

Abstract
We have studied abnormalities in the tangential and radial expansion of the cerebral cortex during fetal development in the trisomy 16 (Ts16) mouse, a model for human trisomy 21 (Down syndrome). Slowed tangential expansion of the neuroepithelium in Ts16 resulted in a reduction of final telencephalic size and is predicted to decrease the number of radial cortical units in the mature brain. In addition, radial growth of the Ts16 cortex was delayed at the time of peak cortical neurogenesis in normal mice, but by embryonic day 18 the cortex reached normal thickness. Because mouse chromosome 16 shares many genes with human chromosome 21, abnormalities in Ts16 brain development may parallel abnormalities in trisomy 21.
Keywords: cerebral cortex, development, histogenesis, Down syndrome, trisomy 16, mental retardation
Human trisomy 21 [Down syndrome (DS)], the most common genetic form of mental retardation, occurs in ∼1 of every 700 live births (Hook, 1978; Adams et al., 1981). The causes of mental retardation in DS remain unknown, although certain neuroanatomical changes have been found in individuals with Down syndrome. Although the cerebral cortex in DS brains is of normal thickness after cortical histogenesis has ended at 18 weeks of gestation, abnormal cortical stratification is observed as early as midgestation or 22 weeksin utero (Schmidt-Sidor et al., 1990). Postnatal histopathology in DS includes anomalies in cellular differentiation (Wisniewski et al., 1984; Golden and Hyman, 1994), hypocellular neocortex (Crome and Stern, 1972; Ross et al., 1984; Wisniewski et al., 1986; Wisniewski, 1990), and abnormal lamination of the cortex (Colon, 1972; Becker, 1991; Golden and Hyman, 1994). DS brains also exhibit abnormal convolutions, namely, shallow primary sulci and absent or missing secondary sulci (Davidoff, 1928; Crome and Stern, 1972), consistent with a decrease in cortical surface area. In addition, cellular proliferation rates are decreased in DS (Mittwoch, 1967; Segal and McCoy, 1974). The relationship, if any, between early fetal DS brain development and later postnatal abnormalities is not known.
Many studies have reported the time of onset and scope of the various postnatal neurological symptoms seen in DS, but because of the scarcity of early fetal brain tissue and the inevitable morphogenetic differences between genetically unrelated individuals of the same genotype, few studies have addressed early cortical development in fetal DS and matched normal brains. An animal model of DS with abnormal cortical development would provide the potential for controlled experimental manipulation and investigation of DS, and could also elucidate mechanisms of normal brain development.
Mouse chromosome 16 (MMU16) is partially syntenic with human chromosome 21 (HSA21) (Reeves et al., 1986; Holtzman and Epstein, 1992). Most of distal HSA21, the region necessary for the DS phenotype, also is on MMU16. Therefore, the trisomy 16 (Ts16) mouse may be a genetic model of DS. In support of this idea, mice trisomic for MMU16 share many phenotypic similarities with human trisomy 21 (Ts21) fetuses. Like Ts21 individuals, Ts16 mice exhibit growth retardation (Grausz et al., 1991;Lacey-Casem and Oster-Granite, 1994), craniofacial and liver abnormalities, immunological defects, and congenital heart defects (Gearhart et al., 1986). In vivo and in vitrostudies (Sweeney et al., 1987; Kiss et al., 1989) have shown that Ts16 brains also exhibit a decrease in the numbers of basal forebrain cholinergic neurons, deficits in choline uptake and acetylcholine release, and decreased activity of choline acetyltransferase. Additional studies have shown higher rates of cell death (Bambrick et al., 1995) and altered electrophysiological properties (Galdzicki et al., 1993) in cultured Ts16 neurons. Unfortunately, Ts16 animals do not survive until birth and, therefore, must be studied during the embryonic and fetal periods.
Histogenesis of the murine cerebral cortex occurs from embryonic day 10 (E10) to E17. It is during the first half of this period that the proliferative neuroepithelium, or ventricular zone (VZ), expands tangentially, producing increased surface area of the telencephalic vesicle (Caviness et al., 1995). This expansion determines the number of cells and ultimately the size of the mature brain. Subsequently, the VZ gives rise to the successive cellular layers of the cerebral cortex as postmitotic neurons migrate radially, outward toward the pia (Rakic, 1972) (for review, see McConnell, 1988; Volpe, 1995). As cells migrate, they form two developmental compartments, the cortical plate (CP) and intermediate zone (IZ), which later become the laminated cortical gray matter and white matter. This relatively short developmental period (which corresponds to gestational weeks 7–18 in the human) (Marin-Padilla, 1988) is ideal for examining critical developmental processes in the mouse and consequences of the genetic abnormalities in Ts16 for these processes.
In the present study, the morphological development of the cerebral cortex in Ts16 was characterized by cross-sectional area, thickness, and cell density measurements of the pallium and the developing cortical layers from E13 to E18. Gross brain size was measured, and the morphogenesis of cortical layers in trisomic mice was compared with that from normal, euploid littermates. The data reveal two discrete defects in Ts16 brain development. Slower neuroblast proliferation is suggested as an explanation for both of these defects.
MATERIALS AND METHODS
Ts16 fetuses were generated by mating males doubly heterozygous for the appropriate Robertsonian chromosome translocation [Rb(6,16)24 LuB × Rb(16,17)7 BNRF1] to C57BL/6J female mice (The Jackson Laboratories, Bar Harbor, ME) as described previously (Gropp et al., 1974). Mating couples were paired at 5 P.M. and separated at 9 A.M. the next morning. The day of separation was designated as E1. Pregnant dams were killed by cervical dislocation, and fetuses were removed, chilled, and decapitated immediately. Fetal heads were immersed in 4% paraformaldehyde/PBS fixative overnight at 4°C, and corresponding fetal livers were used for karyotype analysis to determine the genotype of each fetus. The next day, the fetal heads were dehydrated through a series of ascending ethanol concentrations and left in 100% butanol overnight. The heads were cleared in xylene for 1 hr and then infiltrated for 1.5 hr with paraffin at 60°C under vacuum and paraffin-embedded. Although older trisomic fetuses (≥E16) generally display clear symptoms such as widespread edema and growth retardation, fetal genotype was confirmed by karyotype analysis on corresponding livers.
