

Abstract
The molecular events leading to motoneuronal death are still poorly understood. In mammals, the bcl-2 proto-oncogene, which encodes a membrane-associated protein, has been shown to suppress both developmental motoneuronal death and experimental axotomy-induced motoneuronal death. We assessed a potential protective effect of Bcl-2 on pathological motoneuronal death processes in adult rodents. We took advantage of the murine mutant wobbler, which undergoes progressive degeneration of the spinal and brainstem motoneurons. A hybrid carrying both the wobbler mutation and the humanbcl-2 transgene under the control of the neuron-specific enolase promoter was produced. Although Bcl-2 protected spinal and brainstem motoneurons from developmental death and the postnatal motoneurons of the facial nucleus from axotomy-induced death, the pathological motoneuronal death was not altered in the adult hybrid. These results demonstrate that Bcl-2 sensitivity distinguishes at least two different motoneuronal death pathways in the wobblermutant. They support the hypothesis that experimental and pathological motoneuronal death are dependent on different cellular mechanisms.
Keywords: wobbler, Bcl-2, developmental neuronal death, axotomy-induced cell death, excitotoxicity, neurotrophic factors
An intense effort is being made to understand the mechanisms of motoneuronal death and to identify agents with therapeutic potential in human degenerative motoneuron diseases. Most results have been obtained from two experimental models in rodents: axotomy-induced motoneuron death in neonates and naturally occurring cell death in embryos. These models have proven instrumental in demonstrating the protective effect of several factors, including ciliary neurotrophic factor (CNTF) (Sendtner et al., 1990; Oppenheim et al., 1991), brain-derived neurotrophic factor (BDNF) (Oppenheim et al., 1992; Sendtner et al., 1992; Yan et al., 1992; Koliatsos et al., 1993), and glial cell line-derived neurotrophic factor (Henderson et al., 1994; Oppenheim et al., 1995; Yan et al., 1995); however, extension of the conclusions drawn from these two models to pathological motoneuronal death, such as that observed in mutant mice, has proved difficult. For example, few beneficial effects were observed on motoneuronal survival in the wobbler mutant with CNTF or BDNF, respectively, despite an improvement in the motor function (Ikeda et al., 1995a,b). These relatively negative results raise the question of the existence of different pathways leading to either experimental or pathological motoneuronal death.
One molecule that plays a key role in interfering with mechanisms leading to motoneuronal death in experimental models is Bcl-2 (Dubois-Dauphin et al., 1994; Martinou et al., 1994; Farlie et al., 1995). Bcl-2 interferes with cell apoptosis in a wide variety of cases (for review, see Reed, 1994). There are, however, Bcl-2-insensitive pathways for cell death (Reed, 1994), in particular for some neurons (Allsopp et al., 1993). Moreover, although the neuroprotective effects of some factors are mediated by Bcl-2 (e.g., BDNF), those of others are not (e.g., CNTF) (Allsopp et al., 1995). The neuroprotective efficiency of Bcl-2 thus may represent one way to differentiate pathways for neuronal death.
Whether Bcl-2 is able to interfere with the mechanisms leading to pathological motoneuronal death is yet to be determined. In a recent paper, Sagot et al. (1995) obtained paradoxical results in theprogressive motor neuronopathy (pmn) mutant mouse overexpressing Bcl-2. Facial motoneurons appeared preserved, whereas the number of motor axons was not. The mechanisms leading to neuronal damage in the pmn mouse are poorly understood, however, and this complicates the interpretation of these complex results. Although not fully explored, the wobblermouse, which results from an inherited autosomal recessive mutation leading to the progressive degeneration of spinal and brainstem motoneurons (Duchen and Strich, 1968; La Vail et al., 1987), offers a clearer mechanistic picture. In particular, it has been suggested that the Cu/Zn superoxide dismutase (SOD), an enzyme that shares with Bcl-2 the capacity to protect cells from oxidative stress (Lipton et al., 1993; Kane et al., 1993; Hockenbery et al., 1993; Cadet et al., 1994;Sarafian et al., 1994; Greenlund et al., 1995), exerts a partial protective effect on motoneurons in the murine mutantwobbler (Ikeda et al., 1995c). It seemed appropriate, therefore, to evaluate the ability of Bcl-2 to affect pathological motoneuronal death in this mouse.
To determine whether Bcl-2 could arrest motoneuronal death inwobbler mice as it does in experimental models, we producedwobbler mutants overexpressing high levels of the human Bcl-2 protein in neurons. The results of this study support the hypothesis that experimental and pathological motoneuronal death are dependent on different cellular mechanisms.
MATERIALS AND METHODS
We previously described NZB-elicited wobbler (NEW) mice carrying the double allele wobbler(wr/wr) and allowing genetic diagnosis before the onset of the disease (Des Portes et al., 1994). We now describewobbler and transgene (WAT) mice, derived from NEW mice and carrying the human bcl-2 transgene.
Animals
Production of mice of the WAT lineage. The hybrid NEW mice came from the cross between NZB (GNZB/GNZB; +/+) and C57Bl/6J (GC57/GC57; +/wr) mice carrying thewr mutation. By means of this cross, a genetic polymorphic marker, a microsatellite of the glutamine synthetase gene (glns), was introduced close to the wrgene. The NEW (wr/wr) animals carried the alleles GC57/GC57 of glns, and wild-type animals were either heterozygotes (GC57/GNZB) or homozygotes (GNZB/GNZB) for glns. The scheme of the cross (Fig. 1) shows how the humanbcl-2 transgene was introduced first into mice of the NZB strain and then into heterozygote mice of the NEW lineage. At the third generation, a hybrid carrying the two copies of the mutated alleles ofwr, the two copies of the GC57 alleles ofglns, and one copy of the human bcl-2 transgene was obtained. The cDNA of the human bcl-2 transgene was under the control of the neuron-specific enolase promoter, so that only neuronal cells expressed the human Bcl-2 protein.
