

Abstract
Chronic Δ9-tetrahydrocannabinol (Δ9-THC) administration produces tolerance to cannabinoid effects, but alterations in signal transduction that mediate these changes are not yet known. The present study uses in vitro autoradiography of agonist-stimulated [35S]GTPγS binding to localize cannabinoid receptor-activated G-proteins after chronic Δ9-THC treatment. Cannabinoid (WIN 55212-2)-stimulated [35S]GTPγS binding was performed in brain sections from rats treated chronically with 10 mg/kg Δ9-THC for 21 d. Control animals received saline or an acute injection of Δ9-THC. Acute Δ9-THC treatment had no effect on basal or WIN 55212-2-stimulated [35S]GTPγS binding. After chronic Δ9-THC treatment, net WIN 55212-2-stimulated [35S]GTPγS binding was reduced significantly (up to 70%) in most brain regions, including the hippocampus, caudate-putamen, perirhinal and entorhinal cortex, globus pallidus, substantia nigra, and cerebellum. In contrast, chronic Δ9-THC treatment had no effect on GABAB-stimulated [35S]GTPγS binding. In membranes and brain sections, Δ9-THC was a partial agonist, stimulating [35S]GTPγS by only 20% of the level stimulated by WIN 55212-2 and inhibiting WIN 55212-2-stimulated [35S]GTPγS at high concentrations. Because the EC50 of WIN 55212-2-stimulated [35S]GTPγS binding and the KD of cannabinoid receptor binding were unchanged by chronic Δ9-THC treatment, the partial agonist actions of Δ9-THC did not produce the decrease in cannabinoid-stimulated [35S]GTPγS binding. These results suggest that profound desensitization of cannabinoid-activated signal transduction mechanisms occurs after chronic Δ9-THC treatment.
Keywords: Δ9-THC, [35S]GTPγS autoradiography, cannabinoid receptor, GABAB receptor, G-protein
Marijuana produces behavioral effects via its biologically active constituent Δ9-tetrahydrocannabinol (Δ9-THC) (Gaoni and Mechoulam, 1964; Deadwyler et al., 1995a). Studies using potent synthetic cannabinoid analogs demonstrated that this activity occurs at cannabinoid receptors (Devane et al., 1988). The cloned cannabinoid receptor exhibits seven transmembrane spanning regions characteristic of G-protein-coupled receptors (Matsuda et al., 1990). Cannabinoid receptors act via Gi/o to inhibit adenylyl cyclase (Howlett, 1985; Howlett et al., 1986; Pacheco et al., 1991), alter potassium channel conductance (Hampson et al., 1995b), and decrease calcium channel conductance (Mackie and Hille, 1992). Cannabinoid receptors in the brain (CB1) are numerous compared with other G-protein-coupled receptors and are localized in most brain regions, including the hippocampus, cortex, caudate-putamen, globus pallidus, substantia nigra, and cerebellum (Herkenham et al., 1991b;Jansen et al., 1992). This anatomical distribution is consistent with behavioral effects of cannabinoids, including memory disruption, decreased motor activity, catalepsy, antinociception, and hypothermia (Dewey, 1986; Compton et al., 1993; Deadwyler et al., 1995a).
Chronic Δ9-THC treatment results in the development of behavioral tolerance (Carlini, 1968; Dewey, 1986; Abood et al., 1993;Deadwyler et al., 1995b). Some laboratories have reported a decrease in the Bmax of cannabinoid receptors after chronic Δ9-THC treatment (Oviedo et al., 1993; De Fonseca et al., 1994), whereas others reported no change in cannabinoid receptor density (Westlake et al., 1991; Abood et al., 1993). Moreover, changes in receptor binding may not reflect changes in receptor function, and a measurement of agonist efficacy is necessary to answer this question. In cultured neuroblastoma cells, chronic cannabinoid exposure desensitized cannabinoid-inhibited adenylyl cyclase (Dill and Howlett, 1988), indicating that cannabinoid tolerance may involve alterations in signal transduction. Changes in G-protein activity and G-protein levels after chronic drug treatment have been reported previously for other receptor systems, including μ opioid receptors, which also couple to Gi/o (Nestler et al., 1994; Sim et al., 1996a; Selley et al., 1996a).
Our laboratory has developed a technique for examining receptor-activated G-proteins in brain sections using [35S]GTPγS autoradiography (Sim et al., 1995). This technique is based on agonist-stimulated [35S]GTPγS binding in membranes (Hilf et al., 1989; Traynor and Nahorski, 1995). For [35S]GTPγS autoradiography, sections are first incubated with excess GDP to decrease basal [35S]GTPγS binding and then with [35S]GTPγS in the presence (stimulated) or absence (basal) of a specific agonist. The applicability of this method to chronic drug studies has been demonstrated in the opioid system, where regionally specific changes in μ opioid-stimulated [35S]GTPγS binding were identified after chronic morphine treatment (Sim et al., 1996a). The present study was performed to examine the effect of chronic treatment with Δ9-THC on cannabinoid receptor-activated G-proteins in different brain regions. In addition to demonstrating that chronic Δ9-THC treatment produced large decreases in G-protein activation throughout the brain, these studies also reveal that Δ9-THC is a partial agonist in activating G-proteins in brain, which may have important implications in the mechanism of action of this drug.
MATERIALS AND METHODS
Materials. Male Sprague Dawley rats (200–250 gm) were purchased from Zivic-Miller (Zelienople, PA). [35S]GTPγS (1228 Ci/mmol) was purchased from New England Nuclear (Boston, MA). Baclofen and WIN 55212-2 were obtained from Research Biochemicals International (Natick, MA). Δ9-THC was provided by the National Institute on Drug Abuse. SR141716A was provided by Dr. F. Barth (Sanofi, Montpelier, France). GTPγS and GDP for membrane assays were purchased from Boehringer Mannheim (New York, NY). GDP for autoradiography was obtained from Sigma (St. Louis, MO). Reflections autoradiography film was purchased from New England Nuclear. All other reagent grade chemicals were obtained from Sigma or Fisher Scientific (Houston, TX).
Δ9-THC treatment. Δ9-THC was dissolved in ethanol and prepared for injection as described previously (Heyser et al., 1993). The ethanol solution was suspended in a 1:4:1 ratio with Pluronic F68 detergent in ethanol and saline, and the ethanol was evaporated under a stream of nitrogen gas. The Δ9-THC was diluted to 10 mg/ml in saline for injection. Chronically treated animals received a single daily intraperitoneal injection of 10 mg/kg Δ9-THC for 21 d. Control animals received an equal volume of vehicle. Animals were killed 24 after the last injection. A separate group of animals received a single acute intraperitoneal injection of 10 mg/kg Δ9-THC or vehicle 24 hr before they were killed.
