

Abstract
We investigated actions of somatostatin (Som) on voltage-gated calcium channels in acutely isolated rat amygdaloid neurons. Somatostatin caused a dose-dependent inhibition of the high voltage-activated (HVA) Ca2+ current, with little or no effect on the low voltage-activated (LVA) current. Nifedipine (2–10 μm) reduced the peak current by ∼15% without reducing inhibition of current by Som significantly, ruling out L-type channels as the target of modulation. The modulation appears to involve N- and P/Q-type calcium channels. After pretreatment with ω-conotoxin-GVIA (ω-CgTx) or ω-agatoxin-IVA, the inhibition was reduced but not abolished, whereas the combined application of both toxins nearly abolished the modulation. The Som analog BIM-23060 mimicked the effects of Som, whereas BIM-23058 had no effect, implicating Som type-2 receptors (SSTR-2). The inhibition was voltage-dependent, being minimal for small depolarizations, and was often accompanied by a slowing of the activation time course. Strong depolarizing prepulses partially relieved the inhibition and restored the time course of activation. Intracellular dialysis with GTPγS led to spontaneous inhibition and a slowing of the current like that with Som and occluded the effects of the peptide. Dialysis with GDPβS also diminished the inhibition. A short preincubation with 50 μm of the alkylating agentN-ethylmaleimide (NEM) prevented the action of somatostatin. These results suggest a role for NEM-sensitive G-proteins in the Som inhibition. Application of 8-CPT-cAMP and IBMX did not mimic or prevent the effects of Som.
Keywords: amygdala, neuropeptide, G-protein, NEM, somatostatin, calcium channels, limbic system
Somatostatin (Som), a tetradecapeptide initially described to inhibit release of growth hormone from the pituitary (Brazeau et al., 1973), is also widely expressed in the nervous system and has been shown to have modulatory actions in the brain (for review, see Epelbaum, 1986; Inoue and Yoshii, 1992). Actions of Som include the modulation of potassium (Scharfman and Schwartzkroin, 1988; Schweitzer et al., 1993) and calcium channels in peripheral (Ikeda and Schofield, 1989; Beech et al., 1992; Golard and Siegelbaum, 1993; Shapiro and Hille, 1993; Meriney et al., 1994) and central neurons (Sah, 1990; Wang et al., 1990; Ishibashi and Akaike, 1995). The multiple actions of Som appear to be mediated by a family of G-protein-coupled receptors (for review, see Bell and Reisine, 1993; Hoyer et al., 1995). Of the five classes of Som receptors cloned to date, four are expressed in the CNS and two, SSTR-2 and SSTR-3, are highly expressed within the amygdala (Kong et al., 1994).
The amygdala is formed by a group of nuclei located in the basal forebrain and directly connected with other components of the limbic system: hippocampus and entorhinal cortex. Its functions remain poorly understood, but different experimental approaches suggest crucial roles in emotions (e.g., fear, anxiety) (Adolphs et al., 1994), social behavior, learning, and memory (for review, see Aggleton, 1993; Davis et al., 1994). Dysfunctions of the amygdala have also been linked to neurodegenerative disorders, kindling, and temporal lobe epilepsy. Two main, anatomical classes of neurons have been described in the amygdala. Projection neurons are large, pyramidal or stellate-shaped cells that send their axons to targets outside the amygdala; their axons also branch into numerous collaterals to influence neighboring cells within the amygdala. A second main group of cells is formed by smaller local-circuit interneurons containing GABA and choline acetyltransferase. Many local interneurons are also immunoreactive for various neuropeptides, including Som (consult references in Roberts et al., 1982). Indeed, Som is distributed throughout the amygdala, and some nuclei contain the highest concentration of Som in the brain outside the hypothalamus (Leroux et al., 1993). Som may function as an inhibitory neurotransmitter within the amygdala because its injection can prevent the spread of temporal seizures (Higuchi et al., 1986).
In comparison with other CNS structures, and despite its potential clinical relevance for the treatment of different anxiety disorders (e.g., post-traumatic stress syndrome, panic attacks), little is known about the ionic currents present in amygdala neurons and their possible modulation. Only recently have data emerged on the intrinsic membrane properties of the different classes of neurons within the amygdala (Washburn and Moises, 1992; Sugita et al., 1993). In a brief note, Foehring and Scroogs (1994) described the presence of several pharmacological subtypes of Ca2+ channels in rat amygdala neurons.
Here we characterize the effects of Som on voltage-activated Ca2+ channels in acutely dissociated neurons of the rat amygdala. We discriminate the actions on different classes of Ca2+ channels. We also analyzed the possible role of G-proteins in the modulatory actions of Som and the involvement of specific receptor subtypes.
MATERIALS AND METHODS
Acutely dissociated neurons were obtained from the amygdala complex of 1- to 3-week-old Sprague Dawley rats. Under methoxyflurane anesthesia, rats were rapidly decapitated; the brain was quickly removed, and coronal slices (400–500 μm thick) of the forebrain were cut with a Vibroslicer in a bicarbonate-buffered saline (BBS; 4°C) that contained (in mm): NaCl 130, KCl 3, CaCl21.5, MgCl2 2, NaH2PO4 1.25, NaHCO3 26, glucose 10. Three to four slices were allowed to recover for 1 hr at room temperature in the same BBS saline bubbled with carbogen (95% CO2/5% O2). The amygdala was dissected from the temporal lobe under a binocular microscope (20×) with a home-built micropuncher, consisting of a sharpened 18-gauge stainless steel tube (outer diameter 1.2 mm). One or two tissue pieces containing the amygdala were incubated in an oxygenated Cell-Stir chamber (Wheaton, Millville, NJ) with low-Ca2+HEPES buffer (LCB) containing pronase E (1–2 mg/ml). Histological verification of the punched area confirmed that the tissue was limited to the amygdaloid complex (see Fig.1A), and in no case was the surrounding pyriform cortex involved. The tissue was digested for 20–30 min, rinsed several times in the LCB solution, and triturated by aspiration in a graded series of fire-polished Pasteur pipettes. After calcium (0.5 mm) was restored, 200 μl of the cell suspension was pipetted into four or five recording chambers that were made from 35 mm plastic Petri dishes and a SYLGARD insert to reduce the chamber volume (∼200 μl). One chamber was mounted on the stage of an inverted microscope for recording while the rest were kept under a small hood washed with humidified 100% O2. Dissociated cells were kept for a maximum of 3 hr and, afterward, a new cycle of enzymatic digestion was started.
Fig. 1.
