

Abstract
The molecular basis for the degeneration of neurons and the deposition of amyloid in plaques and in the cerebrovasculature in Alzheimer’s disease (AD) is incompletely understood. We have proposed that one molecule common to these abnormal processes is a fragment of the Alzheimer amyloid precursor protein (APP) comprising the C-terminal 100 amino acids of this molecule (APP-C100). We tested this hypothesis by creating transgenic mice expressing APP-C100 in the brain. We report here that aging (18–28 month) APP-C100 transgenic mice exhibit profound degeneration of neurons and synapses in Ammon’s horn and the dentate gyrus of the hippocampal formation. Of the 106 transgenic mice between 8 and 28 months of age that were examined, all of those older than 18 months displayed severe hippocampal degeneration. The numerous degenerating axonal profiles contained increased numbers of neurofilaments, whorls of membrane, and accumulations of debris resembling secondary lysosomes near the cell body. The dendrites of degenerating granule and pyramidal cells contained disorganized, wavy microtubules. Cerebral blood vessels had thickened refractile basal laminae, and microglia laden with debris lay adjacent to larger venous vessels. Mice transgenic for Flag-APP-C100 (in which the hydrophilic Flag tag was fused to the N terminus of APP-C100) showed a similar degree of neurodegeneration in the hippocampal formation as early as 12 months of age. The 45 control mice displayed only occasional necrotic cells and no extensive cell degeneration in the same brain regions. These findings show that APP-C100 is capable of causing some of the neuropathological features of AD.
Keywords: Alzheimer’s disease, amyloid, neurodegeneration, hippocampus, Flag tag, aging
All individuals with Alzheimer’s disease (AD) experience a progressive and specific loss of cognitive function resulting from a neurodegenerative process characterized classically by granulovacuolar degeneration, the deposition of amyloid in plaques and in the cerebrovasculature, and the formation of neurofibrillary tangles in neurons. Additional pathological hallmarks of AD include degeneration of synapses (Hamos et al., 1989; DeKosky and Scheff, 1990;Terry et al., 1990) and decreases in cell density (Terry et al., 1981) in distinct regions of the brain, lysosomal abnormalities (Benowitz et al., 1989; Cataldo et al., 1994), proliferation of activated microglia (Carpenter et al., 1993), alterations of vascular basement membrane (Kalaria, 1992; Perlmutter et al., 1994), increases in the levels of neurofilament proteins (Vickers et al., 1994), and appearance of the Alz-50 antigen (Wolozin et al., 1986). The molecular events that link these distinct pathological entities remain cryptic.
We have hypothesized that the C-terminal 100-amino acid fragment of the amyloid protein precursor (APP-C100), which includes the 42-amino acid Aβ peptide and 58 adjacent amino acids in the C terminus of APP, is instrumental in causing AD neuropathology. This fragment is toxic to neuronal cells (Yankner et al., 1989; Fukuchi et al., 1993; Sopher et al., 1994). Moreover, simple overexpression of normal APP (as occurs in Down syndrome, which is a predictor of AD neuropathology) can lead to the generation of APP-C100-like C-terminal fragments of APP and the death of the neuronal cells synthesizing these fragments in vitro (Fukuchi et al., 1992; Yoshikawa et al., 1992) and to the deposition of extracellular Aβ in vivo (Quon et al., 1991).
To test the hypothesis that APP-C100 can mediate the development of AD-like neuropathology, we transplanted APP-C100-expressing PC12 cells into mouse brain (Neve et al., 1992) and showed that they caused severe cortical atrophy and abnormal Alz-50 immunostaining. Similarly, Fukuchi et al. (1994) transplanted differentiated P19 cells stably transfected with C100 into mouse brains and observed distortion and shrinkage in the hippocampus around the site of the transplant and β-amyloid immunoreactivity in blood vessel walls and in the neuropil surrounding the site of the transplant. We detected comparable but more extensive pathology in 4.5-month-old transgenic mice expressing APP-C100 in the brain (Kammesheidt et al., 1992).
We now describe the profound degeneration of the hippocampal formation that occurs in the brains of aged transgenic mice expressing APP-C100 in the brain. We observed a progressive deterioration that culminated in massive degeneration of neurons and synapses by 18 months. Mice generated with an additional APP-C100 construct that contains the “Flag” epitope (Prickett et al., 1989) fused to the N terminus of APP-C100 displayed very advanced neurodegeneration by only 12 months of age. The APP-C100 transgenic mice represent the first in vivo model in which synaptic, axonal–dendritic, and neuronal degeneration have been demonstrated systematically and unequivocally in large numbers of animals. The appearance of multiple stereotypical features of AD in these transgenic mice provides support for the hypothesis that APP-C100 may play a critical role in AD neurodegeneration.
