

Abstract
We reported earlier that the levels of Ca2+-dependent metalloproteinases are increased in Alzheimer’s disease (AD) specimens, relative to control specimens. Here we show that these enzymes are forms of the matrix metalloproteinase MMP-9 (EC3.4.24.35) and are expressed in the human hippocampus. Affinity-purified antibodies to MMP-9 labeled pyramidal neurons, but not granular neurons or glial cells. MMP-9 mRNA is expressed in pyramidal neurons, as determined with digoxigenin-labeled MMP-9 riboprobes, and the presence of this mRNA is confirmed with reverse transcriptase PCR. The cellular distribution of MMP-9 is altered in AD because 76% of the total 100 kDa enzyme activity is found in the soluble fraction of control specimens, whereas only 51% is detectable in the same fraction from AD specimens. The accumulated 100 kDa enzyme from AD brain is latent and can be converted to an active form with aminophenylmercuric acetate.
MMP-9 also is detected in close proximity to extracellular amyloid plaques. Because a major constituent of plaques is the 4 kDa β-amyloid peptide, synthetic Aβ1–40 was incubated with activated MMP-9. The enzyme cleaves the peptide at several sites, predominantly at Leu34-Met35 within the membrane-spanning domain. These results establish that neurons have the capacity to synthesize MMP-9, which, on activation, may degrade extracellular substrates such as β-amyloid. Because the latent form of MMP-9 accumulates in AD brain, it is hypothesized that the lack of enzyme activation contributes to the accumulation of insoluble β-amyloid peptides in plaques.
Keywords: matrix metalloproteinases, Alzheimer’s disease, amyloid cleavage, amyloid plaques, gelatinase, protease activation
The amyloid β peptide (Aβ) found in plaques is a 39–42 residue peptide derived from one or several of the membrane-associated precursors (reviewed in Selkoe, 1994). An enzyme termed α-secretase cleaves amyloid precursor protein (APP) within the Aβ sequence to release the extracellular portion of the precursor, along with part of the Aβ sequence (Esch et al., 1990; Sisodia et al., 1990). Because the plaques contain the intact Aβ, it seems that Aβ in plaques originates from APP that has been processed by enzymes other than α-secretase.
The secreted portions of APPs contain inhibitor domains that may regulate the activities of extracellular proteinases. Secreted APP-751 and APP-770, termed protease nexin II, contain a serine proteinase inhibitor domain that inactivates chymotrypsin-like enzymes (Kitaguchi et al., 1988; Ponte et al., 1988; Tanzi et al., 1988; Oltersdorf et al., 1989; Van Nostrand et al., 1989). Another domain of APP inhibits matrix metalloproteinases (MMPs; Miyazaki et al., 1993).
The human hippocampus contains proteinases that are biochemically similar to the Ca2+- and Zn2+-dependent MMP family (Backstrom et al., 1992). Interestingly, the hippocampus of Alzheimer’s disease (AD) individuals contained greater amounts of a 100 kDa enzyme, relative to control individuals, as measured by zymography (Backstrom et al., 1992). This assay is a standard SDS-polyacrylamide gel that contains gelatin (Heussen and Dowdle, 1980). The enzymes that are trapped in polyacrylamide are “renatured” in nonionic detergent and incubated in a detergent-free buffer. Because SDS artificially can activate latent MMPs, an additional biotin-gelatin plate assay was used in the current study to determine whether the 100 kDa enzyme is latent. To characterize this metalloproteinase further, we examined the mass of the enzyme and its activity by using gelatin and Aβ peptide after treatment withp-aminophenylmercuric acetate (APMA).
APMA causes the autocatalytic conversion of the latent MMPs to active forms by a “cysteine switch” mechanism (Sprinman et al., 1990), which promotes the removal of a 10 kDa N-terminal proregion. The active enzyme cleaves types IV and V collagen (Wilhelm et al., 1989), immobilized gelatin (Davis and Martin, 1990), and substance P (Backstrom and Tökés, 1995). The substrate specificity of MMP-9 was examined extensively with peptides on the basis of the cleavage site of gelatin (Netzel-Arnett et al., 1993). Here we demonstrate that APMA causes an increase of gelatinase activity from a soluble brain fraction and a concomitant decrease in mass to 90 kDa, which is consistent with the 100 kDa proteinase being the latent form of MMP-9.
The cleavage sites of Aβ1–40 by activated MMP-9 are documented. To determine the location of MMP-9 and the cellular source of its mRNA, we used affinity-purified polyclonal or specific monoclonal antibodies and riboprobes to label AD and control hippocampus sections. The presence of MMP-9 mRNA in the brain specimens was examined by reverse transcriptase-PCR.
MATERIALS AND METHODS
Tissue specimens. Hippocampal specimens were obtained from the Alzheimer’s Disease Research Center at the University of Southern California. Ten Alzheimer’s patients (3 males and 7 females) and seven normal patients who died of non-neurological disorders (5 males and 2 females) were used in this study. Patients with metastatic cancer to the brain or cerebral hemorrhage were excluded. The ages of the patients ranged from 33 to 91 years; the mean age of Alzheimer’s patients was 76.4 ± 7.9 and 64 ± 19 years for normal patients. The mean postmortem interval (PMI) for Alzheimer’s patients was 4.3 ± 2.7 and 10.3 ± 4.2 hr for normal patients. Previous studies indicated that PMIs <15 hr did not affect the activities of MMPs (Backstrom et al., 1992). All AD patients were selected after a clinical diagnosis of possible AD according to the National Institute of Neurological and Communicative Disorders and Stroke–Alzheimer’s Disease and Related Disorders Association (NINCDS–ADRDA) criteria (McKhann et al., 1984). The diagnosis of AD was confirmed by postmortem neuropathological examination via the diagnostic criteria of the Consortium to Establish a Registry for Alzheimer’s Disease, a modification of the Khachaturian protocol (Khachaturian, 1985). Evaluation of the extent of neurofibrillary tangles, neuritic plaques, and amyloid angiopathy was performed on 8 μm paraffin sections stained with either hematoxylin and eosin, Bielschowsky silver-impregnation method, or thioflavine S. A minimum of three 1 mm2 microscopic fields were evaluated for each section. All AD samples had moderate-to-severe amounts of neurofibrillary tangles and neuritic plaques and occasional amyloid angiopathy. Control patients had trace or no amounts of either neurofibrillary tangles or neuritic plaques. Other factors such as ethnicity, age of onset, number of years of education, family history, and scores from the Mini Mental State Examination, the Clinical Dementia Rating, and the Boston Naming Test are summarized in Table1. Hippocampal specimens were either snap-frozen in liquid nitrogen-chilled isopentane and stored at −70°C for various biochemical experiments or fixed in 4% paraformaldehyde for immunohistochemistry.
Table 1.
| ID# | Age | Sex | PMI | Ethnicity | Age of onset | Autopsy diagnosis | Education | Family history | MMSE | CDR | BNT |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 4 | 63 | M | 11 | Cauc | 49 | AD | 12 | No | 0 | 1 | 1 |
| 41 | 80 | F | 2 | Cauc | 68 | AD | 16 | Mother, sister | 0 | NA | NA |
| 107 | 82 | F | 2.5 | Cauc | 69 | AD | 11 | Cousin | 1 | NA | NA |
| 161 | 73 | M | 7.5 | Cauc | 64 | AD/MID | 12 | Mother, brother (Parkinson’s) | 0 | NA | NA |
| 206 | 81 | F | 4 | Cauc | 65 | AD | 14 | No | 1 | NA | NA |
| 235 | 72 | M | 2.3 | Cauc | 61 | AD | 13 | Two aunts | 0 | NA | NA |
| 342 | 91 | F | 5 | Cauc | 77 | AD | 12 | NO | NA | 3 | NA |
| 538 | 66 | F | 3 | Cauc | 57 | AD | 12 | Mother, grandmother | 9 | 2 | 21 |
| 595 | 75 | F | 3.5 | Cauc | 57 | AD | NA | Mother, uncle | NA | 3 | NA |
| 602 | 81 | F | 2.2 | Cauc | NA | AD | NA | NA | NA | NA | NA |
| 95 | 77 | F | 3.3 | Cauc | Not appl | Norm | 12 | Brother, grandmother, two uncles | 30 | 0 | 52 |
| 343 | 83 | M | 14 | Af-Am | Not appl | Norm | NA | NA | NA | NA | NA |
| 351 | 71 | M | 11.5 | Af-Am | Not appl | Norm | NA | NA | NA | NA | NA |
| 359 | 64 | M | 11 | Cauc | Not appl | Norm | NA | NA | NA | NA | NA |
| 381 | 38 | M | 8.5 | NA | Not appl | Norm | NA | NA | NA | NA | NA |
| 559 | 82 | F | 17 | Cauc | Not appl | Norm | 12 | No | 27 | 0 | 54 |
| 612 | 33 | M | 7 | Af-Am | Not appl | Norm | NA | NA | NA | NA | NA |
PMI, Postmortem interval is given in hours; NA, not available; Not appl, not applicable; MMSE, Mini Mental State Examination [where a score of 22 or less (of a possible 30) indicates dementia]; CDR, Clinical Dementia Rating (where 0 = normal and 3 = moderate dementia); BNT, Boston Naming Test (where the highest score is 60); Cauc, Caucasian; Af-Am, African-American.