Agonist-stimulated [35S]GTPγS autoradiography. Animals were killed by rapid decapitation. Brains were removed and immediately immersed in isopentane at −35°C. Twenty micrometer horizontal sections were cut on a cryostat maintained at −20°C and mounted onto gelatin-subbed slides. Slides were incubated in assay buffer (50 mm Tris-HCl, 3 mmMgCl2, 0.2 mm EGTA, 100 mm NaCl, 0.5% BSA, pH 7.4) at 25°C for 10 min, and then in 2 mmGDP in assay buffer for 15 min at 25°C. Slides were then transferred into assay buffer containing 2 mm GDP and 0.04 nm [35S]GTPγS, with (stimulated) or without (basal) 10 μm WIN 55212-2, and incubated at 25°C for 2 hr. Adjacent sections were incubated with 300 μm baclofen and 0.04 nm [35S]GTPγS to evaluate GABAB receptor activation of G-proteins. Sections from control animals were also processed using 1 or 10 μm WIN 55212-2 and 3 or 10 μm Δ9-THC alone and in combination. Slides were rinsed twice for 2 min each in 50 mm Tris-HCl buffer, pH 7.4, at 4°C, and once in deionized water, dried, and exposed to film for 48 hr. Films were digitized with a Sony XC-77 video camera and analyzed using the National Institutes of Health IMAGE program for Macintosh computers. Images were quantified by densitometric analysis with [14C] standards, and values were corrected to nanocuries/gram [35S] based on a correction factor determined with brain paste standards (Sim et al., 1996a). Data are mean values ± SE of duplicate sections of brains from five animals. Statistical significance was determined by the nonpaired two tailed Student’s t test using JMP (SAS Institute, Cary, NC).
Agonist-stimulated [35S]GTPγS binding in membranes. Cannabinoid-stimulated [35S]GTPγS binding was determined as described previously (Selley et al., 1996b), using membranes from rat cerebellum (15 μg protein). Membranes were incubated at 30°C for 1 hr in assay buffer (50 mmTris-HCl, 3 mm MgCl2, 0.2 mm EGTA, 100 mm NaCl, 0.1 mg/ml BSA, pH 7.4), with the appropriate concentrations of WIN 55212-2 or Δ9-THC, in the presence of 20 μm GDP and 0.05 nm[35S]GTPγS in a 1 ml total volume. Basal binding was measured in the absence of agonist, and nonspecific binding was measured with 10 μm GTPγS. The reaction was terminated by rapid filtration under vacuum through Whatman GF/B filters, followed by three washes with cold Tris buffer. Bound radioactivity was determined by liquid scintillation spectrophotometry, at 95% efficiency for [35S], after overnight extraction in 5 ml Ecolite scintillation fluid. Data are reported as mean ± SE values of three experiments that were performed in triplicate. Nonlinear iterative regression analyses of agonist concentration–effect curves were performed with JMP (SAS, Cary, NC).
RESULTS
Effect of Δ9-THC on cannabinoid-stimulated [35S]GTPγS binding in cerebellar membranes and brain sections
Previous studies (Sim et al., 1995; Selley et al., 1996b; Sim et al., 1996b) have established that cannabinoid agonists stimulate [35S]GTPγS binding in both isolated membranes and brain sections, with a distribution and pharmacology that parallels that of cannabinoid receptor binding. To compare the effect of Δ9-THC and a more potent cannabinoid agonist in this assay system, concentration–effect curves of Δ9-THC- and WIN 55212-2-stimulated [35S]GTPγS binding were generated in rat cerebellar membranes (Fig.1A). Both Δ9-THC- and WIN 55212-2-stimulated [35S]GTPγS binding were concentration-dependent. The maximal stimulation of [35S]GTPγS binding by WIN 55212-2 under these conditions was 270% over basal, with an EC50 value of 0.12 μm. The effect of Δ9-THC was considerably less than that of WIN 55212-2, with an apparent maximal stimulation of only 20% compared with that of WIN 55212-2. If Δ9-THC were a true partial agonist in this system, then high concentrations of Δ9-THC should antagonize the effect of a full agonist like WIN 55212-2 when the two drugs are added together, and the presence of residual Δ9-THC in sections and membranes from chronic Δ9-THC-treated rats could artificially reduce WIN 55212-2-stimulated [35S]GTPγS binding. This inhibitory effect of Δ9-THC was indeed observed in cerebellar membranes (Fig. 1A) when various concentrations of Δ9-THC were added with either 1 μm or 10 μm WIN 55212-2. In the presence of 1 μm WIN 55212-2, 0.3 μmΔ9-THC began to produce significant inhibition of WIN 55212-2-stimulated [35S]GTPγS binding, and 0.8 μm Δ9-THC inhibited 50% of WIN 55212-2-stimulated [35S]GTPγS binding. As would be predicted for a partial agonist, in the presence of a higher concentration (10 μm) of WIN 55212-2, Δ9-THC was less potent, and significant inhibition of WIN 55212-2-stimulated [35S]GTPγS required at least 10 μm Δ9-THC. From these data, it was estimated that >200 μm Δ9-THC would be required to inhibit 50% of WIN 55212-2-stimulated [35S]GTPγS binding in the presence of 10 μm WIN 55212-2. The inhibitory effect of Δ9-THC on WIN 55212-2-stimulated [35S]GTPγS binding was not attributable to a vehicle effect from a combination of the two drugs, because the same concentrations of vehicle (ethanol) had no effect on cannabinoid-stimulated [35S]GTPγS binding (data not shown).
Fig. 1.
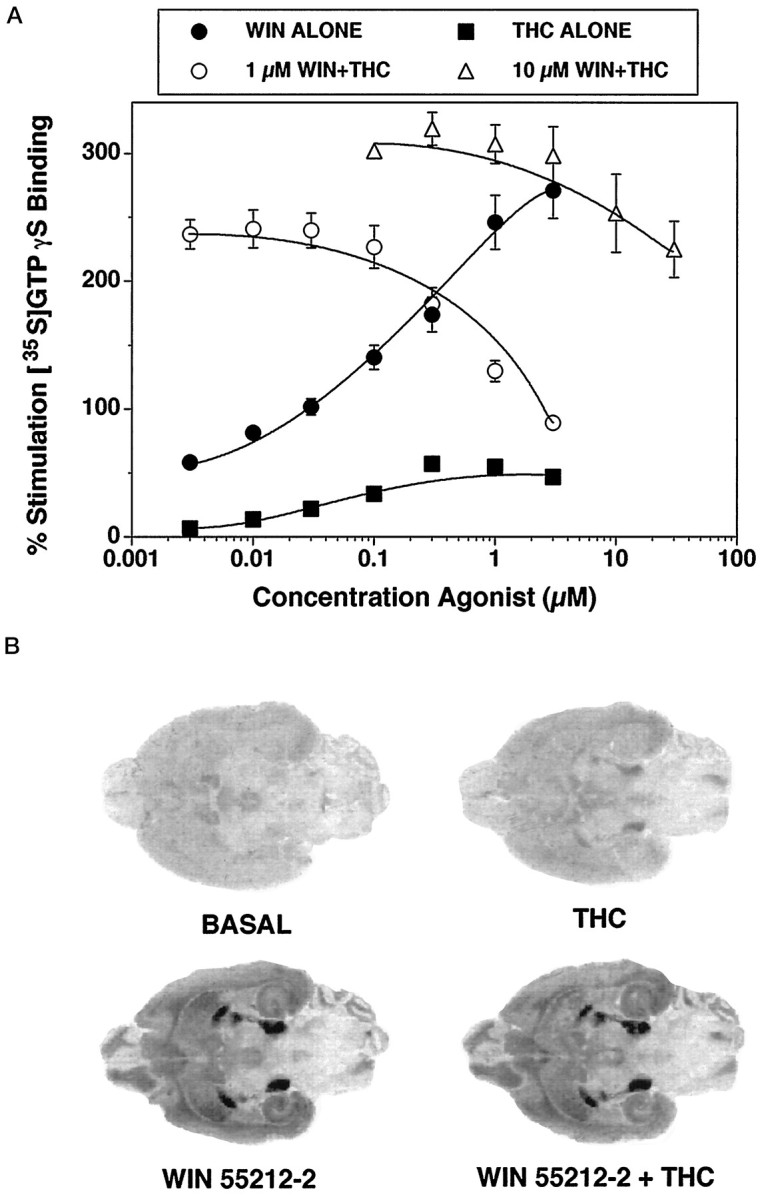
Effect of Δ9-THC and WIN 55212-2 on [35S]GTPγS binding in rat cerebellar membranes (A) and rat brain sections (B). Membranes (A) were incubated with 0.05 nm[35S]GTPγS and 20 μm GDP, as described in Materials and Methods, with various concentrations of either Δ9-THC or WIN 55212-2 alone (closed symbols) or with various concentrations of Δ9-THC in the presence of either 1 μm or 10 μm WIN 55212-2 (open symbols). Data are expressed as percentage basal [35S]GTPγS binding and represent mean values ± SE from three separate experiments. Rat brain sections (B) were incubated with 0.04 nm[35S]GTPγS and 2 mm GDP, as described in Materials and Methods, and represent basal [35S]GTPγS binding, 10 μm Δ9-THC alone, 10 μm WIN 55212-2 alone, and 10 μmWIN 55212-2 + 10 μm Δ9-THC.
