differences and parallels with the more general model of Braak and Braak (1991). The main difference is that they consider a large number of areas, cortical and subcortical, to highlight the presumed longitudinal events taking place in AD. Our grades focus only on the entorhinal cortex. Moreover, they did not consider the presence of NPs in the entorhinal layer III, which is a critical factor for this investigation. Nevertheless, based on the distribution of NFTs in the entorhinal cortex, our grade 1 would seem to correspond to their stage II, our grade 2 to their stages III and IV, our grade 3 to their stage V, and our grade 4 to their stage VI.
The grades of pathology in the entorhinal cortex proposed here are a useful tool to aid in evaluating the effects of pathological changes on parvalbumin cells vulnerability because they have a good correlation with duration of dementia. In contrast, the lack of correlation between the pathology grades and age of the patients suggests that age does not determine the distribution of pathological markers within area 28 (Mizutani and Shimada, 1992), or in other cortical areas for that matter (Arnold et al., 1991).
Grade 1
The main observations were the decrease in the density of parvalbumin neurons in layer II and a preservation of immunostaining in layer IIIb. It is not clear, however, if the decrease seen in layer II was associated with neuronal death because the patches of neurites in the cell islands were still detectable. The decrease in immunostaining may also reflect a decrease in the concentration of parvalbumin by a reduction in activity because their postsynaptic targets contain NFTs. A high correlation between parvalbumin immunostaining and neuronal activity levels has been shown previously (Omidi et al., 1988; Celio, 1990) and AD pathology in layer II is correlated with diminished cytochrome oxidase staining (Chandrasekaran et al., 1994). Interestingly, the presence of amyloid deposits in layer IIIb did not result in a decrease in the density of parvalbumin neurons, a consequence, perhaps, of their efficient buffering of intracellular calcium (Pike and Cotman, 1995).
Grade 2
This grade was characterized by three changes. First, there was a conspicuous increase in NFTs in layer II. Second, neurons containing NFTs made their first appearance in the large modified pyramids that form layer IV. Third, there was a further decrease in immunolabeling in layer II. Obviously, this alone could be accounted for by the increase in destructive pathology to the projection neurons of layer II, but because layer IV neurons provide intrinsic axon collaterals to the superficial layers of the entorhinal cortex, the loss of their contacts and further diminution of neural activity cannot be ruled out as an exacerbating variable.
Grades 3 and 4
These grades were notable for the absence of change in the density of parvalbumin neurons on layer IIIb in the face of increasing NFTs directly affecting this layer. Likewise, there was a preservation of immunostaining in the band that occupies layer IIIb consisting of axons and dendrites, although it shrunk in width by nearly one-half. Were it not for the shrinkage, and the fact that layer II immunoreactivity had already changed dramatically in grades 1 and 2, it would be easy to reach the erroneous conclusion that parvalbumin staining in the entorhinal cortex mimicked the absence of change in AD often reported for the isocortex. This laminar-specific preservation of staining is not immediately explainable, and we can only surmise that it is maintained by plastic responses on the intact processes of surviving neurons similar to those reported in the isocortex (Cellerino et al., 1992; Schmidt-Kastner et al., 1992). Such changes are known to occur in AD in other cortical areas (Hyman et al., 1987; Masliah et al., 1993). On the other hand, and even though as in the isocortex (Arai et al., 1987; Satoh, 1991; Fonseca et al., 1993), some of the parvalbumin neurons remaining had morphological abnormalities in their processes, it is difficult to explain how the density of these cells is maintained in these latest grades of pathology. Differential vulnerability of parvalbumin neurons in cortical layers is not an unusual occurrence in the cerebral cortex. DeFelipe et al. (1993) showed in temporal lobe epilepsy a selective decrease in the density of these cells restricted to layers II, III, and IV. In our case, viability may be preserved if these cells are protected against abnormal increases of intracellular calcium (Chard et al., 1993) and, hence, are not vulnerable to the toxic effects of amyloid (Khachaturian et al., 1989; Mattson, 1994); but that does not explain why they do not degenerate either when the density of NFTs increased. An additional characteristic of these cells could be that their efferent targets and afferent inputs are not affected in the same degree (Arnold et al., 1991), or it could be that in this layer there is a higher concentration of trophic factors (Price et al., 1991; Scott et al., 1995; Zhan et al., 1995).