Assessment of metalloproteinase activities in hippocampal fractions. Matrix proteinase activities were measured by gelatin zymography on hippocampal specimens from 10 AD and 7 control patients (Backstrom et al., 1992). Four representative AD and four control hippocampal tissues were examined for the relative amount of soluble and detergent-extractable gelatinase activities in three fractions. PBS-washed tissues were sonicated in 3 vol (wet weight) of 50 mm Tris and 0.05% NaN3, pH 7.6, containing the proteinase inhibitors diisopropyl fluorophosphate (DIFP; 1.7 mm), 1 mmp-hydroxymercuric benzoate, 5 μg/ml leupeptin, and 5 μg/ml pepstatin A and then spun at 100,000 × g for 1 hr at 4°C. The resulting Tris-soluble fraction was collected and used for subsequent assays. The Tris-insoluble pellet was treated with 1% Triton X-100 in Tris buffer containing proteinase inhibitors, and the resulting soluble fraction was collected. The remaining pellet was sonicated in 6 vol of 1× SDS-PAGE sample buffer (2% SDS) and spun; the resulting supernatant was collected as the SDS-soluble fraction.
For the substrate gel assay, 40 μl of the Tris-, Triton-, and SDS-soluble fractions (containing identical tissue weight-equivalents per fraction) were electrophoresed in gelatin-containing substrate gels as described [Backstrom et al. (1992); the Tris- and Triton-soluble fractions were diluted 1:2 in 2× SDS-PAGE sample buffer]. The gels were incubated for 18 hr at 37°C in 50 mm Tris, 5 mm CaCl2, and 0.05% NaN3, pH 8.0. The gels were stained with Coomassie brilliant blue and scanned with an LKB Ultrascan XL densitometer to quantify the amount of metalloproteinase activities from each fraction.
For the gelatin-biotin assay, Tris-soluble samples were analyzed as described (Davis and Martin, 1990), with minor modifications. Fifty microliters of biotinylated gelatin (5 μg/ml) or control gelatin (5 μg/ml) in 1 m NaCl were added to 96 well plates and incubated for 1 hr at room temperature (RT). Then the wells were blocked with 200 μl of 0.15 m NaCl containing 0.05% Tween-20 for 20 min. The Tris-soluble fractions were treated with 7 μg/ml leupeptin, 7 μg/ml pepstatin A, and 2 mm DIFP to inhibit nonmetalloproteinase activities. Fifty microliters of the inhibitor-treated sample, preincubated in the absence or presence of 1 mm APMA and 5 mm CaCl2 in Tris-buffered saline (TBS), were added to the wells in triplicate. After 1 hr at 37°C, samples in the wells were washed with NaCl-Tween solution and with NaCl-Tween solution containing 1% bovine serum albumin (BSA; Sigma, St. Louis, MO). Then the samples were incubated for an additional 45 min in 1% BSA. One hundred microliters of 1 μg/ml avidin-peroxidase (Sigma) in 1% BSA solution were added to each well and incubated for 0.5 hr. The samples were washed with the 1% BSA solution, and the peroxidase activity was detected with 100 μl of a solution containing 1 mg/ml 0-phenylenediamine hydrochloride and 0.01% H2O2 in a 0.1m citrate-phosphate buffer, pH 5.0. The reaction was allowed to proceed for 15 min and then terminated with the addition of 100 μl of 1N H2S04. The optical absorbance was blanked against the control samples containing gelatin and read at 405 nm. Because a decrease in absorbance corresponds to an increase in activity, experimental values were subtracted from the mean value of the wells containing nonbiotinylated gelatin to make a direct relationship between an increase in activity and an increase in absorbance. The SEM was consistently <10%.
The gelatinase activity of a soluble brain fraction was compared with the activity of α-chymotrypsin in the plate assay to examine relative increases in metalloproteinase activities attributable to treatment with APMA. Because the Tris-soluble fraction was prepared from tissue at 1:3 (w/v) and the final dilution was 1:5 (v/v) in the plate assay, the fractions analyzed in the plate assay (50 μl) represent soluble protein from ≈3.3 mg of wet weight tissue. Therefore, specific activities (nanograms of chymotryptic-equivalent activity in soluble fraction/mg tissue) can be calculated by dividing the activities by 3.3.
For the activation of latent enzymes, the Tris-soluble fractions containing protease inhibitors were collected and treated with 1 mm APMA at 37°C. Aliquots (100 μl) were removed at the appropriate times (0, 6, 12, and 24 hr), treated with 100 μl of 2× sample buffer, and stored overnight at 4°C. Zymography was performed with 7.5% substrate gels.
Purification of MMP-9 from cell cultures. MMP-9 was purified from the conditioned media of the human promyelocytic leukemia cell line HL-60. Stimulation of these cells with phorbol esters causes them to secrete predominantly latent forms of MMP-9 (Davis and Martin, 1990;Moll et al., 1990; Backstrom and Tökés, 1995). MMP-9 was purified with gelatin-agarose affinity chromatography (Hibbs et al., 1985) in the absence of detergent, followed by two steps of gel filtration HPLC in TBS (50 mm Tris, pH 7.6, containing 150 mm NaCl and 0.05% NaN3). The latent form of MMP-9 eluted at 11.4 ± 0.1 min from the 7.5 mm (inner diameter) × 30 cm TSK G3000SW HPLC column (TosoHaas, Montgomeryville, PA) at a flow rate of 0.5 ml/min (Backstrom and Tökés, 1995). After two passes, the enzyme was determined to be >95% pure as judged by silver-stained gels. The <5% contamination is a complexed form of MMP-9 (270 kDa), which is removable by immunodepletion with the use of specific monoclonal antibodies to MMP-9 (J. R. Backstrom and G. P. Lim, unpublished observations).
Immunodepletion of MMP-9 activity. All procedures were performed at 4°C. Brain tissue (150 mg) was cut from snap-frozen specimens and washed 4× in PBS. Three wet-weight volumes of TBS containing a proteinase inhibitor cocktail (50 μg/ml leupeptin, 50 μg/ml pepstatin A, and 50 μg/ml phenylmethylsulfonyl fluoride in 1% DMSO) were added, and the samples were incubated for 15 min. The samples were sonicated at 100 W for two 10 sec intervals and centrifuged for 30 min at 13,000 × g. The supernatant was collected, and the total soluble brain protein concentration was determined. On the basis of our previous observation that a representative sample of 200 μg of brain protein contained ∼10 ng of MMPs (Backstrom, unpublished data), 50 μl samples were incubated with an estimated 40-fold excess of murine monoclonal MMP-9 antibodies (Oncogene Science, Cambridge, MA). Two different monoclonal antibodies were used; Ab-1 recognizes both the latent and active forms of human MMP-9, whereas Ab-2 recognizes only the latent form. The specificity of monoclonal antibodies was established with Western blots and HT1080 cell culture supernatant in which only MMP-9 was recognized (Oncogene Science). The solutions were incubated for 8–12 hr with gentle rocking. Aliquots of protein G-Sepharose beads (Sigma) were added in an estimated 300-fold excess of the amount of antibodies added, and samples were incubated for 10–12 hr. The samples were centrifuged for 30 min at 13,000 × g to spin down the beads, and the supernatant was removed for zymography to determine the remaining enzyme activities. Controls were incubated with only the protein G-Sepharose beads. Supernatant containing 200 μg of brain protein was electrophoresed as described before, and the gels were incubated in 5 μm ZnCl2 for 60 min before the final incubation in 5 mm CaCl2 for 18 hr at 37°C; the Coomassie blue-stained gels were scanned as outlined before.