Karyotype analysis. Individual fetal livers were collected in RPMI 1640 (Life Technologies, Gaithersburg, MD) and triturated at 37°C. Dissociated cells then were incubated in colchicine (0.005% in PBS, 37°C) for 45 min, pelleted, and then resuspended in 75 mm KCl (37°C) for 30–45 min. Cells then were pelleted and resuspended in Carnoy’s fix (1:3, glacial acetic acid/methanol) three times. This cell preparation was dropped onto slides and dried, and the number and configuration of chromosomes were determined. Chromosome spreads from euploid fetuses have 40 chromosomes, 2 of which are in a single metacentric configuration because of the inheritance of one Robertsonian translocation from the father. Trisomic fetuses have 41 chromosomes (2 metacentric pairs), indicating a triplication of MMU16.
Tissue sectioning and histology. Coronal sections (5 μm) from Ts16 and euploid littermate brains were collected on Superfrost Plus slides (Fisher Scientific, Springfield, NJ) and air-dried. Sections were deparaffinized with xylene for 15 min and then rehydrated through a series of descending ethanol concentrations and washed in 0.1 m Tris. Sections were stained with 0.5% cresyl violet, dehydrated, and coverslipped in Permount (Fisher).
Gross brain measurements. Fixed euploid and Ts16 brains were removed from the calvarium and measured without paraffin processing. Calipers calibrated to 0.05 mm (Mitutoyo) were used to make two measurements: the maximum lateral span between the parietal poles and the maximum rostral–caudal distance between the olfactory and occipital poles of the cortex. Four Ts16/euploid littermate pairs were analyzed for each age in this manner.
Cross-sectional area measurements. Cross-sectional areas of the ventricular/subventricular zones (VZ/SVZ) and the subplate/cortical plate (SP/CP) were determined using a Metamorph image analysis system (Universal Imaging, West Chester, PA). Measurements were made in matched Nissl-stained sagittal sections (5 μm thick) from five euploid and Ts16 littermate pairs at E16. The contours of each measured cortical area (see Fig. 1A) were traced using a manually driven cursor. Areas were calculated from the digitized data derived from a series of six sections per animal, the level of which is shown in Figure 2. The data were evaluated by a nested ANOVA to determine whether there were significant differences between Ts16 and euploid cross-sectional cortical areas.
Fig. 1.

Illustrations of the level of coronal section analyzed and method of cross-sectional area analysis. A, Cross-sectional area measurements were made of the VZ, CP, and pallium in Nissl-stained parasagittal sections. The areas of the SP/CP and VZ/SVZ (whited out) and the pallium (between the arrows) were measured.B, Coronal brain sections were taken at a point 200 μm caudal to the interventricular foramen in a portion of the future sensorimotor cortex where the hippocampus is found just dorsal to the thalamus.
Fig. 2.

Low-power photomicrographs of Nissl-stained parasagittal sections from euploid (A) and Ts16 (B) brains. By E16, Ts16 brains clearly are smaller than control brains in thickness and tangential extent of the cortex and cortical layers. Scale bar, 200 μm.
Measurements of cell density and thickness of cortical layers. Total cell density measurements were made at the level of the dorsal hippocampus (or future sensorimotor cortex, caudal aspect) (see Figs. 1B, 3A–C for level of section) from coronal sections taken from six littermate pairs each at E13, E14, E16, and E18. All sections were matched, both within and between groups, to be at a specific telencephalic level starting from 200 μm caudal to the interventricular foramen and extending for 50 μm in the caudal direction. All layers of the pallium, including the VZ/SVZ, IZ, and SP/CP, were counted. Cell density was determined from 5-μm-thick sections counted on a Leitz Ortholux microscope equipped with a square reticle divided into 10 μm boxes in a 10 × 10 array. Cell counts were made at a consistent position on the dorsal pallium, midway between the medial and lateral borders of the lateral ventricle. The cortical area in which cells were counted is demarcated by the rectangle in Figure 3A. All cresyl violet-stained nuclei were counted excluding endothelial cells, located adjacent to blood vessels and identified by long and tapered fusiform somata.
Fig. 3.
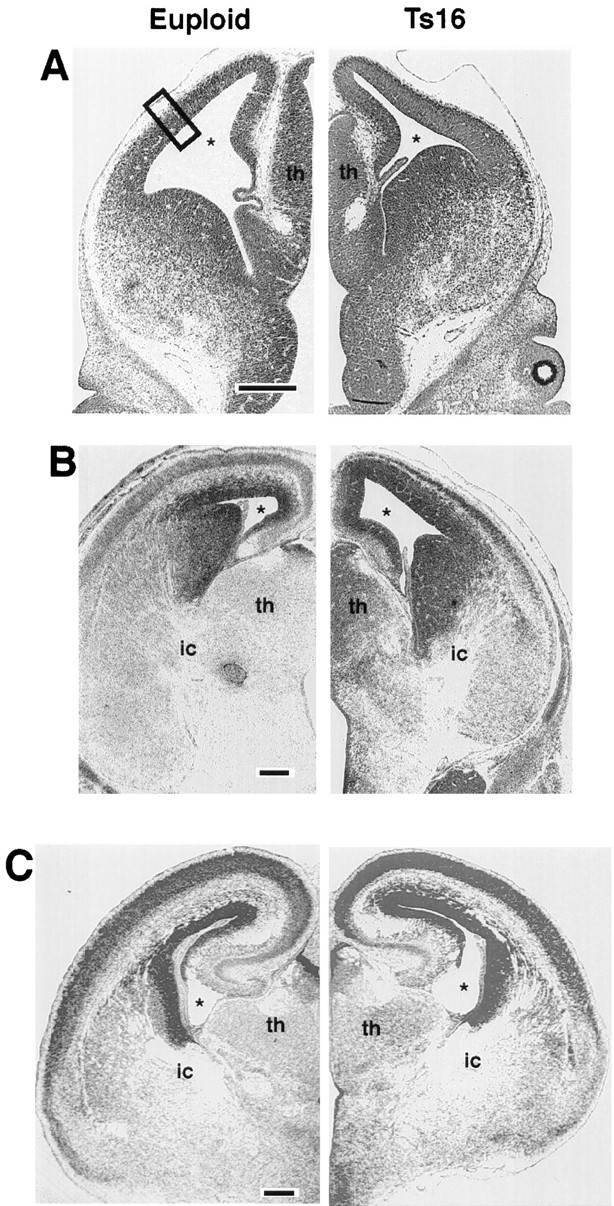
Low-power photomicrographs of Nissl-stained coronal sections from euploid (left) and Ts16 (right) brains at the level of the dorsal hippocampus. E13 brains (A) are indistinguishable in overall size and in cortical thickness. By E16 (B), Ts16 brains are clearly different from control brains in thickness of the pallium, development of cortical layers, and overall size of the brain. By E18 (C), the decrease in thickness of the cortex and its constituent layers has recovered in Ts16 and appears normal with respect to controls. The box in A shows the region of measurements of layer thickness and cell density given in Figures 5 and 6. This boxed area also is shown at higher magnification in Figure 4. th, Thalamus;ic, internal capsule; star, lateral ventricle. Scale bar, 100 μm.