MATERIALS AND METHODS
Production of transgenic mice. We described previously the creation of the line seven APP-C100 transgenic mice (Kammesheidt et al., 1992), in which APP-C100 is expressed under the control of the dystrophin brain promoter. The Flag-APP-C100 transgenic mice are identical except for the fusion of the Flag sequence (Asp-Tyr-Lys-Asp-Asp-Asp-Asp-Lys), preceded by a methionine, to the N terminus of APP-C100. This was accomplished by inserting the following DNA sequence at the 5′ end of the C100 coding region: 5′-ATGGACTACAAAGACGATGACGATAAA-3′. The Flag-APP-C100 transgene, together with the upstream dystrophin promoter and the downstream SV40 splice and polyadenylation sequences, was microinjected into the pronuclei of fertilized F2 eggs from hybrid (C57BL/6J × SJL/J) mice at DNX (Princeton, NJ). The microinjected mouse eggs were transferred into the uterus of pseudopregnant females, where they were implanted and carried to term.
Genomic DNA was extracted from tail tissue of the progeny of these mice using a simplified protocol obtained from Dr. M. Rosenberg (personal communication): the tails were treated with SDS/proteinase K at 55°C overnight, sodium chloride was added to a final concentration of 1.5 m, and the mixture was extracted with chloroform. The DNA was precipitated from the aqueous phase with ethanol and resuspended overnight at 4°C. We used 200 ng of the DNA in the PCR, which was carried out for 33 cycles (94°C, 1 min; 50°C, 1 min; 72°C, 3 min). The 5′ primer sequence (5′-GGAG-ATCTCTGAAGTGAAGATGGATG-3′) was within the APP-C100 cDNA, and the 3′ primer (5′-GTCACACCACAGAAGTAAGGTTCC-3′) represented sequence within the SV40 splice and polyadenylation region. The predicted 600 bp PCR product was detected in the DNA of 8 of 53 potential transgenic founder mice. For Southern blots, 32P-labeled probes containing the SV40 splice and polyadenylation sequence were prepared by the random hexanucleotide priming method. The blots were washed to a maximum stringency of 0.5× SSC at 65°C with 0.1% SDS. We estimated transgene copy number by comparing the intensity of a positively hybridizing transgene restriction fragment with that hybridizing with the cDNA for the endogenous growth-associated protein GAP-43 gene (Neve et al., 1987), which exists as a single copy in the haploid genome. The founder mice and subsequent generations of transgenic mice were backcrossed to C57BL/6J mice.
RT-PCR. We prepared total RNA from 100–500 mg of tissue by a guanidinium thiocyanate procedure that we adapted previously (Neve et al., 1986). In the final step, the RNA was precipitated with 1/2 volume of ethanol, which preferentially precipitates RNA but not DNA. We treated 1 μg of RNA from each tissue with DNase I (0.3 U/μl in a volume of 13.1 μl) at 37°C for 20 min to remove possible contaminating DNA before using it as a template for RT-PCR as described (Ivins et al., 1993). The 5′ primer (5′-TGCTTTCAGGAAGATGACAGAATCAGGAGA-3′) re- presented sequence within the dystrophin promoter region that is transcribed and is part of the 5′ untranslated region of the transgene transcript, whereas the 3′ primer (5′-GTCACACCACAGAAGTAAGGTTCC-3′) represented sequence within the SV40 splice and polyadenylation region. These primers were used to generate the predicted PCR product of 650 bp. We confirmed the identity of the PCR products by a Southern blot, in which the PCR products were subjected to electrophoresis, transferred to a nylon membrane, and probed with a 40 bp antisense oligonucleotide representing sequence internal to the predicted PCR fragment.
Ages of animals examined for neuropathological changes. We examined 160 mice ranging in age from 8 to 28 months. Our controls included 23 C57BL/6J and SJL/J mice as well as 22 age- and sex-matched nontransgenic littermates from line seven. The 113 transgenic mice included 25 mice older than 18 months, of which 2 were the founder mice from which the APP-C100 colony is derived, and 27 Flag-APP-C100 mice (7 line-two mice, 16 line-18 mice, 2 line-17 mice, and 1 mouse each from lines one and six). Approximately equal numbers of each sex were included in the analysis.
Collection of tissues for microscope analysis. We anesthetized the mice by Halothane inhalation and perfused them transcardially with a variety of buffers and fixatives that included 0.1 m cacodylate buffer, pH 7.4 containing 4% sucrose and 4% paraformaldehyde; 3% glutaraldehyde in 0.1 mcacodylate buffer, pH 7.4; 0.l m PBS, pH 7.4; and 4% paraformaldehyde in PBS, pH 7.4, as described previously (Oster-Granite and Herndon, 1976). After perfusion of the animals, we placed their tissues into fresh fixative overnight. The brains were cut into 2 mm slabs using a coronal brain matrix mold, and the slabs were placed in fresh fixative. The right half of each slab was then dissected as appropriate to isolate cortex, hippocampus, striatum, cerebellum, and hypothalamus. These regions were cut into 1 mm slabs, processed through graded alcohols to Polybed 812, and stained en bloc with uranyl acetate. We collected plastic semithin sections (1.0 μm) from each block, selected areas for study, and generated ultrathin sections for examination in a Zeiss 10 electron microscope.