Fig. 1.

Scheme of the cross used to obtain WAT mice. Only selected mice are represented. Mice of the NZB strain were first crossed with a mouse transgenic for the human bcl-2 gene (Martinou et al., 1994). At the first generation, heterozygotes forglns (GNZB/GC57), which carry the human bcl-2 transgene, were selected by PCR (dotted frame) and crossed with mice of the NZB-elicitedwobbler (NEW) colony, heterozygotes for glns(GNZB/GC57) and for wr(+/wr). At the second generation, two types of transgenic mice heterozygous for glns were selected by PCR. One carried a mutated allele of wr (dotted frame), and the other did not (not represented). To identify the one that carried the mutated allele, a backcross with a heterozygous mouse of the NEW colony was necessary. The individual carrying the wr allele was selected ifwobbler mice with evident clinical signs of the disease were present in its progeny. At the third generation, all mice heterozygous for glns were heterozygotes forwr (not represented). Heterozygotes transgenic forbcl-2 were used to perpetuate the colony. All mice homozygous for the allele GC57 of glnscarried the double allele of the wr mutation. WAT mice are represented in the dark frame.
DNA analysis. DNA from tails of 4-d-old mice was obtained using the Genereleaser assay (BioVentures, Murfreesboro, TN). Alleles of both glns and the human bcl-2 transgene were analyzed through a double PCR amplification. Primers amplifying theglns microsatellite and those detecting the presence of the human bcl-2 transgene are described in Table1.
Table 1.
Primer sequences used for double amplification by PCR
| glnsprobe | |
| Forward: | 5′-AGC TTT GGA GAC AAC AAT TAG ATC-3′ |
| Reverse: | 5′-GGA TGG GGA AAT GGT GGT ACA-3′ |
| Humanbcl-2probe | |
| Forward: | 5′-ATG AGC CTT GGG ACT GTG AA-3′ |
| Reverse: | 5′-GAA GAC TCT GCT CAG TTT GG-3′ |
According to the results of the double PCR analysis, we defined four distinct groups of animals on which all experiments were performed (Table 2): (1) wild type (GNZB/?;bcl-2 −); (2) wild type–transgenic (GNZB/?;bcl-2 +); (3) wobbler(GC57/GC57; bcl-2 −); and (4) WAT (GC57/GC57; bcl-2 +)
Table 2.
Groups of animals on which all experiments were performed
| Group | Name | wralleles | glns alleles | Presence of the humanbcl-2transgene |
|---|---|---|---|---|
| 1 | Wild type | +/+ or +/wr | GNZB/GNZB or GNZB/GC57 | − |
| 2 | Wild type transgenic | +/+ or +/wr | GNZB/GNZB or GNZB/GC57 | + |
| 3 | wobbler | wr/wr | GC57/GC57 | − |
| 4 | WAT | wr/wr | GC57/GC57 | + |
Facial nerve transection. Two-day-old mice from the same litter were anesthetized by hypothermia and subjected to a unilateral transection of the right facial nerve. The small branch that innervates the caudal auricular muscle and corresponds to motoneurons in the ventromedial part of the facial nucleus was not sectioned. Mutants and wild-type mice could not be distinguished either clinically or histologically at this age, because pathological degeneration of motoneurons in wobbler mice begins in the third week of life; therefore, they were identified by genetic analysis. Animals were killed 7 d after nerve section. Three pups of each group were anesthetized with sodium pentobarbitone (50 mg/kg) and perfused transcardially with 20 ml of PBS (0.1 m), pH 7.4, followed by 20 ml of 4% (w/v) paraformaldehyde in PBS (0.1 m). The brainstem of each pup was removed, postfixed for 4 hr in the same fixative, and cryoprotected in 20% (w/v) sucrose in PBS at 4°C for 12 hr. Serial coronal 20-μm-thick sections were cut, recovered onto gelatinized slides, and stained with cresyl violet. All sections were observed at the facial nucleus level, and motoneurons with clearly defined nuclei and nucleoli were counted on each section, on both the lesioned and the unlesioned sides. No correction was applied for split nucleoli, because the size of the nucleoli did not vary in the different groups and was very small by comparison with the thickness of the section. No more than one nucleolus was observed per motoneuron. Three animals were studied for each of the four groups described previously. The unpaired Student’s t test was used for statistical analysis.
Grip-strength test. Grip strength was measured by placing mice on a vertical grid and grading as follows. 0, Animals grip but fall in <5 sec; 1, animals are able to grip but not climb; and 2, animals grip and climb to the top of the grid.
Histological procedures
Immunostaining of human Bcl-2 protein. In each of the four groups, four animals (one 2-d-old and three 5-week-old mice) were treated for immunofluorescence detection of the human Bcl-2 protein. Mice received 50 mg/kg sodium pentobarbitone and were perfused transcardially as described above. Brainstems and cervical spinal cords were dissected out immediately, postfixed for 4 hr at 4°C in the same fixative, and cryoprotected with 20% sucrose in PBS. Twenty-micrometer-thick sections were cut on a cryostat and put on gelatin-coated slides. Sections were permeabilized by soaking in −20°C cooled acetone for 5 min, rinsed in PBS, and incubated in BSA (3% in PBS) for 1 hr at room temperature to block nonspecific sites. Sections were then incubated for 2 hr at room temperature with a mouse monoclonal antibody specific for the human Bcl-2 protein (1:80 dilution; Cambridge Research Biochemicals, Cheshire, UK), followed by a 90 min incubation with a fluorescein isothiocyanate-conjugated antibody (F(ab′)2 fragment) raised against mouse IgG (1:200 dilution, Sigma F-8646; Sigma, St. Louis, MO).
