

Abstract
The voltage-gated sodium channel Scn8a is broadly distributed in brain and spinal cord. We have identified a missense mutation in Scn8a that is associated with cerebellar ataxia in the joltingmutant, a mild allele of the “motor endplate disease” locus. Thejolting mutation results in substitution of Thr for an evolutionarily conserved Ala residue in the cytoplasmic S4–S5 linker of domain III. Introduction of the corresponding mutation into the rat brain IIA sodium channel shifted the voltage dependence of activation by 14 mV in the depolarizing direction, without affecting the kinetics of fast inactivation or recovery from inactivation. A shift in the threshold of the Scn8a channel could account for the reduced spontaneous activity of Purkinje cells, reduced inhibitory output from the cerebellum, and loss of motor control observed injolting mice.
Keywords: sodium channel, ataxia, med, mouse, cerebellum, mutation, Scn8a, Purkinje
The inherited cerebellar ataxias are a heterogenous group of disorders characterized by impaired motor function of cerebellar origin. Recent successes in positional cloning have resulted in the identification of several human and mouse genes underlying these disorders. These include two potassium channel genes (Browne et al., 1994; Patil et al., 1995), a retinoic acid receptor (Hamilton et al., 1996), and four genes of unknown function (Orr et al., 1993; D’Arcangelo et al., 1995; Schols et al., 1995; Campuzano et al., 1996). In this report, we demonstrate that a mutation that shifts the voltage dependence of a brain sodium channel is associated with inherited cerebellar ataxia in the mouse.
The mouse mutant jolting arose as a spontaneous mutation (Dickie et al., 1965; Sidman and Eicher, 1979). Genetic mapping and complementation tests demonstrated that jolting is an allele (medjo) of the motor endplate disease locus (gene symbol med) on mouse chromosome 15 (Sidman and Eicher, 1979; Harris and Pollard, 1986). At 3 weeks of age, when normal littermates are highly mobile and have well coordinated motor control, homozygous jolting mice have an unsteady, wide-based gait and a rhythmical tremor of head and neck that is induced by attempted movement (Dick et al., 1986). Electrophysiological studies demonstrated that the ataxia is cerebellar in origin. Extracellular recordings from the cerebellum of medjo mice demonstrated the absence of spontaneous, regular, simple discharges from the Purkinje cells, whereas the complex spikes generated by climbing fiber inputs are normal (Dick et al., 1986). The simple discharges of the Purkinje cells are believed to be generated in the cell soma and are sodium-dependent, although they may require an initial calcium-dependent depolarizing current (Llinas and Sugimori, 1980a). The complex spikes generated by climbing fiber inputs arise in the dendritic tree of the Purkinje cells as a result of an inwardly directed calcium current with a minor sodium component (Llinas and Sugimori, 1980b). These data suggested that the defect injolting mice involved a sodium or calcium channel located on the soma of the Purkinje cells.
Molecular access to the med locus was provided by the identification of an allele caused by random insertion of a transgene (Kohrman et al., 1995). Analysis of the insertion site demonstrated that the transgene had disrupted a novel voltage-gated sodium channel α subunit, Scn8a, that is expressed in brain and spinal cord (Burgess et al., 1995). The rat Scn8a cDNA, originally designated NaCh6, was independently isolated by Schaller et al. (1995), who found that the abundance of transcripts in rat brain is comparable to that of the rat brain sodium channels I, II, and III and that the broad distribution in brain includes cerebellar Purkinje and granule cells, spinal motor neurons, and some types of glia. Complete loss of function of Scn8a in the transgenic mutant is responsible for severe muscle atrophy, progressive paralysis, and juvenile death (Burgess et al., 1995). The spontaneous alleles med andmedJ also contain inactivating mutations that result in progressive paralysis and early death (Kohrman et al., 1996). We now report the molecular basis for the Scn8a mutation injolting mice and its effects on channel function.
MATERIALS AND METHODS
Animals. The medjomutation arose as a spontaneous mutation on the inbred strain DBA/2WyDi in 1958 at The Jackson Laboratory (Bar Harbor, ME) (Dickie, 1965; Sidman and Eicher, 1979). A colony ofmedjo mice was established at the University of Newcastle in 1980 and was transferred to the University of Michigan in 1995. The line is currently maintained by crossing to the inbred strain C56BL/6J.
Northern analysis. Two micrograms of poly(A+) RNA were electrophoresed through 1.2% agarose in the presence of 6% formaldehyde and transferred to a Zetaprobe GT filter (Bio-Rad, Richmond, CA) in 20× SSC. Hybridization with a cDNA probe corresponding to nucleotides 1–771 of the cDNA was carried out as described previously (Burgess et al., 1995), with washing at 65°C to a final stringency of 0.2× SSC. Under these conditions, hybridization is specific for the two Scn8a transcripts, as indicated by the absence of both transcripts in mice homozygous for themedtg allele (Burgess et al., 1995).
RT-PCR. Total RNA was prepared from brains of 1-month-old animals using the Trizol reagent (Life Technologies, Grand Island, NY). First-strand cDNA synthesis and PCR reactions were carried out as described previously (Burgess et al., 1995), except that the forward primer upstream of the translation start site was 5′-GGACG CAGCA TAACT AACGA (primer 1).
Genomic DNA. Genomic DNA was prepared from spleens of mice homozygous for the mutation by protease digestion and phenol/chloroform extraction. The conditions for PCR of genomic DNA were as described above, with 100 ng of DNA as template. The sequencing primer for the data in Figure 2A was 5′-AGGTG CCATA AAGTC CCTTA GGACC (primer 2). The S4–S5 linker region of domain 3 was amplified from genomic DNA with a forward primer based on intron sequence 5′-TGGCA GAACA TGCCA CAGAA GTGTC ATTCC (primer 3) and a reverse primer derived from cDNA sequence 5′-GGAAT TCCTC ACCTC GTCGA TTTCG AACCG (primer 4).