RESULTS
Characterization of transgenic mice expressing APP-C100 and Flag-APP-C100 under control of the dystrophin brain promoter
We described previously the generation of several lines of APP-C100 transgenic mice (Kammesheidt et al., 1992), in which the transgene was expressed under the control of the dystrophin brain promoter. In this report, we extend our analysis of APP-C100 mice from line seven, in which the transgene was present as a single copy and that displayed particularly high expression of APP-C100, to describe age-related neuropathology appearing in these mice. We also made new transgenic lines using Flag-APP-C100, which is identical to APP-C100 except for the fusion of the hydrophilic Flag sequence to the N terminus of APP-C100. We had discovered that addition of the Flag to APP-C100 enhanced its neurotoxicity in vitro without altering its specific binding to a receptor (Kozlowski et al., 1992) (M. Kozlowski and R. Neve, unpublished observations). We reasoned, therefore, that pathology in Flag-APP-C100 transgenic mice might appear earlier than that observed in the APP-C100 transgenics.
We isolated a 4.65 kilobase DNA fragment containing the dystrophin brain promoter-Flag-C100 fusion gene with the SV40 early region splice and polyadenylation sequences (described in Materials and Methods) and microinjected the DNA into the male pronuclei of C57BL/6J × SJL/J F2 hybrid mouse eggs. DNA extracted from the tails of 53 offspring was analyzed for the presence of the Flag-APP-C100 transgene by the PCR, using primers internal to the transgene construct. We found that eight of the mice were positive for the transgene, and six of these eight produced transgene-positive progeny. Southern blot analysis was used to estimate the transgene copy number in each founder line (data not shown). Line 18 appeared to have a single copy of the transgene; line six had ∼10 copies; and the remaining lines had 10–20 copies. In subsequent Southern blot analyses of DNA from F1 and F2 progeny, we observed that the transgene was inherited without rearrangements or changes in copy number.
Expression of the APP-C100 transgene RNA and protein products
We examined RNA from the brains of 6- to 7-month-old Flag-APP-C100 transgenic animals for expression of the transgene. RT-PCR was used to amplify a segment of the RNA that was predicted to be expressed from the transgene. Using a sense primer representing transcribed sequence within the dystrophin promoter region and an antisense primer within the SV40 splice and polyadenylation sequence, we observed the expected 650 bp RT-PCR fragment in all transgenic brain RNAs examined (J. Berger-Sweeney, D. McPhie, J. Arters, J. Greenan, M. Oster-Granite, R. Neve, unpublished observations). Expression of the transgene RNA was highest in the brains of mice from lines 2, 17, and 18.
We had shown previously expression of the Flag-APP-C100 transgene product in the brains of line-two mice (Neve and Boyce, 1996). We used two additional strategies to show expression of the transgene protein product in the brains of line-18 transgenic mice (J. Berger-Sweeney, D. McPhie, J. Alters, J. Greenan, M. Oster-Granite, R. Neve, unpublished observations). Immunoblots of the fractionated proteins from the mouse brains with antibody 369A, an affinity-purified antibody prepared by immunization with the C-terminal 50 amino acids of APP (Buxbaum et al., 1990), revealed the appropriate-sized immunoreactive band in both cytosolic and membrane fractions of the transgenic but not control mice (Berger-Sweeney et al., unpublished observations). These immunoblot results were confirmed by independent immunoprecipitation experiments in which C-terminal derivatives of APP were immunoprecipitated from transgenic brain homogenates using an antibody against the 42-amino acid Aβ fragment (Cummings et al., 1992) and then probed on immunoblots with C4, an antibody to the C-terminal 10 amino acids of APP (Selkoe et al., 1988), to show the expected immunoreactive band in the brains of transgenic but not control mice (J. Berger-Sweeney et al., unpublished observations).
Ages and numbers of mice used for neuropathological studies
We examined 160 mice, ranging in age from 8 to 28 months. These mice comprise 88 APP-C100 transgenic mice (including 25 mice over 18 months of age), 27 Flag-APP-C100 mice (7 from line 2, 16 from line 18, and 2 from line 17, and 1 each from lines one and six), 22 nontransgenic control littermates matched for age and sex, and 23 C57BL/6J and SJL/J age-matched controls. Approximately equal numbers of each sex were examined. We have been selective in our choice of mice to document the findings common to each of the groups in this study, partly because we wish to show that the entire spectrum of pathology that we describe is seen in any one mouse of the older age group. The illustrations in this report are taken from the following seven mice: one 28-month-old male, one 24-month-old female (both F1 progeny of the founder), and one of their 23-month-old male progeny, all from line seven and heterozygous for the APP-C100 transgene; two 14.5-month-old male Flag-APP-C100 mice that are founders for lines 1 and 17, respectively; one representative 22-month-old male C57BL/6J control mouse; and one representative 24-month-old male SJL/J control mouse.
Neurodegeneration in the hippocampal formation of APP-C100 transgenic mice
Numerous pyramidal cells in various states of degeneration could be found throughout the dentate gyrus and Ammon’s horn in the 23-month-old and 28-month-old male APP-C100 mice, respectively (Fig.1A,B), and with lesser magnitude in a 24-month-old female APP-C100 mouse as well (data not shown). Degeneration of variable extent has been detected in all positive transgenic progeny over 18 months of age derived from the mating of these line-seven APP-C100 founders; we have not determined whether homozygous individuals exhibit a comparable degree of pathology at an earlier age than do heterozygous individuals.
Fig. 1.