In summary, it is clear that there is a lack of homogeneity in the fate of parvalbumin cells in the entorhinal cortex in AD. Especially in the first grade of the disease, when the development of NFTs is confined to layer II, it is tempting to think that the most likely candidate determining the viability of these cells is the presence or absence of their synaptic targets. Passive mechanisms such as retrograde degeneration (Nitsch and Frotscher, 1993) are an attractive possibility. Interestingly, Hof and Morrison (1991) have reported a similar correlation between the density of NFTs in layers V–VI of prefrontal cortex and calbindin-containing neurons in AD. An additional candidate could be apoptosis. In a provocative report, Su et al. (1994)showed that in the hippocampus and entorhinal cortex of AD cases, apoptosis is present in neurons, with and without NFTs. Hence, we cannot rule out that some of the neurons showing apoptosis could contain parvalbumin. Another possibility could be aging, as suggested by Heinsen et al. (1994). However, in this study, that may not be the case because the density of parvalbumin neurons in layer II or IIIb of controls was not correlated with age (Trillo and Gonzalo, 1993; West et al., 1994). Finally, another possible mechanism of degeneration could be the presence of toxic factors produced as consequence of inflammatory reactions around the NPs (Kalaria, 1993; McGeer et al., 1993). This does not seem to be the case either because in layer IIIb, in grades 1 and 2, where NPs are the only lesions, the density and number of parvalbumin cells is unchanged.
In summary, our findings reveal that a clear up or down statement cannot be made about the vulnerability of parvalbumin-containing neurons in AD. Viability is contingent on the type of cortex in which these neurons are located and the specific layer within which they reside. Moreover, in some instances, their fate seems linked also to the fate of the projection neurons with which they associate, and some partners may be better than others. Finally, it seems likely that parvalbumin neurons themselves may vary greatly in their ability to cope with destruction of neuropil around them. All considered, however, it is clear that the disrupted mechanisms that lead to NFTs in projection neurons are likely not shared by parvalbumin neurons and that their vulnerability is governed by yet unidentified factors ongoing in the disease process itself.
Footnotes
This research was supported by National Institutes of Health NS 14944 and PO NS 19632. We thank Dr. P. Davies, Albert Einstein University, and Abbott Pharmaceutical Co. for the Alz-50 antibody; D. B. Schenk, Athena Neuroscience for the 10D5 antibody; and Drs. B. D. Hyman, M. N. Hart, and R. J. Traub for their valuable comments. We also thank Mr. P. Reimann for photography, Mrs. P. Frantz for help with this manuscript, and Mr. L. Spence for brain acquisition.
Correspondence should be addressed to Ana Solodkin, Department of Anatomy/BSB, University of Iowa College of Medicine, Iowa City, IA 52242.