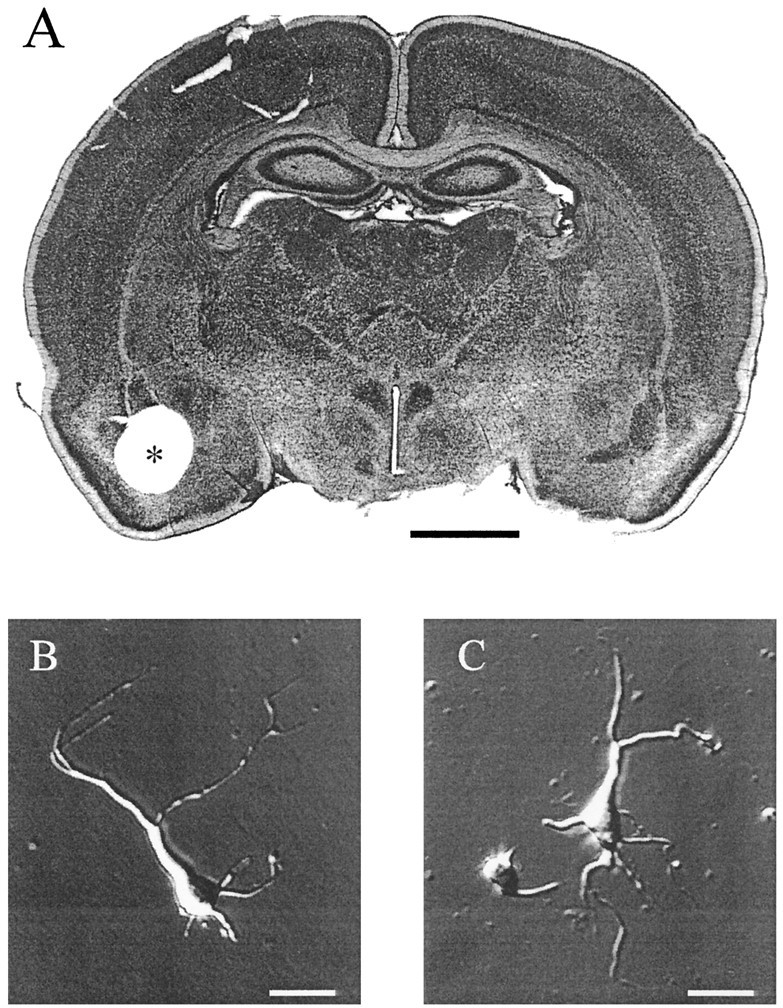
Dissociation of neurons from the rat amygdaloid region. A, Photomicrograph of a Nissl-stained section of the rat forebrain, corresponding approximately to plate 27 of the atlas of Paxinos and Watson (1986). The area punched out for isolation of neurons is marked with an asterisk and is centered on the amygdala complex. The 80-μm-thick section was obtained after fixing and resectioning a freshly cut 500-μm-thick slice. Calibration bar, 1 mm. B, Digitized phase-contrast photomicrograph of a typical pyramidal-shaped neuron obtained after mechanical dissociation of the amygdala tissue. C, An example of a stellate shape neuron from the amygdala region. Scale bars, 30 μm.
Current recordings (20–25°C) were obtained in the standard whole-cell configuration (Hamill et al., 1981) using a List EPC-7 clamp amplifier. Capacity currents were canceled, and series-resistance compensation (40–60%) was normally used. A PC/486 clone computer running BASIC-FASTLAB (Indec Systems, Capitola, CA) was used to generate voltage pulses and to acquire and analyze data. Depolarizations were applied every 5–10 sec, depending on the protocol. Patch pipettes (2–4 MΩ) were pulled from 1.5 mm hematocrit glass (VWR), SYLGARD-coated and fire-polished before use. Recording patch pipettes were filled with (in mm): di-Tris-PO4 70, Tris-base 41, MgCl2 4, EGTA 10, Tris-ATP 4, Tris-GTP 0.3, phosphocreatine 20, and creatine phosphokinase 50 U/ml (pH-adjusted to 7.2 with Tris-OH). Gigaseals between the patch pipette and the cell membrane were obtained in a modified external solution containing (in mm): NaCl 130, KCl 3, MgCl2 2, CaCl2 5, glucose 10, HEPES 10 (pH-adjusted to 7.3 with NaOH). To isolate currents in calcium channels, the extracellular solution was exchanged after seal formation to one containing (in mm): TEA-Cl 150, BaCl2 5, TTX 0.0005, EGTA 0.1, glucose 10, HEPES 10, and albumin 0.2 mg/ml (pH-adjusted to 7.3 with TEA-OH). After patch rupture, the size of barium currents gradually increased for a variable period of time. Experimental tests were not initiated until the size of the current stabilized (usually ∼5 min). The solutions to the chamber were exchanged (1–2 ml/min) through a gravity-fed system consisting of eight reservoirs controlled by solenoid valves. Potentials in the text and figures have not been corrected for a measured junction potential of −5 mV.
The amplitude of the whole-cell Ba2+ current was defined as that sensitive to block by 200–400 μm Cd2+and, unless indicated, all figures display Cd2+-subtracted traces. In a few cases, the leak current was estimated from scaled hyperpolarizing voltage steps (10–20 mV) and subtracted. To avoid complications introduced by the change in current time course induced by the peptides, “peak” current measurements after application of agonists were made in a time window (8–12 msec after the beginning of a step) isochronal to measurements of the control current.
All reagents were of laboratory grade and obtained as follows. Somatostatin was purchased from Peninsula Laboratories (Belmont, CA). The somatostatin analogs BIM-23058 and BIM-23060 were a gift of Dr. R. Murphy (Tulane University). ATP and GTP were purchased from Pharmacia (Piscataway, NJ); pronase E and N-ethylmaleimide (NEM) were from Sigma (St. Louis, MO); guanosine 5′-(γ-thio)triphosphate (GTPγS) and guanosine 5′-O-(2-thiodiphosphate) (GDPβS) were from Boehringer Mannheim (Indianapolis, IN); ω-CgTx was from Peninsula Laboratories and Bachem (King of Prussia, PA); and ω-Aga-IVA was from Peptide Institute Kyoto and Bachem.
All values are given as mean ± SEM and, where appropriate, Student’s t test was used to determine statistical significance (p < 0.05).
RESULTS
The effects of Som were investigated in >100 neurons from the amygdala of young rats. As shown in the stained histological section of Figure 1A, the source of neurons was a small (∼1 mm diameter) chunk of tissue punched out from the temporal pole of freshly cut coronal brain slices. It included several nuclei of the amygdaloid complex but not surrounding cortical regions such as piriform cortex. From various cell shapes obtained after trituration, we selected only large- or medium-size neurons with a pyramidal or stellate morphology. No obvious differences in properties of these two cell types were observed, and the results were pooled. The distribution of whole-cell capacitances was Gaussian with a mean cell capacitance of 16.5 ± 0.5 pF (n = 75). Figure 1, B andC, shows phase-contrast photomicrographs of two typical neurons obtained after mechanical trituration of the tissue.