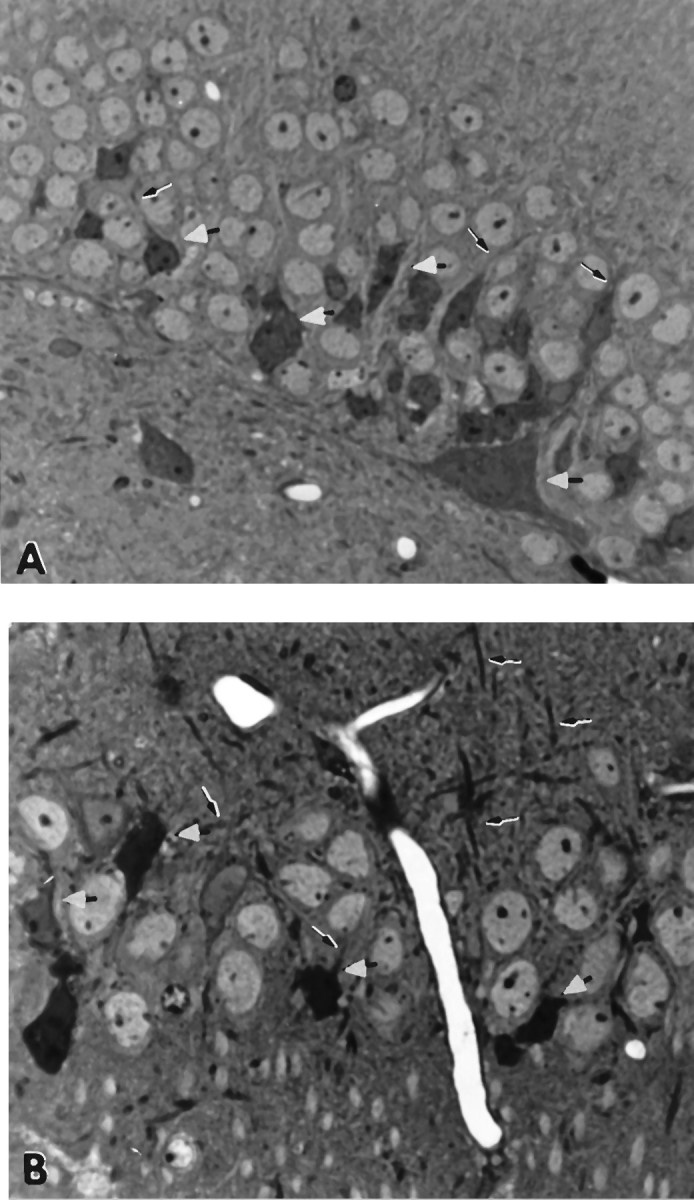
A, Granule cell layer of a transgene-positive 23-month-old male progeny from two F1 heterozygous APP-C100 line-seven mice. The sections are stained with toluidine blue. Note the numerous degenerating neurons in the cell layer (some indicated with white arrowheads) and in the neuropil below. Some dystrophic dendrites are evident (2800× magnification).B, Pyramidal cell layer of 28-month-old heterozygous male APP-C100 line-seven founder mouse. Note the numerous degenerating neurons in the pyramidal layer (white arrowheads). Degenerating dystrophic dendrites are also evident (arrows) (2800× magnification).
The pyramidal cell layers in the line-one and the line-17 14.5-month-old Flag-APP-C100 transgenic male mice are shown in Figure2, A and B. Numerous degenerating pyramidal cells with dystrophic dendrites were readily detected in Ammon’s horn of the line-one Flag-APP-C100 transgenic mouse (Fig.2A), whereas a similar but less severe degeneration was visible in the line-17 Flag-APP-C100 mouse (Fig.2B).
Fig. 2.