Fig. 2.

Nucleotide substitution in thejolting mRNA. A, Sequence chromatograms of Ala(1071)Thr mutation. RT-PCR products amplified from brain RNA of DBA/2J and jolting homozygous mice were gel-purified and directly sequenced as described in Materials and Methods.B, Predicted structure of Scn8a, based on sequence similarity with other voltage-gated sodium channel α subunits (Catterall, 1992). The position of the Thr substitution in the S4–S5 loop of domain III is marked with an asterisk.
DNA sequencing. After gel electrophoresis and visualization by ethidium bromide fluorescence, PCR products were excised and purified with Qiaex beads (Qiagen, Hilden, Germany) according to the manufacturer’s instructions. DNA was sequenced directly by dideoxynucleotide incorporation using Taq polymerase and analyzed with an ABI373A automated sequencer. Databases were searched using the BLAST algorithm (Altschul et al., 1990) via a WWW server. MacVector 3.5 (Kodak/IBI) software was used for sequence alignment.
Site-directed mutagenesis. The plasmid pVA2580 contains the rat brain IIA (RIIA) sodium channel coding region downstream from a T7 RNA polymerase promoter (Auld et al.,1990). The domain III S4–S5 region of RIIA, as defined by the unique BglII andBstEII restriction sites in pVA2580, was subcloned into M13 mp18A, a version of M13 mp18 with a polylinker region that was modified to contain unique restriction sites in the RIIA coding region. M13 single-strand mutagenesis was used to create the mutation as described previously (Kunkel, 1985). Uracil-containing, single-stranded DNA was isolated from phage grown in the dut−ung− Escherichia coli strain RZ1032 in YT media supplemented with 250 μg/ml uridine. The mutagenic oligonucleotide primer GCCTAAGAGAGTGTTTACAACAACC was phosphorylated with T4 polynucleotide kinase and annealed to the template at a 10:1 molar ratio. The primer was extended with T7 DNA polymerase (2.5 U) in the presence of T4 DNA ligase (6 Weiss units) in a 100 μl reaction containing 10 mm MgCl2, 0.5 mmdNTPs, 1 mm ATP, 2 mm dithiothreitol, and 20 mm Tris-HCl, pH 7.5. The reaction mixture was incubated first on ice for 5 min, then at room temperature for 5 min, and finally at 37°C for 2 hr. XL-1 bacteria were transformed with the heteroduplex DNA by electroporation and plated with XL-1 lawn cells. Single-stranded DNA was isolated from plaque-purified phage, and mutant clones were identified by dideoxynucleotide sequencing. To generate a full-length coding region containing the mutated sequence, aBglII–BstEII fragment containing the mutation was isolated from double-stranded RF DNA and ligated into pVA2580.
Oocyte expression and electrophysiological analysis. RNA transcripts were synthesized from NotI-linearized DNA templates using a T7 RNA polymerase transcription kit (Ambion). Stage V oocytes were removed from adult female Xenopus laevis frogs and prepared as described previously (Goldin, 1991) and incubated in ND-96 media (96 mm NaCl, 2 mm KCl, 1.8 mm CaCl2, 1 mm MgCl2, and 5 mm HEPES, pH 7.5), supplemented with 0.1 mg/ml gentamicin, 0.55 mg/ml pyruvate, and 0.5 mm theophylline. Sodium channel RNA and sodium channel β1 subunit RNA were injected into oocytes to attain current levels of 1–4 μA 1–3 d after injection. After incubation at 20°C in ND-96 for 1–3 d, sodium currents were recorded at room temperature using a two-electrode voltage clamp (Patton and Goldin, 1991) or a cut-open oocyte clamp (Taglialatela et al., 1992) as described previously. For the two-electrode voltage clamp, the bath recording solution consisted of ND-96. For the cut-open oocyte clamp, the internal solution consisted of 88 mm KSO4, 10 mmNaSO4, 10 mm EGTA, and 10 mm HEPES, pH 7.5, and the external solution consisted of 120 mmNa-Mes, 1.8 mm Ca-Mes, and 10 mm HEPES, pH 7.5. Currents were measured and analyzed using pClamp 6.0.2 software (Axon Instruments, Foster City, CA).
The cut-open oocyte voltage clamp was used to determine the kinetics of inactivation and the voltage dependence of activation. Oocytes were clamped at a holding potential of −100 mV, and currents were elicited by 35 msec depolarizations ranging from −90 to +55 mV in 5 mV increments using P/4 subtraction. Inactivation time constants were obtained by using the Chebyshev method to fit the current traces with a double-exponential equation,A1 · exp{−(t − K)/τ1} + A2 · exp{−(t − K)/τ2} + C, whereA1 and A2 represent the proportion of channels inactivating with time constants τ1 and τ2, respectively, K is the time shift, and C is the steady-state asymptote. Conductance values were calculated using the formula G = I/(V − Vr), whereG is conductance, I is current amplitude,V is the depolarized membrane potential, andVr is the reversal potential for sodium. Reversal potentials were individually estimated for each data set by fitting the I–V data as described previously (Kontis and Goldin, 1993). Conductance values were fit with a two-state Boltzmann equation, G = 1/(1 + exp[−0.03937 · z · (V − V1/2)]), where z is the apparent gating charge, V is the command potential, andV1/2 is the voltage required for half-maximal activation.