Preparation of tissue sections. Tissue sections were fixed in 4% paraformaldehyde (J. T. Baker Chemical Company, Phillipsburg, NJ) in PBS for 1–7 d. The samples were rinsed in PBS for 1 d and cryoprotected in 5% sucrose in PBS for 1 d, followed by 15% sucrose in PBS for 1–2 d. Tissue specimens were frozen in the vapor phase of liquid nitrogen on cryostat blocks with Tissue-Tek O.C.T. (Miles, Elkhart, IN). Sections were cut (8 μm) on a Reichert Histostat at −14°C, mounted on Superfrost/Plus slides (Fisher Scientific, Pittsburgh, PA), and stored at −20°C.
Immunolocalization of metalloproteinases. Rabbit anti-MMP-9, a generous gift of Dr. Margaret Hibbs (Veterans Administration Medical Center, Newington, CT), was affinity-purified with enzymes that were partially purified from cell cultures. The antibodies were purified against the activated form of MMP-9. Material from the gelatin-agarose column (50 μg of protein from the first step of purification; see above) was activated with APMA, electrophoresed in preparatory SDS-polyacrylamide gels, and then Western-blotted to nitrocellulose. The paper was incubated in 0.1% Ponceau S in 5% acetic acid to locate and cut out the activated and stained 84 kDa region. The nitrocellulose strip was incubated in a Tris buffer, pH 7.6, containing 3% BSA for 2 hr at RT, and then incubated with antisera diluted 1:10–1:50 in TBS containing 1% BSA. After 1–2 hr at RT, the nitrocellulose was washed with TBS, and the antibodies were desorbed with 4 ml of a 0.05 m glycine buffer, pH 3.0. The buffer that contained the antibodies was neutralized with 40 μl of 1.0 m Tris, pH 9.0, and then concentrated and exchanged for TBS in Centricon-30 units (30,000 molecular weight cutoff; Amicon, Denvers, MA). The affinity-purified antibodies were stored at 4°C. The specificity of the antibodies was established with Western blots that used HL-60 cell culture supernatant in which only the latent and active forms of MMP-9 were recognized (data not illustrated).
Sections from five AD and three control hippocampal specimens were treated with anti-MMP-9 to determine the location of the enzymein situ. Sections were rinsed three times for 10 min with PBS and then incubated with PBS containing 10% ethanol and 1% H202 for 30 min. After three rinses with PBS, the sections were incubated for 15 min in a blocking buffer (PBS containing 5% BSA and 1% normal goat serum; Dako, Carpinteria, CA). Subsequently, the sections were incubated for 1 hr at RT with anti-MMP-9 diluted to a final concentration of 6 μg/ml in blocking buffer. After several rinses with PBS, the sections were processed with the peroxidase ABC kit according to the manufacturer’s recommendations (Vector Laboratories, Burlingame, CA). Negative controls consisted of sections incubated in solutions of preimmune rabbit serum (Dako). To identify plaques, we stained sections by a modified Bielschowsky stain.
To determine the percentage of MMP-9-labeled cells, we counted the numbers of positively and negatively stained pyramidal neurons on three sections from two representative AD patients (average age of 78 years). A 20× objective lens was used to review the slides, and any pyramidal cell >25 μm diameter present within the field was assessed. Representative areas from each CA region were counted in duplicate.
In situ hybridization of metalloproteinase mRNA. The matrix metalloproteinase-9 gene (Wilhelm et al., 1989) was used to construct a subclone that consists of the 391 bpXbaI/BamHI fragment, which contains the 5′ UTR and sequences that encode the pre- and pro-domains of the enzyme. The fragment was cloned into the respective sites in pBluescript II KS(+) (Stratagene, La Jolla, CA). The identity of the insert was confirmed by restriction mapping and partial sequencing. The plasmid was column-purified (Qiagen, Chatsworth, CA) and digested withBssHII (Boehringer Mannheim, Indianapolis, IN); the fragment containing the polymerase sites was gel-purified (Bio-101, La Jolla, CA). Riboprobes were generated with T3 or T7 polymerases (Promega, Madison, WI) and an NTP mix containing digoxigenin-UTP (Boehringer Mannheim). The concentrations of the riboprobes were quantified as per the manufacturer’s recommendations (Boehringer Mannheim), and the integrity of the probes was checked in agarose gels.
In situ hybridizations were performed with AD and control hippocampal tissues that also were evaluated by immunohistochemistry. The sections were treated with 0.25% acetic anhydride in 0.1% triethanolamine, pH 8.0, for 0.5 hr. The tissues were prehybridized for 2 hr at 37°C in Northern prehybridization buffer (5 Prime–3 Prime, Boulder, CO) containing 200 μg/ml salmon sperm DNA and 200 μg/ml yeast tRNA, 45% formamide (Sigma), and 5% vanadate ribonucleotide complex. The riboprobes were diluted to 0.1 ng/μl in Northern hybridization buffer (5 Prime–3 Prime), which contained the same additives as the prehybridization buffer, added to the sections, and incubated for 18 hr at 37°C. The slides were washed extensively with 4× SSC (20× SSC = 3 m NaCl and 0.3 msodium citrate, pH 7.0) and then with 1× SSC at RT. The sections were treated with 1% normal sheep serum (Dako) in TBS for 1 hr at RT. Alkaline phosphatase-labeled anti-digoxigenin (Boehringer Mannheim) was added to the sections at a dilution of 1:500 for 1 hr at RT. The washed slides were incubated for 12 hr at RT with nitro blue tetrazolium, 5-bromo-4-chloro-3-indolyl phosphate, and 0.25 mg/ml levamisole prepared in phosphatase buffer containing (in mm): 50 Tris-HCl, 100 NaCl, and 5 MgCl2, pH 9.5.
Detection of MMP-9 mRNA with RT-PCR. The isolation of mRNA from AD hippocampal specimens was based on the method of Chomczynski and Sacchi (1987). RNA was isolated and converted to cDNA with random hexamers. Primers 5′-CTGGTGCGCTACCACCTCGAAC-3′ (bases 1132–1156) and 5′-GTGCCGGATGCCATTCACGTCGTC-3′ (bases 1318–1342) of the human type IV collagenase cDNA sequence (Wilhelm et al., 1989), covering a 211 bp fragment of the active site region of MMP-9, were used. The cycling program consisted of 35 cycles of a denaturing step at 96°C for 1 min, an annealing step at 60°C for 1 min, and an extension step at 72°C for 2 min for 35 cycles. The PCR-amplified products were run in a 6% polyacrylamide gel together with a 275 bp fragment of the human thymidylate synthase gene, a 252 bp fragment of the human β-actin gene from tumor specimens (Horikoshi et al., 1993), and a DNA ladder suitable for determining the size of DNA from 123–3075 bp (Life Technologies, Grand Island, NY).
A pool of normal human hippocampal poly(A+) RNA (aged 16–72 years, Clontech, Palo Alto, CA) was reverse-transcribed with random hexamers and Superscript II transcriptase according to the manufacturer’s instructions (Life Technologies). After digestion with RNase H, cDNA was PCR-amplified with Taq polymerase (Perkin-Elmer, Norwalk, CT), which used previously described primers (Devarajan et al., 1992). The primers (5′-ATGA GCCTCTGGCAGCCCCTG-3′ and 5′-CTGGGACCCCGCACCGTGG-3′) amplified nucleotides 20–326 of the cDNA (Wilhelm et al., 1989). This region is present in the 391 bp MMP-9 subclone, which was used to generate the riboprobes (see above). Primers were chosen from two separate exons to exclude possible contamination by DNA fragments, because their amplified fragments would yield higher molecular masses. The PCR conditions consisted of 30 cycles of 1 min at 96°C, 1 min at 65°C, 1 min at 72°C, and a final 7 min extension at 72°C. The amplified fragments were electrophoresed in agarose gels with a 124–1114 bp size standard (Boehringer Mannheim). The DNA sequence of the gel-purified PCR product was confirmed by automated sequencing (Applied Biosystems model 373A, Foster City, CA).