Similar experiments were performed in brain sections to determine the appropriate concentration of WIN 55212-2 to use in [35S]GTPγS autoradiography in Δ9-THC-treated animals (Fig. 1B). The addition of 10 μm WIN 55212-2 produced high levels of stimulated [35S]GTPγS binding in the substantia nigra, entopeduncular nucleus, and globus pallidus, with moderate levels of activation in hippocampus and cortex. A maximally effective concentration of Δ9-THC alone (10 μm) produced little stimulation of [35S]GTPγS binding. This concentration of Δ9-THC had no significant effect on 10 μm WIN 55212-2-stimulated [35S]GTPγS binding in the substantia nigra (<5% decrease by densitometric analysis) (Fig. 1B). In agreement with the membrane assays, however, this concentration of Δ9-THC visibly inhibited [35S]GTPγS binding stimulated by 1 μm WIN 55212-2 (data not shown). From these data, a concentration of 10 μm WIN 55212-2 was used in autoradiographic experiments to minimize potential effects of residual Δ9-THC on the cannabinoid-stimulated [35S]GTPγS autoradiographic signal.
Effects of chronic and acute Δ9-THC administration on WIN 55212-2-stimulated [35S]GTPγS binding
To compare the effect of acute and chronic Δ9-THC administration on cannabinoid receptor activation of G-proteins, rats were treated with either a single acute dose of 10 mg/kg Δ9-THC or were administered daily injections of 10 mg/kg Δ9-THC for 21 d. The effect of Δ9-THC administration on cannabinoid-stimulated [35S]GTPγS binding was examined at four brain levels in horizontal sections from acute and chronic Δ9-THC-treated and control animals. Sections were analyzed at the level of (1) cerebellum, (2) caudate-putamen/septum, (3) globus pallidus, and (4) substantia nigra. At the most dorsal level, cannabinoid-stimulated [35S]GTPγS binding in control sections was observed in the cerebellum, hippocampus, and cortex. Labeling in all of these areas was visibly reduced in sections from chronic Δ9-THC-treated rats. At a more ventral level (Fig.2, top), cannabinoid-stimulated [35S]GTPγS binding was most evident in the cortex, hippocampus, caudate-putamen, septum, and periaqueductal gray (PAG), and was significantly reduced in sections from chronic Δ9-THC-treated rats. At the next level (Fig. 2,middle), the reduction in cannabinoid-stimulated [35S]GTPγS binding in the sections from chronic Δ9-THC-treated rats was most evident throughout the cortex, hippocampus, caudate-putamen, and globus pallidus. In the most ventral sections (Fig. 2, bottom), the dense cannabinoid-stimulated [35S]GTPγS labeling in substantia nigra was significantly reduced in sections from chronic Δ9-THC-treated animals. Thus, visual inspection of autoradiograms demonstrated clear reductions in WIN 55212-2-stimulated [35S]GTPγS binding in virtually every region where significant cannabinoid stimulation of [35S]GTPγS binding was observed.
Fig. 2.
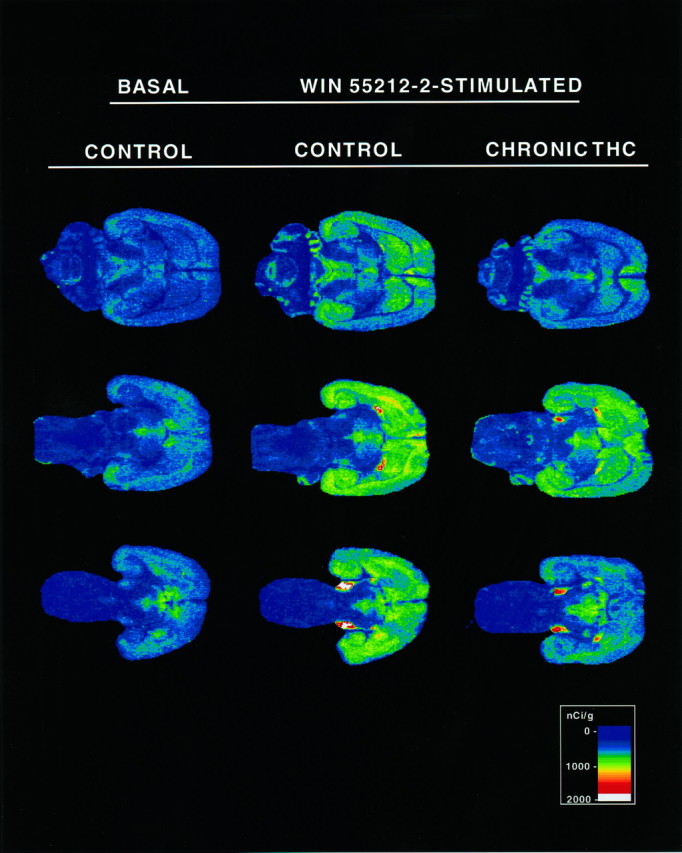
Autoradiograms of brain sections comparing basal and cannabinoid-stimulated [35S]GTPγS binding in control and chronic Δ9-THC-treated rats. Sections were incubated with 2 mm GDP and then with [35S]GTPγS (0.04 nm) and 2 mmGDP in the presence and absence of 10 μm WIN 55212-2. Basal binding (assessed in the absence of agonist) is shown on theleft column. Sections from control (middle column) and chronic Δ9-THC-treated (right column) rats are shown at the appropriate levels to show (1) caudate-putamen and PAG (top row), (2) caudate-putamen and globus pallidus (middle row), and (3) substantia nigra (bottom row). Cannabinoid-stimulated [35S]GTPγS binding in the cortex and hippocampus is seen in sections at all three levels.