The reticle’s grid was positioned such that the first row of 10 boxes was aligned with the ventricular surface. The cells contained within each successive bin of the entire reticle were counted beginning at the ventricular surface (row 1) up to the pial surface. Cells were considered to be within a certain row if they were contained by the medial and dorsal lines of the row but not if they touched the lateral or ventral lines. Endothelial cells were excluded from these measurements. In cortices thicker than 100 μm, the superficial cells were counted by repositioning the reticle such that the ventral border of row 1 was aligned with the dorsal border of the previous row 10. Four alternating sections from each brain were counted in this manner, and the data were analyzed by ANOVA to determine the significance of differences between Ts16 and euploid cell counts. Measurements of the thickness of each layer of the developing cortex were made in the same region of each section as in the cell density measurements. These data were analyzed by ANOVA to assess the significance of differences in the thickness of cortical layers.
Although both the SP and the SVZ have been shown to be distinct cortical layers (Angevine et al., 1970; Kostovic and Molliver, 1974), the precise boundaries delineating these zones are difficult to pinpoint. Therefore, measurements reported here combine these layers with their functionally related neighbors. We use the term “VZ/SVZ” to identify the area encompassing these two proliferative zones as extending from the surface of the lateral ventricle to the outermost abventricular mitotic figure. After E13, SP/CP refers to the area bounded by the large, pale SP cells and the border between the CP and ML.
RESULTS
Gross abnormalities of Ts16 brain development
Throughout the period of cortical development that we examined, the Ts16 brains were significantly smaller than those of euploid littermates in both the medial–lateral and the rostral–caudal dimensions (Table 1). An examination of Nissl-stained sagittal sections also revealed significant abnormalities in Ts16 (Fig.2). At E16, the Ts16 brain (B) is smaller than the euploid brain (A) in both the rostral–caudal and the radial dimensions. This decrease in rostral–caudal development is associated with a reduction in the tangential extent of the entire palium including the VZ/SVZ and SP/CP.
Table 1.
Gross brain measurements (mm)
| Euploid | Ts16 | % Difference | |
|---|---|---|---|
| E14 | |||
| R–C | 3.15 ± 0.08 | 2.83 ± 0.12* | 11.2 |
| P–P | 3.28 ± 0.05 | 2.85 ± 0.15* | 14.1 |
| E16 | |||
| R–C | 4.20 ± 0.13 | 3.63 ± 0.24** | 14.6 |
| P–P | 4.63 ± 0.14 | 4.12 ± 0.08* | 11.0 |
| E18 | |||
| R–C | 5.13 ± 0.15 | 4.22 ± 0.10** | 18.7 |
| P–P | 5.68 ± 0.12 | 5.24 ± 0.20** | 8.7 |
Mean values ± SEM for telencephalic dimensions in the rostral–caudal (R–C) and parietal–parietal (P–P) axes of euploid and Ts16 mice at E14, E16, and E18 (n = 4 pairs for all ages). *p < 0.05; **p < 0.005.
Although the gross dimensions of the Ts16 cortex remained smaller throughout development, examination of coronal sections revealed that the decrease in radial thickness was limited to a 3–4 d period. At E13 (Fig. 3A), Ts16 and euploid brains appeared similar. By E14, differences in the thickness of the cortex became evident (data not shown), and by E16 (Fig. 3B), a marked size difference had developed between the Ts16 and euploid cortices. The overall thickness of the Ts16 pallium was diminished at E16, as were the individual layers intervening between the VZ and the pial surface. The IZ and SP/CP layers were significantly smaller at E14 and E16. By E18, however, the thickness of the Ts16 cortex had increased considerably and was not significantly different with respect to the radial thickness of the pallium and its constituent cortical layers (Figs.3C, 4C, 5D).
Fig. 4.