Digestion of Aβ1–40 with MMP-9. Amyloid protein Aβ1–40 (Sigma or Peninsula, Belmont, CA) was solubilized with 0.1% trifluoroacetic acid (TFA; Pierce, Rockford, IL) and desalted with reverse-phase HPLC. The peptide was injected into a C18 column (0.46 cm × 25 cm; 218TP54, Vydac, Hesperia, CA), and the column was washed with 8% acetonitrile in 0.1% TFA for 10 min. Then the column was developed with an 8–80% acetonitrile gradient from 10 to 54 min. The desalted peptides were aliquoted into 40 μg portions and dried in a speed vac. The HPLC-purified latent form of MMP-9 was treated with a final concentration of 1 mm APMA and 5 mmCaCl2 in TBS for 6 hr at 37°C to activate the enzyme chemically. The active enzyme (1 μg in 7 μl) was added to the dried peptides in a final volume of 100 μl of TBS containing 5 mm CaCl2 and incubated at 37°C for 3 hr. The reactions were terminated with the addition of 0.1% TFA. Then the reaction mixtures were applied to a reverse-phase HPLC column as described above. The digested fragments were collected in microfuge tubes and dried in a speed vac. The mass and sequence of each peptide were determined by electrospray ionization mass spectroscopy (Sciex API III). The Sciex software package program was used for sequence determination, and identities of the peptides were confirmed by N-terminal sequencing (Applied Biosystems).
RESULTS
To examine the subcellular distribution of the metalloproteinase activities in the human hippocampus, we fractionated four representative AD and four control tissues sequentially into Tris-soluble, Triton-soluble, and then SDS-soluble fractions. The three fractions were electrophoresed in substrate gels and then incubated in a buffer overnight to allow the enzymes in the polyacrylamide gel to digest the gelatin substrate. Table 2 shows that the distribution of the 100 kDa metalloproteinase differs between AD and control tissues. The majority (76 ± 11%) of extractable activity from normal aged tissues was found in the Tris-soluble fraction, and the remaining 24% of the total activity was located in the SDS-soluble fraction. In contrast, 51 ± 16% of the activity from the AD samples was partitioned to the Tris-soluble fraction. No significant activity was found in the Triton-soluble fraction. In contrast to the 100 kDa activity, no differences between AD and control tissues were observed in the three fractions for the 70 kDa activity (data not illustrated).
Table 2.
Distribution of the 100 kDa form of MMP-9 activities from hippocampus fractions
| Fraction1-a | |||
|---|---|---|---|
| Tris-soluble | Triton-soluble | SDS-soluble | |
| Alzheimer | 51 (61)1-b | 1 (1) | 48 (16)1-b |
| Control | 76 (11)1-b | 0 (0) | 24 (11)1-b |
Four Alzheimer (Patient ID Nos. 4, 41, 161, 235) and four control tissues (Patient ID Nos. 343, 351, 359, 381) were fractionated sequentially into Tris-soluble, 1% Triton-soluble, and SDS-soluble fractions (see Materials and Methods for experimental details). The average ages were 72 and 64 years for the AD and control patients, respectively. All four AD patients had severe-to-moderate neurofibrillary tangles and neuritic plaques. Of the control patients, only one (No. 343) had moderate neurofibrillary tangles, but no neuritic plaques; all others had mild or no tangles and no plaques.
The percentage of activity in each fraction for the 100 kDa form of MMP-9. The percentage of SD is listed in parentheses.
The difference in MMP-9 activity from Alzheimer and control samples was significant at p < 0.05 (t test).
The Tris-soluble brain fractions were treated with APMA to determine whether the 100 kDa enzyme is latent or active. In the presence of APMA, the enzyme showed a time-dependent decrease in both molecular mass and in activity of the 100 kDa latent form (Fig.1A). A time-dependent increase was observed in the 90 kDa form of the activated enzyme. A decrease in the molecular mass of the 130 kDa latent form to 120 kDa also was observed.
Fig. 1.

Activation of latent MMP-9 from the hippocampus.A, Densitometric scan of a substrate gel with gelatin as the substrate. Aliquots of the inhibitor-treated soluble brain fraction were incubated in the absence (−) or presence (+) of 1 mmAPMA for 0, 6, 12, and 24 hr at 37°C. B, Gelatinase activity of a soluble brain fraction with biotinylated gelatin as the substrate in a plate assay. Aliquots of the inhibitor-treated sample were preincubated with 1 mm APMA for 1 or 3 hr at 37°C (arrows). The control for endogenous metalloproteinase activity included a sample incubated in the absence of APMA (0 hr, arrow). Chymotryptic activities were determined in the plate assay with the indicated nanogram amounts of α-chymotrypsin. The experiments were performed three times in triplicate with a 10% SEM. See Materials and Methods for the calculation of specific activities. C, Immunodepletion of enzyme activities with a specific monoclonal antibody to the latent form of MMP-9 (Ab-2, Oncogene Science). Densitometric scans of substrate electrophoretic gels with samples incubated with or without specific monoclonal antibodies to MMP-9. (All experiments were performed on specimens from three AD patients, and samples from patient 206 were used for the illustration.)
To measure the increase in MMP activity after APMA activation, we performed the gelatin-biotin plate assay with the Tris-soluble brain fraction from a representative AD sample. Approximately 0.5 ng of chymotryptic-equivalent gelatinase activity was observed before activation (Fig. 1B). At the addition of APMA for 1 and 3 hr (optimum activation condition), the activity increased to 26 and 35 ng, respectively. This represents a 70-fold increase in activity because of APMA activation. An APMA-activated sample was treated with the metal ion chelator 1,10-phenanthroline (1.4 mm final concentration) and then added to wells containing gelatin. After a 3 hr incubation, the activity was reduced by 93%, confirming that the gelatinase activity was divalent ion-dependent (data not illustrated).
Immunodepletion experiments established that the 100 kDa enzyme activity was removed selectively with two different specific monoclonal antibodies to MMP-9 without any decrease in the 70 kDa activity (Fig.1C). The 130 kDa activity also was removed, indicating that this enzyme represents a complexed form of MMP-9. The 70 kDa enzyme activity was removed with specific monoclonal antibodies to MMP-2 with no effect on the 100 kDa activity (G. P. Lim et al., unpublished data). This is consistent with the observation that the 70 kDa enzyme is MMP-2, of glial origin, in ALS brain and spinal cord specimens (Lim et al., 1996).
Human hippocampus sections from AD and control tissues were stained with affinity-purified antibodies and riboprobes to determine the location of MMP-9 in situ. Although anti-MMP-9 did not label cells and perivascular areas in control sections (Fig.2A), the antibodies labeled pyramidal neurons from AD sections (Fig. 2B). Prominent cytoplasmic staining appeared granular and did not accumulate in the nuclei (Fig. 2C). However, the stained material extended into the neurites (Fig. 2D). Significant levels of immunoreactive MMP-9 were not detected either in the granule cell neurons in the dentate gyrus, glial cells, or in perivascular regions (data not illustrated). An identical staining pattern was observed with MMP-9-specific murine monoclonal antibodies. Figure 3summarizes the distribution of anti-MMP-9 staining in the human hippocampus. The number of positively stained neurons varied by region. The percentage of immunoreactive pyramidal neurons in the CA1, 2, 3, and 4 regions were 78.7 ± 12.1, 61.6 ± 10.1, 52.6 ± 9.7, and 21.8 ± 3.2%, respectively.
Fig. 2.

Localization of immunoreactive MMP-9 in the human hippocampus. A, Control section demonstrating that reactivity was below the level of detection (200×). B, AD section illustrating reactivity in pyramidal cells (200×). Glial cells and perivascular areas were unstained. C, Higher magnification of pyramidal neuron from AD section showing granular accumulation of immunoreactive MMP-9 in the cytoplasm.D, Pyramidal neuron showing stained material extending into the neurite. E, Senile plaque illustrating positively stained cellular process (arrowheads).F, Neuritic processes labeled with Bielschowsky stain. (Samples from AD patients 107, 342, 538, 595, and 602 and from control patients 95, 559, and 612 were investigated. Specimens from 595 and 559 are used for illustration.)