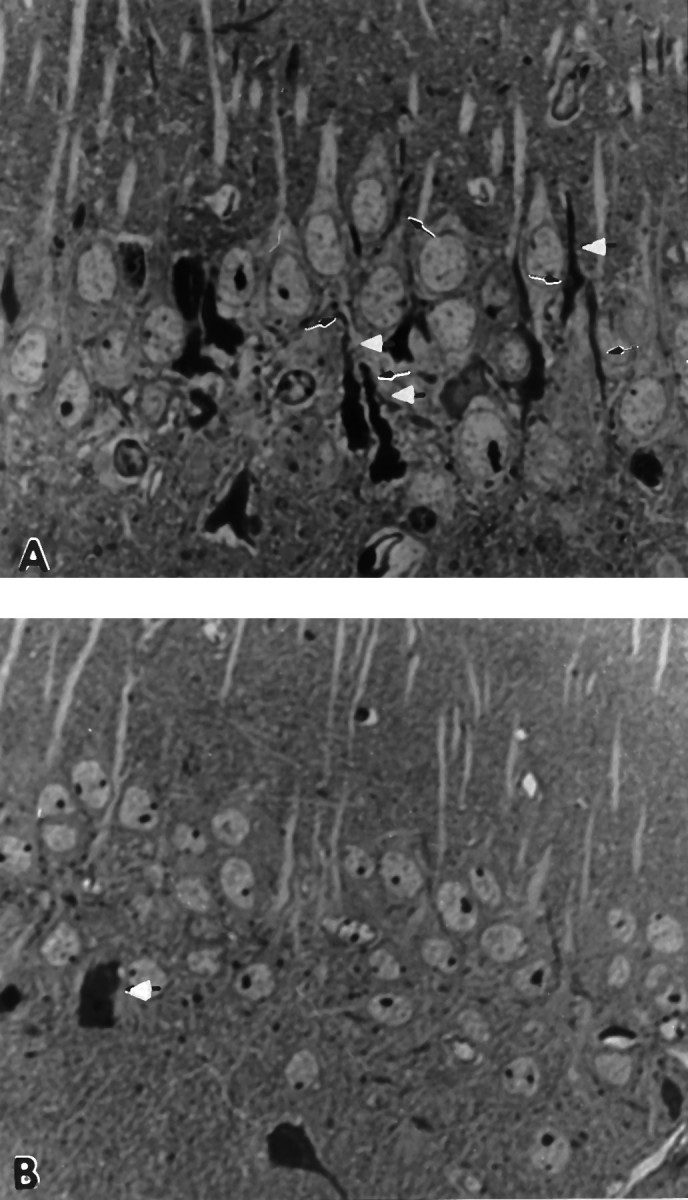
A, Pyramidal cell layer of male 14.5-month-old Flag-APP-C100 line-one founder mouse. Observe the reduced cell density and the numerous dystrophic dendrites and degenerating neurons in the pyramidal cell layer (white arrowheads) (2800× magnification). B, Pyramidal cell layer of 14.5-month-old male Flag-APP-C100 line-17 founder mouse. Observe the degenerating neurons in the pyramidal cell layer and underlying neuropil. Degenerating dystrophic dendrites are also evident (arrows) (2800× magnification).
When we examined the hippocampal formations of the control aged C57BL/6J and SJL/J mice, we observed only occasional necrotic pyramidal cells throughout Ammon’s horn at 22 and 24 months, respectively (Fig.3A,B). The pyramidal cells of both control strains were of approximately the same size (Fig.3A,B) and seemed to be more densely packed than those of the aged transgenic animals.
Fig. 3.
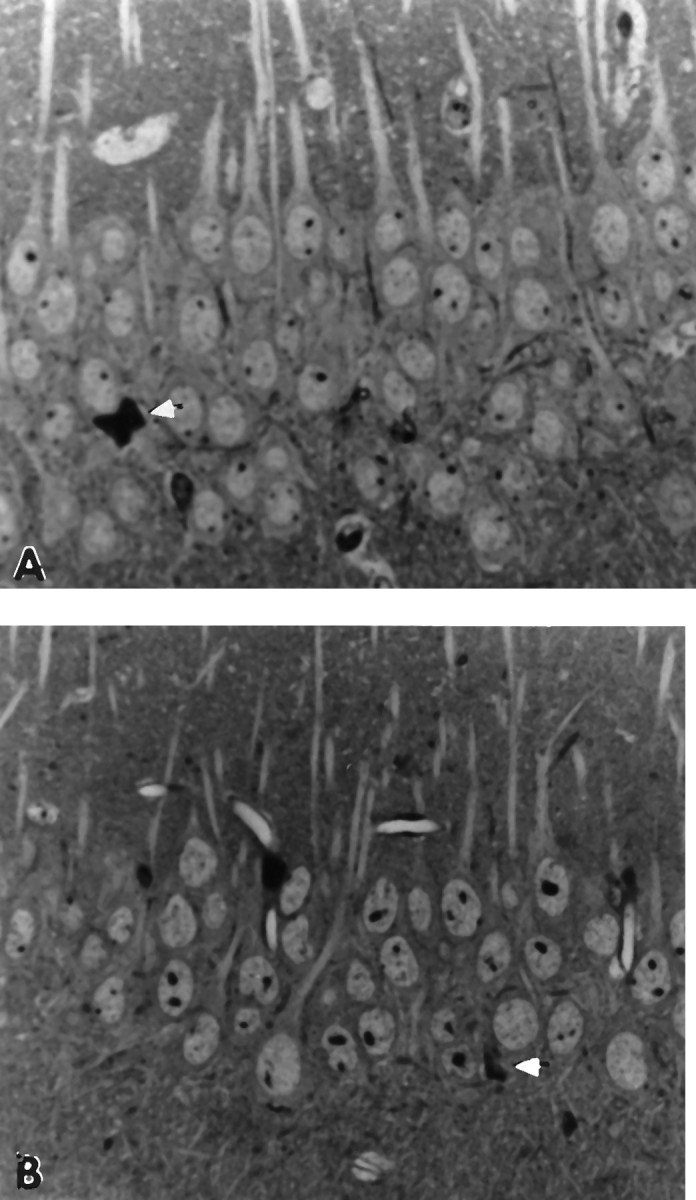
A, Pyramidal cell layer of 22-month-old male C57BL/6J mouse. Note that the cell bodies are similar in size to those in B (SJL/J mouse). White arrow indicates a degenerating neuron in the pyramidal layer, but otherwise the field contains healthy neurons (2800× magnification). B, Pyramidal cell layer in 24-month-old male SJL/J mouse. Note that cell body size is similar to that shown inA, and the cells appear healthy. White arrow indicates degenerating neuron in pyramidal cell layer (2800× magnification).