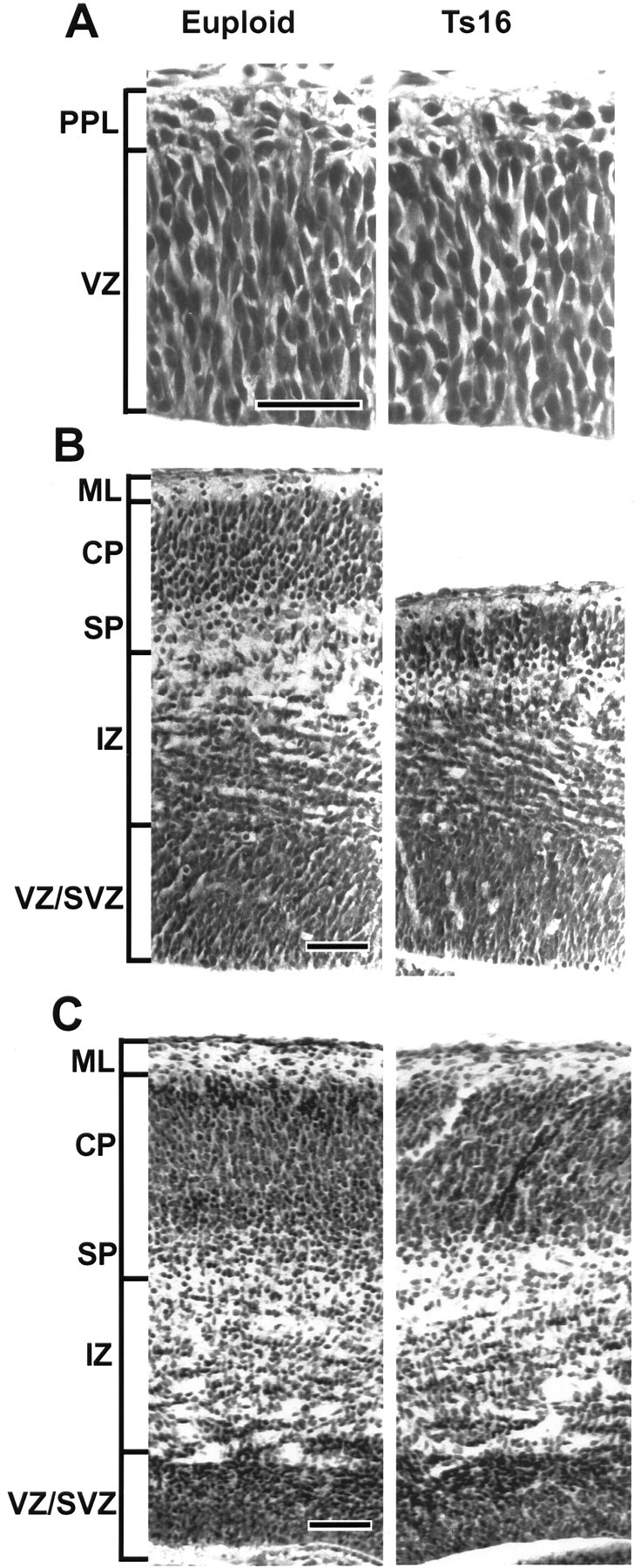
High-magnification photomicrographs of euploid (left) and Ts16 (right) cortices taken from the midpoint of the pallium, as shown in Figure 3A. At E13 (A), euploid and Ts16 cortices appeared similar in thickness of the VZ and PPL. At E16 (B), the size of the SP/CP, IZ, and pallium was smaller in Ts16 than in euploid brains. Moreover, the most dorsal cell-sparse region of the euploid IZ (immediately subjacent to the SP) was absent from the Ts16 cortex. By E18 (C), however, the thickness of all cortical layers appeared normal in Ts16 brains. PPL, Preplate;VZ/SVZ, ventricular zone/subventricular zone;ML, marginal layer; SP, subplate; CP, cortical plate; IZ, intermediate zone. Scale bar, 50 μm.
Fig. 5.

Thickness of the VZ/SVZ (A), IZ (B), SP/CP (C), and pallium (D) in Ts16 (filled circles) and euploid (open squares) cortices. Data represent mean ± SEM (n = 6). Error bars are shown when larger than dimensions of symbols. In all areas examined, there were no differences at E13 (the IZ was not present in Ts16 or euploid brains at E13). However, abnormalities in thickness of the SP/CP, pallium, and IZ were apparent at E14 and were marked by E16. The Ts16 VZ/SVZ exhibited a slight increase at E14. By E18, all cortical layers were of normal thickness. *p < 0.05, **p < 0.001.
At higher magnification, the appearance of the VZ and establishment of the superficial preplate (PPL) were similar in euploid and trisomic brains at E13 (Fig. 4A). The thickness and cellular composition of these layers also appeared to be similar. At E14 and E16, the Ts16 pallium was markedly smaller than normal in the radial dimension. Within the pallium, the thickness of the SP/CP and the IZ compartments was severely reduced at E14 (data not shown) and E16 (Fig. 4B). In contrast, the Ts16 cortex was normal with respect to the thickness of the pallium and each of the cortical layers 2 d later at E18. It is important to note that although the overall thickness of the Ts16 pallium was diminished at E14 and E16, the cellular density of each cortical layer was similar in Ts16 and euploid brains throughout development (see below).
Thickness measurements
To determine the extent to which each layer in a radial unit of the cortex is reduced in Ts16, measurements of the thickness of the cortical layers were made at the level of the future sensorimotor cortex (Fig. 5A–D). The thickness of cortical layers in Ts16 was not different from normal brains at E13. In normal brains, the thickness of the VZ/SVZ declined after E13 (Fig.5A). The Ts16 VZ/SVZ also declined in thickness throughout the period studied, although between E13 and E14 there was a transient increase in VZ thickness of ∼15%.
Between E14 and E16 the thickness of the IZ (Fig. 5B), the SP/CP (Fig. 5C), and the pallium (Fig. 5D) was reduced significantly in Ts16 brains. The SP/CP, IZ, and overall pallium were reduced in thickness by 36, 75, and 23% at E14 and by 35, 17, and 20% at E16, respectively. The thickness of cortical layers in Ts16 fetuses was not markedly abnormal at E18, the SP/CP, IZ, and pallium being reduced only by 4, 0.3, and 2%, respectively.
Cross-sectional area measurements
The cross-sectional area of each cortical layer in the sagittal plane also was different in trisomic and euploid littermates (Table2). At E16, cross-sectional area was reduced by 12% (p < 0.05) in the VZ, 48% (p < 0.0005) in the CP, and 31% (p < 0.00005) in the pallium of Ts16 fetuses.
Table 2.
Cross-sectional area (mm2)
| Euploid | Ts16 | % Difference | |
|---|---|---|---|
| VZ | 0.32 ± 0.08 | 0.28 ± 0.08* | 12 |
| CP | 0.34 ± 0.09 | 0.18 ± 0.01** | 48 |
| Pallium | 1.26 ± 0.06 | 0.87 ± 0.02*** | 31 |
Mean values ± SEM of the VZ, CP, and entire pallium in fetal control and Ts16 mice (n = 5, each group) in parasagittal sections at E16. *p < 0.005; **p < 0.0001; ***p < 0.00001.
Cellular density measurements
To determine whether alterations in cell packing density accompanied the reduction in laminar thickness in Ts16 mice, cell density measurements were made throughout the pallium at the level of the future sensorimotor cortex (Fig. 6). At each of the three ages examined, euploid and Ts16 brains had similar mean cell densities within corresponding cortical layers.
Fig. 6.