Under conditions optimized to isolate whole-cell Ba2+currents, steps to −10 mV from a holding potential of −90 mV revealed a net inward current that was completely blocked by application of 400 μm Cd2+ (Fig. 2). Typically, during the first minute of recording the peak inward current grew with a roughly exponential time course (τ = 107 ± 14 sec;n = 14). Application of 200 nm Som produced a reversible inhibition of the peak Ca2+-channel current (Figs. 3B, 5). Som was effective in the great majority of neurons, but the maximal inhibition ofIBa showed considerable variability. In 70 of 72 amygdala neurons, the inhibition of peak IBa was >5%, with a mean of 19.6% ± 1.2. The extent of inhibition was not correlated with the age of the rat (r = −0.18;p > 0.10). The response to long applications (180 sec) of Som showed only a modest desensitization (19.5 ± 1.3%;n = 4), and a second application of the same concentration gave a similar inhibition (data not shown). During brief applications (<1 min), the inhibition could be elicited repeatedly and showed modest tachyphylaxis. In most cells, inhibition of peakIBa was accompanied by a significant slowing of the current activation kinetics (Figs. 3B, 4, 5), characterized by a slow rising phase and no subsequent decay during a 50 msec depolarizing voltage step (Bean, 1989).
Fig. 2.

Cd2+-sensitive barium currents in amygdala. A, Inward currents obtained in 5 mm external Ba2+ (thick traces) by stepping for 100 msec to −40, −10, and 10 mV (topto bottom) from a holding potential of −90 mV. Thesmaller amplitude traces were obtained after application of 400 μm Cd2+ to the bath. B, Current–voltage relationship for the peak barium current in the same neuron.
Fig. 3.

Som inhibition of IBais dose-dependent. A, Effect of increasing concentrations of Som (0.2, 20, and 200 nm) on peakIBa (•) and on the early-current (8–12 msec) to late-current (44 to 48 msec) ratio (▵) during 50 msec depolarizing pulses to −10 mV from a holding potential of −90 mV.B, Representative current traces of same experiment as in A, obtained in control and three concentrations of Som. C, Concentration–response curve for Som inhibition of IBa in amygdala neurons. Inhibition was measured on currents evoked during a depolarizing step to −10 mV from a holding potential of −90 mV. Each point represents the mean ± SEM for 3–14 neurons. The continuous line represents a least-squares fit to the Michaelis–Menten equation (%I = Imax/{EC50 + [Som]}) with parameters: Imax = 23.7% and EC50 = 7.8 nm.
Fig. 5.

Som inhibition of voltage-activated calcium channels in amygdala neurons is voltage-dependent. A, Inward currents elicited by 100 msec voltage steps from a holding potential of −90 mV to three different potentials (−40, −10, and +20 mV) before (thick traces) and during the application of 200 nm Som (thin traces). B, Current–voltage (I–V) curves of the barium currents in control (•) and Som (○) (same experiment asA). The points represent the mean current during a time window 8–12 msec after the onset of the depolarizing pulse. Records in A and B are difference currents after subtraction from currents recorded in 400 μm Cd2+. C, Inhibition of the barium current by Som at different test potentials. Data represent the mean ± SEM of 8 neurons.
Fig. 4.

Som inhibition involves Som type-2 receptors (SSTR-2). Peak current amplitudes elicited by 50 msec pulses to −10 mV delivered every 10 sec from a holding potential of −90 mV. Consecutive applications of the selective SSTR-2 agonist BIM-23060 (20 nm) caused a reversible reduction of the peak current and a slowing of the rate of activation (left inset). After current recovery, application of the SSTR-3 agonist BIM-23058 (20 nm) caused a modest inhibition of the peak current. Subsequent application of som (200 nm) reversibly inhibited the peak current (right inset) to the same degree as 20 nm BIM-23060.
In Figure 3A, three brief applications of increasing concentrations of Som to the same neuron established that the inhibition was dose-dependent. As can be observed in the current traces in Figure 3B, the largest inhibition is also accompanied by the greatest slowing of activation, producing the smallest early-current to late-current ratio (open triangles in Fig.3A; note inverted scale). Figure 3C shows the average dose–response of IBa inhibition to Som. The data points were fitted to the Michaelis–Menten equation, with a Som concentration of ∼8 nm producing half-maximal inhibition. Except where noted, all subsequent experiments were done with 200 nm Som.
The modulation involves Som type-2 receptors (SSTR-2)
To discriminate which receptor subtype(s) is involved inIBa modulation, we tested the effect of peptide analogs that are relatively specific for the two Som receptors most highly expressed in the amygdala. Figure 4 shows that application of the SSTR-2 agonist BIM-23060 (20 nm) produced a marked, reversible inhibition of the peakIBa, with a clear slowing of activation kinetics (left inset). In five neurons tested, the inhibition by 20 nm BIM-23060 was 19.2 ± 6.2%. Subsequent application of BIM-23058 (20 nm), which is ∼1000-fold more selective for SSTR-3 (Raynor et al., 1993a), caused a modest inhibition of the peak current (3.5 ± 1.1%; n = 5). Finally, application of Som reversibly inhibited the peak current (right inset) to the same degree as 20 nmBIM-23060, although the recovery of the response to BIM-23060 was always slower than for Som.
Modulation by Som varies with voltage
Som did not reduce IBa equally at all voltages. Shown in Figure 5A are Ba2+ currents in response to three different voltage steps in control (thick lines) and during application of Som (thin lines). The corresponding current–voltage relationship is illustrated in Figure 5B using current measured at 10 msec. At negative potentials (e.g., −40 mV), the current was small and the effect of Som minimal, consistent with a small effect on the low voltage-activated (LVA) Ca2+channels (see below). The inhibition seemed largest in the voltage range from −20 to 0 mV and smaller again at positive potentials. Figure 5C summarizes the voltage dependence of the Som inhibition for 8 neurons.
High voltage-activated (HVA) calcium channels are the primary target of inhibition
In many central neurons, the modulatory effects of agonists are restricted to the HVA component of Ca2+ current, sparing the LVA fraction (Anwyl, 1991; Allen and Brown, 1993). To examine whether Som selectively inhibited the HVA component ofIBa, we used a three-pulse protocol to isolate the two current components. In many cells, steps to −30 mV evoke a mostly transient IBa from a holding potential of −90 mV and a smaller, nearly noninactivatingIBa from a holding potential of −60 mV (Fig.6A, left). We defined the LVA current as the difference between these two current traces (Fig.6A, left). Finally, an isolated HVAIBa was obtained by a voltage step from a holding potential of −60 to 0 mV (Fig. 6A,right). Figure 6B is the full time course of the same experiment measuring LVA and HVA currents. The insets are traces of LVA and HVA current before and during application of Som. It is clear that the inhibition by Som is primarily on the HVA component of current. Using this protocol, the LVA current was inhibited by 9.2 ± 2% (n = 9), whereas in the same neurons the HVA current was inhibited by 25 ± 2.7%.