To quantify these effects, autoradiograms from all four groups of animals (acute and chronic Δ9-THC treated, and controls) were analyzed densitometrically. Figure 3 shows data from a number of brain regions in both acute and chronic groups, expressed as net nanocuries [35S] per gram tissue (obtained by subtracting basal binding from WIN 55212-2-stimulated [35S]GTPγS binding values in each brain section). The results from the chronic Δ9-THC-treated rats are shown in Figure 3A. These results demonstrated significant reductions in net binding in sections from chronic Δ9-THC-treated rats compared with controls, with >50% reduction in a number of regions, including the perirhinal cortex, entorhinal cortex, hippocampus, and cerebellum. Smaller but significant reductions in net agonist-stimulated [35S]GTPγS binding were observed in the septum and caudate-putamen. The only region in which chronic Δ9-THC failed to significantly reduce net stimulated [35S]GTPγS binding was the PAG; in this region, the level of net binding was reduced but failed to reach statistical significance. Figure 3B, shows net WIN 55212-2-stimulated [35S]GTPγS binding data from the same regions of acute Δ9-THC-treated animals, along with their respective controls. In contrast to the results observed with the chronic treatment group, acute Δ9-THC administration had no significant effect on net stimulated [35S]GTPγS binding in any region measured.
Fig. 3.
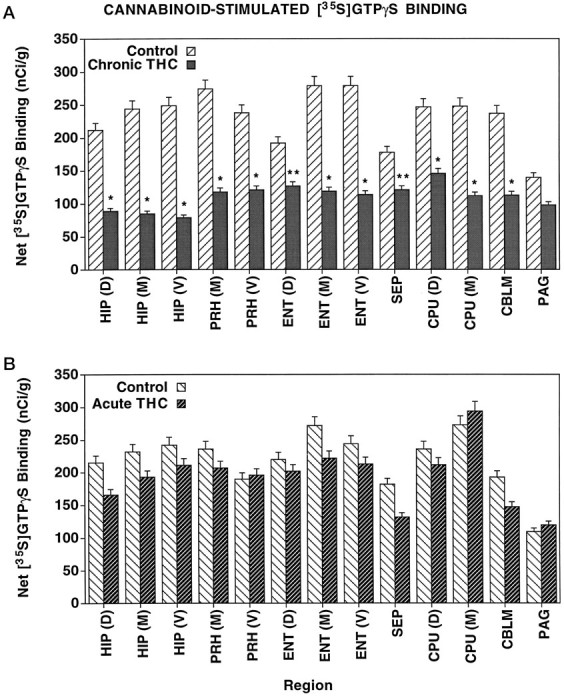
Net cannabinoid-stimulated [35S]GTPγS binding in brain regions from chronic (A) and acute (B) Δ9-THC-treated and control rats. Sections were incubated with 2 mm GDP, and then with [35S]GTPγS (0.04 nm) and 2 mm GDP in the presence and absence of 10 μm WIN 55212-2. Net stimulated [35S]GTPγS binding was determined by subtracting basal [35S]GTPγS binding from WIN 55212-2-stimulated [35S]GTPγS binding. The levels of sections correspond to the images shown in Figure 2: dorsal, Figure 2, top; middle, Figure 2, middle; and ventral, Figure 2, bottom. *p < 0.005, **p < 0.05.CBLM, Cerebellum; CPU, caudate-putamen;ENT, entorhinal cortex; HIP, hippocampus;PAG, periaqueductal gray; PRH, perirhinal cortex; SEP, septum.
Because the levels of net stimulated [35S]GTPγS binding in the globus pallidus and substantia nigra were much greater than those of the other regions, data from these regions are presented separately (Fig. 4). As observed for other regions, net agonist-stimulated [35S]GTPγS binding was significantly reduced in chronic Δ9-THC-treated animals compared with controls, with 45% and 20% reductions in the globus pallidus and substantia nigra, respectively. On the other hand, no significant differences were found comparing net [35S]GTPγS binding in these regions from acute Δ9-THC-treated and control rats (Fig. 4).
Fig. 4.
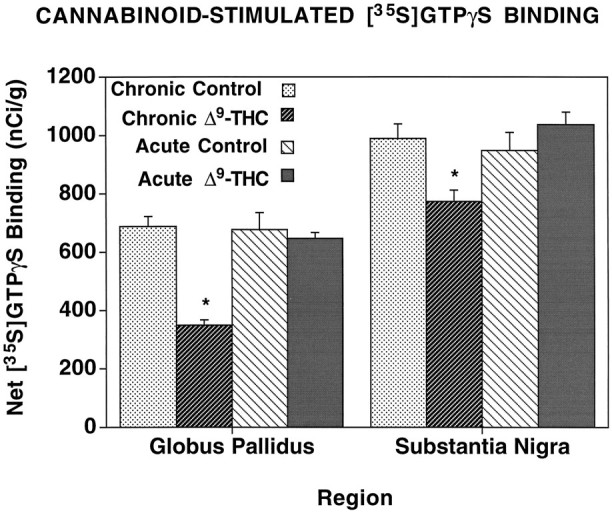
Net cannabinoid-stimulated [35S]GTPγS binding in the globus pallidus and substantia nigra. Sections were incubated with 2 mm GDP, and then with [35S]GTPγS (0.04 nm) and 2 mm GDP in the presence and absence of 10 μmWIN 55212-2. Net stimulated binding was determined by subtracting basal [35S]GTPγS binding from WIN 55212-2-stimulated [35S]GTPγS binding. *p < 0.005.
To determine the effect of acute and chronic Δ9-THC treatment on basal [35S]GTPγS binding, Tables 1 and 2 present the densitometric data as percentages of the control basal values for each group. Table 1 shows the results comparing control and chronic Δ9-THC-treated animals, whereas Table 2 shows the results from the acute Δ9-THC-treated and control groups. In most regions studied, there was no significant effect of either chronic or acute Δ9-THC treatment on basal [35S]GTPγS binding. In the chronic group (Table 1), the only exceptions were small decreases in basal binding in ventral caudate-putamen (15%) and globus pallidus (27%), and a small increase in basal binding in cerebellum (20%). As seen previously in Figures 3 and 4, however, there were significant decreases in WIN 55212-2-stimulated [35S]GTPγS binding in most brain regions in sections from chronic Δ9-THC-treated animals (Table 1). For the acute group (Table 2), no significant changes in either basal or WIN 55212-2-stimulated [35S]GTPγS binding were observed compared with controls.
Table 1.