Density plots across Ts16 (filled circles) and euploid (open squares) cortices. Data represent mean ± SEM (n = 6). At E13 (A), the plots overlap throughout the cortex. At E14 (B), Ts16 and euploid cell density plots overlap up to the IZ (see brackets underneath plot), but the Ts16 plot deviates from euploid through the IZ and SP/CP because the Ts16 cortex is smaller. At E16 (C), the density plots overlap through the Ts16 IZ. Relative cell densities of the VZ/SVZ, IZ, and SP/CP are not different between Ts16 and euploid at any age.
At E13, the cell density plots of Ts16 and euploid brains overlapped along the entire radial extent of the cortex (Fig.6A). At E14, the density plots overlapped from the ventricular surface out to 100 μm into the pallium, beyond which the Ts16 plot diverged as a result of the thinner trisomic cortex (Fig.6B). Although individual cortical layers were abnormally thin in Ts16 brains at E16 (except the VZ/SVZ), the cell densities of euploid and Ts16 cortices were similar within each layer (Fig. 6C). In addition, we found normal cell densities in corresponding Ts16 and euploid cortical layers at E18 (data not shown).
DISCUSSION
Our study of cerebral cortical development in Ts16 brains has revealed two separate abnormalities. (1) The initial tangential expansion of the proliferative zones, before the beginning of the radial growth of cortical layers, is abnormal in Ts16. This early defect results in persistent size deficits in Ts16 telencephalic dimensions and in the cross-sectional area of each pallial zone. (2) The subsequent radial expansion of the Ts16 pallium from E13 to E16 is delayed. This delay results in decreases in thickness of the IZ and CP between E14 and E16. By E18, however, all cortical compartments had attained the same thickness in both groups.
Although radial development of cortical layers and overall telencephalic size was abnormal in Ts16, these brains were normal in several important ways. Ts16 and control brains were indistinguishable morphologically in coronal sections up to E13, a stage in development at which the preplate, the first postmitotic layer of neurons, already has formed (Fig. 4A). Our results also demonstrate that none of the layers of the fetal pallium was absent in Ts16. Moreover, even during the period of delayed development (from E13 to E18), the decreased thickness of the Ts16 cortex was not accompanied by a decrease in relative density at any point within the pallium. This result, which is in agreement with findings from human DS brains (Golden and Hyman, 1994), suggests that cellular density within the developing cortex is a crucial and conserved factor. Our results also suggest that the well described rostral–caudal and lateral–medial spatiotemporal gradients in cortical development (Smart and Smart, 1982) are preserved in Ts16 fetuses; the size of pallial zones in rostral and lateral portions of the cortex were larger and more advanced developmentally than in caudal and medial areas (see Figs. 2,3), and morphometry of Ts16 cellular layers at the more rostral level of the interventricular foramen revealed increased radial thickness of the SP/CP and IZ compared to the caudal level of the dorsal hippocampus (data not shown).
Possible mechanisms for the developmental abnormalities in Ts16
Several theories have been proposed to explain the developmental mechanisms of formation of the mammalian cerebral cortex. One such theory stems from lineage tracing results (Sanes et al., 1986; Price et al., 1987; Kornack and Rakic, 1995; Rakic, 1995) that indicate that some, if not all, cortical cells in discrete architectonic regions of the cortex arise from the proliferative pools directly underlying the developing CP. This “radial unit hypothesis” (Rakic, 1988) states that each segment of clonally related proliferative cells gives rise to a precisely ordered and defined ontogenetic population of postmitotic neurons in the overlying cortex (the “radial unit”) and thus forms a radial “column,” a basic functional unit of the cortex (Mountcastle, 1957; Rakic, 1982).
The number of these radial units in the cortex is thought to be specified during the period of rapid symmetrical proliferation early in development when neuronal progenitor cell division is followed by the reentry of both daughter cells back into the cell cycle. This period of symmetrical division results in an exponential tangential expansion of the VZs and of the telencephalic wall. Hence, this early epoch of cell division produces an increase in the population of proliferative neuroepithelial cells, thereby increasing the potential number of resulting radial units.
The early proliferative period then is followed by a second epoch of division as one or both daughter cells become postmitotic after each cell cycle and migrate outward to form the superficial cortical layers (Takahashi et al., 1994; Kornack and Rakic, 1995). Therefore, the switch from tangential VZ expansion to radial layer formation has to occur sufficiently late in fetal development to ensure the generation of the correct number of radial cortical units required for normal histogenesis, synaptogenesis, and patterning of projections.
One explanation for the reductions in cross-sectional area in Ts16 is a reduction in the rate of the early symmetrical neuroblast proliferation that might lead to decreased numbers of cells and produce fewer radial units during this period. In addition, the end of the initial phase of exponential growth is delineated temporally by the onset of the second proliferative epoch. A decreased pool of precursors at this stage would be depleted further during the ensuing radial expansion of the cortex. This radial cortical growth results in the commitment of daughter cells to fates outside the proliferative zones. Therefore, the effects of decreased early tangential cortical expansion would be a lasting reduction in the numbers of radial units and, consequently, in cortical surface area.
In contrast to tangential expansion, radial expansion of the cortex may not be strictly limited to a fixed time period. Rather, radial expansion of the cortex may be dependent only on the remaining proliferative capacity of the neuroblasts in the VZ. Therefore, a delay in radial expansion of the cortex caused only by slower neuroblast proliferation would be expected to be ameliorated with time. Our observation that the Ts16 pallium thickness “catches up” with normal brains is consistent with this idea. Thus, slower neuroblast proliferation could explain both the reduction in cortical area in Ts16 and the delayed development of the IZ and SP/CP within each radial unit.
Slower neuroblast proliferation, if found, could be explained by several different effects of an extra chromosome in Ts16 cells. First, the simple burden of the added genetic material could alter the rates of the DNA synthetic and/or mitotic phases of the cell cycle. Second, a recent report in cortical slice cultures (LoTurco et al., 1995) suggests that glutamate may regulate the duration of the DNA synthetic phase of the cell cycle in the fetal germinal zones. Overexpression of the AMPA/kainate ionotropic glutamate receptor subunit GluR5, which is among the genes of the DS obligate region on MMU16, could therefore affect proliferation in the fetal Ts16 cortex.