The two-electrode voltage clamp was used to record currents for the voltage dependence of inactivation and recovery from inactivation. All analyses were performed after subtraction of currents recorded in the presence of 400 nm TTX. The voltage dependence of inactivation was determined using 198.4 msec conditioning prepulses from −100 to 15 mV in 5 mV increments, followed by a test pulse to −5 mV for 24.8 msec. Peak current amplitudes measured during the test depolarization were normalized to the maximum current amplitude and are plotted as a function of the conditioning prepulse potential. The data were fit with a two-state Boltzmann equation, I = 1/(1 + exp[(V − V1/2)/a]), where I is current, V is the prepulse depolarization potential,a is the slope factor, and V1/2 is the voltage depolarization required for half-maximal inactivation. To measure recovery from inactivation, oocytes were held at −100 mV. A 40 msec prepulse to −10 mV was followed by a variable recovery period from 30 to 1 msec, followed by a 12 msec test pulse to 0 mV for the mutant or −10 mV for wild-type (the peak of theI–V curve for each channel). The recovery interval was at a potential of −120, −100, or −80 mV. Peak current amplitudes during the test pulse were normalized to the corresponding peak current amplitudes during the prepulse and plotted as a function of the duration of the recovery interval. The data were fit to the single-exponential equation, 1 − (a · exp(−t/τ1), or the double-exponential equation, 1 − [(a · exp(−t/τ1) + b · exp(−t/τ2)], wherea and b represent the proportion of recovery with time constants τ1 and τ2, respectively, andt is the recovery interval.
RESULTS
Scn8a transcripts inmedjo brain
Northern analysis of brain poly(A+) RNA was carried out using an Scn8a cDNA probe. Two transcripts of ∼10 and 12 kb are present in normal C57BL/6J brain (Burgess et al., 1995) (Fig.1). Both transcripts are also present in RNA from homozygous mutant mice (Fig. 1). Amplification by RT-PCR using six primer pairs that span the 5.2 kb coding region (Burgess et al., 1995;Kohrman et al., 1996) did not detect any minor differences in the length of normal and mutant transcripts (data not shown).
Fig. 1.
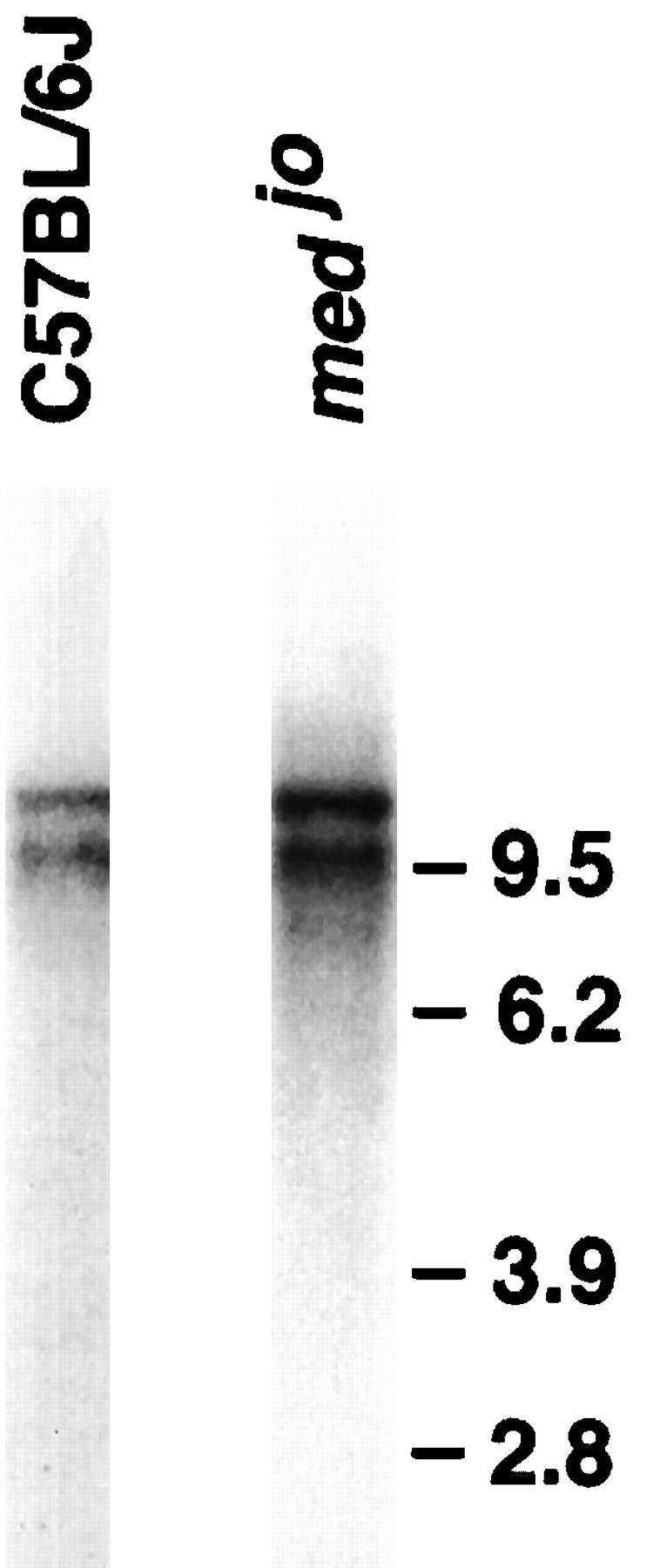
Scn8a transcripts are present injolting brain. Two micrograms of poly(A+) RNA from homozygous mutant and wild-type brain were analyzed by Northern blotting using an Scn8a cDNA probe, as described in Materials and Methods. The wild-type C57BL/6J lane fromBurgess et al. (1995) is reproduced here for comparison with the mutant.
Sequence of the Scn8a transcript frommedjo
RT-PCR products spanning the coding region of Scn8a were purified, and both strands were sequenced and compared with the previously reported wild-type cDNA (Burgess et al., 1995). The nucleotide transition G(3211)A, which is predicted to replace the Ala at residue 1071 with Thr, was identified in the mutant transcript (Fig.2A). The mutation was confirmed by amplifying and sequencing the corresponding exon from genomic DNA as described in Materials and Methods.