Effect of chronic Δ9-THC treatment on basal and WIN 55212-2-stimulated [35S]GTPγS binding in the rat brain
| Region | Basal | Stimulated | ||
|---|---|---|---|---|
| Control | Chronic Δ9-THC | Control | Chronic Δ9-THC | |
| Hippocampus (D) | 100 ± 6% | 92 ± 4% | 181 ± 9% | 125 ± 7%† |
| Hippocampus (M) | 100 ± 8% | 101 ± 6% | 188 ± 11% | 132 ± 10%† |
| Hippocampus (V) | 100 ± 7% | 99 ± 4% | 196 ± 13% | 129 ± 8%‡ |
| Perirhinal cortex (M) | 100 ± 6% | 94 ± 3% | 178 ± 11% | 127 ± 6%* |
| Perirhinal cortex (V) | 100 ± 10% | 83 ± 2% | 166 ± 10% | 117 ± 6%† |
| Entorhinal cortex (D) | 100 ± 5% | 96 ± 8% | 156 ± 7% | 133 ± 7%* |
| Entorhinal cortex (M) | 100 ± 6% | 100 ± 2% | 174 ± 9% | 132 ± 3%‡ |
| Entorhinal cortex (V) | 100 ± 10% | 108 ± 4% | 189 ± 14% | 144 ± 5%* |
| Septum | 100 ± 7% | 104 ± 6% | 157 ± 12% | 144 ± 2% |
| Caudate-putamen (D) | 100 ± 6% | 81 ± 9% | 191 ± 14% | 134 ± 4%† |
| Caudate-putamen (M) | 100 ± 5% | 85 ± 2%* | 163 ± 12% | 118 ± 7%‡ |
| Globus pallidus | 100 ± 8% | 73 ± 4%* | 362 ± 18% | 206 ± 19%‡ |
| Substantia nigra | 100 ± 6% | 85 ± 8% | 484 ± 12% | 349 ± 11%‡ |
| Cerebellum | 100 ± 5% | 123 ± 4%* | 185 ± 13% | 167 ± 12% |
| PAG | 100 ± 8% | 92 ± 10% | 122 ± 4% | 114 ± 9% |
Sections were incubated with 2 mm GDP, and then with [35S]GTPγS (0.04 nm) and 2 mmGDP, with and without 10 μm WIN 55212-2. Data are expressed as percentage of control basal binding and represent mean values ± SE of duplicate sections from five animals. The level of the sections measured are dorsal (D), Figure 2 (top); midlevel (M), Figure2 (middle); and ventral (V), Figure 2 (bottom).
p < 0.05;
p < 0.01;
p < 0.005.
Table 2.
Effect of acute Δ9-THC treatment on basal and WIN 55212-2-stimulated [35S]GTPγS binding in the rat brain
| Region | Basal | Stimulated | ||
|---|---|---|---|---|
| Control | Acute Δ9-THC | Control | Acute Δ9-THC | |
| Hippocampus (D) | 100 ± 4% | 92 ± 2% | 161 ± 12% | 140 ± 6% |
| Hippocampus (M) | 100 ± 5% | 116 ± 5% | 172 ± 24% | 176 ± 5% |
| Hippocampus (V) | 100 ± 14% | 123 ± 2% | 183 ± 13% | 194 ± 8% |
| Perirhinal cortex (M) | 100 ± 5% | 112 ± 6% | 161 ± 17% | 166 ± 8% |
| Perirhinal cortex (V) | 100 ± 1% | 101 ± 3% | 145 ± 5% | 147 ± 8% |
| Entorhinal cortex (D) | 100 ± 6% | 97 ± 1% | 155 ± 10% | 148 ± 4% |
| Entorhinal cortex (M) | 100 ± 7% | 120 ± 6% | 170 ± 12% | 176 ± 6% |
| Entorhinal cortex (V) | 100 ± 16% | 112 ± 4% | 157 ± 10% | 164 ± 7% |
| Septum | 100 ± 7% | 97 ± 10% | 141 ± 6% | 127 ± 5% |
| Caudate-putamen (D) | 100 ± 10% | 100 ± 3% | 190 ± 19% | 181 ± 2% |
| Caudate-putamen (M) | 100 ± 10% | 112 ± 2% | 183 ± 11% | 201 ± 4% |
| Globus pallidus | 100 ± 7% | 109 ± 6% | 382 ± 23% | 379 ± 10% |
| Substantia nigra | 100 ± 8% | 116 ± 1% | 393 ± 21% | 437 ± 12% |
| Cerebellum | 100 ± 6% | 115 ± 5% | 194 ± 21% | 189 ± 6% |
| PAG | 100 ± 6% | 102 ± 7% | 122 ± 6% | 124 ± 5% |
Sections were incubated with 2 mm GDP, and then with [35S]GTPγS (0.04 nm) and 2 mmGDP, in the presence and absence of 10 μm WIN 55212-2. Data are expressed as percentage of control basal binding and represent mean values ± SE of duplicate sections from three animals. The level of the sections measured are dorsal (D), midlevel (M), and ventral (V).
Several independent methods were used to determine whether residual Δ9-THC in sections was potentially affecting the results of the study (data not shown). First, another set of brain sections from these animals was incubated with 1 μm SR141716A, the cannabinoid antagonist. Results revealed that SR141716A blocked WIN 55212-2-stimulated binding in sections from both control and chronic Δ9-THC-treated rats but had no effect on basal [35S]GTPγS binding in sections from either group. Therefore, there was not sufficient residual Δ9-THC acting as an agonist in these sections to affect the results. Another experiment assayed cannabinoid receptor binding in cerebellar membranes from both control and chronic Δ9-THC-treated rats, using the specific antagonist [3H]SR141716A as a radioligand. Results (not shown) revealed that chronic Δ9-THC treatment had no effect on the KD of [3H]SR141716A binding (0.50 nm in control vs 0.42 nm in chronic) but did decreaseBmax values (11.1 pmol/mg in control vs 8.5 pmol/mg in chronic) of [3H]SR141716A binding sites in cerebellar membranes. These results argue against the presence of residual Δ9-THC, because it would increase theKD value of [3H]SR141716A binding. Finally, in the same cerebellar membranes, theEmax of WIN 55212-2-stimulated [35S]GTPγS binding was decreased by 32%, whereas the potency of WIN 55212-2 was not affected at all in membranes from chronic Δ9-THC-treated rats (IC50 values of 0.2 μm WIN 55212-2 in control vs 0.25 μmWIN 55212-2 in chronic). If residual Δ9-THC were present in these membranes and acted as a partial agonist, it would shift the WIN 55212-2 concentration–effect curve to the right and increase the EC50 value of the full agonist (see Fig. 1). All of these results indicate that residual Δ9-THC was not present at the receptor in levels high enough to produce changes in receptor-stimulated [35S]GTPγS binding.
Effects of Δ9-THC administration on GABAB-stimulated [35S]GTPγS autoradiography
Our previous studies (Sim et al., 1995) showed that GABAB-stimulated [35S]GTPγS binding could be localized autoradiographically with use of baclofen as an agonist. Because cannabinoid receptors have been colocalized with GABAB receptors on the same neurons in cerebellum (Pacheco et al., 1993), and because [35S]GTPγS autoradiography allows for the opportunity to assay multiple receptor systems on adjacent brain sections, it was of interest to determine the effect of chronic Δ9-THC treatment on GABAB activation of G-proteins in sections from the same brains that demonstrated significant loss in WIN 55212-2-stimulated [35S]GTPγS binding. In these experiments, GABAB stimulation of [35S]GTPγS binding was performed using 300 μm baclofen in sections adjacent to those used for WIN 55212-2-stimulated [35S]GTPγS autoradiography. The resulting autoradiograms (not shown) revealed that chronic Δ9-THC treatment had no visible effects on baclofen-stimulated [35S]GTPγS binding in any region examined. When data from these autoradiograms were quantified (Fig.5), no significant changes in baclofen-stimulated [35S]GTPγS binding were observed in the cortex, hippocampus, or cerebellum of chronic Δ9-THC-treated animals compared with sections from control animals.
Fig. 5.