REFERENCES
- 1.Akiyama H, McGeer PL, Itagaki S, McGeer EG, Kaneko T. Loss of glutaminase-positive cortical neurons in Alzheimer’s disease. Neurochem Res. 1989;14:353–358. doi: 10.1007/BF01000038. [DOI] [PubMed] [Google Scholar]
- 2.Andressen C, Blümcke Celio MR. Calcium binding proteins: selective markers of nerve cells. Cell Tissue Res. 1993;271:181–208. doi: 10.1007/BF00318606. [DOI] [PubMed] [Google Scholar]
- 3.Arai H, Emson PC, Mountjoy CQ, Carrasco LH, Heizmann CW. Loss of parvalbumin immunoreactive neurons from cortex in Alzheimer’s disease dementia. Brain Res. 1987;418:164–169. doi: 10.1016/0006-8993(87)90974-7. [DOI] [PubMed] [Google Scholar]
- 4.Arnold SE, Hyman BT, Flory J, Damasio AR, Van Hoesen GW. The topographical and neuroanatomical distribution of neurofibrillary tangles and neuritic plaques in the cerebral cortex of patients with Alzheimer’s disease. Cereb Cortex. 1991;1:103–116. doi: 10.1093/cercor/1.1.103. [DOI] [PubMed] [Google Scholar]
- 5.Auchus AP, Green RC, Nemeroff CB. Cortical and subcortical neuropeptides in Alzheimer’s disease. Neurobiol Aging. 1994;15:589–595. doi: 10.1016/0197-4580(94)90102-3. [DOI] [PubMed] [Google Scholar]
- 6.Baimbridge JG, Celio MR, Rogers JH. Calcium-binding proteins in the nervous system. Trends Neurosci. 1992;15:303–308. doi: 10.1016/0166-2236(92)90081-i. [DOI] [PubMed] [Google Scholar]
- 7.Beall MJ, Lewis DA. Heterogeneity of layer II neurons in human entorhinal cortex. J Comp Neurol. 1992;321:241–266. doi: 10.1002/cne.903210206. [DOI] [PubMed] [Google Scholar]
- 8.Braak H, Braak E. Neuropathological staging of Alzheimer-related changes. Acta Neuropathol. 1991;82:239–259. doi: 10.1007/BF00308809. [DOI] [PubMed] [Google Scholar]
- 9.Braak H, Braak E. The human entorhinal cortex: normal morphology and laminar-specific pathology in various diseases. Neurosci Res. 1992;15:6–31. doi: 10.1016/0168-0102(92)90014-4. [DOI] [PubMed] [Google Scholar]
- 10.Braak E, Strotkamp B, Braak H. Parvalbumin-immunoreactive structures in the hippocampus of the human adult. Cell Tissue Res. 1991;264:33–48. doi: 10.1007/BF00305720. [DOI] [PubMed] [Google Scholar]
- 11.Celio MR. Parvalbumin in most gamma-aminobutyric-acid-containing neurons of the cat cerebral cortex. Science. 1986;231:995–997. doi: 10.1126/science.3945815. [DOI] [PubMed] [Google Scholar]
- 12.Celio MR. Calbindin D-28k and parvalbumin in the rat nervous system. Neuroscience. 1990;35:375–475. doi: 10.1016/0306-4522(90)90091-h. [DOI] [PubMed] [Google Scholar]
- 13.Cellerino A, Siciliano R, Domenici L, Maffei L. Parvalbumin immunoreactivity: a reliable marker for the effects of monocular deprivation in the rat visual cortex. Neuroscience. 1992;51:749–753. doi: 10.1016/0306-4522(92)90514-3. [DOI] [PubMed] [Google Scholar]
- 14.Chandrasekaran K, Giordano T, Brady DR, Stoll J, Martin LJ, Rapoport SI. Impairment in mitochondrial cytochrome oxidase gene expression in Alzheimer’s disease. Mol Brain Res. 1994;24:336–340. doi: 10.1016/0169-328x(94)90147-3. [DOI] [PubMed] [Google Scholar]