Quantification of facial motoneurons in 5-week-oldwobbler mice. Five-week-old WAT and wobblernontransgenic mice were perfused transcardially with 80 ml PBS (0.1 m), pH 7.4, followed by 80 ml of 4% (w/v) paraformaldehyde. Brainstems were removed, and a protocol similar to the one described above was followed. Motoneurons of one facial nucleus per animal that displayed no morphological sign of the disease were counted. In addition, vacuolized motoneurons were quantified. Three animals were analyzed in the wobbler nontransgenic group, and four animals were analyzed in the WAT group. The unpaired Student’s t test was used for statistical analysis.
Quantification of axons in the brachial musculocutaneous nerve. In each of the four groups, five mice (5 week old) were used to quantify axons in the brachial musculocutaneous nerve. Nerves were fixed in situ for 5 min with 2.5% glutaraldehyde in PBS (0.1 m) and dissected out. The main branch of the brachial musculocutaneous nerves was taken just before its division into two branches; extreme precautions were taken to remove the same portion of the nerve in each case. After removal, nerves were fixed in 2.5% glutaraldehyde for 1 hr at 4°C and conserved in 0.6% glutaraldehyde. Before and after postfixation in 2% osmium tetroxide for 1 hr at room temperature, nerves were rinsed three times in a wash solution containing 8% glucose in phosphate buffer (0.1 m), pH 7.2. Nerves were dehydrated in graded ethanol solutions and embedded in Embed 812 Epoxy resin (TAAB) for 24 hr at 55°C. One-micrometer-thick semithin sections were collected, and myelinated axons were stained with 1% toluidine blue. Axons were pointed manually with use of a camera lucida and counted. To establish the reliability of the methodology, pilot experiments were performed in wild-type animals to determine that the number of axons on different sections of the same portion of nerve was identical and that the number of axons was similar in sections of different wild-type animals. Axons of one section per nerve were counted. The unpaired Student’st test was used for statistical analysis.
RESULTS
Four generations of mice were produced, and animals from the third and fourth generations (30 litters) were analyzed. A large number ofwobbler mice (transgenic or not) died between the fourth and sixth week after birth, as already described for mutants of the NEW strain (Des Portes et al., 1994). This high death rate justified the need for a large production of mice so that a significant number of 5-week-old animals could be analyzed.
Genetic analysis
Gel electrophoresis showing the products of the double PCR amplification is shown in Figure 2. It revealed one to three bands. The 180 base pair (bp) band and the 200 bp band corresponded to the GC57 and GNZB alleles, respectively, of glns. The 400 bp band was specific for the human bcl-2 transgene. The presence of the single 180 bp band was characteristic of homozygotes for thewr/wr mutation, whereas the presence of the single 200 bp band characterized wild-type homozygotes (+/+). The two co-amplified bands of 180 bp and 200 bp defined heterozygotes (+/wr). Because wild-type homozygotes and heterozygotes presented the same normal clinical characteristics (the mutation is recessive), they have been grouped together and defined as wild-type. The presence or the absence of the 400 bp band allowed separation of transgenic from nontransgenic mice. On the basis of these results, animals were divided into four groups (Table 2): (1) wild type (GNZB/?; +/?; bcl-2 −); (2) wild type–transgenic (GNZB/?; +/?; bcl-2 +); (3)wobbler (GC57/GC57;wr/wr; bcl-2 −); and (4) WAT (GC57/GC57; wr/wr;bcl-2 +).
Fig. 2.

Example of electrophoresis on a 2% agarose gel of the double PCR amplification products used for the genetic identification of animals at the third generation. M, Molecular weight markers. Lane 1, control; lanes 2, 4, 7, 10, wild-type mice (group 1). Note one 200 bp band or two 180/200 bp bands representative of the GNZB and GC57/GNZB alleles of glns, respectively. Lanes 5, 9, wild-type mice (group 2). Theglns bands are the same as described previously. A band at 400 bp shows the presence of the human bcl-2transgene. Lane 8, wobbler mice (group 3). The single 180 bp band is characteristic of the GC57allele of glns and therefore of homozygotes for thewobbler mutation. Lanes 3,6, WAT mice (group 4) characterized by a 180 bp and a 400 bp band.
Functional expression of the human bcl-2 transgene
The human Bcl-2 protein was detected by immunofluorescence in both the ventral and dorsal horn neurons of 5-week-old transgenic mice carrying, or not carrying, the wobbler mutation (Fig.3). No signal was observed in neurons of nontransgenic animals (inset in Fig. 3). The human Bcl-2 immunostaining was observed as early as postnatal day 2, as tested in the brainstem of transgenic mice (data not shown).
Fig. 3.

Human Bcl-2 immunostaining in the cervical spinal cord of 5-week-old wild-type animals. Note the strong immunostaining in large neurons of the ventral horn of transgenic mice.Inset, No immunostaining was present in nontransgenic mice. This photomicrograph was taken at high magnification because the field is completely dark at low magnification. Scale bars: 76 μm in the figure; 53 μm in the inset.
The functionality of the human Bcl-2 protein was assayed by evaluating its effect on neonatal axotomy-induced facial motoneuron death. The examination of facial nuclei of nontransgenic mice (wild type andwobbler) 7 d after transection of the nerve revealed a dramatic reduction in the number of cells (Figs.4C,E). Seventy-five percent of facial motoneurons were lost (Fig. 5). The number of motoneurons was strongly affected, except in the ventromedial part of the nucleus (dark arrowhead), which corresponds to the nonsectioned caudal auricular branch of the facial nerve. The 25% of cells remaining corresponded mainly to the motoneurons in this ventromedial region. In contrast, in wild type-transgenic and WAT mice, the number of motoneurons on the lesioned side was similar to that on the unlesioned side (Figs. 4B,D,F, 5). The facial nucleus on the side of the lesion, however, presented a dense aspect and a reduced size as compared with the intact nucleus (Fig.4B,D,F), apparently resulting from a reduced size of the soma of the motoneurons.
Fig. 4.