The jolting mutation arose in inbred DBA/2WyDi mice (Dickie, 1965), a substrain closely related to DBA/2J. The RT-PCR product of brain RNA from DBA/2J contains the wild-type G(3211) nucleotide (Fig.2A), indicating that the substitution was not present on the progenitor chromosome. The wild-type G(3211) was also detected in eight other mouse strains (data not shown).
Three additional nucleotide substitutions were identified in the coding region of the jolting transcript. Two of these, T(2622)C and A(4947)G, are silent at the amino acid level. The G(13)C substitution changes the Leu at residue 5 to Val. Val was also identified at residue 5 in normal mice of strain DBA/2J and in the wild-type human SCN8A gene (our unpublished observations), indicating that this is a neutral polymorphism. No other differences were observed between the Scn8a transcript in jolting and wildtype mice.
The jolting mutation is located in the evolutionarily conserved S4–S5 linker of domain III
The Ala(1071)Thr substitution is located in a region of the ion channel that is predicted to be the cytoplasmic linker between integral membrane segments S4 and S5 in domain III (Fig. 2B). This linker region is highly conserved in sodium channels from vertebrates and invertebrates (Table 1). The Ala1071 residue is invariant among the voltage-gated sodium channels that have been sequenced, with the single exception of a sodium channel from jellyfish. The functional importance of this region is also demonstrated by the association of mutations in the III S4–S5 linker of SCN4A with inherited muscle disorders (McClatchey et al., 1992; Ptacek et al., 1994) and the altered channel properties resulting from mutagenesis of the voltage-gated potassium channel (Isacoff et al., 1991; McCormack et al., 1991).
Table 1.
Evolutionary conservation of the S4–S5 linker in domain 3 of the voltage-gated sodium channel α subunit gene family

The position of the Thr substitution in the joltingmutant is marked with a “T.”
Electrophysiological analysis of mutant and wild-type channels
To analyze the electrophysiological effects of thejolting mutation, the Ala→Thr substitution in the III S4–S5 linker was introduced by site-directed mutagenesis into the rat brain type IIA sodium channel (RIIA) (Auld et al., 1990). RIIA was used in these experiments because attempts to clone the full-length Scn8a channel have thus far been unsuccessful. The close evolutionary relationship between RIIA and Scn8a is indicated by their amino acid identity at 16/17 residues in the III S4–S5 linker (Table 1), 56/59 residues in the intracellular loop between domains III and IV, and 462/496 residues in the 24 transmembrane segments. The Ala at position 1071 of Scn8a corresponds to the Ala at position 1329 of RIIA. RIIA RNA containing the A1329T mutation was generated as described in Materials and Methods, transcribed in vitro, and coexpressed with sodium channel β1 RNA (Isom et al., 1992) inXenopus oocytes. Electrophysiological properties of the channel were examined using the two-electrode voltage clamp and the cut-open oocyte voltage clamp. The two channel properties that were considered most likely to be altered by the mutation, based on previous studies of other channels, were the kinetics of inactivation and the voltage dependence of channel gating.
Inactivation kinetics are not altered by the Ala→Thr mutation
To determine whether the Ala→Thr mutation altered the kinetics of sodium channel inactivation, macroscopic currents were recorded using a cut-open oocyte voltage clamp. This clamp has a faster time resolution than the two-electrode voltage clamp and, therefore, it is a more sensitive system to detect differences in kinetics. Representative current traces for depolarizations to −10 mV for the A1329T mutant and wild-type channels are shown in Figure 3A. Inactivation time constants were determined by fitting the current traces to a double-exponential equation, as described in Materials and Methods. No significant differences between mutant and wild-type channels were detected in the fast component of inactivation (Fig.3B, open symbols), which accounted for ∼75% of channel inactivation. Approximately 25% of inactivation occurred with a slower time constant (Fig. 3B, closedsymbols), and only a slight difference was observed at more negative potentials between the mutant and wild-type channels. Recovery from inactivation was examined using the two-electrode voltage clamp as described in Materials and Methods. Recovery from inactivation was similar for the mutant and wild-type channels at recovery potentials of either −100 or −80 mV (Fig. 3C). Recovery was also similar at −120 mV (data not shown). These results indicate that the Ala→Thr substitution does not significantly alter sodium channel inactivation kinetics.
Fig. 3.

Inactivation kinetics of wild-type and mutant channels. A, Representative current traces for mutant and wild-type channels at a depolarization of −10 mV. The cut-open voltage clamp was used to record currents from a holding potential of −100 mV to a depolarizing potential of −10 mV, using P/4 subtraction.B, Inactivation time constants are shown for mutant (squares) and wild-type (circles) channels. Current traces were fit with a double-exponential as described in Materials and Methods. Approximately 75% of inactivation occurred with a fast time constant (open symbols), and ∼25% of inactivation occurred with a slow time constant (closed symbols). Symbols represent the means, and bars indicate SDs from 5 oocytes. C, Recovery from inactivation at a recovery potential of either −100 mV (open symbols) or −80 mV (closed symbols) is shown for mutant (squares) and wild-type (circles) channels. The two-electrode voltage clamp was used to record currents for recovery from inactivation as described in Materials and Methods. The data were fit with either a single [1 − (a · exp(−t/τ))] or double (1 − [(a1 · exp(−t/τ1) + a2 · exp(−t/τ2)]) exponential equation, as described in Materials and Methods. Symbols represent the means, and bars indicate SDs from 5 oocytes. The parameters of the fit at −100 mV are: mutant,a1 = 0.90, τ1 = 3.0 msec,a2 = 0.10, τ2 = 24.3 msec; wild-type, a1 = 0.93, τ2 = 3.0 msec, a2 = 0.07, τ2 = 64.6 msec. The parameters at −80 mV are: mutant, a = 1.0, τ = 8.5 msec; wild-type, a1 = 0.91, τ1 = 7.24 msec, a2 = 0.09, τ2 = 25 sec.