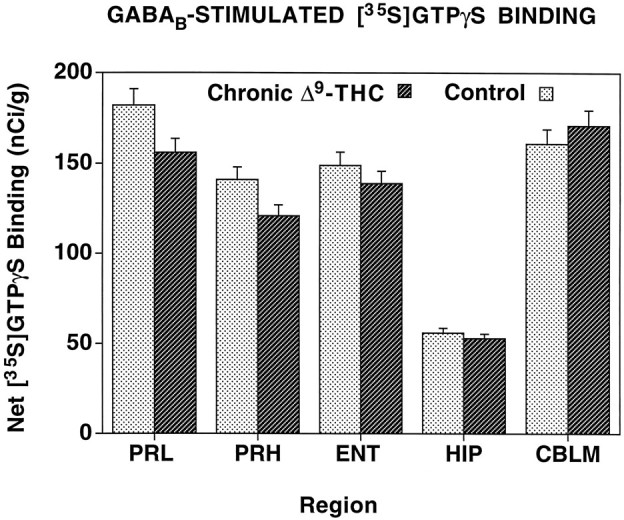
Net GABAB-stimulated [35S]GTPγS binding in brain regions from chronic Δ9-THC-treated and control rats. Sections were incubated with 2 mm GDP, and then with [35S]GTPγS (0.04 nm) and 2 mm GDP in the presence and absence of 300 μm baclofen. Net stimulated binding was determined by subtracting basal [35S]GTPγS binding from baclofen-stimulated [35S]GTPγS binding.CBLM, Cerebellum; ENT, entorhinal cortex;HIP, hippocampus; PRH, perirhinal cortex;PRL, prelimbic cortex.
DISCUSSION
Chronic treatment of rats with Δ9-THC resulted in decreased WIN 55212-2-stimulated [35S]GTPγS binding throughout the brain. This decrease in net WIN 55212-2-stimulated [35S]GTPγS binding was dramatic, with losses >50% in many regions. Any experiment administering chronic doses of Δ9-THC must be interpreted with caution, however, because the lipophilic nature of the drug may cause residual Δ9-THC to remain bound to sections and produce an artifactual result. The results of Figure 1, showing that Δ9-THC behaved as a partial agonist/antagonist in this system, provide a possible rationale for such an artifact; i.e., the presence of high concentrations of residual Δ9-THC could act as an antagonist to artificially reduce WIN 55212-2-stimulated [35S]GTPγS binding. This was not likely for several reasons. First, the concentration of Δ9-THC required to inhibit >50% of 10 μm WIN 55212-2-stimulated [35S]GTPγS binding was >200 μm (Fig. 1), a level that is unlikely to be attained in these brain sections. Second, preliminary data from cannabinoid receptor binding studies, using the antagonist [3H]SR141716A under the same conditions as the [35S]GTPγS binding assay, showed no change in KD value for [3H]SR141716A in membranes from chronic Δ9-THC-treated rats. Finally, Table 2 shows that acute Δ9-THC injection had no effect on WIN 55212-2-stimulated [35S]GTPγS binding in any brain region examined. Therefore, the finding that Δ9-THC is an agonist/antagonist in brain cannabinoid signal transduction systems may be important in regard to its mechanism of action, but this is unlikely to explain the large decrease in cannabinoid activation of G-proteins observed after chronic Δ9-THC treatment.
Because receptor activation of G-proteins is the critical step in the signal transduction pathway that determines agonist efficacy (Kenakin, 1993), the loss in agonist activity observed in this study is analogous to desensitization. Previous studies examining the effects of chronic Δ9-THC treatment have reported both decreased (Oviedo et al., 1993; De Fonseca et al., 1994) and unchanged (Westlake et al., 1991; Abood et al., 1993) cannabinoid receptor binding after chronic Δ9-THC treatment. Thus, effects on receptor function may occur without consistent detectable changes in the number of receptor binding sites. This is consistent with the established concept that desensitization and downregulation are separate processes, and that desensitization (uncoupling of G-proteins) precedes downregulation. In preliminary studies (C. Breivogel, L. Sim, and S. Childers, unpublished observations), a significant decrease in Bmaxvalues of [3H]SR141716A binding was observed in cerebellar membranes from chronic Δ9-THC-treated rats; thus, both desensitization and downregulation may be occurring after this chronic drug treatment. Detailed time course studies are now being performed to differentiate between these two processes. Another important consideration is the agonist used to develop cannabinoid tolerance. Chronic treatment with CP 55,940 (a full agonist) has been reported to decrease cannabinoid receptor number in mouse cerebellum (Fan et al., 1996), despite earlier reports that chronic Δ9-THC treatment had no effect on cannabinoid receptor number in mouse brain (Abood et al., 1993). Chronic Δ9-THC treatment also had no effect on cannabinoid receptor mRNA (Abood et al., 1993), although chronic CP-55,940 treatment decreased cannabinoid receptor mRNA in the caudate-putamen (Rubino et al., 1994) and increased it in the cerebellum (Fan et al., 1996).
Interestingly, the effects of chronic Δ9-THC treatment on cannabinoid-activated G-proteins contrast with the effects of chronic morphine treatment on μ opioid receptor function (Sim et al., 1996a). In that study, decreased μ opioid-stimulated [35S]GTPγS binding was found in only specific brainstem nuclei after chronic morphine treatment. Although both studies revealed that chronic agonist treatment decreased receptor-coupled G-protein activity, there are fundamental differences between the results of the two studies. The changes in μ opioid-stimulated [35S]GTPγS binding after chronic morphine treatment were anatomically discrete and relatively small in magnitude. In contrast, large, widespread decreases in cannabinoid-stimulated [35S]GTPγS binding were identified throughout the brain after chronic Δ9-THC treatment. One explanation for this difference may be the catalytic efficiency of these two receptors; for example, in the striatum, each μ opioid receptor activates approximately seven times the number of G-proteins as each cannabinoid receptor (Sim et al., 1996b). This difference in catalytic amplification may have consequences during chronic agonist treatment, resulting in greater adaptive changes in receptor–G-protein coupling in the less efficiently coupled system. The observation that chronic Δ9-THC treatment produces a dramatic decrease in [35S]GTPγS binding throughout the brain, whereas chronic morphine administration has a much more subtle effect, also suggests that effector events downstream from the G-protein influence the development of tolerance.
The effects in the cerebellum are of particular interest because of the known relationship between cannabinoid and GABAB receptors. Previous studies have shown that although cannabinoid and GABAB receptors are located on the same cerebellar neurons and act via the same pool of adenylyl cyclase, these receptors are coupled to distinct G-proteins (Pacheco et al., 1993). The finding that chronic Δ9-THC treatment decreased cannabinoid-stimulated [35S]GTPγS binding, but had no effect on GABAB-stimulated [35S]GTPγS binding in adjacent sections, suggests that this chronic effect of Δ9-THC is best described as homologous desensitization. Furthermore, these results confirm that although these receptors may share common effectors, the receptors are coupled to different populations of G-proteins.
The large decreases in cannabinoid-activated G-proteins in the nigrostriatal system may be relevant to the motor effects of cannabinoids. Cannabinoid receptors and cannabinoid-stimulated [35S]GTPγS binding are particularly high in the caudate-putamen, globus pallidus, and substantia nigra (Herkenham et al., 1991b; Jansen et al., 1992; Sim et al., 1995). Previous anatomical studies have shown that cannabinoid receptors are located on striatal neurons projecting to globus pallidus and substantia nigra (Herkenham et al., 1991a). Decreased cannabinoid-stimulated [35S]GTPγS binding in the caudate-putamen, globus pallidus, and substantia nigra indicates that all of the components of this system are affected by chronic Δ9-THC treatment. Changes in these regions may be particularly relevant to the cannabinoid withdrawal syndrome elicited when tolerant animals are administered the antagonist SR 141716A (Aceto et al., 1995; Tsou et al., 1995). This syndrome is characterized by patterns of motor behaviors that may be mediated via the nigrostriatal system.