Abnormal accumulations of secondary lysosomes in the APP-C100 transgenic mice
A previously reported feature of the APP-C100 transgenic mice, detected as early as 4.5 months of age, was the appearance of oddly shaped secondary lysosomes that were immunoreactive with antibodies directed against portions of APP-C100 (Kammesheidt et al., 1992). These inclusions strongly resembled those that we had identified earlier in affected regions of AD brain (Benowitz et al., 1989). In the older groups of transgenic animals, these structures took the form of abundant deposits of dense, granular material in many of the pyramidal cells of the hippocampal formation (Fig. 1B, however, note that the accumulations are not obvious at this low magnification). The granular accumulations are more prominent in neurons in the molecular layer of Ammon’s horn (Fig.4B,C).
Fig. 4.
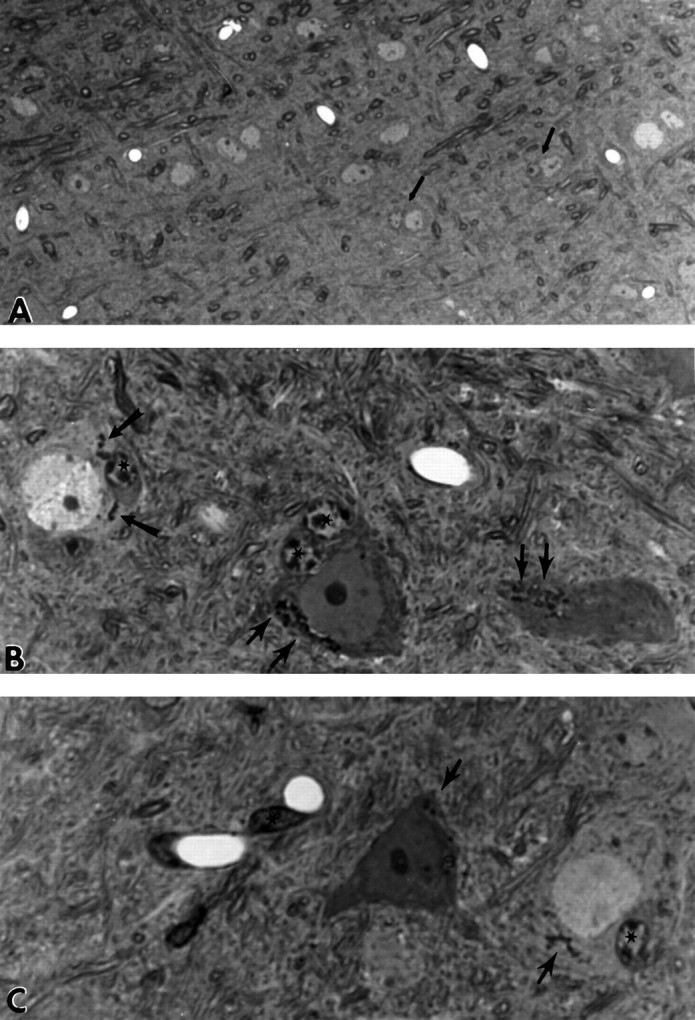
A, Neuropil of hippocampal formation of 24-month-old SJL/J male mouse. Note that the neurons are well spaced and few occur as duets with cells that appear to be glial in origin, such as those shown by the black arrows(2800× magnification). B, Neuropil of hippocampal formation of 24-month-old female APP-C100 line-seven F1 mouse. Two degenerating neurons and one healthy neuron make up this field. Observe the cytoplasmic accumulations of dense granular material that represent the aberrant secondary lysosomes that accumulate in these neurons as the mice age (black arrows). Cells adjacent to the neurons appear to have many characteristics of glial or microglial cells (star) (7000× magnification). C, Neuropil of hippocampal formation of 24-month-old female APP-C100 line-seven F1 mouse. Note the glial cells in apposition to degenerating and healthy neurons (star) and the dense cells (star) that lie close to blood vessels and resemble pericytes. Ultrastructural examination of these cells reveals that they contain degenerating debris and have a characteristic nuclear morphology that suggests they are microglial cells. Black arrows indicate the secondary lysosomal accumulations evident in the two neurons (7000× magnification).
Both degenerating and relatively healthy-looking neurons contained numerous accumulations of these secondary lysosomal structures, which are illustrated most effectively in the neuropil of a 24-month-old female APP-C100 founder (Fig. 4B,C). Further, and in contrast to the neurons in the aged normal mice, both normal and degenerating neurons in the transgenic mice had, in close apposition, cells strikingly reminiscent in their nuclear morphology of microglial cells (Fig.4B,C). In addition, dense cells, which on ultrastructural examination were shown to have the morphological characteristics of microglia laden with debris, lay next to blood vessels (Fig. 4C) in the transgenic mouse brains. In contrast, we detected few degenerating neurons or duets of neurons with adjacent microglial cells in the neuropil of the aged SJL mice (Fig.4A; note the lower magnification to document the scarcity of abnormal inclusions and degeneration in the control mice).