Voltage dependence of activation and inactivation is shifted in the depolarizing direction by the Ala→Thr mutation
To examine the effects of the Ala→Thr mutation on the voltage dependence of sodium channel activation, the cut-open oocyte voltage clamp was used to measure peak current at test potentials ranging from −100 to 30 mV. The peak current was then converted to conductance, as described in Materials and Methods, and the relationship between normalized conductance and voltage was fit with a Boltzmann distribution (Fig. 4A). TheV1/2 for the wild-type channel was −23.9 ± 1.9 mV (circles). The A1329T mutant demonstrated a 14 mV shift in the depolarizing direction, with V1/2 = −9.8 ± 1.5 mV (squares). There was no significant difference in the slope for the two channels (z = 4.4 ± 0.8 for the wild-type channel; z = 4.1 ± 0.6 for the mutant channel). This shift in the voltage dependence of activation would result in a significant decrease in the percentage of mutant channels that open in response to a given depolarization compared to wild-type.
Fig. 4.
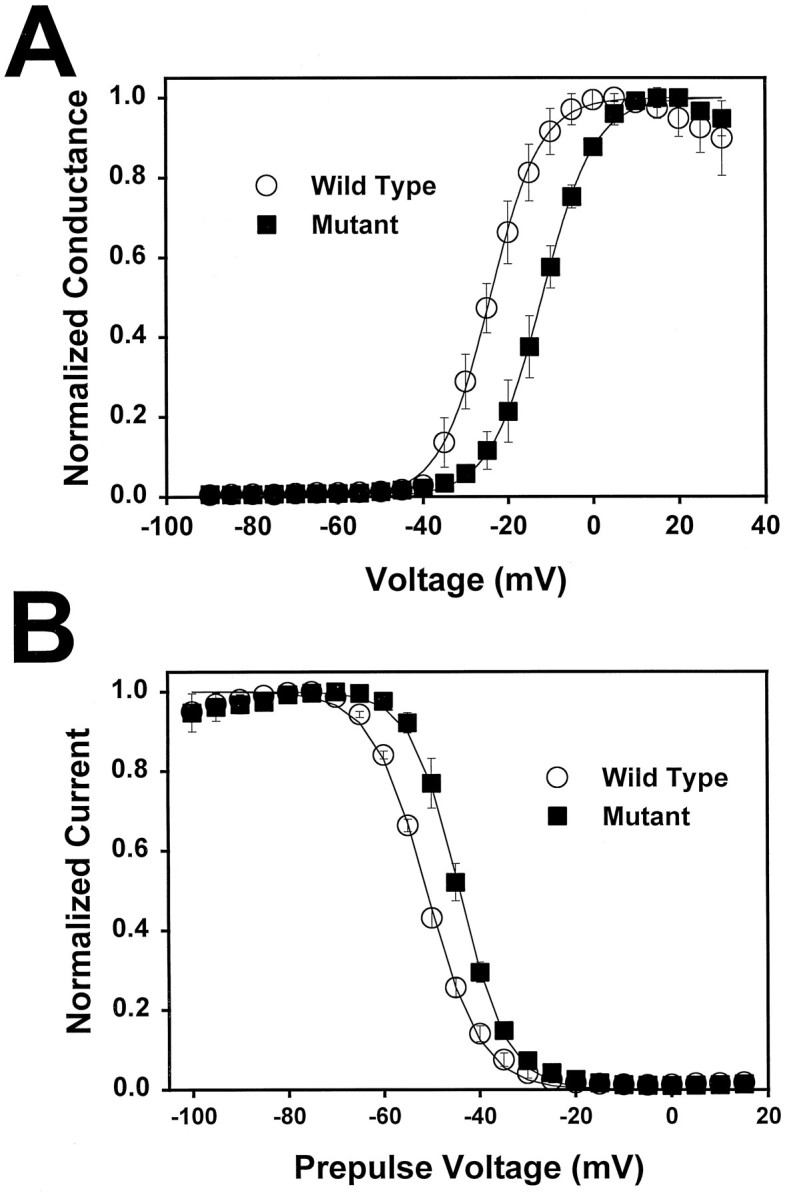
Voltage dependence of activation and inactivation of wild-type and mutant channels. A, Conductance values for mutant (squares) and wild-type (circles) channels were determined and plotted as a function of depolarizing voltage, and the data were fit with a two-state Boltzmann equation, as described in Materials and Methods. The mutant channels (V1/2 = −9.8 ± 1.5 mV) demonstrated a 14 mV shift in the voltage dependence of activation compared to the wild-type (V1/2 = −23.9 ± 1.9 mV). Symbols represent the means, and bars indicate SDs from 5 oocytes. B, The voltage dependence of inactivation was determined using a 198.4 msec inactivating prepulse, as described in Materials and Methods. Peak current amplitudes for mutant (squares) and wild-type (circles) channels were measured during the test depolarization, normalized to the maximum amplitude, and plotted as a function of the prepulse potential. The data were fit with a two-state Boltzmann equation as described in Materials and Methods. The mutant channels (V1/2 = −44.3 ± 0.9 mV) demonstrated a 7 mV shift in the depolarizing direction compared to the wild-type channels (V1/2 = −51.1 ± 0.6 mV). Symbols represent the means, and bars indicate SDs from 5 oocytes.