Fig. 6.

Som inhibits only the high voltage-activated component of current. A, Three-pulse protocol used for the isolation of the LVA and HVA barium current components. The neuron was held at −90 mV for 5 sec and stepped to −30 mV. After 5 sec holding at −60 mV it was stepped again to −30 mV. The two voltage steps and currents elicited are superimposed on the left side of A. The LVA current was defined as the difference between currents during steps from −90 and from −60 mV. After maintaining a holding of −60 mV for 5 sec, a step to 0 mV elicited a sustained component of current (right side ofA) that represents the HVA current in isolation.B, Effect of Som on the currents elicited by the same three-pulse protocol. During application of Som, the transient LVA component of current did not change (left inset), but the HVA barium current was reversibly reduced (right inset). This reduction was accompanied by a slowing of activation.
Subtypes of calcium channel modulated by Som
Foehring and Scroogs (1994) established that the HVA calcium current in rat amygdala neurons comprises primarily L-, N-, and P/Q-type currents. To determine which component of HVA current is modulated by Som, we tested the ability of specific channel antagonists to block the modulation. In the presence of 2–10 μmnifedipine, Som inhibition of IBa was still robust (Fig. 7A), suggesting that L-type calcium channels are not the target of Som modulation. On average, Som inhibited IBa by 19.2 ± 1.8% (n = 7) after application of nifedipine (Fig.7B). After the combined application of nifedipine and 2 μm ω-CgTx-GVIA, the inhibition ofIBa was reduced but not abolished (9.5 ± 2.2%; n = 6). Figure 7C shows that the P/Q-channel blocker ω-Aga-IVA (100 nm) only partially removed the Som-mediated inhibition. In the same neuron, coapplication of ω-Aga-IVA and the N-channel blocker ω-CgTx-GVIA (2 μm) nearly completely eliminated the Som modulation ofIBa. On average, Som inhibition ofIBa was only 1.0 ± 0.5% (n = 4) after inhibition of P/Q- and N-type channels (Fig. 7B). Thus, N- and P/Q-type channels seem to be the primary targets of modulation.
Fig. 7.

Inhibition by Som involves N- and P/Q-type channels. A, Peak current amplitudes during steps to −10 mV (holding potential, −90 mV) plotted as a function of time. Nifedipine (nif, 2 μm), Som (som, 200 nm), and Cd2+ (400 μm) were applied at times indicated bybars. Application of nifedipine reduced the peak current and changed the time course (see inset) but did not prevent a further reduction by Som. B, The bars represent mean ± SEM inhibition of IBaby Som in control conditions and after application of combinations of the calcium channel blockers nifedipine (2 μm), ω-CgTx-IVA (2 μm), and ω-Aga-IVA (100 nm). The percent inhibition has been calculated referenced to the amplitude of IBa before application of any blocker. C, Peak current amplitudes during a pulse protocol identical to A. The applications of Som (200 nm), ω-Aga-IVA (100 nm), ω-CgTx-IVA (2 μm), nifedipine (2 μm), and Cd2+ (400 μm). The insets show sample traces before Som (thick lines) and during Som (thin lines) at the times indicated. Calibration bars (A, C): 20 msec, 500 pA.
Facilitation of IBa by prepulses
To explore further the voltage dependence of Som effects onIBa, we used a standard double-pulse protocol with a facilitating prepulse before the second pulse (Fig.8B, inset). In neurons recorded with normal pipette solution (0.3 mm GTP), facilitation induced by the 20 msec prepulse to +70 mV was minimal, so the ratio between peak IBa in pulse P2 and peakIBa in pulse P1 was close to 1 (1.07 ± 0.01; n = 12) (open circles in Fig.8A). During Som applicationIBa, tested during pulse P1, was reduced and slowed. However, the 20 msec prepulse to +70 mV partially restored the peak IBa during pulse P2 and sped its time course. Thus, the facilitation ratio increased significantly to 1.26 ± 0.02 (n = 12; p < 0.0001) (Fig. 9B).
Fig. 8.

Som inhibition is voltage-dependent and involves G-proteins. A, Currents elicited in a control cell (300 μm GTP in recording pipette). The voltage protocol is illustrated in B. The filled circlesrepresent peak current amplitudes (measured at 10 msec) elicited by 50 msec pulses to −10 mV delivered every 10 sec, from a holding potential of −90 mV (pulse P1). After 5 sec at −90 mV, a second, identical pulse was applied (pulse P2) but preceded by a 25 msec prepulse to +70 mV and a 10 msec repolarization to −90 mV to close calcium channels. The open circles represent the ratio peak current amplitude elicited by pulse P2 and pulse P1 (facilitation ratio). Application of 200 nm Som (som) caused a strong reduction of the peak current and a slowing of the activation time course in the control pulse (pulseP1). Current inhibition and slowing were substantially reduced when depolarization was preceded by a brief prepulse (pulseP2). B, Currents elicited in a neuron recorded with an electrode containing 50 μm GTPγS (and 300 μm GTP). Identical protocol as in A. Note the substantial facilitation of the current elicited by the prepulse in control conditions (facilitation ratio > 1). Som inhibition of peak current was very modest (left andright insets), and Som did not induce a further facilitation of the current by the prepulse. Calibration bars, 20 msec and 1 nA.
Fig. 9.

GDPβS and GTPγS in the pipette attenuate the response to Som. A, Bars represent the mean inhibition of peak current by Som (som) with the control pipette solution (300 μm GTP) or with 50 μm GTPγS (300 μm GTP), or with 2 mm GDPβS (0 GTP). To minimize errors induced by variable responsiveness to Som in different dissociations, control and test pipettes were alternated.B, Effects of Som on the facilitation ratio in cells recorded with control pipettes (see above) or GTPγS-containing pipettes.