- 15.Chard PS, Bleakman D, Christakos S, Fullmer CS, Miller RJ. Calcium buffering properties of calbindin D28K and parvalbumin in rat sensory neurons. J Physiol (Lond) 1993;472:341–357. doi: 10.1113/jphysiol.1993.sp019950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Davies P, Maloney AJR. Selective loss of central cholinergic neurons in Alzheimer’s disease. Lancet. 1976;2:1403. doi: 10.1016/s0140-6736(76)91936-x. [DOI] [PubMed] [Google Scholar]
- 17.De Jong GI, Van der Zee EA, Bohus B, Luiten PGM. Reversed alterations of hippocampal parvalbumin and protein kinase C-γ immunoreactivity after stroke in spontaneously hypertensive stroke-prone rats. Stroke. 1993;24:2082–2086. doi: 10.1161/01.str.24.12.2082. [DOI] [PubMed] [Google Scholar]
- 18.DeFelipe J, Hendry SHC, Jones EG. Visualization of chandelier cell axons by parvalbumin immunoreactivity in the monkey cerebral cortex. Proc Natl Acad Sci USA. 1989;86:2093–2097. doi: 10.1073/pnas.86.6.2093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.DeFelipe J, Garcia Sola R, Marco P, del Rio MR, Pulido P, Ramon y Cajal S. Selective changes in the microorganization of the human epileptogenic neocortex revealed by parvalbumin immunoreactivity. Cereb Cortex. 1993;3:39–48. doi: 10.1093/cercor/3.1.39. [DOI] [PubMed] [Google Scholar]
- 20.Demeulemeester H, Arckens L, Vandesande F, Orban GA, Heizmann CW, Pochet R. Calcium-binding proteins and neuropeptides as molecular markers of GABAergic interneurons in the cat visual cortex. Exp Brain Res. 1991;84:538–544. doi: 10.1007/BF00230966. [DOI] [PubMed] [Google Scholar]
- 21.Ferrer I, Soriano E, Tuñon T, Fonseca M, Guionnet N. Parvalbumin immunoreactive neurons in normal human temporal neocortex and in patients with Alzheimer’s disease. J Neurol Sci. 1991;106:135–141. doi: 10.1016/0022-510x(91)90250-b. [DOI] [PubMed] [Google Scholar]
- 22.Ferrer I, Casas R, Rivera R. Parvalbumin-immunoreactive cortical neurons in Creutzfeldt-Jakob’s disease. Ann Neurol. 1993;34:864–866. doi: 10.1002/ana.410340617. [DOI] [PubMed] [Google Scholar]
- 23.Fonseca M, Soriano E, Ferrer I, Martinez A, Tuñon T. Chandelier cell axons identified by parvalbumin-immunoreactivity in the normal human temporal cortex and in Alzheimer’s disease. Neuroscience. 1993;55:1107–1116. doi: 10.1016/0306-4522(93)90324-9. [DOI] [PubMed] [Google Scholar]
- 24.Geula C, Mesulam M-M. Cortical cholinergic fibers in aging and Alzheimer’s disease: a morphometric study. Neuroscience. 1989;33:469–481. doi: 10.1016/0306-4522(89)90399-0. [DOI] [PubMed] [Google Scholar]
- 25.Giacobini E. Cholinergic receptors in human brain: effects of aging and Alzheimer’s disease. J Neurosci Res. 1990;27:548–560. doi: 10.1002/jnr.490270416. [DOI] [PubMed] [Google Scholar]
- 26.Goedert M, Spillantini MG, Jakes R. Localization of the Alz-50 epitope in recombinant human microtubule-associated protein Tau. Neurosci Lett. 1991;126:149–154. doi: 10.1016/0304-3940(91)90541-z. [DOI] [PubMed] [Google Scholar]
- 27.Hansen LA, DeTeresa R, Davies P, Terry RD. Neocortical morphometry, lesion counts and choline acetyltransferase levels in the age spectrum of Alzheimer’s disease. Neurology. 1988;38:48–54. doi: 10.1212/wnl.38.1.48. [DOI] [PubMed] [Google Scholar]