Defects in any of several processes of cortical neurogenesis such as programmed cell death and migration might possibly account for the disorders of radial cortical development seen in Ts16. Several reports have shown that apoptotic death occurs in the normal cortex near or after birth in rodents (Ferrer et al., 1990; Spreafico et al., 1995). Moreover, a recent report suggests that more cortical cells may be dying during gestation as well (Blaschke et al., 1996). The 35% decrease in the CP, therefore, could be the result of higher rates of neuronal death in the Ts16 cortex. In preliminary experiments at E14 and E16, however, we found no evidence of apoptosis (pyknotic or terminal dUTP nick end-labeled nuclei) at any position within Ts16 or euploid cortices (data not shown). These results indicate that rates of cell death in the Ts16 cerebral cortex are not higher than in the euploid cortex.
Abnormal neuronal migration might also explain reduced numbers of cells in the CP. However, if proliferation rates were normal, delayed or failed migration in Ts16 would be expected to give rise to the substantial overpopulation and enlargement of one or more laminae. Our analysis has not detected definitive evidence of defective migration, although the transient increase in VZ thickness at E14 (Fig.5A) could reflect a slower departure of postmitotic neurons from the VZ at that time. A transient build-up of cells in this region might reflect abnormalities in the molecular mechanisms of neuronal migration. Conversely, this might also be attributable to an inability of the existing migrational substrates to support the relative increase in the production of cells during this period which expands the SP/CP to normal levels by E18.
Potential repercussions of abnormal development in Ts16 brains
Several different and possibly interrelated processes of development may be affected by the abnormalities in tangential and radial cortical expansion we have found in the Ts16 mouse. The results presented in Tables 1 and 2 indicate that these mice have smaller brains at all fetal ages studied. A reduction in cell numbers and telencephalic size of the Ts16 cortex would be expected to result from slower tangential growth and could have long-term consequences. Although the cortex may reach normal thickness in any given area, a reduction in the total number of radial columns is likely to produce a decrease in cortical–cortical association connections. Integrative cortical function is crucially dependent on association connections, and a decrease in these cortical–cortical connections can be predicted to impair higher level cognitive function.
The delays in radial development of the SP/CP may have adverse effects on the time course or extent of afferent projections through the IZ to the CP. For example, projections from the ventral posterior thalamus establish a pattern of afferentation immediately before invading the somatosensory cortex (Erzurumlu and Jhaveri, 1990; Schlaggar and O’Leary, 1991), indicating that the cells of the CP or SP may play an early role in the organization of these afferents. In addition, SP cells have been shown to pioneer efferent axon pathways into the thalamus before CP axons (Bicknese et al., 1994; DeCarlos and O’Leary, 1994; McConnell et al., 1994). The absence of CP or SP neurons at critical times may adversely affect the formation of synapses and axon pathways.
There are indications that axonal projections may be abnormal in Ts16. The decreased thickness of the IZ between E14 and E16 (Fig.5B) may result from a reduction in the number of axons arising from CP, SP, or thalamic neurons because this zone contains projections to and from the cortex. Although the analysis presented here was based on Nissl staining and was limited in that primarily cell somata were stained, this stain did allow several different observations of this relatively acellular compartment. Our results showed that the deepest level of the IZ, that cellular portion that also contains laterally emanating efferent projections that create a striated pattern above the VZ/SVZ, appears normal with respect to thickness and cellular composition at E16 (Figs. 4B). In contrast, the Ts16 IZ immediately subjacent to the SP (the relatively sparse area seen in the E16 control brain in Fig.4B) was absent. This area of the IZ, called the external sagittal stratum, normally contains a fiber bundle that originates from the thalamus (Caviness and Frost, 1980). The absence of this area may be evidence for missing or retarded afferent projections in Ts16 at a time when corticothalamic and thalamocortical connections are normally made. Therefore, although these brains appear normal by E18 in terms of radial thickness of the different developmental compartments, the delay in their formation may alter the timing and number of afferent and efferent synaptic contacts.
Late prenatal and postnatal brain abnormalities in DS have been studied rather extensively. It has been found that although neonatal DS brains are reduced in total volume and surface area, the radial size and lamination of the cortex is normal at birth. The decrease in cortical volume and surface area in DS may be analogous to the decrease in volume and cross-sectional area of the Ts16 mouse pallium described here. Moreover, the thickness of the Ts16 pallium is normal late in gestation and therefore also parallels DS development. Our results indicate that the normal appearance of the Ts16 cortex late in gestation is preceded by a significant delay in the formation of pallial zones. Studies have not been done early enough to ascertain whether the same delay occurs in DS but, like the overall decrease in the number of cortical units, a temporal disturbance in layer formation could adversely affect later cortical development in DS and potentially contribute to cognitive deficits such as mental retardation.
Footnotes
This work was supported by National Institutes of Health Grants AG10686 (B.K.K.), NS29167 (M.E.B.), HD20461 (M.E.B.), P01-HD19920 (M.E.M.), NS15199 (M.E.M.), the Strategic Research Fund, Zeneca Pharmaceuticals (B.K.K.), and the Special Research Initiative Fund, University of Maryland (P.J.Y.). T.F.H. was supported by National Institutes of Health Training Grant T32GM08181. We thank Jordan M. Denner at the Baltimore VA Medical Center Medical Media Service for his help on the figures, and we thank Dat Chu, Thu Pham, and McRae Williams for their efforts in karyotyping.
Correspondence should be addressed to Tarik F. Haydar, Department of Physiology, University of Maryland School of Medicine, 655 West Baltimore Street, Baltimore, MD 21201.