Involvement of G-proteins
We tested for the involvement of G-proteins in several ways. First, we compared the inhibition of cells dialyzed with a pipette solution containing either 0.3 mm GTP (see above) or 2 mm GDPβS, an antagonist of G-protein activation (Eckstein et al., 1979; Holz et al., 1986; Wanke et al., 1987). Cells were recorded alternately with each solution. As summarized in Figure9A, mean inhibition by Som was reduced from 18.2 ± 3.4% (n = 6) in cells recorded with GTP to 7.9 ± 1.0% (n = 6) in those cells recorded with GDPβS. This difference was significant (p < 0.05). Second, we tested the effect of Som in cells dialyzed with GTPγS, a poorly hydrolyzable activator of G-proteins. Most cells dialyzed with mixtures of GTP and GTPγS in molar ratios that varied from 3:1 to 12:1 (keeping GTP constant at 0.3 mm) developed a spontaneous inhibition and slowing of IBa, with almost no further modulation by Som. On average, inhibition by Som in cells dialyzed with GTPγS was only 6.7 ± 0.6% (n = 7), significantly smaller than in cells from the same preparations dialyzed with GTP (p < 0.0001) (Fig. 9A). Figure 8B presents a typical example of a neuron recorded with GTPγS. It is clear that the control IBa is slow (pulse P1) and smaller than the facilitated current (pulse P2). This gives rise to a facilitation ratio > 1. On average, the facilitation ratio was significantly larger (p < 0.005) in cells dialyzed with GTPγS (1.19 ± 0.03; n = 7) than in cells recorded with GTP (1.07 ± 0.01; n = 12; Fig. 9B). In cells recorded with GTPγS, application of Som did not increase the facilitation ratio further (p > 0.1). The spontaneous inhibition caused by GTPγS presumably is attributable to a high spontaneous turnover rate of GDP/GTP exchange, even in the absence of activation by bound receptors. However, a few cells showed an irreversible inhibition ofIBa after application of Som (data not shown).
A final test for involvement of G-proteins was the action of NEM. NEM, a sulfhydryl-alkylating agent, has been shown to block G-protein–effector interactions by alkylating the α-subunits of pertussis toxin (PTX)-sensitive GTP-binding G-proteins (Winslow et al., 1987; Nakajima et al., 1990; Shapiro et al., 1994). In the neuron presented in Figure 10A, application of Som caused a 23% inhibition of peak IBa and slowing of activation (left inset). After bath application of 50 μm NEM for 3 min, the inhibition ofIBa by Som was abolished. In paired protocols, Som inhibition was reduced from 22 ± 3 to 1.7 ± 0.9% (n = 8; Fig. 10B). This difference was highly significant (p < 0.001). In most neurons, NEM also enhanced the rate of current rundown.
Fig. 10.

NEM treatment blocks Som inhibition of barium current. A, Time course of peak current amplitude during step to −10 mV (holding potential, −90 mV). The insetsshow the currents before (thick lines) and during Som application (thin lines) at the times indicated. Som (10 and 200 nm) and NEM (50 μm) were applied as indicated by bars. B, Bars show mean ± SEM (n = 8) inhibition ofIBa during paired applications of 200 nm Som with or without intervening exposure to 50 μm NEM for 180 sec.
The modulation of IBa by Som is independent of cAMP
Several Som receptors are known to be coupled to inhibition of adenylyl cyclase, as is found, for example, in cultured neocortical neurons (Wang et al., 1989). To test whether the effects of Som onIBa were mediated by a reduction in cAMP levels, we determined the actions of Som in cells preexposed for 5 min to 8-CPT-cAMP, a membrane-permeant and poorly hydrolyzable cAMP analog. This treatment changed neither IBa nor the inhibitory effects of Som (n = 4). In two additional cells, the exposure to 8-CPT-cAMP was combined with intracellular dialysis with the phosphodiesterase inhibitor IBMX. Again, we observed no effect on the inhibitory effects of Som onIBa.
DISCUSSION
We have tested the effects of Som and Som analogs on voltage-activated Ca2+ currents in rat amygdala neurons. We find that Som selectively inhibits the HVA component of the Ca2+ current, and that the G-protein-coupled Som receptor subtype SSTR-2 mediates the inhibition of IBa. The inhibition involves ω-CgTx-GVIA- and ω-Aga-IVA-sensitive components of the Ca2+ current. The inhibition is voltage-dependent and mediated by NEM-sensitive G-proteins.
The inhibitory effects of Som on IBa were rapid, reversible and concentration-dependent. Compared to the effects of Som on ICa in rat sympathetic neurons (Ikeda and Schofield, 1989; Shapiro and Hille, 1993), the inhibition ofIBa in the amygdala showed less desensitization. The inhibition was associated with a slowing of activation kinetics and was partially relieved by strong depolarizing prepulses. A similar marked voltage dependence of Som inhibition and change in gating has been demonstrated in sympathetic neurons (Ikeda and Schofield, 1989;Golard and Siegelbaum, 1993; Shapiro and Hille, 1993), spinal cord neurons (Bean, 1989), and pituitary cells (Luini et al., 1986). In amygdala there seems to be almost no voltage-dependent inhibition of Ca2+ channels in the absence of receptor agonists, and the facilitation of current in the absence of agonist is small. Bean (1989)has proposed that the voltage dependence of the inhibition by many neurotransmitters on ICa reflects alterations in gating (shift to a reluctant mode) caused by direct interactions between the calcium channel and the G-protein. In sympathetic neurons and in cells transfected with the α subunits of N- and P/Q-type calcium channels, this inhibitory effect appears to be mediated by the βγ subunits of G-proteins (Herlitze et al., 1996; Ikeda, 1996).
Specific antagonists of Som receptors are completely lacking, and highly selective agonists are not available for all of the Som receptors. However, with some caution we can assign the inhibitory actions of Som on IBa to SSTR-2 in these cells. The effects of BIM-23060 are not consistent with activation of SSTR-1, SSTR-5, and SSTR-3 because receptor-binding assays on cloned Som receptors indicate that BIM-23060 is not an effective agonist for these three receptors (Raynor et al., 1993a,b). BIM-23060 does not discriminate between SSTR-2 and SSTR-4, but BIM-23058, which was ineffective, has a 40-fold higher selectivity for SSTR-4 over SSTR-2 (Raynor et al., 1993a,b). Like all other Som receptors, SSTR-2 is coupled to intracellular effectors through GTP-binding regulatory proteins. Autoradiographic studies have shown that the amygdala expresses high levels of SSTR-2 mRNA (Kong et al., 1994). The lack of effect of cAMP on IBa inhibition is also consistent with the possible involvement of SSTR-2 because this Som receptor lacks functional coupling to adenylyl cyclase (Law et al., 1993).
Recordings with the nonhydrolyzable GTP analog GTPγS caused a gradual decrease in the amplitude of IBa and a gradual slowing of activation kinetics. These effects were identical to, and occluded, those produced by Som. This suggests that in amygdala neurons GTPγS can exchange for GDP on a G-protein in the absence of apparent receptor stimulation. Similar results have been reported for the transmitter inhibition of N- and P/Q-type Ca2+ channels in other central and peripheral neurons (Dolphin and Scott, 1987; Wanke et al., 1987; Mintz and Bean, 1993). The inhibitory effects of GDPβS were more modest than those reported in some systems (e.g., Holz et al., 1986) but still stronger than in rat sympathetic neurons (Ikeda and Schofield, 1989; Shapiro and Hille, 1993).