- 28.Heinsen H, Henn R, Eisenmenger W, Gotz M, Bohl J, Bethke B, Lockemann U, Püschel K. Quantitative investigations on the human entorhinal cortex areas: left-right asymmetry and age-related changes. Anat Embryol. 1994;190:181–194. doi: 10.1007/BF00193414. [DOI] [PubMed] [Google Scholar]
- 29.Heizmann CW, Braun K. Changes in Ca2+-binding proteins in human neurodegenerative disorders. Trends Neurosci. 1992;15:259–264. doi: 10.1016/0166-2236(92)90067-i. [DOI] [PubMed] [Google Scholar]
- 30.Hendry SHC, Jones EG, Emson PC, Lawson DEM, Heizmann CW, Streit P. Two classes of cortical GABA neurons defined by differential calcium-binding protein immunoreactivities. Exp Brain Res. 1989;76:467–472. doi: 10.1007/BF00247904. [DOI] [PubMed] [Google Scholar]
- 31.Hof PR, Morrison JH. Neocortical neuronal subpopulations labeled by a monoclonal antibody to calbindin exhibit differential vulnerability in Alzheimer’s disease. Exp Neurol. 1991;111:293–301. doi: 10.1016/0014-4886(91)90096-u. [DOI] [PubMed] [Google Scholar]
- 32.Hof PR, Cox K, Young WG, Celio MR, Rogers J, Morrison JH. Parvalbumin-immunoreactive neurons in the neocortex are resistant to degeneration in Alzheimer’s disease. J Neuropathol Exp Neurol. 1991;50:451–462. doi: 10.1097/00005072-199107000-00006. [DOI] [PubMed] [Google Scholar]
- 33.Hof PR, Nimchinsky EA, Celio MR, Bouras C, Morrison JH. Calretinin-immunoreactive neocortical interneurons are unaffected in Alzheimer’s disease. Neurosci Lett. 1993;152:145–149. doi: 10.1016/0304-3940(93)90504-e. [DOI] [PubMed] [Google Scholar]
- 34.Hyman BT, Gomez-Isla T. Alzheimer’s disease is a laminar, regional and neural system specific disease, not a global brain disease. Neurobiol Aging. 1994;15:353–354. doi: 10.1016/0197-4580(94)90031-0. [DOI] [PubMed] [Google Scholar]
- 35.Hyman BT, Van Hoesen GW, Damasio AR, Barnes CL. Alzheimer’s disease: cell specific pathology isolates the hippocampal formation. Science. 1984;225:1168–1170. doi: 10.1126/science.6474172. [DOI] [PubMed] [Google Scholar]
- 36.Hyman BT, Van Hoesen GW, Damasio AR. Alzheimer’s disease: glutamate depletion in the hippocampal perforant pathway zone. Ann Neurol. 1987;22:37–40. doi: 10.1002/ana.410220110. [DOI] [PubMed] [Google Scholar]
- 37.Inaguma Y, Shinohara H, Inagaki T, Kato K. Immunoreactive parvalbumin concentrations in patients with Alzheimer’s disease. J Neurol Sci. 1992;110:57–61. doi: 10.1016/0022-510x(92)90009-a. [DOI] [PubMed] [Google Scholar]
- 38.Insausti R, Tuñon T, Sobreviela T, Insausti AM, Gonzalo LM. The human entorhinal cortex: a cytoarchitectonic analysis. J Comp Neurol. 1995;355:171–198. doi: 10.1002/cne.903550203. [DOI] [PubMed] [Google Scholar]
- 39.Iwamoto N, Emson PC. Demonstration of neurofibrillary tangles in parvalbumin-immunoreactive interneurons in the cerebral cortex of Alzheimer-type dementia brain. Neurosci Lett. 1991;128:81–84. doi: 10.1016/0304-3940(91)90764-k. [DOI] [PubMed] [Google Scholar]
- 40.Jaarsma D, Veenma-van der Duin L, Korf J. N -acetylaspartate and N -acetylaspatylglutamate grades in Alzheimer’s disease post-mortem brain tissue. J Neurol Sci. 1994;127:230–233. doi: 10.1016/0022-510x(94)90077-9. [DOI] [PubMed] [Google Scholar]