Ultrastructural analysis of the inclusions in the pyramidal cells of the aged APP-C100 mice revealed that these characteristic inclusions so prominent in the thin sections were oddly shaped secondary lysosomes, more abundant in the cell cytoplasm surrounding the nucleus than in dendritic or axonal processes of the cells. These lysosomes had a distinctly granular appearance and often contained material that was clear and lipid-like (Fig.5A).
Fig. 5.
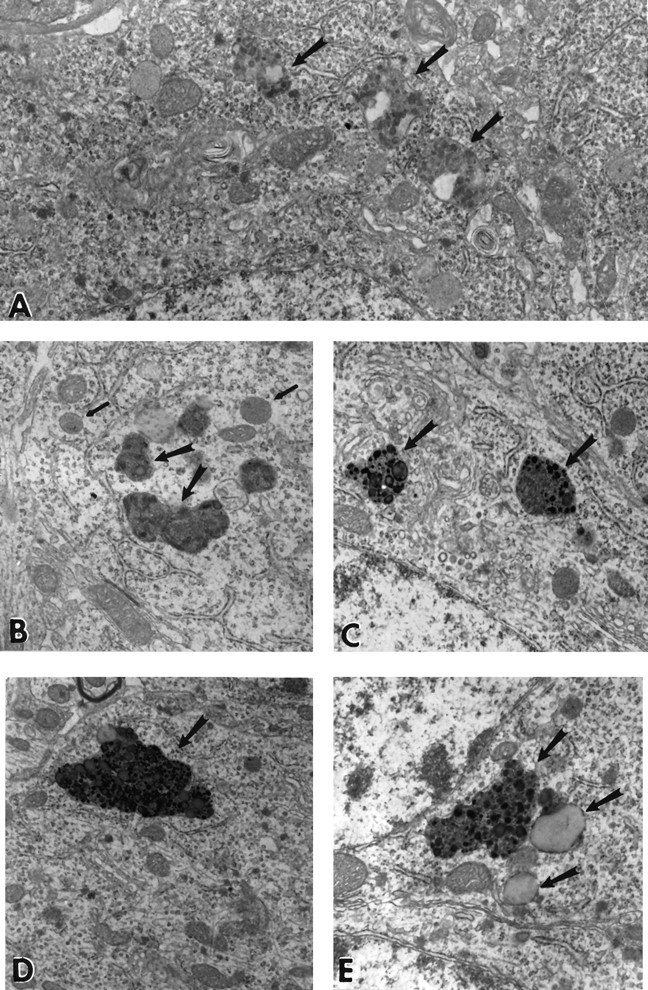
A, Aberrant secondary lysosomal inclusions of 28-month-old male APP-C100 line-seven F1 mouse. Note the heterogeneous granular accumulations that characterize the secondary inclusions (black arrows). Clear areas are interpreted to represent lipid-like accumulations. Observe that the cytoplasm of this reasonably healthy-looking pyramidal neuron contains whorls of membrane that may be associated in part with smooth endoplasmic reticulum (16,500× magnification). B, Secondary lysosomes in 22-month-old male C57BL/6J mouse. Note that the heterogeneous nature of the inclusions present in this mouse differs from that of the inclusions shown in A. In these inclusions (large black arrows), membranous whorls of material with few granular densities and little accumulation of lipid-like material are present. Nearby lie more typical lysosomal inclusions (small black arrows) that are much more abundant in younger normal animals of this strain (18,000× magnification). C, Secondary lysosomes in 24-month-old male SJL/J mouse. Secondary lysosomes in this mouse strain are characterized by membranous accumulations with dense heterogeneous granules within the lysosomal granule (large arrows). Nearby lie more typical lysosomal inclusions that resemble those shown in B (18,000× magnification. D, Secondary lysosomes in 14.5-month-old male line-one founder mouse. In this mouse, it is easier to find inclusions that are larger in size, but that are dense and heterogeneous, in the cytoplasm of an otherwise normal pyramidal cell (13,000× magnification). E, Secondary lysosomes in 14.5-month-old male line-17 founder mouse. Smaller inclusions than in D were found in this mouse. They have a density and heterogeneity similar to that of secondary lysosomes in aged SJL/J mice; however, vacuoles containing lipid-like material are more frequently associated with these secondary lysosomes than they are in SJL/J mice (21,600× magnification).
The lysosomal accumulations of the Flag-APP-C100 mice, in general, were more densely granular in appearance in the less severely affected mice, such as the line-17 founder (Fig. 5D) and more heterogeneous in appearance in the line-one founder mouse (Fig.5E). The morphology of these inclusions is fairly distinct within each line; however, the inclusions in Flag-C100-APP mice are more heterogeneous than those in the APP-C100 mice and occur in abundance at much earlier ages than do the inclusions of the APP-C100 mice. In turn, the inclusions in the APP-C100 mice are as abundant at 1 year of age as are the inclusions in aged normal C57BL/6J (Fig.5B) and SJL/J (Fig. 5C) mice at 2 years of age. Thus, these secondary lysosomes occur to some extent during normal aging of C57BL/6J and SJL/J mice but are morphologically abnormal and more abundant at earlier ages in the transgenic mice.