Perhaps the most dramatic effect of chronic Δ9-THC treatment on cannabinoid-stimulated [35S]GTPγS binding was detected in hippocampus, consistent with behavioral effects of cannabinoids on short-term memory tasks (Heyser et al., 1993). In delayed-to-match sample (DMS) tasks, acute exposure to Δ9-THC disrupted performance in a manner analogous to that of a hippocampal lesion (Hampson et al., 1995a), except that the cannabinoid effect was fully reversible (Heyser et al., 1993). These acute effects were selectively associated with blockade of hippocampal cell activation during the information “encoding” phase in the DMS trial; hence, there was a delay-dependent deficit in performance under the influence of the drug (Heyser et al., 1993; Hampson et al., 1995a).
The uncoupling of the receptor from the G-protein by chronic drug treatment would probably lead to a cessation of the “disruptive acute” effects of Δ9-THC on DMS performance. The 21 d chronic treatment regimen and dose of Δ9-THC used in the present study were chosen on the basis of a previous study (Deadwyler et al., 1995b) in which significant (75%) tolerance to Δ9-THC effects was found at 21 d and complete tolerance was achieved after 30–35 d. The results of the present study, showing profound reductions in hippocampal cannabinoid receptor–G-protein coupling during the same treatment period associated with marked recovery from the acute effects of Δ9-THC on DMS performance, strongly suggest that the two phenomena are closely related. These findings also indicate an adaptive significance to the uncoupling of the receptor and G-protein after repeated drug exposure, especially in circumstances where acute cannabinoid receptor stimulation leads to a decrease in accuracy of performance.
The mechanism by which cannabinoids disrupt hippocampal function has been examined in studies on the effects of cannabinoids on cAMP modulation of voltage-gated potassium “A” current in cultured hippocampal neurons (Deadwyler et al., 1993). The net effect of cannabinoid receptor stimulation is to decrease cAMP-mediated reductions in potassium A current at a given membrane potential. It has been hypothesized (Deadwyler et al., 1995a) that if cannabinoid receptor-linked potassium channels occupied a strategic position on the terminals of the perforant path projection from the entorhinal cortex to the molecular layer of the dentate gyrus [a region reported to have high densities of cannabinoid receptors (Herkenham et al., 1991b)], they could regulate transmitter release via changes in steady-state potassium conductances in perforant path terminals. The demonstrated receptor–G-protein uncoupling after chronic Δ9-THC treatment would therefore result in a relative increase (i.e., lack of cannabinoid-induced terminal hyperpolarization) in neurotransmitter release at the perforant path-to-dentate granule cell synapse. Because effects on potassium A channels (and indeed on all other effectors linked to these Gi/o-coupled cannabinoid receptors) occur downstream from the receptor–transducer coupling, the result of decreased activation of G-proteins after chronic agonist treatment should have profound implications at the effector level.
Footnotes
These studies were supported by Public Health Service Grants DA-06784 and DA-07625 from the National Institute on Drug Abuse. Doug Byrd, Joanne Konstantopoulis, and Ruoyu Xiao provided excellent technical assistance. Dr. Dana E. Selley, Dr. Linda Porrino, and Christopher S. Breivogel provided helpful discussions.
Correspondence should be addressed to Dr. Steven R. Childers, Department of Physiology and Pharmacology, Bowman Gray School of Medicine, Wake Forest University, Medical Center Boulevard, Winston-Salem, NC 27157.
REFERENCES
- 1.Abood ME, Sauss C, Fan F, Tilton CL, Martin BR. Development of behavioral tolerance to Δ9-THC without alteration of cannabinoid receptor binding or mRNA levels in whole brain. Pharmacol Biochem Behav. 1993;46:575–579. doi: 10.1016/0091-3057(93)90546-6. [DOI] [PubMed] [Google Scholar]
- 2.Aceto MD, Scates SM, Lowe JA, Martin BR. Cannabinoid precipitated withdrawal by the selective cannabinoid receptor antagonist, SR 141716A. Eur J Pharmacol. 1995;282:R1–R2. doi: 10.1016/0014-2999(95)00447-s. [DOI] [PubMed] [Google Scholar]
- 3.Carlini EA. Tolerance to chronic administration of cannabis sativa (marijuana) in rats. Pharmacology. 1968;1:135–142. doi: 10.1159/000135954. [DOI] [PubMed] [Google Scholar]
- 4.Compton DR, Rice KC, DeCosta BR, Razdan RK, Melvin LS, Johnson MR, Martin BR. Cannabinoid structure-activity relationships: correlation of receptor binding and in vivo activities. J Pharmacol Exp Ther. 1993;265:218–226. [PubMed] [Google Scholar]
- 5.De Fonseca FR, Gorriti MA, Fernandez-Ruiz JJ, Palomo T, Ramos JA. Downregulation of rat brain cannabinoid binding sites after chronic Δ9-tetrahydrocannabinol treatment. Pharmacol Biochem Behav. 1994;47:33–40. doi: 10.1016/0091-3057(94)90108-2. [DOI] [PubMed] [Google Scholar]
- 6.Deadwyler SA, Hampson RE, Bennett BA, Edwards TA, Mu J, Pacheco MA, Ward SJ, Childers SR. Cannabinoids modulate potassium current in cultured hippocampal neurons. Receptors Channels. 1993;1:121–134. [PubMed] [Google Scholar]
- 7.Deadwyler SA, Hampson RE, Childers SR. Functional significance of cannabinoid receptors in brain. In: Pertwee RG, editor. Cannabinoid receptors. Academic; New York: 1995a. pp. 206–231. [Google Scholar]
- 8.Deadwyler SA, Heyser CJ, Hampson RE. Complete adaptation to the memory disruptive effects of delta-9-THC following 35 days of exposure. Neurosci Res Commun. 1995b;17:9–18. [Google Scholar]
- 9.Devane WA, Dysarz FAI, Johnson MR, Melvin LS, Howlett AC. Determination and characterization of a cannabinoid receptor in rat brain. Mol Pharmacol. 1988;34:605–613. [PubMed] [Google Scholar]
- 10.Dewey WL. Cannabinoid pharmacology. Pharmacol Rev. 1986;48:151–174. [PubMed] [Google Scholar]
- 11.Dill JA, Howlett AC. Regulation of adenylate cyclase by chronic exposure to cannabimimetic drugs. J Pharmacol Exp Ther. 1988;244:1157–1163. [PubMed] [Google Scholar]
- 12.Fan F, Tao Q, Abood ME, Martin BR. Cannabinoid receptor down-regulation without alteration of the inhibitory effect of CP 55,940 on adenylyl cyclase in the cerebellum of CP 55,940-tolerant mice. Brain Res. 1996;706:13–20. doi: 10.1016/0006-8993(95)01113-7. [DOI] [PubMed] [Google Scholar]