- 41.Johansen FF, Tonder N, Zimmer J, Baimbridge KG, Diemer NH. Short term changes of parvalbumin and calbindin immunoreactivity in the rat hippocampus following cerebral ischemia. Neurosci Lett. 1990;120:171–174. doi: 10.1016/0304-3940(90)90030-d. [DOI] [PubMed] [Google Scholar]
- 42.Kalaria RN. The immunopathology of Alzheimer’s disease and some related disorders. Brain Pathol. 1993;3:333–347. doi: 10.1111/j.1750-3639.1993.tb00761.x. [DOI] [PubMed] [Google Scholar]
- 43.Khachaturian ZS. Diagnosis of Alzheimer’s disease. Arch Neurol. 1985;42:1097–1105. doi: 10.1001/archneur.1985.04060100083029. [DOI] [PubMed] [Google Scholar]
- 44.Khachaturian ZS, Cotman CW, Pettegrew JW. Calcium, membranes, aging and Alzheimer’s disease. Ann NY Acad Sci. 1989;568:1–292. doi: 10.1111/j.1749-6632.1989.tb12485.x. [DOI] [PubMed] [Google Scholar]
- 45.Kosaka T, Katsamaru H, Kawaguchi Y, Hama K, Wu JY, Heizmann CW. GABAergic neurons containing the Ca2+-binding protein parvalbumin in the rat hippocampus and dentate gyrus. Brain Res. 1987;419:119–130. doi: 10.1016/0006-8993(87)90575-0. [DOI] [PubMed] [Google Scholar]
- 46.Lewis DA, Lund JS. Heterogeneity of chandelier neurons in monkey neocortex: corticotropin-releasing factor and parvalbumin-immunoreactive populations. J Comp Neurol. 1990;293:599–615. doi: 10.1002/cne.902930406. [DOI] [PubMed] [Google Scholar]
- 47.Lorente de Nó R. Studies on the structure of the cerebral cortex. J Psychol Neurol. 1933;45:381–438. [Google Scholar]
- 48.Lowe SL, Bowen DM, Francis PT, Neary D. Ante mortem cerebral amino acid concentrations indicate selective degeneration of glutamate-enriched neurons in Alzheimer’s disease. Neuroscience. 1990;38:571–577. doi: 10.1016/0306-4522(90)90051-5. [DOI] [PubMed] [Google Scholar]
- 49.Masliah E, Mallory M, DeTeresa R, Alford M, Hansen L. Differing patterns of aberrant neuronal sprouting in Alzheimer’s disease with and without Lewy bodies. Brain Res. 1993;617:258–266. doi: 10.1016/0006-8993(93)91093-8. [DOI] [PubMed] [Google Scholar]
- 50.Mattson MP. Calcium and neuronal injury in Alzheimer’s disease: contributions of beta-amyloid precursor protein mismetabolism, free radicals and metabolic compromise. Ann NY Acad Sci. 1994;747:50–76. [PubMed] [Google Scholar]
- 51.McGeer PL, Kawamata T, Walker DG, Akiyama H, Tooyama I, McGeer EG. Microglia in degenerative neurological disease. Glia. 1993;7:84–92. doi: 10.1002/glia.440070114. [DOI] [PubMed] [Google Scholar]
- 52.Mirra SS, Heyman A, McKeel D, Sumi SM, Crain BJ, Brownlee LM, Vogel FS, Hughes JP, van Belle G, Berg L. The consortium to establish a registry for Alzheimer’s disease (CERAD). II. Standardization of the neuropathologic assessment of Alzheimer’s disease. Neurology. 1991;41:479–486. doi: 10.1212/wnl.41.4.479. [DOI] [PubMed] [Google Scholar]
- 53.Mizutani T, Shimada H. Neuropathological background of twenty-seven centenarian brains. J Neurol Sci. 1992;108:168–177. doi: 10.1016/0022-510x(92)90047-o. [DOI] [PubMed] [Google Scholar]