Fig. 3.

Summary of anti-MMP-9 staining in the human hippocampus. Pyramidal neurons that were positively stained with anti-MMP-9 (filled triangles) were found in the CA1–CA3 subfields from AD sections. Increasing numbers of unstained neurons (open triangles) were seen from the CA1–CA3 regions. Neurons in the CA4 subfield, as well as granule neurons (open circles) in the dentate gyrus, were unstained.PRES, Presubiculum; PROS, prosubiculum;SUB, subiculum; PARA, parahippocampal gyrus. (Specimens from AD patients 107, 595, and 602 were used for the studies.)
MMP-9 immunoreactivity also was detected near the extracellular amyloid plaques (Fig. 2E). The antibodies consistently labeled the cellular processes of classical and diffuse senile plaques throughout Ammon’s horn (Fig. 2E, arrow), but not the dense amyloid core lesions. Bielschowsky-stained sections confirmed that these structures were the neuritic portions of the plaques (Fig.2F).
The results from the in situ hybridization experiments that used MMP-9 riboprobes correlated with the results from immunohistology. The MMP-9 subclone containing the 5′ 391 bp sequence was used to generate digoxigenin-labeled riboprobes. The pyramidal neurons in the CA1–CA4 subfields of AD sections were labeled with the antisense probe (Fig. 4B, CA3 region), but not with the sense probe (Fig. 4A). Staining was not detected in the granule cell neurons or in glial cells. Staining with the antisense riboprobe was below the level of detection in the control hippocampal specimens.
Fig. 4.

The antisense MMP-9 riboprobe labels pyramidal neurons in the human hippocampus. The Alzheimer sections were treated with sense (A) or antisense (B) riboprobes. The CA3 region of the hippocampus is illustrated. (Specimens from patient 107 were used for the illustration.)
Attempts to perform Northern blot analysis for MMP-9 mRNA revealed substantial RNA degradation in the postmortem brain specimens. Consequently, two sets of RT-PCR experiments were performed. In the first set of experiments, the 211 bp fragment of the MMP-9 active site region was PCR-amplified successfully from AD hippocampus specimens (Fig. 5A). Positive controls included the 275 bp fragment of human thymidylate synthase and the 252 bp fragment of human β-actin genes from tumors (Horikoshi et al., 1993). In the second set of experiments, RT-PCR was used to confirm the presence of MMP-9 mRNA from a pool of normal hippocampus poly(A+) RNA. A 306 bp product derived from the cDNA was detected in agarose gels (Fig. 5B, lane 2) and after purification and reamplification (Fig. 5B, lane 3). Digestion of the 306 bp PCR product with PvuII generated the expected fragments of 177 and 129 bp (Fig. 5B, lane 8). The 306 bp product was sequenced and found to be identical to the previously reported sequence of MMP-9 (Wilhelm et al., 1989).
Fig. 5.
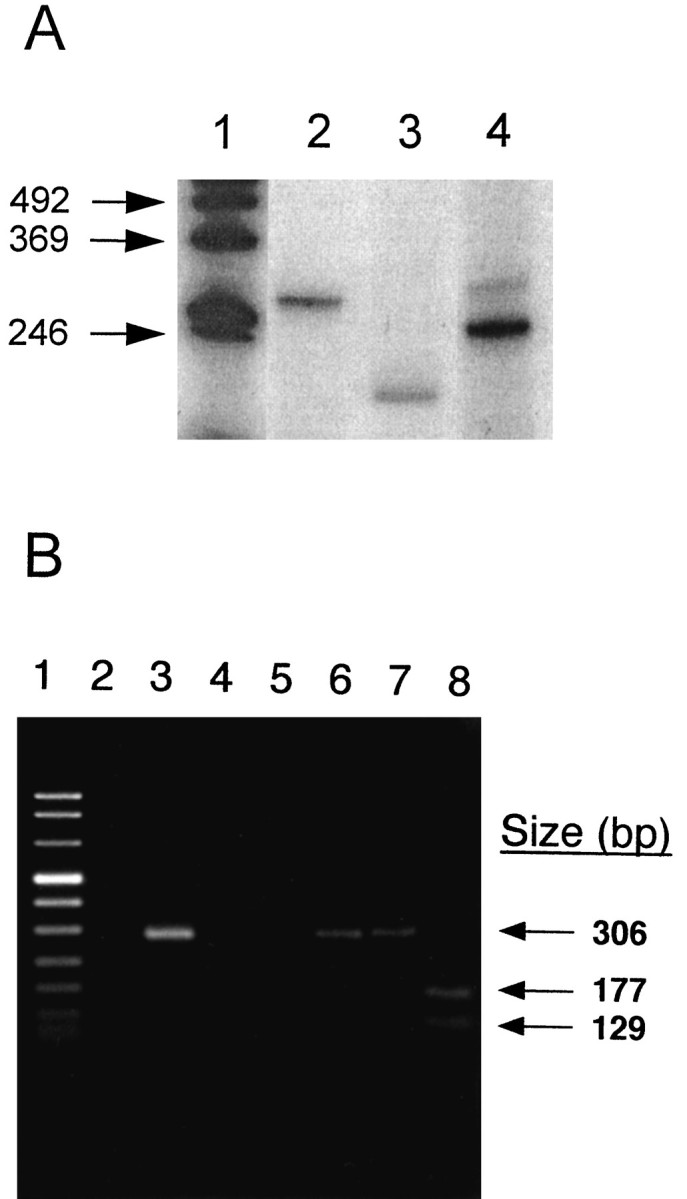
Reverse transcriptase-PCR of hippocampus RNA.A, PCR amplification of the MMP-9 active site from an AD sample (No. 206). Lane 1, Size standards; lanes 2, 4, 275 bp fragment of the human thymidylate synthase gene and 252 bp fragment of the human β-actin gene, used as control amplifications, respectively. Lane 3, 211 bp fragment of the MMP-9 active site. B, PCR amplification of the 5′ region of MMP-9 from a normal human cDNA pool. Lane 1, Size standards; lane 2, amplification of the hippocampus cDNA. The remaining portion of the sample was electrophoresed in a separate gel, and the 306 bp DNA was removed, purified, and PCR-amplified. Lane 3 illustrates the reamplification of this 306 bp product. Lanes 4,5, Water controls for the first and second PCR amplifications, respectively. Lane 6, Gel-purified PCR fragment; lanes 7, 8, the gel-purified fragment treated with ApaI (negative control) andPvuII, respectively. The arrows indicate the positions of the 306 bp fragment and the 177 and 129 bp digestion fragments from PvuII.
Because endogenous MMP-9 was immunolocalized to amyloid plaques (Fig.2E), the purified and activated enzyme was incubated with synthetic Aβ1–40 to determine whether amyloid core peptides can serve as a substrate. The latent enzyme purified from cell cultures was converted to an active enzyme with APMA and then incubated with the peptide at 37°C. After a 3 hr incubation to determine the major cleavage sites or a 20 hr incubation to determine the additional minor sites, the reaction mixture was subjected to reverse-phase HPLC. The peptides, corresponding to the peaks at 215 nm, were collected and analyzed by mass spectroscopy and amino acid sequencing. Five characteristic peptides were identified with m/z ratios of 560.7, 1954.5, 3390.6, 3786.2, and 4328.9. Amino acid sequences of the peptides were determined with the Sciex Software program and confirmed by N-terminal sequencing. A summary of the major and minor cleavage sites is illustrated in Figure 6. The major cleavage site of the amyloid peptide was at the Leu34-Met35 bond, and the minor sites were at the Ala30-Ile31, Gly37-Gly38, and Lys16-Leu17 bonds.
Fig. 6.

Summary of the results from the digestion of Aβ1–40 by MMP-9. Reverse-phase HPLC was used to separate the peptides, and the sequences of the digestion products were determined by mass spectroscopy and amino acid sequencing. The major cleavage site (arrow above line, Leu34-Met35) and minor cleavage sites (arrows below line, Lys16-Leu17, Ala30-Ile31, and Gly37-Gly38) are indicated. Theboxed amino acids represent the region of Aβ within the membrane. The m/z ratios were 4328.9 (Aβ1–40), 3786.2 (Aβ1–34), 560.7 (Aβ35–40), 3390.6 (Aβ1–30), and 1954.5 (Aβ1–16).