The Som-induced inhibition of IBa in amygdala neurons was completely prevented by brief preincubations with NEM. Using a similar protocol in rat sympathetic neurons, which have several well characterized G-protein-coupled signaling pathways (Hille, 1994),Shapiro et al. (1994) found that the actions of NEM were specific for modulatory pathways involving PTX-sensitive G-proteins. In cultured cortical neurons (Wang et al., 1990) and in sympathetic neurons, the inhibition of ICa by Som is wholly PTX-sensitive (Ikeda and Schofield, 1989; Shapiro and Hille, 1993). Similarly, Som inhibition of IBa in the amygdala is likely to involve PTX-sensitive G-protein(s). In contrast, Som-induced activation of a K+ conductance in rat septal neurons was not sensitive to PTX (Twery et al., 1991). Two PTX-sensitive G-proteins, Giα3 and Goα, selectively associate with the Som receptor subtype SSTR-2 (Law et al., 1993), and studies byKleuss et al. (1991) indicate that Goα2 mediates the inhibitory effects of Som on ICa in pituitary GH3 cells. It remains to be established which specific G-protein(s) mediates the effects of Som on calcium channels in the amygdala. Our results were obtained under strong chelation of intracellular calcium (10 mm EGTA). Therefore, we cannot exclude modulatory actions that are disrupted by a high concentration of Ca2+ chelators (Bernheim et al., 1991). Nevertheless, unlike results obtained with other neurotransmitters, modulation of calcium channels by Som in rat sympathetic neurons involves only a fast, membrane-delimited pathway that is not affected by calcium chelators (Shapiro and Hille, 1993).
We have found that both N- and P/Q-type components ofIBa are inhibited by Som. Inhibition of these two Ca2+ currents by a single transmitter has been demonstrated in other neurons such as spinal interneurons, inhibited by activation of GABAB receptors (Mintz and Bean, 1993), brainstem motoneurons, inhibited by serotonin (Bayliss et al., 1995), and neostriatal neurons, inhibited by dopamine (Surmeier et al., 1995). Other transmitters, in particular adenosine, can have differential effects on N- and P/Q-type calcium channels. For example, in CA3 pyramidal cells, activation of A1 receptors by adenosine inhibits N-type channels, whereas activation of A2breceptors potentiates P/Q-type channels (Mogul et al., 1993). In contrast to the effects of Som in the amygdala, in rat CA1 pyramidal neurons Som inhibited only the N-type calcium current (Ishibashi and Akaike, 1995).
Functional implications
In rats the Som gene is well expressed before birth (Burgunder, 1994), suggesting a functional role in the immature brain. Many nonpyramidal neurons of the amygdala are immunoreactive for Som (McDonald and Pearson, 1989). In addition, the amygdala is richly innervated by numerous afferent nerve terminals immunoreactive for Som (Roberts et al., 1982). Electron microscopic studies indicate that some of these terminals synapse on somata and dendrites of amygdala neurons (Aylward and Totterdell, 1993). Som inhibition ofICa may influence the excitability of amygdala neurons postsynaptically, by modulating the amplitude of the Ca2+-dependent afterdepolarization and afterhyperpolarization conductances that follow the action potential of amygdala pyramidal neurons (Rainnie et al., 1993; Sugita et al., 1993). In addition, liberation of Som near axonal terminals of amygdala pyramidal neurons could modulate N- and P/Q-type calcium channels, reducing presynaptic entry of Ca2+ and thus inhibiting transmitter release.
Footnotes
This work was supported by National Institutes of Health Grant NS-08174 to B.H. and a postdoctoral fellowship from the W. M. Keck Foundation to F.V. We thank Drs. J. S. Isaacson, K. P. Mackie, B. Nilius, and M. S. Shapiro for comments on this manuscript, and D. Anderson, L. Gibbs, L. Miller, and C. Taylor for technical assistance. The somatostatin analogs were a generous gift from Dr. W. A. Murphy (Tulane University).
Correspondence should be addressed to Dr. Félix Viana, Laboratory of Physiology KU Leuven, Campus Gasthuisberg, B-3000 Leuven, Belgium.
REFERENCES
- 1.Adolphs R, Tranel D, Damasio H, Damasio A. Impaired recognition of emotion in facial expressions following bilateral damage to the human amygdala. Nature. 1994;372:669–672. doi: 10.1038/372669a0. [DOI] [PubMed] [Google Scholar]
- 2.Allen TGJ, Brown DA. M2 muscarinic receptor-mediated inhibition of the Ca2+ current in rat magnocellular cholinergic basal forebrain neurones. J Physiol (Lond) 1993;466:173–189. [PMC free article] [PubMed] [Google Scholar]
- 3.Aggleton JP. The contribution of the amygdala to normal and abnormal emotional states. Trends Neurosci. 1993;16:328–333. doi: 10.1016/0166-2236(93)90110-8. [DOI] [PubMed] [Google Scholar]
- 4.Anwyl R. Modulation of vertebrate neuronal calcium channels by transmitters. Brain Res Rev. 1991;16:265–281. doi: 10.1016/0165-0173(91)90010-6. [DOI] [PubMed] [Google Scholar]
- 5.Aylward RL, Totterdell S. Neurons in the ventral subiculum, amygdala and entorhinal cortex which project to the nucleus accumbens: their input from somatostatin-immunoreactive boutons. J Chem Neuroanat. 1993;6:31–42. doi: 10.1016/0891-0618(93)90005-o. [DOI] [PubMed] [Google Scholar]
- 6.Bayliss DA, Umemiya M, Berger AJ. Inhibition of N- and P-type calcium currents and the after-hyperpolarization in rat motoneurones by serotonin. J Physiol (Lond) 1995;485:635–647. doi: 10.1113/jphysiol.1995.sp020758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bean BP. Neurotransmitter inhibition of neuronal calcium currents by changes in channel voltage dependence. Nature. 1989;340:153–156. doi: 10.1038/340153a0. [DOI] [PubMed] [Google Scholar]