- 54.Morrison JH, Lewis DA, Campbell MJ, Huntley GW, Benson DL, Bouras C. A monoclonal antibody to non-phosphorylated neurofilament protein marks the vulnerable cortical neurons in Alzheimer’s disease. Brain Res. 1987;416:331–336. doi: 10.1016/0006-8993(87)90914-0. [DOI] [PubMed] [Google Scholar]
- 55.Mufson EJ, Brandabur MM. Sparing of NADPH-diaphorase striatal neurons in Parkinson’s and Alzheimer’s diseases. NeuroReport. 1994;5:705–708. doi: 10.1097/00001756-199402000-00011. [DOI] [PubMed] [Google Scholar]
- 56.Nitsch R, Frotscher M. Transneuronal changes in dendrites of GABAergic parvalbumin-containing neurons of the rat fascia dentata following entorhinal lesion. Hippocampus. 1993;3:481–490. doi: 10.1002/hipo.450030409. [DOI] [PubMed] [Google Scholar]
- 57.Omidi K, Hendry SHC, Jones EG, Emson PC. Organization of plasticity of GABA neuronal subpopulation in monkey area 17, defined by differential coexistence of calcium-binding proteins. Soc Neurosci Abstr. 1988;14:780. [Google Scholar]
- 58.Palmer AM, DeKosky ST. Monoamine neurons in aging and Alzheimer’s disease. J Neural Transm [GenSect] 1993;91:135–159. doi: 10.1007/BF01245229. [DOI] [PubMed] [Google Scholar]
- 59.Perry EK, Perry RH, Blessed G, Tomlison BE. Necropsy evidence of central cholinergic deficits in senile dementia. Lancet. 1977;1:89. doi: 10.1016/s0140-6736(77)91780-9. [DOI] [PubMed] [Google Scholar]
- 60.Pike CJ, Cotman CW. Calretinin-immunoreactive neurons are resistant to β-amyloid toxicity in vitro. Brain Res. 1995;671:293–298. doi: 10.1016/0006-8993(94)01354-k. [DOI] [PubMed] [Google Scholar]
- 61.Price DL, Koliatsos VE, Sisodia SS, Koo EH, Martin LJ, Walker LC, Applegate MD, Cork LC (1991) Amyloid-related proteins and nerve growth factor in Alzheimer’s disease and animal models. Clin Neuropharmacol 14[Suppl 1]:S9–S14. [DOI] [PubMed]
- 62.Satoh J, Tabira T, Sano M, Nakayama H, Tateishi J. Parvalbumin-immunoreactive neurons in the human central nervous system are decreased in Alzheimer’s disease. Acta Neuropathol. 1991;81:388–395. doi: 10.1007/BF00293459. [DOI] [PubMed] [Google Scholar]
- 63.Schmidt-Kastner R, Meller D, Eysel UT. Immunohistochemical changes of neuronal calcium-binding proteins parvalbumin and calbindin-D-28k following unilateral differentiation in the rat visual system. Exp Neurol. 1992;117:230–246. doi: 10.1016/0014-4886(92)90132-a. [DOI] [PubMed] [Google Scholar]
- 64.Scott SA, Mufson EJ, Weingartner JA, Skau KA, Crutcher KA. Nerve growth factor in Alzheimer’s disease: increased levels throughout the brain coupled with declines in nucleus basalis. J Neurosci. 1995;15:6213–6221. doi: 10.1523/JNEUROSCI.15-09-06213.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Solodkin A, Van Hoesen GW. Calcium-binding proteins on the human parahippocampal gyrus and their alterations in Alzheimer’s disease. Soc Neurosci Abstr. 1993;19:191. [Google Scholar]
- 66.Solodkin A, Van Hoesen GW. The entorhinal cortex modules of the human brain. J Comp Neurol. 1996;365:1–18. doi: 10.1002/(SICI)1096-9861(19960219)365:4<610::AID-CNE8>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- 67.Srivastava R, Brouillet E, Beal MF, Storey E, Hyman BT. Blockade of 1-methyl-4-phenylpyridinium ion (MPP+) nigrat toxicity in the rat by prior decortication of MK-801 treatment: a stereological estimate of neuronal loss. Neurobiol Aging. 1993;14:295–301. doi: 10.1016/0197-4580(93)90114-q. [DOI] [PubMed] [Google Scholar]