Because activation and inactivation are coupled in the voltage-gated sodium channel, it was likely that there would be a similar shift in the voltage dependence of inactivation in the mutant channel. The voltage dependence of inactivation was recorded using a two-electrode voltage clamp, and a similar shift in the depolarizing direction was observed (Fig. 4B). The V1/2for the mutant channel was −44.3 ± 0.9 mV (squares) compared to −51.1 ± 0.6 mV for the wild-type channel (circles). As with the voltage dependence of activation, there was no significant difference in the slope of the curves (a = 5.7 ± 0.3 mV for the wild-type channel;a = 5.0 ± 0.3 mV for the mutant channel). These results indicate that the voltage dependence of both activation and inactivation is shifted by the Ala→Thr mutation in the S4–S5 region of domain 3.
DISCUSSION
The single nucleotide substitution that changes the nonpolar Ala at residue 1071 to a polar Thr residue in Scn8a appears to be sufficient to account for the mutant phenotype inmedjo mice. This Ala residue is invariant in all of the mouse strains examined, including the progenitor strain DBA/2, and is conserved in voltage-gated sodium channel α subunits from evolutionarily distant organisms. The S4–S5 region surrounding this position is also highly conserved. The S4–S5 region has been predicted to form part of the docking site for the fast inactivation particle in potassium (Isacoff et al., 1991) and sodium (E. Smith and A. Goldin, unpublished data) channels. It seemed likely, therefore, that the Ala→Thr mutation in medjo mice would alter the kinetics of sodium channel inactivation. However, we have shown that the Ala→Thr substitution does not affect the kinetics of inactivation or recovery from inactivation. Rather, this substitution alters the voltage dependence of both activation and inactivation, resulting in a depolarizing shift in theV1/2 for activation and inactivation with no significant change in the slope. These results indicate that the mutant channel is more stable in the closed state than is the wild-type channel, so that a stronger depolarization across the cell membrane is necessary to elicit opening of the mutant channel. This decrease in voltage sensitivity results in a reduction in the number of channels opening at negative membrane potentials.
Previous in vitro studies on jolting cerebellar slices demonstrated that cerebellar Purkinje cells could be excited antidromically by stimulating the Purkinje cell axon, either directly via an intracellular electrode or indirectly via stimulation of the climbing fibers in the cerebellar white matter (Harris et al., 1992). These data suggested that the primary abnormality inmedjo mice was the inability of Purkinje cells to generate spontaneously a repetitive Na+-dependent action potential. The demonstration that the genetic abnormality is a mutant Scn8a sodium channel, and that this mutation causes an increase of ∼14 mV in the activation threshold, is entirely consistent with this model. Such an abnormality would reduce the probability of Purkinje cell firing without necessarily affecting the ability of the cell to generate an action potential if appropriately stimulated.
Reduction in Purkinje cell firing would cause a loss of the major electrical output from the cerebellum, which depends on the repetitive and continuous discharge of Purkinje cells. The frequency of discharge is determined by the spontaneous endogenous activity of the Purkinje cell soma and modulation by the numerous inputs received from climbing fibers and the parallel fibers of the cerebellar granule cells. The effects of Purkinje cell discharge are inhibitory in nature. A decrease in the inhibitory signal from the cerebellum results in loss of control of rapid movements and poor correlation between posture and movement (Ito, 1982). In the jolting mutant, the predicted increase in voltage threshold for Scn8a could account for the reduced spontaneous activity of the Purkinje cells, reduced inhibitory output from the cerebellum, and loss of motor control characteristic of cerebellar ataxia.
The loss of Scn8a by null mutations results in failure of evoked neurotransmitter release at the neuromuscular junction and functional denervation of skeletal muscles (Duchen and Stefani, 1971; Harris and Pollard, 1986; Burgess et al., 1995; Kohrman et al., 1996). In contrast, the missense mutation in jolting mice does not affect the neuromuscular junction, although Purkinje cell activity is reduced (Harris and Pollard, 1986). These observations suggest that in motor neurons Scn8a functions primarily in the propagation of action potentials, whereas in the cerebellum it is also required for initiation of action potentials. The “all-or-none” action potentials in the motor nerve axons are apparently unaffected by the small change in the voltage dependence of sodium channel activation. In Purkinje cells, however, electrical signaling results from subtle temporal and spatial summations of incoming signals (Llinas and Walton, 1990). The higher-voltage activation threshold of the mutant sodium channel may reduce the likelihood of initiation of an action potential in response to the low-amplitude inputs from the mossy fiber/granule cell system.
Most of the previously identified mutations in the muscle sodium channel SCN4A alter the kinetics of inactivation and produce dominantly inherited disorders (for review, see Cannon, 1996), whereas the ataxia in jolting mice is a recessively inherited trait. This difference can be explained by the predicted effects of the various mutant channels on the activity of wild-type channels in heterozygous cells. Incomplete inactivation of the mutant SCN4A channels after a depolarization results in a continued inward current that is predicted to increase the resting membrane potential of the cell (Cannon et al., 1993). In heterozygous cells, the increased resting membrane potential would inactivate the wild-type channels, preventing channel opening in response to subsequent signals. The shift in the voltage dependence of activation of the jolting channel, on the other hand, would not prevent the normal activation of wild-type channels in the same cells. The normal input to the Purkinje cell, therefore, would be adequate to open the wild-type channels in heterozygotes, which would further depolarize the membrane, resulting in opening of the mutant channels; there would be no defect in the ability of the Purkinje cell to reach the activation threshold necessary to initiate an action potential. In jolting homozygotes, a larger input from the mossy fiber/granule cell system would be necessary to elicit an action potential. The Purkinje cells, therefore, would be less excitable, resulting in reduced output and cerebellar ataxia. We have not ruled out the possibility of additional in vivo effects of the missense mutation on protein folding, stability, or subcellular localization.