DISCUSSION
We have demonstrated previously that the activities of a 100 kDa metalloproteinase from AD tissues were increased, relative to control tissues (Backstrom et al., 1992). Because the enzyme has similar biochemical properties to MMP-9, experiments were performed to confirm the identity of the 100 kDa enzyme and to investigate its cellular location in the hippocampus. Immunodepletion studies, APMA-activation data, and RT-PCR experiments confirm that the 100 kDa metalloproteinase is a latent form of MMP-9 (EC3.4.24.35). This metalloproteinase is expressed by neurons, and, when activated, it is capable of degrading the Aβ1–40 peptide.
Monoclonal antibodies to MMP-9 (Ab-1 and Ab-2) specifically removed the 100 kDa activity from AD brain extracts. These experiments also establish that the enzyme is in the latent form, because Ab-2 selectively binds to only the inactive proenzyme. Furthermore, activation studies confirm that the 100 kDa enzyme is latent. The APMA treatment of soluble brain fractions reduced the molecular mass by 10 kDa and increased the gelatinase activity 70-fold (Fig. 1). Furthermore, the presence of a chelating agent, 1,10-phenanthroline, inhibited >90% of the enzyme activity obtained after APMA treatment, indicating that divalent metal ions were essential for activity. These observations are consistent with previous reports for MMP-9 (Wilhelm et al., 1989; Davis and Martin, 1990). In addition, the immunodepletion of the 130 kDa activity and its shift to 120 kDa at APMA activation further demonstrates that this enzyme is a complexed form of MMP-9, as reported by Lim et al., 1996.
Evidence for the expression of MMP-9 in the hippocampus comes from two sets of RT-PCR experiments. A fragment of 211 bp consisting of the active site region of MMP-9 from position 1132 to 1342 (Wilhelm et al., 1989) was PCR-amplified from AD hippocampus (Fig. 5A). In addition, the fragment representing nucleotides 20–326 also was amplified from a pool of normal human hippocampal specimens. The fragment was sequenced and found to be identical to the previously published sequence of the human type IV collagenase cDNA (MMP-9;Wilhelm et al., 1989). These results establish that this gene is expressed in the human hippocampus.
Other metalloproteinases recently have been identified from human brain. McDermott and Gibson (1991) purified an active endopeptidase (EC3.4.24.11) from cerebral cortex. Unlike MMP-9, it is membrane-bound, inhibited by Zn2+, Cd2+, and Ni2+, stimulated by Mn2+, and has a molecular mass of 105 to 120 kDa. Metalloproteinases (84 and 43 kDa) with a high homology to rat endopeptidase (EC3.4.24.15) were purified from AD brain. These enzymes cleave the amyloid precursor protein (APP) at the Met-Asp bond and generate a 15 kDa amyloidogenic fragment (Papstoitsis et al., 1994). In contrast to MMP-9, these proteinases are devoid of caseinolytic and gelatinase activity. A third novel metalloproteinase has been characterized partially from AD brain (Schönlein et al., 1994). This 100 kDa enzyme is active, Mg2+-dependent, and highly inhibited by Zn2+ (0.5 mm Zn2+). Gelatinase A (MMP-2) was found in the white matter microglial cells and in Schwann cells of neurologically normal, lacunar stroke, AD, amyotrophic lateral sclerosis (ALS), and myasthenia gravis cases (Yamada et al., 1995). It has been suggested that this enzyme also may function as an α-secretase to produce secretory forms of APP (Roher et al., 1994). In our recent study, we observed that MMP-2 was localized to astrocytes, and the enzyme activities essentially were unchanged between ALS and control CNS specimens. The motor neurons in ALS patients expressed significantly higher levels of MMP-9, suggesting a role in motor neuron degeneration (Lim et al., 1996).
To determine the cellular location of MMP-9 in the human hippocampus, we performed immunohistology and in situ hybridization experiments. In the AD hippocampus, anti-MMP-9 labeled pyramidal neurons in the CA1–CA3 fields, but not granular neurons in the dentate gyrus or glial cells (Fig. 2B–D). The distribution of MMP-9 staining corresponds to the hippocampal regions that are most affected in AD (Davies et al., 1992). In addition to the cellular staining of MMP-9, antibodies also labeled neuritic portions of classical and diffuse plaques (Fig. 2E,F). The location and dimension of the labeled neurites suggest that they are dendritic extensions of the stained pyramidal neurons in Ammon’s horn.In situ hybridizations with MMP-9 riboprobes confirmed that pyramidal neurons are the major source of the enzyme in AD hippocampus (Fig. 4). Sections from control specimens were not labeled with the antisense riboprobe to MMP-9, an observation that is consistent with the results from Northern blot analysis, which demonstrated that the amount of mRNA was below the level of detection (Devarajan et al., 1992).
The results of tissue fractionation experiments were in agreement with the immunohistochemical staining. The majority (76%) of the 100 kDa enzyme activity from control samples was partitioned in the Tris-soluble fraction, which is consistent with an enzyme that is present in a secretory form. Only 51% of the activity from the Alzheimer-affected tissue was present in the same soluble form. Less than 2% of the activity was extracted into a Triton-soluble fraction, signifying that the enzyme was not membrane-associated. Nonionic detergents were able to solubilize other metalloproteinases, such as the membrane-associated enkephalinases (Fulcher and Kenny, 1983; Matsas et al., 1983) and the human endopeptidase (EC3.4.24.11; McDermott and Gibson, 1991). Twice as much enzyme activity was observed in the SDS-soluble fraction in AD, as compared with control specimens: 48 versus 24%, respectively (see Table 1). This fraction represents enzymes tightly associated with possible particulate compartments, such as the amyloid plaques. The altered distribution seems to be unique for MMP-9, because the distribution of the 70 kDa metalloproteinase was not different between AD and control samples (data not illustrated).
Because MMP-9 was detected in the regions near plaques (Fig.2E), we questioned whether the active form of the enzyme could process the major plaque component, β-amyloid. Purified and APMA-activated MMP-9 cleaved the soluble amyloid peptide Aβ1–40 primarily between the Leu34-Met35 bond and, to a lesser extent, at Lys16-Leu17, Ala30-Ile31, and Gly37-Gly38 (Fig. 6). Three of these cleavage sites correspond to a region in the membrane-spanning domain of the amyloid precursors (reviewed in Selkoe, 1994). These cleavages are significant because they can eliminate the neurotoxic β-sheet-forming capacity of the amyloid peptide (Simmons et al., 1994). Seubert et al. (1992) purified endogenous amyloid fragments from human CSF and human fetal mixed brain cultures. They did not report a peptide that terminated at the Leu34 residue, as our study would suggest, but such peptides would not be detected if other peptidases subsequently process the amyloid fragments. The observations that MMP-9 is localized near plaques and that latent enzyme is shifted to a more particulate location in AD raise the possibility that the enzyme is synthesized in response to Aβ. If activated, it would further degrade the Aβ peptide in vivo and would reduce the probability of accumulation of the peptide in the plaques.
Secreted, latent MMP-9 can be processed proteolytically to an active form by serine proteinases such as elastase and cathepsin G, metalloproteinases, and superoxide anions such as HOCl (Murphy et al., 1980; Peppin and Weiss, 1986; Shah et al., 1987; Vissers and Winterbourn, 1988; Goldberg et al., 1992; Morodomi et al., 1992; Ogata et al., 1992; Okada et al., 1992). It is significant that levels of α1-antichymotrypsin (ACHY) are increased in AD specimens relative to controls (Abraham et al., 1988). ACHY is an inflammatory protein that accumulates in amyloid plaques (Abraham et al., 1988) where MMP-9 also is observed (Fig. 2E). ACHY can bind to a region of the amyloid peptide that resembles a serine proteinase domain (Potter et al., 1991). ACHY bound to this site interferes with the α-secretase activity at Lys16-Leu17(Potter et al., 1991) and also might interfere with the processing near the Leu34-Met35 site. Increased local concentrations of the soluble region of APPs, which contains an inhibitor of matrix metalloproteinases (Miyazaki et al., 1993), also would interfere with MMP-9 activity. ACHY is a known inhibitor of serine proteinases, which may be involved in the in vivoactivation of MMP-9 (Ennis and Matrisian, 1994). Inflammatory reactions have been implicated in AD (Aisen and Davis, 1994; Breitner et al., 1994). An inverse association of anti-inflammatory treatments and the onset of AD was revealed in a co-twin control study (Breitner et al., 1994). Thus, it is conceivable that the inflammatory process and the accumulation of the inflammatory protein ACHY contribute to the latency of MMP-9. Consequently, the proteolytic processing of soluble Aβ peptides by MMP-9 would be reduced, leading to their accumulation in senile plaques. This hypothesis is being tested now in our laboratories.