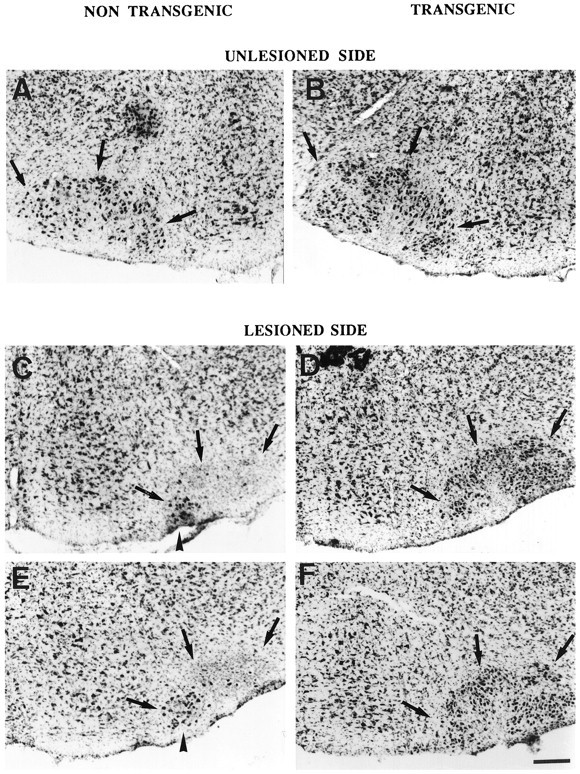
Photomicrographs showing the facial nuclei (dark arrows) in representative 9-d-old animals of each of the four groups after transection of the right facial nerve. Unlesioned side: (A) wild type, (B) wild type–transgenic. Lesioned side: (C) wild type, (D) wild type–transgenic, (E)wobbler, (F) WAT. The black arrowhead points to the ventromedial facial motoneurons, the axons of which are preserved by the transection. Scale bar, 180 μm.
Fig. 5.
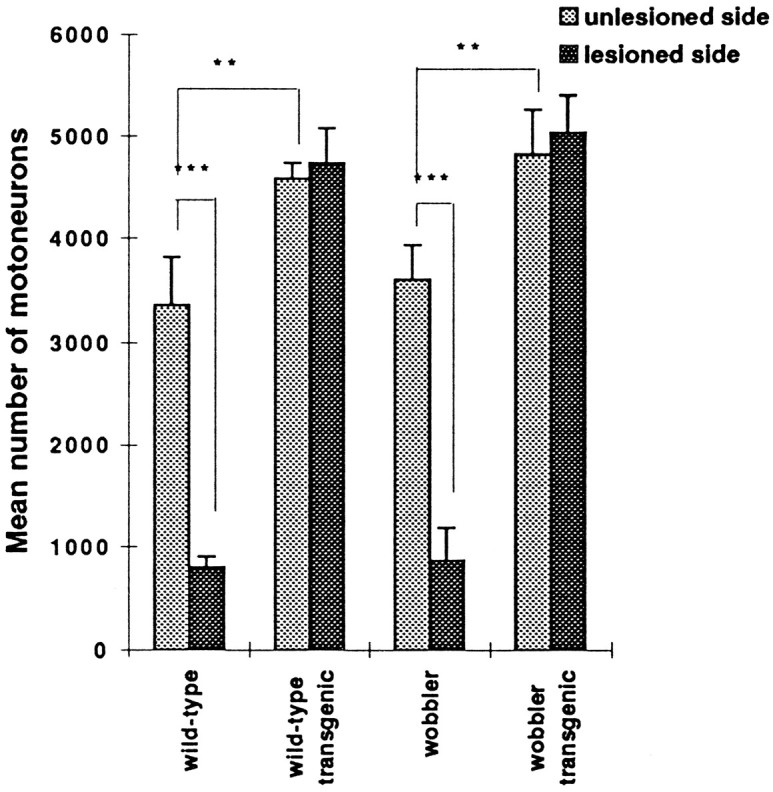
Quantification of facial motoneurons on the unlesioned and lesioned sides after nerve transection in the four genetically differentiated groups. Data are the mean ± SD of three 9-d-old animals in each group. ***p < 0.001; **p < 0.02; Student’s t test analysis.
The quantification of motoneurons also revealed an increased number of cells on the unlesioned side in transgenic mice as compared with nontransgenic mice (Figs. 4A,B, 5).
Phenotypic differences among mice of the four groups
Clinical characteristics
Transgenic individuals could not be recognized by clinical examination, because Bcl-2 overexpression did not modify their behavior compared with that of paired nontransgenic mice. Wild-type–transgenic mice bred normally. In the wobbler mutants, the first clinical signs of the disease appeared 3–4 weeks after birth. The mutants were characterized by a reduced body weight, an unsteadiness, and a diminished muscular strength. The age of onset of the disease and the time course of its evolution seemed to be similar in WAT as compared with wobbler nontransgenic mice. When they were weighed (Fig. 6) or assayed for muscular strength, the two groups of 5-week-old animals were indistinguishable. Allwobbler animals were graded 0 (fall in <5 sec), whereas all non-wobbler animals were graded 2 (climb to the top) in the grip-strength test.
Fig. 6.

Comparison of the weight of three 5-week-old animals in each of the four groups. ****p < 0.0001; ***p < 0.001.
Histological characteristics
Histological analysis of the facial nucleus and of the cervical spinal cord of 5-week-old animals revealed that overexpression of the human bcl-2 gene did not alter the morphology of the motoneurons, as revealed by cresyl violet staining (data not shown). Five-week-old WAT mice displayed histological characteristics identical to those of wobbler nontransgenic mice. Numerous motoneurons, although expressing the human Bcl-2 protein, were filled with vacuoles, a landmark of the wobbler disease (Fig.7), and a strong reactive astrogliosis was present throughout the gray matter, as revealed by glial fibrillary acidic protein immunoreactivity (data not shown).
Fig. 7.