- 13.Gaoni Y, Mechoulam R. Isolation, structure, and partial synthesis of an active constituent of hashish. J Am Chem Soc. 1964;86:1646–1647. [Google Scholar]
- 14.Hampson RE, Byrd D, Konstantopolous J, Bunn T, Jarrard LE, Deadwyler SA. Proactive interference and short term memory during performance of a DNMS task in normal rats and rats with hippocampus removed. Soc Neurosci Abstr. 1995a;21:1215. [Google Scholar]
- 15.Hampson RE, Evans GJO, Mu J, Zhuang S, King VC, Childers SR, Deadwyler SA. Role of cyclic AMP dependent protein kinase in cannabinoid receptor modulation of potassium “A-current” in cultured rat hippocampal neurons. Life Sci. 1995b;56:2081–2088. doi: 10.1016/0024-3205(95)00192-9. [DOI] [PubMed] [Google Scholar]
- 16.Herkenham M, Lynn AB, de Costa BR, Richfield EK. Neuronal localization of cannabinoid receptors in the basal ganglia of the rat. Brain Res. 1991a;547:267–274. doi: 10.1016/0006-8993(91)90970-7. [DOI] [PubMed] [Google Scholar]
- 17.Herkenham M, Lynn AB, Johnson MR, Melvin LS, de Costa BR, Rice KC. Characterization and localization of cannabinoid receptors in rat brain: a quantitative in vitro autoradiographic study. J Neurosci. 1991b;11:563–583. doi: 10.1523/JNEUROSCI.11-02-00563.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Heyser CJ, Hampson RE, Deadwyler SA. Effects of delta-9-tetrahydrocannabinol on delayed match to sample performance in rats: alterations in short-term memory associated with changes in task specific firing of hippocampal cells. J Pharmacol Exp Ther. 1993;264:294–307. [PubMed] [Google Scholar]
- 19.Hilf G, Gierschik P, Jakobs KH. Muscarinic acetylcholine receptor-stimulated binding of guanosine 5′-O-(3-thiotriphosphate) to guanine-nucleotide-binding proteins in cardiac membranes. Eur J Biochem. 1989;186:725–731. doi: 10.1111/j.1432-1033.1989.tb15266.x. [DOI] [PubMed] [Google Scholar]
- 20.Howlett AC. Cannabinoid inhibition of adenylate cyclase: biochemistry of the response in neuroblastoma cell membranes. Mol Pharmacol. 1985;27:429–436. [PubMed] [Google Scholar]
- 21.Howlett AC, Qualy JM, Khachatrian LL. Involvement of Gi in the inhibition of adenylate cyclase by cannabimimetic drugs. Mol Pharmacol. 1986;29:307–313. [PubMed] [Google Scholar]
- 22.Jansen EM, Haycock DA, Ward SJ, Seybold VS. Distribution of cannabinoid receptors in rat brain determined with aminoalkylindoles. Brain Res. 1992;575:93–102. doi: 10.1016/0006-8993(92)90428-c. [DOI] [PubMed] [Google Scholar]
- 23.Kenakin T. Efficacy. In: Kenakin T, editor. Pharmacologic analysis of drug-receptor interaction. Raven; New York: 1993. pp. 249–277. [Google Scholar]
- 24.Mackie K, Hille B. Cannabinoids inhibit N-type calcium channels in neuroblastoma-glioma cells. Proc Natl Acad Sci USA. 1992;89:3825–3829. doi: 10.1073/pnas.89.9.3825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Matsuda LA, Lolait SJ, Brownstein MJ, Young AL, Bonner TI. Structure of a cannabinoid receptor and functional expression of the cloned cDNA. Nature. 1990;346:561–564. doi: 10.1038/346561a0. [DOI] [PubMed] [Google Scholar]
- 26.Nestler EJ, Alreja M, Aghajanian GK. Molecular and cellular mechanisms of opiate action: studies in the locus coeruleus. Brain Res Bull. 1994;35:521–528. doi: 10.1016/0361-9230(94)90166-x. [DOI] [PubMed] [Google Scholar]
- 27.Oviedo A, Glowa J, Herkenham M. Chronic cannabinoid administration alters cannabinoid receptor binding in rat brain: a quantitative autoradiographic study. Brain Res. 1993;616:293–302. doi: 10.1016/0006-8993(93)90220-h. [DOI] [PubMed] [Google Scholar]
- 28.Pacheco M, Childers SR, Arnold R, Casiano F, Ward SJ. Aminoalkylindoles: actions on specific G-protein-linked receptors. J Pharmacol Exp Ther. 1991;257:170–183. [PubMed] [Google Scholar]
- 29.Pacheco M, Ward SJ, Childers SR. Identification of cannabinoid receptors in cultures of rat cerebellar granule cells. Brain Res. 1993;603:102–110. doi: 10.1016/0006-8993(93)91304-b. [DOI] [PubMed] [Google Scholar]
- 30.Rubino T, Massi P, Patrini G, Venier I, Giagnoni G, Parolaro D. Chronic CP-55,940 alters cannabinoid receptor mRNA in the rat brain: an in situ hybridization study. NeuroReport. 1994;5:2493–2496. doi: 10.1097/00001756-199412000-00022. [DOI] [PubMed] [Google Scholar]
- 31.Selley DE, Nestler EJ, Breivogel CS, Childers SR (1996a) Opioid receptor-coupled G-proteins in rat locus coeruleus membranes: decrease in activity after chronic morphine treatment. Brain Res, in press. [DOI] [PubMed]
- 32.Selley DE, Stark S, Sim LJ, Childers SR. Cannabinoid receptor stimulation of guanosine-5′-O-(3-[35S]thio)triphosphate binding in rat brain membranes. Life Sci. 1996b;59:659–668. doi: 10.1016/0024-3205(96)00347-5. [DOI] [PubMed] [Google Scholar]
- 33.Sim LJ, Selley DE, Childers SR. In vitro autoradiography of receptor-activated G-proteins in rat brain by agonist-stimulated guanylyl 5′-[γ-[35S]thio]-triphosphate binding. Proc Natl Acad Sci USA. 1995;92:7242–7246. doi: 10.1073/pnas.92.16.7242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sim LJ, Selley DE, Dworkin SI, Childers SR. Effects of chronic morphine administration on mu opioid receptor-stimulated [35S]GTPγS autoradiography in rat brain. J Neurosci. 1996a;16:2684–2692. doi: 10.1523/JNEUROSCI.16-08-02684.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sim LJ, Selley DE, Xiao R, Childers SR. Differences in G-protein activation by mu and delta opioid, and cannabinoid, receptors in rat striatum. Eur J Pharmacol. 1996b;307:95–107. doi: 10.1016/0014-2999(96)00211-7. [DOI] [PubMed] [Google Scholar]
- 36.Traynor JR, Nahorski SR. Modulation by μ-opioid agonists of guanosine-5′-O-(3-[35S]thio)triphosphate binding to membranes from human neuroblastoma SH-SY5Y cells. Mol Pharmacol. 1995;47:848–854. doi: 10.1016/S0026-895X(25)08634-1. [DOI] [PubMed] [Google Scholar]
- 37.Tsou K, Patrick SL, Walker JM. Physical withdrawal in rats tolerant to Δ9-tetrahydrocannabinol precipitated by a cannabinoid receptor antagonist. Eur J Pharmacol. 1995;280:R13–R15. doi: 10.1016/0014-2999(95)00360-w. [DOI] [PubMed] [Google Scholar]
- 38.Westlake TM, Howlett AC, Ali SF, Paule MG, Scallet AC, Slikker W. Chronic exposure to Δ9-tetrahydrocannabinol fails to irreversibly alter brain cannabinoid receptors. Brain Res. 1991;544:145–149. doi: 10.1016/0006-8993(91)90897-5. [DOI] [PubMed] [Google Scholar]