Footnotes
This work was supported by National Institute on Aging Grant R01-AG09681 to Z.T. We thank Dr. Audree Fowler, Protein Microsequencing Facility, and Dr. Kym Faull, Center for Molecular and Medical Sciences Mass Spectroscopy, University of California at Los Angeles, for their assistance in protein sequencing and mass spectroscopy analyses, respectively. The assistance of Dr. Peter Danenberg, Kathleen Danenberg, and Dr. Heinz-Josef Lenz with the RT-PCR studies of MMP-9 active site region is greatly appreciated.
Correspondence should be addressed to Dr. Zoltán A. Tökés, Cancer Research Laboratories, 1303 North Mission Road, Los Angeles, CA 90033.
REFERENCES
- 1.Abraham CR, Selkoe DJ, Potter H. Immunochemical identification of the serine proteinase inhibitor, alpha-1-antichymotrypsin, in the brain amyloid deposit of Alzheimer’s disease. Cell. 1988;52:487–501. doi: 10.1016/0092-8674(88)90462-x. [DOI] [PubMed] [Google Scholar]
- 2.Aisen PS, Davis KL. Inflammatory mechanisms in Alzheimer’s disease: implications for therapy. Am J Psychiatry. 1994;151:1105–1113. doi: 10.1176/ajp.151.8.1105. [DOI] [PubMed] [Google Scholar]
- 3.Backstrom JR, Tökés ZA. The 84 kDa form of human matrix metalloproteinase-9 (MMP-9) degrades substance P and gelatin. J Neurochem. 1995;64:1312–1318. doi: 10.1046/j.1471-4159.1995.64031312.x. [DOI] [PubMed] [Google Scholar]
- 4.Backstrom JR, Miller CA, Tökés ZA. Characterization of neutral proteinases from Alzheimer-affected and control brain specimens: identification of calcium-dependent metalloproteinases from the hippocampus. J Neurochem. 1992;58:983–992. doi: 10.1111/j.1471-4159.1992.tb09352.x. [DOI] [PubMed] [Google Scholar]
- 5.Breitner JCS, Gau BA, Welsh KA, Plassman BL, McDonald WM, Helms MJ, Anthony JC. Inverse association of anti-inflammatory treatments and Alzheimer’s disease: initial results of a co-twin control study. Neurology. 1994;44:227–232. doi: 10.1212/wnl.44.2.227. [DOI] [PubMed] [Google Scholar]
- 6.Chomczynski P, Sacchi N. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal Biochem. 1987;162:156–159. doi: 10.1006/abio.1987.9999. [DOI] [PubMed] [Google Scholar]
- 7.Davies DC, Horwood N, Isaacs SL, Mann DM. The effect of age and Alzheimer’s disease on pyramidal neuron density in the individual fields of the hippocampal formation. Acta Neuropathol (Berl) 1992;83:510–517. doi: 10.1007/BF00310028. [DOI] [PubMed] [Google Scholar]
- 8.Davis GE, Martin BM. A latent Mr 94,000 gelatin-degrading metalloproteinase induced during differentiation of HL-60 promyelocytic leukemia cells: a member of the collagenase family of enzymes. Cancer Res. 1990;50:1113–1120. [PubMed] [Google Scholar]
- 9.Devarajan P, Johnston JJ, Ginsberg SS, Van Wart HE, Berliner N. Structure and expression of neutrophil gelatinase cDNA. J Biol Chem. 1992;267:25228–25232. [PubMed] [Google Scholar]
- 10.Ennis BW, Matrisian LM. Matrix degrading metalloproteinases. J Neurooncol. 1994;18:105–109. doi: 10.1007/BF01050416. [DOI] [PubMed] [Google Scholar]
- 11.Esch FS, Keim PS, Beattie EC, Blacher RW, Cultwell AR, Olsterdorf T, McClure D, Ward PJ. Cleavage of amyloid β peptide during constitutive processing of its precursor. Science. 1990;248:1122–1124. doi: 10.1126/science.2111583. [DOI] [PubMed] [Google Scholar]
- 12.Fulcher IS, Kenny AJ. Proteins of the kidney microvillar membrane. The amphipathic form of endopeptidase purified from pig kidneys. Biochem J. 1983;211:743–753. doi: 10.1042/bj2110743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Goldberg GI, Strongin A, Collier IA, Genrich LT, Marmer BL. Interaction of 92 kDa type IV collagenase with the tissue inhibitor of metalloproteinases prevents dimerization, complex formation with interstitial collagenase, and activation of the proenzyme with stromelysin. J Biol Chem. 1992;267:4583–4591. [PubMed] [Google Scholar]
- 14.Heussen C, Dowdle EB. Electrophoretic analysis of plasminogen activators in polyacrylamide gels containing sodium dodecyl sulfate and copolymerized substrates. Anal Biochem. 1980;102:196–202. doi: 10.1016/0003-2697(80)90338-3. [DOI] [PubMed] [Google Scholar]
- 15.Hibbs MS, Hasty KA, Seyer JM, Kang AH, Mainardi CL. Biochemical and immunological characterization of the secreted forms of human neutrophil gelatinase. J Biol Chem. 1985;260:2493–2500. [PubMed] [Google Scholar]
- 16.Horikoshi T, Danenberg K, Volkenandt M, Stadlbauer T, Danenberg PV. Quantitative measurement of relative gene expression in human tumors. In: White BA, editor. Methods in molecular biology, Vol 15. Humana; Totowa, NJ: 1993. pp. 177–188. [DOI] [PubMed] [Google Scholar]
- 17.Khachaturian ZS. Diagnosis of Alzheimer’s disease. Arch Neurol. 1985;42:1097–1105. doi: 10.1001/archneur.1985.04060100083029. [DOI] [PubMed] [Google Scholar]
- 18.Kitaguchi N, Takahashi Y, Tokushima Y, Shiojiri S, Ito H. Novel precursor of Alzheimer’s disease amyloid protein shows protease inhibitory activity. Nature. 1988;331:530–532. doi: 10.1038/331530a0. [DOI] [PubMed] [Google Scholar]
- 19.Lim GP, Backstrom JR, Cullen MJ, Miller CA, Atkinson RD, Tökés ZA. Matrix metalloproteinases in the neocortex and spinal cord of amyotrophic lateral sclerosis (ALS) patients. J Neurochem. 1996;66:251–259. doi: 10.1046/j.1471-4159.1996.67010251.x. [DOI] [PubMed] [Google Scholar]
- 20.Matsas R, Fulcher IS, Kenny AJ, Turner AJ. Substance P and [leu]enkephalin are hydrolyzed by an enzyme in pig caudate synaptic membranes that is identical with the endopeptidase of kidney microvilli. Proc Natl Acad Sci USA. 1983;80:3111–3115. doi: 10.1073/pnas.80.10.3111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.McDermott JR, Gibson AM. The processing of Alzheimer A4/β-amyloid protein precursor: identification of a human brain metallopeptidase which cleaves -Lys-Leu- in a model peptide. Biochem Biophys Res Commun. 1991;179:1148–1154. doi: 10.1016/0006-291x(91)91691-5. [DOI] [PubMed] [Google Scholar]
- 22.McKhann G, Drachman D, Folstein M, Katzman R, Price D, Stadlan EM. Clinical diagnosis of Alzheimer’s disease: report of the NINCDS–ADRDA work group under the auspices of Department of Health and Human Services Task Force on Alzheimer’s disease. Neurology. 1984;34:939–944. doi: 10.1212/wnl.34.7.939. [DOI] [PubMed] [Google Scholar]
- 23.Miyazaki K, Hasegawa M, Funahashi K, Umeda M. A metalloproteinase inhibitor domain in Alzheimer amyloid protein precursor. Nature. 1993;362:839–841. doi: 10.1038/362839a0. [DOI] [PubMed] [Google Scholar]