Vacuolized motoneurons in the cervical spinal cord of 5-week-old WAT mice. (A) Human Bcl-2 immunostaining. Note that motoneurons exhibiting numerous vacuoles (arrows) show a positive immunostaining for the human Bcl-2 protein. (B) Cresyl violet staining showing vacuolized motoneuron (arrows) surrounded by glial cells. Scale bar, 20 μm.
As shown in table 3, the same proportion of motoneurons was lost or exhibited morphological signs of the disease in 5-week-oldwobbler mice in the nontransgenic and transgenic groups. A loss of 15% and 13%, respectively, of motoneurons was observed, as compared with the number of cells present before the onset of the degenerative process.
Table 3.
Quantification of facial motoneurons in wobblertransgenic and wobbler nontransgenic mice at a time that precedes (9-d-old) and during (5-week-old) the course of the disease
| wobblernontransgenic | wobblertransgenic | |||
|---|---|---|---|---|
| 9-d-old | 5-week-old | 9-d-old | 5-week-old | |
| Number of facial motoneurons | 3607 ± 188 | 3073 ± 69 | 4814 ± 252 | 4171 ± 11 |
| Number of vacuolized motoneurons | 111 ± 34 | 71 ± 5 | ||
Data are the mean counts of motoneurons displaying any intact, or else affected (vacuolized), morphology in three animals in thewobbler nontransgenic group and four animals in thewobbler transgenic group (±SEM). Comparison was made between 5-week-old and 9-d-old mice using the Student’s ttest: p = 0.05 in the wobbler nontransgenic group; p = 0.03 in the wobbler transgenic group. The number of vacuolized motoneurons was not statistically different between wobbler nontransgenic and wobblertransgenic mice.
Examination of sections of the musculocutaneous nerve showed a loss of myelinated axons, mainly large fibers, in the two groups ofwobbler mice, compared with the two groups of wild-type mice (Fig. 8). This loss was 23% in nontransgenic mice and 17% in transgenic mice (Fig. 9). The transgenic mice always possessed more axons than the nontransgenic: 832 ± 44 for wild-type–transgenic versus 709 ± 33 for wild-type nontransgenic and 696 ± 63 for WAT versus 551 ± 27 for wobblernontransgenic mice.
Fig. 8.
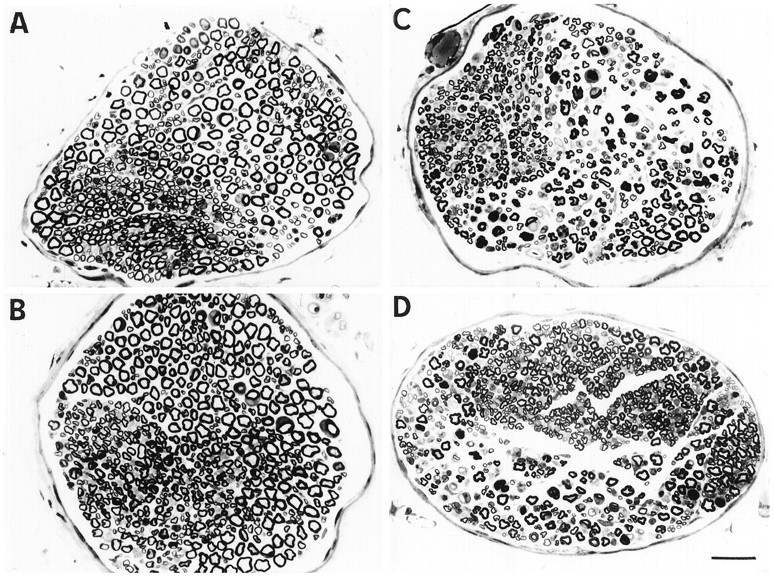
Photomicrographs showing semithin sections of the musculocutaneous nerve in representative 5-week-old animals of each group. (A) Wild-type mice; (B) wild-type–transgenic mice; (C) wobblermice; (D) WAT mice. Scale bar, 25 μm.
Fig. 9.

Quantification of myelinated axons in musculocutaneous nerves of 5-week-old animals of the four genetically differentiated groups. Results are the mean ± SD of five distinct animals in each group. ***p < 0.001; **p < 0.01; Student’s t test analysis.
DISCUSSION
To assess the ability of the Bcl-2 protein to interfere with motoneuronal pathology in the wobbler mutant,wobbler mice overexpressing the human bcl-2transgene were generated by specific crossing and compared with relevant controls. Our results show that Bcl-2 neither arrests nor delays the pathological degeneration of motoneurons, although it protects these same cells against both naturally occurring cell death and degeneration induced by neonatal axotomy. These results favor the existence of a Bcl-2-insensitive pathway leading to pathological motoneuronal death in the wobbler mutant, and they point to major mechanistic differences between this pathological death and death induced experimentally by axotomy; therefore, caution should be exercised when extrapolation to the former is on the basis of results obtained with the latter.
Pathological motoneuronal death in the wobbler mutant is Bcl-2-insensitive
It has been shown recently that the administration of the Cu/Zn SOD, an antioxidant enzyme, to the wobbler mouse slows the degeneration of its motoneurons (Ikeda et al., 1995c). The fact that Bcl-2 shares with SOD the capacity to protect cells from an oxidative stress (Hockenbery et al., 1993; Kane et al., 1993; Lipton et al., 1993; Cadet et al., 1994; Sarafian et al., 1994; Greenlund et al., 1995), and thus seems to act along the same pathway of neuroprotection, suggested that Bcl-2 might also rescue motoneurons of thewobbler mutant. The results here show thatwobbler mice overexpressing the human bcl-2 gene present clinical and histological characteristics identical to those ofwobbler nontransgenic mice, including age of onset of the disease, time course of its evolution, vacuolar degeneration of motoneurons, loss of motoneurons and of myelinated axons, and astrogliosis. Thus, the pathological motoneuronal death in WAT mice is neither stopped nor delayed.