- 68.Su JH, Anderson AJ, Cummings BJ, Cotman CW. Immunohistochemical evidence for apoptosis in Alzheimer’s disease. NeuroReport. 1994;5:2529–2533. doi: 10.1097/00001756-199412000-00031. [DOI] [PubMed] [Google Scholar]
- 69.Terry RD, Masliah E, Salmon DP, Butters N, De Teresa R, Hill R, Hansen LA, Katzman R. Physical basis of cognitive alterations in Alzheimer’s disease: synapse loss is the major correlate of cognitive impairment. Ann Neurol. 1991;30:572–580. doi: 10.1002/ana.410300410. [DOI] [PubMed] [Google Scholar]
- 70.Tortosa A, Ferrer I. Parvalbumin immunoreactivity in the hippocampus of the gerbil after transient forebrain ischemia: a qualitative and quantitative study. Neuroscience. 1993;55:33–43. doi: 10.1016/0306-4522(93)90452-l. [DOI] [PubMed] [Google Scholar]
- 71.Trillo L, Gonzalo LM. Ageing of the human entorhinal cortex and subicular complex. Histol Histopathol. 1993;7:17–22. [PubMed] [Google Scholar]
- 72.Tuñón T, Insausti R, Ferrer I, Sobreviela T, Soriano E. Parvalbumin and calbindin D-28K in the human entorhinal cortex: an immunohistochemical study. Brain Res. 1992;589:24–32. doi: 10.1016/0006-8993(92)91157-a. [DOI] [PubMed] [Google Scholar]
- 73.VanBrederode JF, Mulligan KA, Hendrickson AE. Calcium-binding proteins as markers for subpopulations of GABAergic neurons in monkey striate cortex. J Comp Neurol. 1990;298:1–22. doi: 10.1002/cne.902980102. [DOI] [PubMed] [Google Scholar]
- 74.Van Hoesen GW, Solodkin A. Some modular features of temporal cortex in humans as revealed by pathological changes in Alzheimer’s disease. Cereb Cortex. 1993;3:465–475. doi: 10.1093/cercor/3.5.465. [DOI] [PubMed] [Google Scholar]
- 75.Van Hoesen GW, Solodkin A. Cellular and systems neuroanatomical changes in Alzheimer’s disease. In: Disterhoft JF, Khachaturian ZS, editors. Calcium hypothesis of aging and dementia, Vol. 747. New York Academy of Science; New York: 1994. pp. 12–35. [DOI] [PubMed] [Google Scholar]
- 76.West MJ. Regionally specific loss of neurons in the human aging hippocampus. Neurobiol Aging. 1994;14:287–293. doi: 10.1016/0197-4580(93)90113-p. [DOI] [PubMed] [Google Scholar]
- 77.West MJ, Coleman PD, Flood DG, Troncoso JC. Differences in the pattern of hippocampal neuronal loss in normal ageing and Alzheimer’s disease. Lancet. 1994;344:769–772. doi: 10.1016/s0140-6736(94)92338-8. [DOI] [PubMed] [Google Scholar]
- 78.Whitehouse PJ, Price DL, Clark AW, Coyle JT, DeLong MR. Alzheimer’s disease: evidence for a selective loss of cholinergic neurons in the nucleus basalis. Ann Neurol. 1981;10:122–126. doi: 10.1002/ana.410100203. [DOI] [PubMed] [Google Scholar]
- 79.Zhan SS, Kamphorst W, Van Nostrand WE, Eikelenboom P. Distribution of neuronal growth factor-promoting factors and cytoskeletal proteins in altered neurites in Alzheimer’s disease and non-demented elderly. Acta Neuropathol. 1995;89:356–362. doi: 10.1007/BF00309629. [DOI] [PubMed] [Google Scholar]