- 8.Beech DJ, Bernheim L, Hille B. Pertussis toxin and voltage dependence distinguish multiple pathways modulating calcium channels of rat sympathetic neurons. Neuron. 1992;8:97–106. doi: 10.1016/0896-6273(92)90111-p. [DOI] [PubMed] [Google Scholar]
- 9.Bernheim L, Beech DJ, Hille B. A diffusible second messenger mediates one of the pathways coupling receptors to calcium channels in rat sympathetic neurons. Neuron. 1991;6:859–867. doi: 10.1016/0896-6273(91)90226-p. [DOI] [PubMed] [Google Scholar]
- 10.Bell GI, Reisine T. Molecular biology of somatostatin receptors. Trends Neurosci. 1993;16:34–38. doi: 10.1016/0166-2236(93)90050-v. [DOI] [PubMed] [Google Scholar]
- 11.Brazeau P, Vale W, Burgus R, Ling N, Butcher M, Rivier J, Guillemin R. Hypothalamic polypeptide that inhibits the secretion of immunoreactive pituitary growth hormone. Science. 1973;179:77–79. doi: 10.1126/science.179.4068.77. [DOI] [PubMed] [Google Scholar]
- 12.Burgunder JM. Ontogeny of somatostatin gene expression in rat forebrain. Brain Res Dev Brain Res. 1994;78:109–122. doi: 10.1016/0165-3806(94)90015-9. [DOI] [PubMed] [Google Scholar]
- 13.Davis MD, Rainnie D, Cassell M. Neurotransmission in the rat amygdala related to fear and anxiety. Trends Neurosci. 1994;17:208–214. doi: 10.1016/0166-2236(94)90106-6. [DOI] [PubMed] [Google Scholar]
- 14.Dolphin AC, Scott RH. Calcium channel currents and their inhibition by (−)-baclofen in rat sensory neurons: modulation by guanine nucleotides. J Physiol (Lond) 1987;386:1–17. doi: 10.1113/jphysiol.1987.sp016518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Eckstein F, Cassel D, Levkovitz H, Lowe M, Selinger Z. Guanosine 5′-O -(2-thiodiphosphate): an inhibitor of adenylate cyclase stimulation by guanine nucleotides and fluoride ions. J Biol Chem. 1979;254:9829–9834. [PubMed] [Google Scholar]
- 16.Epelbaum J. Somatostatin in the central nervous system: physiology and pathological modifications. Prog Neurobiol. 1986;27:63–100. doi: 10.1016/0301-0082(86)90012-2. [DOI] [PubMed] [Google Scholar]
- 17.Foehring RC, Scroggs RS. Multiple high-threshold calcium currents in acutely isolated rat amygdaloid pyramidal cells. J Neurophysiol. 1994;71:433–436. doi: 10.1152/jn.1994.71.1.433. [DOI] [PubMed] [Google Scholar]
- 18.Golard A, Siegelbaum SA. Kinetic basis for the voltage-dependent inhibition of N-type calcium current by somatostatin and norepinephrine in chick sympathetic neurons. J Neurosci. 1993;13:3884–3894. doi: 10.1523/JNEUROSCI.13-09-03884.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hamill OP, Marty A, Neher E, Sakmann B, Sigworth FJ. Improved patch-clamp techniques for high-resolution current recording from cells and cell-free membrane patches. Pflügers Arch. 1981;391:85–100. doi: 10.1007/BF00656997. [DOI] [PubMed] [Google Scholar]
- 20.Herlitze S, Garcia DE, Mackie K, Hille B, Scheuer T, Catterall WA. Modulation of Ca2+ channels by G-protein βγ subunits. Nature. 1996;380:258–262. doi: 10.1038/380258a0. [DOI] [PubMed] [Google Scholar]
- 21.Higuchi T, Yamazaki O, Takazawa A, Kato N, Watanabe N, Minatogawa Y, Yamazaki J, Oshima H, Nagaki S, Igarashi Y, Noguchi T. Effects of carbamazepine and valproic acid on brain immunoreactive somatostatin and γ-aminobutyric acid in amygdaloid kindled rats. Eur J Pharmacol. 1986;125:169–175. doi: 10.1016/0014-2999(86)90024-5. [DOI] [PubMed] [Google Scholar]
- 22.Hille B. Modulation of ion-channel function by G-protein-coupled receptors. Trends Neurosci. 1994;17:531–536. doi: 10.1016/0166-2236(94)90157-0. [DOI] [PubMed] [Google Scholar]
- 23.Holz GG, Rane SG, Dunlap K. GTP-binding proteins mediate transmitter inhibition of voltage-dependent calcium channels. Nature. 1986;329:670–672. doi: 10.1038/319670a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hoyer D, Bell GI, Berelowitz M, Epelbaum J, Feniuk W, Humphrey PPA, O’Carroll AM, Patel YC, Schonbrunn A, Taylor JE, Reisine T. Classification and nomenclature of somatostatin receptors. Trends Pharmacol Sci. 1995;16:86–88. doi: 10.1016/s0165-6147(00)88988-9. [DOI] [PubMed] [Google Scholar]
- 25.Ikeda SR. Voltage-dependent modulation of N-type calcium channels by G-protein βγ subunits. Nature. 1996;380:255–258. doi: 10.1038/380255a0. [DOI] [PubMed] [Google Scholar]
- 26.Ikeda SR, Schofield GG. Somatostatin blocks a calcium current in rat sympathetic ganglion neurones. J Physiol (Lond) 1989;409:221–240. doi: 10.1113/jphysiol.1989.sp017494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Inoue M, Yoshii M. Modulation of ion channels by somatostatin and acetylcholine. Prog Neurobiol. 1992;38:203–230. doi: 10.1016/0301-0082(92)90040-l. [DOI] [PubMed] [Google Scholar]
- 28.Ishibashi H, Akaike N. Somatostatin modulates high-voltage-activated Ca2+ channels in freshly dissociated rat hippocampal neurons. J Neurophysiol. 1995;74:1028–1036. doi: 10.1152/jn.1995.74.3.1028. [DOI] [PubMed] [Google Scholar]
- 29.Kleuss C, Hescheler J, Ewel C, Rosenthal W, Schultz G, Wittig B. Assignment of G-protein subtypes to specific receptors inducing inhibition of calcium currents. Nature. 1991;353:43–48. doi: 10.1038/353043a0. [DOI] [PubMed] [Google Scholar]
- 30.Kong H, DePaoli AM, Breder CD, Yasuda K, Bell GI, Reisine T. Differential expression of messenger RNAs for somatostatin receptor subtypes SSTR1, SSTR2 and SSTR3 in adult rat brain: analysis by RNA blotting and in situ hybridization histochemistry. Neuroscience. 1994;59:175–184. doi: 10.1016/0306-4522(94)90108-2. [DOI] [PubMed] [Google Scholar]
- 31.Law SF, Yasuda K, Bell GI, Reisine T. Giα3 and Goα selectively associate with the cloned somatostatin receptor subtype SSTR2. J Biol Chem. 1993;268:10721–10727. [PubMed] [Google Scholar]
- 32.Leroux P, Weissmann D, Pujol JF, Vaudry H. Quantitative autoradiography of somatostatin receptors in the rat limbic system. J Comp Neurol. 1993;331:389–401. doi: 10.1002/cne.903310308. [DOI] [PubMed] [Google Scholar]