One could assert that this lack of neuroprotection was attributable to the fact that the introduction of the human bcl-2 transgene into wobbler mice did not lead to the expression of the transgene in relevant motoneurons nor to the synthesis of an efficient Bcl-2 protein. Two explanations argue against this hypothesis, however, and support the validation of our experimental model.
First, the human Bcl-2 protein was immunodetected in facial motoneurons as early as 2 d after birth, a time that clearly precedes the appearance of degenerative motoneurons in the third week of life. Moreover, it was also detected in vacuolized motoneurons of 5-week-old WAT mice, showing that even degenerative motoneurons synthesize the human Bcl-2 protein.
Second, we demonstrated the functionality of the human Bcl-2 protein in transgenic mice of the WAT lineage using the same experimental models of motoneuronal death as those used to demonstrate its functionality in the original strain of transgenic mice, namely axotomy-induced cell death (Dubois-Dauphin et al., 1994) and developmental cell death (Martinou et al., 1994).
Altogether, these results demonstrate that the human Bcl-2 protein synthesized in transgenic mice of the WAT lineage is functional. It is possible, however, that this functionality is limited to motoneurons in the perinatal period. Indeed, one major difference between models used to demonstrate the functionality of the Bcl-2 protein and thewobbler model is the age of mice at the time of the occurrence of motoneuronal degeneration. It might be argued that factors necessary for Bcl-2 protection are present only at specific times during development and are not present or not present in sufficient amounts in the adult. This seems unlikely, because cortical neurons in adult mice have been shown to be protected by Bcl-2 from experimental ischemia-induced death (Martinou et al., 1994). To go one step further, motoneurons might specifically lose Bcl-2 sensitivity in the adult. Again, this hypothesis does not seem warranted, because Bcl-2 overexpression protects adult motoneurons in pmn mice (Sagot et al., 1995).
These observations indicate that adult motoneurons, as is the case for motoneurons during the perinatal period, possess all of the metabolic machinery necessary for the action of Bcl-2. Thus, our results argue in favor of the existence of a Bcl-2-insensitive pathological death pathway in the wobbler mutant.
Elements of the excitotoxic cascade, such as glutamate (Behl et al., 1993; Zhong et al., 1993a), reactive oxygen species (Hockenbery et al., 1993; Kane et al., 1993), and calcium influx (Miyashita and Reed, 1992;Zhong et al., 1993b) lead to a Bcl-2-sensitive death. The existence of a Bcl-2-insensitive mechanism for motoneuronal death in thewobbler mouse suggests that excitotoxicity is not implicated in the wobbler pathology. This conclusion is in agreement with that of Krieger et al. (1992), who showed that application of an NMDA receptor antagonist does not improve the wobblermutant.
Existence of different pathways that lead to motoneuronal death
Our results show that motoneurons in the wobbler mouse can die by taking two different pathways. One is Bcl-2-sensitive, exemplified by axotomy-induced cell death and developmental cell death, and the other is Bcl-2-insensitive, exemplified by pathological death. The demonstration of the existence of different pathways leading to motoneuronal death suggests that motoneurons could respond differently to therapeutic agents depending on the specific mechanisms involved.
Evidence for the existence of different pathways leading to neuronal death has been provided by Allsopp et al. (1993), who showed that the same cells, neurons of the trigeminal mesencephalic nucleus, placed under different experimental conditions could go down either a Bcl-2-sensitive or a Bcl-2-insensitive death pathway. Martinou et al. (1994) also showed the existence of two motoneuronal death pathways during developmental cell death, because a proportion of motoneurons were unprotected by Bcl-2.
The demonstration that axotomy-induced cell death, to date the most frequently used to evaluate the effect of therapeutic agents on the survival of motoneurons, and pathological death in thewobbler mutant result from different mechanisms calls for caution to avoid overgeneralization of results obtained with the first model and application of those results to a pathological model such aswobbler or in general to other experimental or pathological motoneuronal degeneration.
In line with this idea, it is interesting to note that BDNF, whose neuroprotective effects seem to be mediated by Bcl-2 (Allsopp et al., 1995), was protective for axotomized motoneurons but did not prove efficient for the survival of wobbler motoneurons. Inwobbler, BDNF treatment slowed the rate of axonal depletion but did not prevent the death of anterior horn cells (Ikeda et al., 1995b). Because neurotrophin 4/5 (NT4/5) shares with BDNF the common trkB receptor (Soppet et al., 1991) and the same survival effect on motoneurons in vitro (Henderson et al., 1993) and in vivo (Koliatsos et al., 1994), we postulated that NT-4/5 would not protect motoneurons of the wobbler mutant. This seemed to be the case, because in preliminary studies administered NT4/5 did not protect motoneurons in wobbler mice (our unpublished observations).
The recent demonstration showing that Bcl-2 is able to protect the cell bodies of motoneurons but not the axons in pmn mice (Sagot et al., 1995) is yet another type of effect of Bcl-2 on motoneurons that differs from both that observed after axotomy and that observed inwobbler. This adds to the caution that must be taken before results obtained in one model are generalized and applied to another model and, even more so, before they are applied to diseases.
In conclusion, our data underline the fact that in vivo asin vitro, two pathways for neuronal death can be distinguished on the basis of their sensitivity to Bcl-2. This mechanistic difference, which is probably one of many, clearly calls for caution when the results obtained in a particular model are extrapolated to another.
Footnotes
This work was supported by grants from the Institut National de la Santé et de la Recherche Médicale and the Association Française contre les Myopathies (AFM) (M.P., P.A.D.) and a fellowship from AFM (M.C.). We are greatly indebted to Dr. J. C. Martinou for providing Bcl-2 transgenic mice. We thank E. Parrish for revision of this manuscript.
Correspondence should be addressed to Patrick A. Dreyfus, Institut National de la Santé et de la Recherche Médicale U421, IM3, 8 Rue du Général Sarrail, 94010 Créteil Cedex, France.