The identification of the Scn8a mutation in jolting mice brings to eight the number of genes associated with inherited cerebellar ataxias. The heterogeneity in clinical phenotypes is reflected in the variety of genetic mechanisms underlying these disorders. Mutations of the voltage-gated potassium channel gene KCN1A that alter voltage dependence and gating kinetics were identified inpatients with periodic ataxia (Browne et al., 1994). A missense mutation in a pore-forming domain of the inwardly rectifying potassium channel GIRK2 is responsible for abnormal development of the cerebellum in the mouse mutant weaver (Patil et al., 1995; Slesinger et al., 1996). A deletion in the retinoic acid receptor gene RORα has been associated with cerebellar dysfunction in the mousestaggerer mutant (Hamilton et al., 1996). Human spinocerebellar ataxia types I and III (SCAI and SCAIII) result from expansion of triplet repeat sequences in the coding regions of genes of unknown function (Orr et al., 1993; Schols et al., 1995). Mutations of genes of unknown function have also been associated with the mouse mutant reeler (D’Arcangelo et al., 1995) and the human disorder Friedrich’s ataxia (Campuzano et al., 1996). The human SCN8A gene, which we mapped to chromosome 12q13 (Burgess et al., 1995), is a likely candidate for neurological disorders that may be mapped to this location in the future.
The identification of a sodium channel mutation inmedjo mice associates Scn8a with the simple spike discharges in Purkinje cells. Evidence from this study suggests that the cerebellar ataxia in jolting mice is caused by a shift in the voltage dependence of activation of the sodium channels responsible for initiating action potentials in Purkinje cells. Intracellular recording from Purkinje cells, granule cells, and the cerebellar nuclei of jolting mice could provide further insight into the role of Scn8a in cerebellar function. The widespread distribution of Scn8a in the brain (Schaller et al., 1995) suggests that additional functional defects may be identified injolting mice. These mice also provide a model for evaluating the therapeutic potential of pharmacological agents that alter the voltage dependence of channel gating, such as β scorpion toxin, and for assessment of the role of Scn8a in various forms of cerebellar learning.
Footnotes
This work was supported by National Institutes of Health Grants GM24872 (M.H.M.), NS34509 (M.H.M.), and NS26729 (A.L.G.), and by the National Multiple Sclerosis Society (A.L.G.). A.L.G. is an Established Investigator of the American Heart Association. We thank Dr. Leslie Sprunger for helpful discussions and Jane Santoro for expert assistance with manuscript preparation.
Correspondence should be addressed to Dr. Miriam Meisler, Department of Human Genetics, 4708 Medical Science II, University of Michigan, Ann Arbor, MI 48109-0618.
REFERENCES
- 1.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 2.Auld VJ, Goldin AL, Krafte DS, Catterall WA, Lester HA, Davidson N, Dunn RJ. A neutral amino acid change in segment IIS4 dramatically alters the gating properties of the voltage-dependent sodium channel. Proc Natl Acad Sci USA. 1990;87:323–327. doi: 10.1073/pnas.87.1.323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Browne DL, Gancher ST, Nutt JG, Brunt ER, Smith EA, Kramer P, Litt M. Episodic ataxia/myokymia syndrome is associated with point mutations in the human potassium channel gene KCN1A . Nature Genet. 1994;8:136–140. doi: 10.1038/ng1094-136. [DOI] [PubMed] [Google Scholar]
- 4.Burgess DL, Kohrman DC, Galt J, Plummer NW, Jones JM, Spear B, Meisler MH (1995) Mutation of a new sodium channel gene,Scn8a, in the mouse mutant “motor endplate disease.” Nature Genet 10:461–465. [DOI] [PubMed]
- 5.Campuzano V, Montermini L, Molto MD, Pianese L, Cossee M, Cavalcanti F, Monros E. Friedreich’s ataxia: autosomal recessive disease caused by an intronic GAA triplet repeat expansion. Science. 1996;271:1423–1427. doi: 10.1126/science.271.5254.1423. [DOI] [PubMed] [Google Scholar]
- 6.Cannon SC. Ion-channel defects and aberrant excitability in myotonia and periodic paralysis. Trends Neurosci. 1996;19:3–10. doi: 10.1016/0166-2236(96)81859-5. [DOI] [PubMed] [Google Scholar]
- 7.Cannon SC, Brown RH, Jr, Corey DP. Theoretical reconstruction of myotonia and paralysis caused by incomplete inactivation of sodium channels. Biophys J. 1993;65:270–288. doi: 10.1016/S0006-3495(93)81045-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Catterall WA. Cellular and molecular biology of voltage-gated sodium channels. Physiol Rev. 1992;72:S15–S48. doi: 10.1152/physrev.1992.72.suppl_4.S15. [DOI] [PubMed] [Google Scholar]
- 9.D’Arcangelo G, Miao GG, Chen SC, Soares HD, Morgan JI, Curran T. A protein related to extracellular matrix proteins deleted in the mouse mutant reeler. Nature. 1995;374:719–23. doi: 10.1038/374719a0. [DOI] [PubMed] [Google Scholar]
- 10.Dick DJ, Boakes RJ, Harris JB. A cerebellar abnormality in the mouse with motor end-plate disease. Neuropathol Appl Neurobiol. 1985;11:141–147. doi: 10.1111/j.1365-2990.1985.tb00011.x. [DOI] [PubMed] [Google Scholar]
- 11.Dickie MM. Jolting. Mouse News Lett. 1965;32:44. [Google Scholar]