REFERENCES
- 1.Adams MM, Erickson JD, Layde PM, Oakley GP. Down’s syndrome: recent trends in the United States. J Am Med Assoc. 1981;246:758–760. doi: 10.1001/jama.246.7.758. [DOI] [PubMed] [Google Scholar]
- 2.Angevine JB, Jr, Bodian D, Coulombre AJ, Edds MV, Jr, Hamburger V, Jacobson M, Lyser KM, Prestige MC, Sidman RL, Varon S, Weiss PA. Embryonic vertebrate central nervous system: revised terminology. Anat Rec. 1970;166:257–262. doi: 10.1002/ar.1091660214. [DOI] [PubMed] [Google Scholar]
- 3.Bambrick LL, Yarowsky PJ, Krueger BK. Glutamate as a hippocampal neuron survival factor: an inherited defect in the trisomy 16 mouse. Proc Natl Acad Sci USA. 1995;92:9692–9696. doi: 10.1073/pnas.92.21.9692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Becker LE. Synaptic dysgenesis. Can J Neurol Sci. 1991;18:170–180. doi: 10.1017/s0317167100031644. [DOI] [PubMed] [Google Scholar]
- 5.Bicknese AR, Sheppard AM, O’Leary DDM, Pearlman AL. Thalamocortical axons extend along a chondroitin sulfate proteoglycan-enriched pathway coincident with the neocortical subplate and distinct from the efferent path. J Neurosci. 1994;14:3500–3510. doi: 10.1523/JNEUROSCI.14-06-03500.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Blaschke AJ, Staley K, Chun J. Widespread programmed cell death in proliferative and postmitotic regions of the fetal cerebral cortex. Development. 1996;122:1165–1174. doi: 10.1242/dev.122.4.1165. [DOI] [PubMed] [Google Scholar]
- 7.Caviness VS, Jr, Frost DO. Tangential organization of thalamic projections to the neocortex in the mouse. J Comp Neurol. 1980;194:335–367. doi: 10.1002/cne.901940205. [DOI] [PubMed] [Google Scholar]
- 8.Caviness VS, Jr, Takahashi T, Nowakowski RS. Numbers, time and neocortical neurogenesis: a general developmental and evolutionary model. Trends Neurosci. 1995;18:379–388. doi: 10.1016/0166-2236(95)93933-o. [DOI] [PubMed] [Google Scholar]
- 9.Colon EJ. The structure of the cerebral cortex in Down’s syndrome: a quantitative analysis. Neuropadiatrie. 1972;3:362–376. [Google Scholar]
- 10.Crome L, Stern J. Churchill Livingstone; Edinburgh: 1972. Down syndrome. Pathology of mental retardation (Crome L, Stern J, eds), pp 200–224. . [Google Scholar]
- 11.Davidoff LM. The brain in mongolian idiocy. Arch Neurol Psychiatry. 1928;20:1229–1257. [Google Scholar]
- 12.DeCarlos JA, O’Leary DDM. Growth and targeting of subplate axons and establishment of major cortical pathways. J Neurosci. 1994;12:1194–1211. doi: 10.1523/JNEUROSCI.12-04-01194.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Erzurumlu RS, Jhaveri S. Thalamic axons confer a blueprint of the sensory periphery onto the developing rat somatosensory cortex. Dev Brain Res. 1990;56:229–234. doi: 10.1016/0165-3806(90)90087-f. [DOI] [PubMed] [Google Scholar]
- 14.Ferrer I, Soriano JA, Del Rio T, Alcantara S, Fonseca M. Naturally occurring cell death in the cerebral cortex of the rat and removal of dead cells by transitory phagocytes. Neuroscience. 1990;39:451–458. doi: 10.1016/0306-4522(90)90281-8. [DOI] [PubMed] [Google Scholar]
- 15.Galdzicki Z, Coan E, Rapaport SI. Cultured hippocampal neurons from trisomy 16 mouse, a model for Down’s syndrome, have an abnormal action potential due to a reduced inward sodium current. Brain Res. 1993;604:69–78. doi: 10.1016/0006-8993(93)90353-o. [DOI] [PubMed] [Google Scholar]
- 16.Gearhart JD, Davisson MT, Oster-Granite ML. Autosomal aneuploidy in mice: generation and developmental consequences. Brain Res Bull. 1986;16:789–801. doi: 10.1016/0361-9230(86)90075-4. [DOI] [PubMed] [Google Scholar]
- 17.Golden JA, Hyman BT. Development of the superior temporal neocortex is anomalous in trisomy 21. J Neuropathol Exp Neurol. 1994;53:513–520. doi: 10.1097/00005072-199409000-00011. [DOI] [PubMed] [Google Scholar]
- 18. Grausz H, Richtsmeier JT, Oster-Granite ML. Morphogenesis of the brain and craniofacial complex in trisomy 16 mice. Epstein CJ. The morphogenesis of Down syndrome 1991. 168 188 Wiley; New York-Liss. [PubMed] [Google Scholar]
- 19.Gropp A, Giers D, Kolbus U. Trisomy in the fetal backcross progeny of male and female metacentric heterozygotes in the mouse. Cytogenet Cell Genet. 1974;13:511–535. doi: 10.1159/000130304. [DOI] [PubMed] [Google Scholar]
- 20.Holtzman DM, Epstein CJ. The molecular genetics of Down syndrome. In: Friedmann T, editor. Molecular genetic medicine, Vol. 2. Academic; San Diego: 1992. pp. 105–120. [DOI] [PubMed] [Google Scholar]
- 21.Hook EB. Down syndrome: its frequency in human populations and some factors pertinent to variations in rates. In: de la Cruz FF, Gerald PS, editors. Trisomy 21 (Down syndrome): research perspectives. University Park; Baltimore: 1978. pp. 3–68. [Google Scholar]
- 22.Kiss J, Schlumpf M, Balazs R. Selective retardation of the development of the basal forebrain cholinergic and pontine catecholaminergic nuclei in the brain of trisomy 16 mouse, an animal model of Down’s syndrome. Dev Brain Res. 1989;50:251–264. doi: 10.1016/0165-3806(89)90201-0. [DOI] [PubMed] [Google Scholar]
- 23.Kornack DR, Rakic P. Radial and horizontal deployment of clonally related cells in the primate neocortex: relationship to distinct mitotic lineages. Neuron. 1995;15:311–321. doi: 10.1016/0896-6273(95)90036-5. [DOI] [PubMed] [Google Scholar]
- 24.Kostovic I, Molliver ME. A new interpretation of the laminar development of cerebral cortex: synaptogenesis in different layers of neopallium in the human fetus. Anat Rec. 1974;178:395. [Google Scholar]