REFERENCES
- 1.Allsopp T, Wyatt S, Paterson H, Davies A. The proto-oncogene bcl-2 can selectively rescue neurotrophic factor-dependent neurons from apoptosis. Cell. 1993;73:295–307. doi: 10.1016/0092-8674(93)90230-n. [DOI] [PubMed] [Google Scholar]
- 2.Allsopp T, Kiselev S, Wyatt S, Davies A. Role of Bcl-2 in the brain-derived neurotrophic factor survival response. Eur J Neurosci. 1995;7:1266–1272. doi: 10.1111/j.1460-9568.1995.tb01116.x. [DOI] [PubMed] [Google Scholar]
- 3.Behl C, Hovey L, III, Krajewski S, Schubert D, Reed JC. BCL-2 prevents killing of neuronal cells by glutamate but not by amyloid beta protein. Biochem Biophys Res Commun. 1993;197:949–956. doi: 10.1006/bbrc.1993.2571. [DOI] [PubMed] [Google Scholar]
- 4.Cadet JL, Sheng P, Ali S, Rothman R, Carlson E, Epstein C. Attenuation of metamphetamine-induced neurotoxicity in copper/zinc superoxide dismutase transgenic mice. J Neurochem. 1994;62:380–383. doi: 10.1046/j.1471-4159.1994.62010380.x. [DOI] [PubMed] [Google Scholar]
- 5.Des Portes V, Coulpier M, Melki J, Dreyfus P. Early detection of mouse wobbler mutation: a model of pathological motoneurone death. NeuroReport. 1994;5:1861–1864. doi: 10.1097/00001756-199410000-00005. [DOI] [PubMed] [Google Scholar]
- 6.Dubois-Dauphin M, Frankowski H, Tsujimoto Y, Huarte J, Martinou JC. Neonatal motoneurons overexpressing the Bcl-2 protooncogene in transgenic mice are protected from axotomy-induced cell death. Proc Natl Acad Sci USA. 1994;91:2459–2463. doi: 10.1073/pnas.91.8.3309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Duchen LW, Strich SJ. An hereditary motor neuron disease with progressive denervation of muscle in the mouse: the mutant “wobbler”. J Neurol Neurosurg Psychiatry. 1968;31:535–542. doi: 10.1136/jnnp.31.6.535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Farlie P, Dringen R, Rees S, Kannourakis G, Bernard O. Bcl-2 transgene expression can protect neurons against developmental and induced cell death. Proc Natl Acad Sci USA. 1995;92:4397–4401. doi: 10.1073/pnas.92.10.4397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Greenlund LJS, Deckwerth TL, Johnson EM. Superoxide dismutase delays neuronal apoptosis: a role for reactive oxygen species in programmed neuronal death. Neuron. 1995;14:303–315. doi: 10.1016/0896-6273(95)90287-2. [DOI] [PubMed] [Google Scholar]
- 10.Henderson C, Camu W, Mettling C, Gouin A, Poulsen K, Karihaloo M, Rullamas J, Evans T, Mc Mahon SB, Armanini MP, Berkemeier L, Phillip HS, Rosenthal A. Neurotrophins promote motor neuron survival and are present in embryonic limb bud. Nature. 1993;363:266–270. doi: 10.1038/363266a0. [DOI] [PubMed] [Google Scholar]
- 11.Henderson CE, Phillips HS, Pollock RA, Davies AM, Lemeulle C, Armanini M, Simpson LC, Moffet B, Vandlen RA, Koliatsos VE, Rosenthal A. GDNF: a potent survival factor for motoneurons present in peripheral nerve and muscle. Science. 1994;266:1062–1064. doi: 10.1126/science.7973664. [DOI] [PubMed] [Google Scholar]
- 12.Hockenbery DM, Oltavi ZN, Yin XM, Milliman CL, Korsmeyer SJ. Bcl-2 functions in an antioxidant pathway to prevent apoptosis. Cell. 1993;75:241–251. doi: 10.1016/0092-8674(93)80066-n. [DOI] [PubMed] [Google Scholar]
- 13.Ikeda K, Wong V, Holmlund T, Green T, Cedarbaum M, Lindsay RM, Mitsumoto H. Histometric effects of ciliary neurotrophic factor in wobbler mouse motor neuron disease. Ann Neurol. 1995a;37:47–54. doi: 10.1002/ana.410370110. [DOI] [PubMed] [Google Scholar]
- 14.Ikeda K, Klinkosz B, Green T, Cedarbaum JM, Wong V, Lindsay RM, Mitsumoto H. Effects of brain-derived neurotrophic factor on motor dysfunction in wobbler mouse motor neuron disease. Ann Neurol. 1995b;37:505–511. doi: 10.1002/ana.410370413. [DOI] [PubMed] [Google Scholar]
- 15.Ikeda K, Kinoshita M, Iwasaki Y, Tagaya N, Shiojima T. Lecithinized superoxide dismutase retards wobbler mouse motoneuron disease. Neuromuscul Disord. 1995c;5:383–390. doi: 10.1016/0960-8966(95)00003-6. [DOI] [PubMed] [Google Scholar]
- 16.Kane DJ, Sarafian TA, Anton R, Hahn H, Gralla EB, Valentine JS, Ord T, Bredesen D. Bcl-2 inhibition of neural death: decreased generation of reactive oxygen species. Science. 1993;262:1274–1276. doi: 10.1126/science.8235659. [DOI] [PubMed] [Google Scholar]
- 17.Koliatsos V, Clatterbuck RE, Winslow JW, Cayouette MH, Price D. Evidence that brain-derived neurotrophic factor is a trophic factor for motor neurons in vivo. Neuron. 1993;10:359–367. doi: 10.1016/0896-6273(93)90326-m. [DOI] [PubMed] [Google Scholar]