- 33.Luini A, Lewis D, Guild S, Schofield G, Weight F. Somatostatin, an inhibitor of ACTH secretion, decreases cytosolic free calcium and voltage-dependent calcium current in a pituitary cell line. J Neurosci. 1986;6:3128–3132. doi: 10.1523/JNEUROSCI.06-11-03128.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.McDonald AJ, Pearson JC. Coexistence of GABA and peptide immunoreactivity in non-pyramidal neurons of the basolateral amygdala. Neurosci Lett. 1989;100:53–58. doi: 10.1016/0304-3940(89)90659-9. [DOI] [PubMed] [Google Scholar]
- 35.Mintz IM, Bean BP. GABAB receptor inhibition of P-type Ca2+ channels in central neurons. Neuron. 1993;10:889–898. doi: 10.1016/0896-6273(93)90204-5. [DOI] [PubMed] [Google Scholar]
- 36.Mogul DJ, Adams ME, Fox AP. Differential activation of adenosine receptors decreases N-type but potentiates P-type Ca2+ current in hippocampal CA3 neurons. Neuron. 1993;10:327–334. doi: 10.1016/0896-6273(93)90322-i. [DOI] [PubMed] [Google Scholar]
- 37.Meriney SD, Gray DB, Pilar GR. Somatostatin-induced inhibition of neuronal Ca2+ current modulated by cGMP-dependent protein kinase. Nature. 1994;369:336–339. doi: 10.1038/369336a0. [DOI] [PubMed] [Google Scholar]
- 38.Nakajima T, Irisawa H, Giles W. N -ethylmaleimide uncouples muscarinic receptors from acetylcholine-sensitive potassium channels in bullfrog atrium. J Gen Physiol. 1990;96:887–903. doi: 10.1085/jgp.96.4.887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Paxinos G, Watson C. Academic; San Diego: 1986. The rat brain in stereotaxic coordinates. . [DOI] [PubMed] [Google Scholar]
- 40.Rainnie DG, Asprodini EK, Shinnick-Gallagher P. Intracellular recordings from morphologically identified neurons of the basolateral amygdala. J Neurophysiol. 1993;69:1350–1362. doi: 10.1152/jn.1993.69.4.1350. [DOI] [PubMed] [Google Scholar]
- 41.Raynor K, Murphy WA, Coy DH, Taylor JE, Moreau JP, Yasuda K, Bell GI, Reisine T. Cloned somatostatin receptors: identification of subtype-selective peptides and demonstration of high affinity binding of linear peptides. Mol Pharmacol. 1993a;43:838–844. [PubMed] [Google Scholar]
- 42.Raynor K, O’Carroll AM, Kong H, Yasuda K, Mahan LC, Bell GI, Reisine T. Characterization of cloned somatostatin receptors SSTR4 and SSTR5. Mol Pharmacol. 1993b;44:385–392. [PubMed] [Google Scholar]
- 43.Roberts GW, Woodhams PL, Polak JM, Crow TJ. Distribution of neuropeptides in the limbic system of the rat: the amygdaloid complex. Neuroscience. 1982;7:99–131. doi: 10.1016/0306-4522(82)90156-7. [DOI] [PubMed] [Google Scholar]
- 44.Sah DWY. Neurotransmitter modulation of calcium current in rat spinal cord neurons. J Neurosci. 1990;10:136–141. doi: 10.1523/JNEUROSCI.10-01-00136.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Schweitzer P, Madamba S, Champagnat J, Siggins GR. Somatostatin inhibition of hippocampal CA1 pyramidal neurons: mediation by arachidonic acid and its metabolites. J Neurosci. 1993;13:2033–2049. doi: 10.1523/JNEUROSCI.13-05-02033.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Shapiro MS, Hille B. Substance P and somatostatin inhibit calcium channels in rat sympathetic neurons via different G protein pathways. Neuron. 1993;10:11–20. doi: 10.1016/0896-6273(93)90237-l. [DOI] [PubMed] [Google Scholar]
- 47.Shapiro MS, Wollmuth LP, Hille B. Modulation of Ca2+ channels by PTX-sensitive G-proteins is blocked by N -ethylmaleimide in rat sympathetic neurons. J Neurosci. 1994;14:7109–7116. doi: 10.1523/JNEUROSCI.14-11-07109.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Scharfman HE, Schwatzkroin PA. Further studies of the effects of somatostatin and related peptides in area CA1 of rabbit hippocampus. Cell Mol Neurobiol. 1988;8:411–429. doi: 10.1007/BF00711226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sugita S, Tanaka E, North RA. Membrane properties and synaptic potentials of three types of neurone in rat lateral amygdala. J Physiol (Lond) 1993;460:705–718. doi: 10.1113/jphysiol.1993.sp019495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Surmeier DJ, Bargas J, Hemmings HC, Jr, Nairn AC, Greengard P. Modulation of calcium currents by a D1 dopaminergic protein kinase/phosphatase cascade in rat neostriatal neurons. Neuron. 1995;14:385–397. doi: 10.1016/0896-6273(95)90294-5. [DOI] [PubMed] [Google Scholar]
- 51.Twery MJ, Wong LA, Gallagher JP. Somatostatin induced hyperpolarization of septal neurons is not blocked by pertussis toxin. Eur J Pharmacol. 1991;192:287–291. doi: 10.1016/0014-2999(91)90054-t. [DOI] [PubMed] [Google Scholar]
- 52.Wang HL, Bogen C, Reisine T, Dichter M. Somatostatin-14 and somatostatin-28 induce opposite effects on potassium currents in rat neocortical neurons. Proc Natl Acad Sci USA. 1989;86:9616–9620. doi: 10.1073/pnas.86.23.9616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wang HL, Reisine T, Dichter M. Somatostatin-14 and somatostatin-28 inhibit calcium currents in rat neocortical neurons. Neuroscience. 1990;38:335–342. doi: 10.1016/0306-4522(90)90032-y. [DOI] [PubMed] [Google Scholar]
- 54.Wanke E, Ferroni A, Malgaroli A, Ambrosini A, Pozzan T, Meldolesi J. Activation of a muscarinic receptor selectively inhibits a rapidly inactivated Ca2+ current in rat sympathetic neurons. Proc Natl Acad Sci USA. 1987;84:4313–4317. doi: 10.1073/pnas.84.12.4313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Washburn MS, Moises HC. Electrophysiological and morphological properties of rat basolateral amygdaloid neurons in vitro . J Neurosci. 1992;12:4066–4079. doi: 10.1523/JNEUROSCI.12-10-04066.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Winslow JW, Bradley JD, Smith JA, Neer EJ. Reactive sulphydryl groups of α39, a guanine nucleotide-binding protein from brain: location and function. J Biol Chem. 1987;262:4501–4507. [PubMed] [Google Scholar]