- 24.Moll UM, Youngleib GL, Rosinski KB, Quigley JP. Tumor promoter-stimulated Mr 92,000 gelatinase secreted by normal and malignant human cells: isolation and characterization of the enzyme from HT1080 tumor cells. Cancer Res. 1990;50:6162–6170. [PubMed] [Google Scholar]
- 25.Morodomi T, Ogata Y, Sasaguri Y, Morimatsu M, Nagase H. Purification and characterization of matrix metalloproteinase 9 from U937 monocytic leukaemia and HT1080 fibrosarcoma cells. Biochem J. 1992;285:603–611. doi: 10.1042/bj2850603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Murphy G, Bretz U, Baggiolini M, Reynolds JJ. The latent collagenase and gelatinase of human polymorphonuclear neutrophil leucocytes. Biochem J. 1980;192:517–525. doi: 10.1042/bj1920517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Netzel-Arnett S, Sang Q-X, Moore WGI, Navre M, Birkedal-Hansen H, Van Wart HE. Comparative sequence specificities of human 72- and 92-kDa gelatinases (type IV collagenases) and PUMP (matrilysin). Biochemistry. 1993;32:6427–6432. doi: 10.1021/bi00076a016. [DOI] [PubMed] [Google Scholar]
- 28.Ogata Y, Enghild JJ, Nagase H. Matrix metalloproteinase 3 (stromelysin) activates the precursor for the human matrix metalloproteinase 9. J Biol Chem. 1992;267:3581–3584. [PubMed] [Google Scholar]
- 29.Okada Y, Gonoji Y, Naka K, Tomita K, Nakanishi I, Iwata K, Yamashita K, Hayakawa T. Matrix metalloproteinase 9 (92 kDa gelatinase/type IV collagenase) from HT1080 human fibrosarcoma cells. J Biol Chem. 1992;267:21712–21719. [PubMed] [Google Scholar]
- 30.Oltersdorf T, Fritz LC, Schenk DB, Lieberburg I, Johnson-Wood KL, Beattie EC, Ward PJ, Blacher RW, Dovey HF, Sinha S. The secreted form of the Alzheimer’s amyloid precursor protein with the Kunitz domain is protease nexin-II. Nature. 1989;341:144–147. doi: 10.1038/341144a0. [DOI] [PubMed] [Google Scholar]
- 31.Papstoitsis G, Siman R, Scott R, Abraham CR. Identification of a metalloprotease from Alzheimer’s disease brain able to degrade the β-amyloid precursor protein and generate amyloidogenic fragments. Biochemistry. 1994;33:192–199. doi: 10.1021/bi00167a025. [DOI] [PubMed] [Google Scholar]
- 32.Peppin GJ, Weiss SJ. Activation of the endogenous metalloproteinase, gelatinase, by triggered human neutrophils. Proc Natl Acad Sci USA. 1986;83:4322–4326. doi: 10.1073/pnas.83.12.4322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ponte P, Gonzalez-DeWhitt P, Schilling J, Miller J, Hsu D, Greenberg B, Davis K, Wallace W, Lieberburg I, Fuller F, Cordell B. A new A4 amyloid mRNA contains a domain homologous to serine proteinase inhibitors. Nature. 1988;331:525–527. doi: 10.1038/331525a0. [DOI] [PubMed] [Google Scholar]
- 34.Potter H, Abraham CR, Dressler D. The Alzheimer amyloid components α1-antichymotrypsin and β-protein form a stable complex in vitro. In: Iqbal K, McLachlan DRC, Winblad B, Wisniewski HM, editors. Alzheimer’s disease: basic mechanisms, diagnosis, and therapeutic strategies. Wiley; New York: 1991. pp. 275–279. [Google Scholar]
- 35.Roher AE, Kasunic TC, Woods AS, Cotter RJ, Ball MJ, Fridman R. Proteolysis of Aβ peptide from Alzheimer disease brain by gelatinase A. Biochem Biophys Res Commun. 1994;205:1755–1761. doi: 10.1006/bbrc.1994.2872. [DOI] [PubMed] [Google Scholar]
- 36.Schönlein C, Löffler J, Huber G. Purification and characterization of a novel metalloprotease from human brain with the ability to cleave substrates derived from the N terminus of β-amyloid protein. Biochem Biophys Res Commun. 1994;201:45–53. doi: 10.1006/bbrc.1994.1667. [DOI] [PubMed] [Google Scholar]
- 37.Selkoe DJ. Normal and abnormal biology of the β-amyloid precursor protein. Annu Rev Neurosci. 1994;17:489–517. doi: 10.1146/annurev.ne.17.030194.002421. [DOI] [PubMed] [Google Scholar]
- 38.Seubert P, Vigo-Pelfrey C, Esch F, Lee M, Dovey H, Davis D, Sinha S, Schlossmacher C, McCormack R, Wolfert R, Selkoe D, Lieberburg I, Schenk D. Isolation and quantitation of soluble Alzheimer’s β-peptide from biological fluids. Nature. 1992;359:325–327. doi: 10.1038/359325a0. [DOI] [PubMed] [Google Scholar]
- 39.Shah SV, Baricos WH, Basci A. Degradation of human glomerular basement membrane by stimulated neutrophils. J Clin Invest. 1987;79:25–31. doi: 10.1172/JCI112790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Simmons LK, May PC, Tomaselli KJ, Rydel RE, Fuson KS, Brigham EF, Wright S, Lieberburg I, Becker GW, Brems DN, Li WY. Secondary structure of amyloid beta peptide correlates with neurotoxic activity in vitro. Mol Pharmacol. 1994;45:373–379. [PubMed] [Google Scholar]
- 41.Sisodia SS, Koo EH, Bayreuther K, Unterbeck A, Price DL. Evidence that β-amyloid in Alzheimer’s disease is not derived by normal processing. Science. 1990;248:492–495. doi: 10.1126/science.1691865. [DOI] [PubMed] [Google Scholar]
- 42.Sprinman EB, Angleton EL, Birkedal-Hansen H, Van Wart HE. Multiple modes of activation of latent human fibroblast collagenase: evidence for the role of a Cys73 active-site zinc complex in latency and a “cysteine switch” mechanism for activation. Proc Natl Acad Sci USA. 1990;87:364–368. doi: 10.1073/pnas.87.1.364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tanzi RE, McClatchey AI, Lamperti ED, Villa-Komaroff L, Gusella JF, Neve RL. Protease inhibitor domain encoded by an amyloid protein precursor mRNA associated with Alzheimer’s disease. Nature. 1988;331:528–530. doi: 10.1038/331528a0. [DOI] [PubMed] [Google Scholar]
- 44.Van Nostrand WE, Wagner SL, Suzuki M, Choi BH, Farrow JS, Geddes JW, Cotman CW, Cunningham DD. Protease nexin-II, a potent antichymotrypsin, shows identity to amyloid β-protein precursor. Nature. 1989;341:546–549. doi: 10.1038/341546a0. [DOI] [PubMed] [Google Scholar]
- 45.Vissers CM, Winterbourn CC. Activation of human neutrophil gelatinase by endogenous serine proteinases. Biochem J. 1988;249:327–331. doi: 10.1042/bj2490327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wilhelm SM, Collier IE, Marmer BL, Eisen AZ, Grant GA, Goldberg GI. SV40-transformed human lung fibroblasts secrete a 92 kDa type IV collagenase which is identical to that secreted by normal human macrophages. J Biol Chem. 1989;264:17213–17221. [PubMed] [Google Scholar]
- 47.Yamada T, Miyazaki K, Koshikawa N, Takahashi M, Akatsu H, Yamamoto T. Selective localization of gelatinase A, an enzyme-degrading beta-amyloid protein, in white matter microglia and in Schwann cells. Acta Neuropathol (Berl) 1995;89:199–203. doi: 10.1007/BF00309334. [DOI] [PubMed] [Google Scholar]