- 18.Koliatsos V, Cayouette MH, Berkemeier LR, Clatterbuck RE, Price D, Rosenthal A. Neurotrophin 4/5 is a trophic factor for mammalian facial motor neurons. Proc Natl Acad Sci USA. 1994;91:3304–3308. doi: 10.1073/pnas.91.8.3304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Krieger C, Perry TL, Hansen S, Mitsumoto H, Honoré T. Excitatory amino acid receptor antagonist in murine motoneuron disease (the wobbler mouse). Can J Neurol Sci. 1992;19:462–465. [PubMed] [Google Scholar]
- 20.La Vail JH, Koo EH, Dekker NP. Motoneuron loss in the abducens nucleus of wobbler mice. Brain Res. 1987;404:127–132. doi: 10.1016/0006-8993(87)91363-1. [DOI] [PubMed] [Google Scholar]
- 21.Lipton SA, Choi Y-B, Pan Z-H, Sizheng ZL, Chen H-SV, Sucher NJ, Loscalzo J, Singel DJ, Stamler JS. A redox-based mechanism for the neuroprotective and the neurodestructive effects of nitric oxide and related nitroso-compounds. Nature. 1993;364:626–632. doi: 10.1038/364626a0. [DOI] [PubMed] [Google Scholar]
- 22.Martinou JC, Dubois-Dauphin M, Staple J, Rodriguez I, Frankowsky H, Missotten M, Albertini P, Talabot D, Catsicas S, Pietra C, Huarte J. Overexpression of Bcl-2 in transgenic mice protects neurons from naturally occurring cell death and experimental ischemia. Neuron. 1994;13:1017–1030. doi: 10.1016/0896-6273(94)90266-6. [DOI] [PubMed] [Google Scholar]
- 23.Miyashita T, Reed JC. bcl-2 gene transfer increases relative resistance of S49.1 and WEH17.2 lymphoid cells to cell death and DNA fragmentation induced by glucocorticoids and multiple chemotherapeutic drugs. Cancer Res. 1992;52:5407–5411. [PubMed] [Google Scholar]
- 24.Oppenheim RW, Prevette D, Yin Q-W, Collins F, Mac Donald J. Control of embryonic motoneuron survival in vivo by ciliary neurotrophic factor. Science. 1991;257:1616–1618. doi: 10.1126/science.2011743. [DOI] [PubMed] [Google Scholar]
- 25.Oppenheim RW, Yin Q-W, Prevette D, Yan Q. Brain-derived neurotrophic factor rescues developing avian motoneurons from cell death. Nature. 1992;360:755–757. doi: 10.1038/360755a0. [DOI] [PubMed] [Google Scholar]
- 26.Oppenheim RW, Houenou L, Johnson JE, Lin LH, Li L, Lo AC, Newsome AL, Prevette D, Wang S. Developing motor neurons rescued from programmed and axotomy-induced cell death by GDNF. Nature. 1995;373:344–346. doi: 10.1038/373344a0. [DOI] [PubMed] [Google Scholar]
- 27.Reed JC. Bcl-2 and the regulation of programmed cell death. J Cell Biol. 1994;124:1–6. doi: 10.1083/jcb.124.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sagot Y, Dubois-Dauphin SA, Tan SA, Debilbao F, Aebischer P, Martinou JC, Kato AC. Bcl-2 overexpression prevents motoneuron cell body loss but not axonal degeneration in a mouse model of a neurodegenerative disease. J Neurosci. 1995;15:7727–7733. doi: 10.1523/JNEUROSCI.15-11-07727.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sarafian TA, Vartavarian L, Kane DJ, Bredesen DE, Verity MA. bcl-2 expression decreases methyl mercury-induced free-radical generation and cell killing in a neural cell line. Toxicol Lett. 1994;74:149–155. doi: 10.1016/0378-4274(94)90093-0. [DOI] [PubMed] [Google Scholar]
- 30.Sendtner M, Kreutzberg GW, Thoenen H. Ciliary neurotrophic factor prevents the degeneration of motor neurons after axotomy. Nature. 1990;345:440–441. doi: 10.1038/345440a0. [DOI] [PubMed] [Google Scholar]
- 31.Sendtner M, Holtmann B, Kolbeck R, Thoenen H, Barde YA. Brain-derived neurotrophic factor prevents the death of motoneurons in newborn rats after nerve section. Nature. 1992;360:757–759. doi: 10.1038/360757a0. [DOI] [PubMed] [Google Scholar]
- 32.Soppet D, Escandon E, Maragos J, Middlemas D, Reid S, Blair J, Burton L, Stanton B, Kaplan D, Hunter T, Nikolics K, Parada L. The neurotrophic factors brain-derived neurotrophic factor and neurotrophin-3 are ligands for the trkB tyrosine kinase receptor. Cell. 1991;65:895–903. doi: 10.1016/0092-8674(91)90396-g. [DOI] [PubMed] [Google Scholar]
- 33.Yan Q, Elliot J, Snider WD. Brain-derived neurotrophic factor rescues spinal motor neurons from axotomy-induced cell death. Nature. 1992;360:753–755. doi: 10.1038/360753a0. [DOI] [PubMed] [Google Scholar]
- 34.Yan Q, Matheson C, Lopez OT. In vivo neurotrophic effects of GDNF on neonatal and adult facial motor neurons. Nature. 1995;373:341–344. doi: 10.1038/373341a0. [DOI] [PubMed] [Google Scholar]
- 35.Zhong L, Kane DJ, Bredesen DE. Bcl-2 blocks glutamate toxicity in neural cell lines. Brain Res. 1993a;19:353–355. doi: 10.1016/0169-328x(93)90139-g. [DOI] [PubMed] [Google Scholar]
- 36.Zhong L, Sarafian T, Kane DJ, Charles AC, Mah SP, Edwards RH, Bredesen DE. Bcl-2 inhibits death of central neural cells induced by multiple agents. Proc Natl Acad Sci USA. 1993b;90:4533–4537. doi: 10.1073/pnas.90.10.4533. [DOI] [PMC free article] [PubMed] [Google Scholar]