- 25.Lacey-Casem ML, Oster-Granite ML. The neuropathology of the trisomy 16 mouse. Crit Rev Neurobiol. 1994;8:293–322. [PubMed] [Google Scholar]
- 26.LoTurco JJ, Owens DF, Heath MJS, Davis MBE, Kriegstein AR. GABA and glutamate depolarize cortical progenitor cells and inhibit DNA synthesis. Neuron. 1995;15:1287–1298. doi: 10.1016/0896-6273(95)90008-x. [DOI] [PubMed] [Google Scholar]
- 27.Marin-Padilla M. Early ontogenesis of the human cerebral cortex. In: Peters A, Jones EG, editors. Cerebral cortex, Vol. 7. Plenum; New York: 1988. pp. 1–34. [Google Scholar]
- 28.McConnell SK. Development and decision making in the mammalian cerebral cortex. Brain Res Rev. 1988;13:1–23. doi: 10.1016/0165-0173(88)90002-1. [DOI] [PubMed] [Google Scholar]
- 29.McConnell SK, Ghosh A, Shatz CJ. Subplate pioneers and the formation of descending connections from cerebral cortex. J Neurosci. 1994;14:1892–1907. doi: 10.1523/JNEUROSCI.14-04-01892.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mittwoch U. DNA synthesis in cells grown in tissue culture from patients with mongolism. Ciba Foundation Study Group. 1967;25:51–61. [Google Scholar]
- 31.Mountcastle VB. Modality and topographic properties of single neurons of cat’s somatic sensory cortex. J Neurophysiol. 1957;20:408–434. doi: 10.1152/jn.1957.20.4.408. [DOI] [PubMed] [Google Scholar]
- 32.Price J, Turner D, Cepko C. Lineage analysis in the vertebrate nervous system by retrovirus-mediated gene transfer. Proc Natl Acad Sci USA. 1987;84:156–160. doi: 10.1073/pnas.84.1.156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rakic P. Mode of cell migration to the superficial layers of the fetal monkey neocortex. J Comp Neurol. 1972;145:61–84. doi: 10.1002/cne.901450105. [DOI] [PubMed] [Google Scholar]
- 34.Rakic P. Early developmental events: cell lineages, acquisition of neuronal positions, and areal and laminar development. Neurosci Res Prog Bull. 1982;20:439–451. [PubMed] [Google Scholar]
- 35.Rakic P. Specification of cerebral cortical areas. Science. 1988;241:170–176. doi: 10.1126/science.3291116. [DOI] [PubMed] [Google Scholar]
- 36.Rakic P. Radial versus tangential migration of neuronal clones in the developing cerebral cortex. Proc Natl Acad Sci USA. 1995;92:11323–11327. doi: 10.1073/pnas.92.25.11323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Reeves RH, Gearhart JD, Littlefield JW. Genetic basis for a mouse model of Down syndrome. Brain Res Bull. 1986;16:803–814. doi: 10.1016/0361-9230(86)90076-6. [DOI] [PubMed] [Google Scholar]
- 38.Ross MH, Galaburda AM, Kemper TL. Down’s syndrome: is there a decreased population of neurons? Neurology. 1984;34:909–916. doi: 10.1212/wnl.34.7.909. [DOI] [PubMed] [Google Scholar]
- 39.Sanes JR, Rubenstein JCR, Nicolas J-F. Use of a recombinant retrovirus to study post-implantation cell lineage in mouse embryos. EMBO J. 1986;5:3133–3142. doi: 10.1002/j.1460-2075.1986.tb04620.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schlaggar BL, O’Leary DDM. Potential of visual cortex to develop an array of functional units unique to somatosensory cortex. Science. 1991;252:1556–1560. doi: 10.1126/science.2047863. [DOI] [PubMed] [Google Scholar]
- 41.Schmidt-Sidor B, Wisniewski KE, Shepard TH, Sersen EA. Brain growth in Down syndrome subjects 15 to 22 weeks of gestational age and birth to 60 months. Clin Neuropathol. 1990;9:181–190. [PubMed] [Google Scholar]
- 42.Segal DJ, McCoy E. Studies on Down’s syndrome in tissue culture: growth rates and protein counts of fibroblast cultures. J Cell Physiol. 1974;83:85–90. doi: 10.1002/jcp.1040830112. [DOI] [PubMed] [Google Scholar]
- 43.Smart IHM, Smart M. Growth patterns in the lateral wall of the mouse telencephalon. I. Autoradiographic studies of the histogenesis of the isocortex and adjacent areas. J Anat. 1982;134:273–298. [PMC free article] [PubMed] [Google Scholar]
- 44.Spreafico R, Frassoni C, Arcelli P, Selvaggio M, De Biasi S. In situ labeling of apoptotic cell death in the cerebral cortex and thalamus of rats during development. J Comp Neurol. 1995;363:281–295. doi: 10.1002/cne.903630209. [DOI] [PubMed] [Google Scholar]
- 45.Sweeney JE, Hohmann CF, Oster-Granite ML, Coyle JT. Neurogenesis of the basal forebrain in normal and trisomy 16 mice: an animal model for developmental disorders in Down syndrome. Neuroscience. 1987;31:413–425. doi: 10.1016/0306-4522(89)90384-9. [DOI] [PubMed] [Google Scholar]
- 46.Takahashi T, Nowakowski RS, Caviness VS., Jr Mode of cell proliferation in the developing mouse neocortex. Proc Natl Acad Sci USA. 1994;91:375–379. doi: 10.1073/pnas.91.1.375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Volpe JJ. Neuronal proliferation, migration, organization, and myelination. In: Volpe JJ, editor. Neurology of the newborn, 3rd Ed. Saunders; Philadelphia: 1995. pp. 43–92. [Google Scholar]
- 48.Wisniewski KE. Down syndrome children often have brain with maturation delay, retardation of growth and cortical dysgenesis. Am J Med Genet. 1990;7:274–281. doi: 10.1002/ajmg.1320370755. [DOI] [PubMed] [Google Scholar]
- 49.Wisniewski KE, Laure-Kamionowska M, Wisniewski HM. Evidence of arrest of neurogenesis and synaptogenesis in brains of patients with Down’s syndrome. N Engl J Med. 1984;18:1187–1188. doi: 10.1056/NEJM198411013111818. [DOI] [PubMed] [Google Scholar]
- 50.Wisniewski KE, Laure-Kamionowska M, Connell F, Wen GY. Neuronal density and synaptogenesis in the postnatal stage of brain maturation in Down syndrome. In: Epstein CJ, editor. Neurobiology of Down syndrome. Raven; New York: 1986. pp. 29–44. [Google Scholar]