- 12.Duchen LW, Stefani E. Electrophysiological studies of neuromuscular transmission in hereditary “motor endplate disease” of the mouse. J Physiol (Lond) 1971;212:535–548. doi: 10.1113/jphysiol.1971.sp009340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Goldin AL. Expression of ion channels by injection of mRNA into Xenopus oocytes. Methods Cell Biol. 1991;36:487–509. doi: 10.1016/s0091-679x(08)60293-9. [DOI] [PubMed] [Google Scholar]
- 14.Hamilton BA, Frankel WN, Kerrebrock AW, Hawkins TL, FitzHugh W, Kusumi K, Russell LB, Mueller KL, van Berkel V, Birren BW, Kruglyak L, Lander ES. Disruption of the nuclear hormone receptor RORα in staggerer mice. Nature. 1996;379:736–739. doi: 10.1038/379736a0. [DOI] [PubMed] [Google Scholar]
- 15.Harris JB, Pollard SL (1986) Neuromuscular transmission in the murine mutants “motor end-plate disease” and “jolting.” J Neurol Sci 76:239–253. [DOI] [PubMed]
- 16.Harris JB, Boakes RJ, Court JA (1992) Physiological and biochemical studies on the cerebellar cortex of the murine mutants “jolting” and “motor end-plate disease.” J Neurol Sci 110:186–194. [DOI] [PubMed]
- 17.Isacoff EY, Jan YN, Jan LY. Putative receptor for the cytoplasmic inactivation gate in the Shaker K+channel. Nature. 1991;353:86–90. doi: 10.1038/353086a0. [DOI] [PubMed] [Google Scholar]
- 18.Ito M. Raven; New York: 1982. The cerebellum and neural control. . [Google Scholar]
- 19.Kohrman DC, Plummer NW, Schuster T, Jones JM, Jang W, Burgess DL, Galt J, Spear BT, Meisler MH. Insertional mutation of the motor endplate disease (med ) locus on mouse chromosome 15. Genomics. 1995;26:171–177. doi: 10.1016/0888-7543(95)80198-u. [DOI] [PubMed] [Google Scholar]
- 20.Kohrman DC, Harris JB, Meisler MH. Mutation detection in the med and medj alleles of the sodium channel Scn8a : unusual splicing due to a minor class AT–AC intron. J Biol Chem. 1996;271:17576–17581. doi: 10.1074/jbc.271.29.17576. [DOI] [PubMed] [Google Scholar]
- 21.Kontis KJ, Goldin AL. Site-directed mutagenesis of the putative pore region of the rat IIA sodium channel. Mol Pharmacol. 1993;43:635–644. [PubMed] [Google Scholar]
- 22.Kunkel TA. Rapid and efficient site-specific mutagenesis without phenotypic selection. Proc Natl Acad Sci USA. 1985;82:488–492. doi: 10.1073/pnas.82.2.488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Llinas R, Sugimori M. Electrophysiological properties of in vitro Purkinje cell somata in mammalian cerebellar slices. J Physiol (Lond) 1980a;305:171–195. doi: 10.1113/jphysiol.1980.sp013357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Llinas R, Sugimori M. Electrophysiological properties of in vitro Purkinje cell dendrites in mammalian cerebellar slices. J Physiol (Lond) 1980b;305:197–203. doi: 10.1113/jphysiol.1980.sp013358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.McClatchey AI, Lin CS, Wang J, Hoffman EP, Rojas C, Gusella JF. The genomic structure of the human skeletal muscle sodium channel gene. Hum Mol Genet. 1992;1:521–527. doi: 10.1093/hmg/1.7.521. [DOI] [PubMed] [Google Scholar]
- 26.McCormack K, Tanouye MA, Iverson LE, Lin J-W, Ramaswami M, McCormack T, Campanelli JT, Mathew MK, Rudy B. A role for hydrophobic residues in the voltage-dependent gating of Shaker K+ channels. Proc Natl Acad Sci USA. 1991;88:2931–2935. doi: 10.1073/pnas.88.7.2931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Patil N, Cox DR, Bhat D, Faham M, Myers RM, Peterson AS. A potassium channel mutation in weaver mice implicates membrane excitability in granule cell differentiation. Nature Genet. 1995;11:126–129. doi: 10.1038/ng1095-126. [DOI] [PubMed] [Google Scholar]
- 28.Patton DE, Goldin AL. A voltage-dependent gating transition induces use-dependent block by tetrodotoxin of rat IIA sodium channels expressed in Xenopus oocytes. Neuron. 1991;7:637–647. doi: 10.1016/0896-6273(91)90376-b. [DOI] [PubMed] [Google Scholar]
- 29.Ptácek LJ, Tawil R, Griggs RC, Meola G, McManis P, Barohn RJ, Mendell JR, Harris C, Spitzer R, Santiago F, Leppert MF. Sodium channel mutations in acetazolamide-responsive myotonia congenita, paramyotonia congenita, and hyperkalemic periodic paralysis. Neurology. 1994;44:1500–1503. doi: 10.1212/wnl.44.8.1500. [DOI] [PubMed] [Google Scholar]
- 30.Schaller KL, Krzemien DM, Yarowsky PJ, Krueger BK, Caldwell JH. A novel, abundant sodium channel expressed in neurons and glia. J Neurosci. 1995;15:3231–3242. doi: 10.1523/JNEUROSCI.15-05-03231.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sidman RL, Cowen JS, Eicher EM. Inherited muscle and nerve diseases in mice: a tabulation and commentary. Ann NY Acad Sci. 1979;317:497–505. doi: 10.1111/j.1749-6632.1979.tb56567.x. [DOI] [PubMed] [Google Scholar]
- 32.Slesinger PA, Patil N, Liao J, Jan YN, Jan LY, Cox DR. Functional effects of the mouse weaver mutation on G protein-gated inwardly rectifying K+channels. Neuron. 1996;16:321–331. doi: 10.1016/s0896-6273(00)80050-1. [DOI] [PubMed] [Google Scholar]
- 33.Taglialatela M, Toro L, Stefani E. Novel voltage clamp to record small, fast currents from ion channels expressed in Xenopus oocytes. Biophys J. 1992;61:78–82. doi: 10.1016/S0006-3495(92)81817-9. [DOI] [PMC free article] [PubMed] [Google Scholar]