Cytoskeletal and synaptic degeneration in the APP-C100 transgenic mice
Examination of the pyramidal cell layer of the aged APP-C100 mice revealed the presence of numerous degenerating synapses (Fig.6A,B), axons containing increased numbers of neurofilaments relative to those of control mice (Fig. 6A,B), and membranous whorls in axonal and dendritic processes (Fig. 6B). In addition, we frequently observed secondary lysosomal inclusions in both dendritic and axonal processes near the cell body in numerous neurons in the pyramidal cell layer of these animals (data not shown). Further, microglia laden with debris could be found readily adjacent to large venous vessels that contained thickened basement membranes (Fig.6C).
Fig. 6.
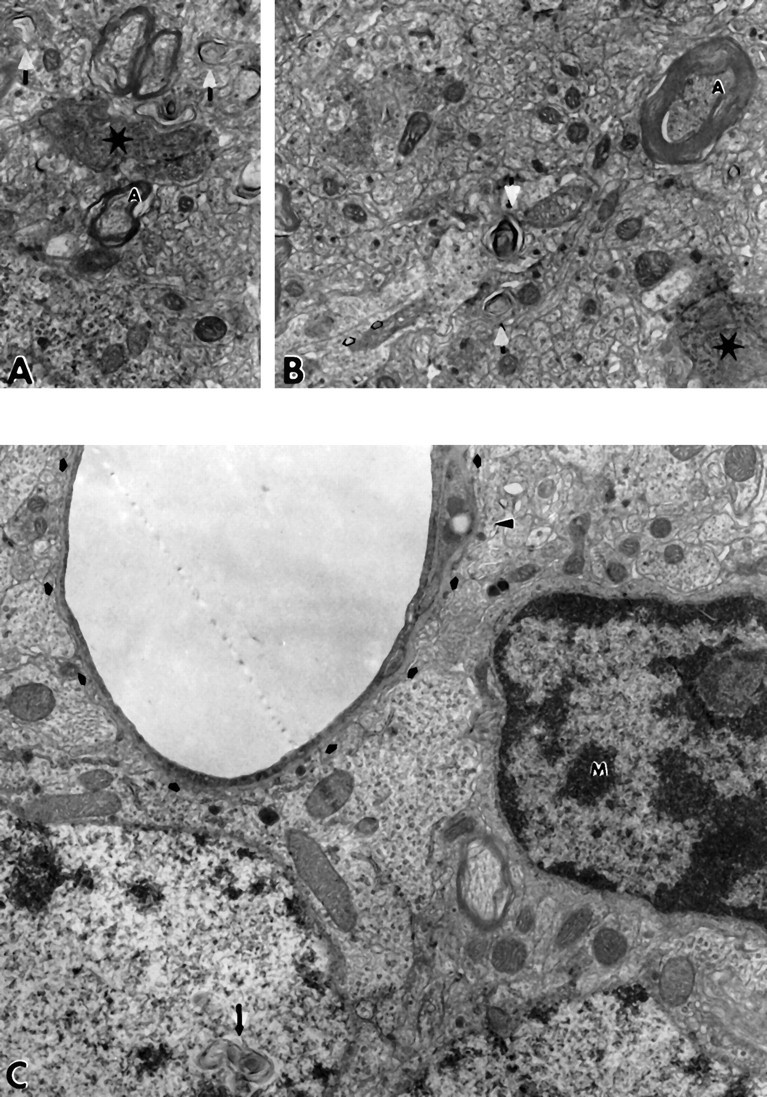
A, Degenerating synapses in neuropil of 28-month-old male APP-C100 line-seven F1 mouse. Note the degenerating dendritic process in this synaptic complex (star). Also, observe whorls of membrane in cell processes, both axonal and dendritic (white arrows), and axons that contain large numbers of neurofilaments (A) (15,000× magnification). B, Degenerating neuropil from another region of the hippocampal formation of the same 28-month-old male APP-C100 line-seven F1 mouse. Again, axons with reasonable amounts of myelin contain densely packed neurofilaments (A), whorls of membrane (white arrows) are seen in dendrites (open black arrows), and a synapse in which both axonal and dendritic processes appear to be degenerating is present (star) (15,000× magnification). C, Neuropil of the underlying dentate gyrus of the hippocampal formation of the same 28-month-old male APP-C100 line-seven F1 mouse. The dentate granule cell for which the nucleus lies adjacent to the capillary contains unusual whorls of membranous material (black arrow). The densely heterochromatic nucleus with large active nucleolus that has dense cytoplasm and lies adjacent to the blood vessel as well is interpreted to be a microglial cell (M). Observe that the basement membrane of the capillary (small black arrows) is thickened and that a cell process laden with debris (black arrowhead) lies within the thickened basement membrane.
The line-one Flag-APP-C100 transgenic contained an abundance of necrotic pyramidal cells, microglial cells laden with debris, and degenerating synaptic complexes (data not shown). In other Flag-APP-C100 transgenic mice that we examined, the extent of the degenerative process seemed to correlate with the degree of expression of the transgene. Even in the less severely affected Flag transgenics, however, the degree of degeneration in Ammon’s horn at 14.5 months of age was very similar to that we observed in the aged APP-C100 transgenic mice at 28 months. We observed a similar magnitude of degeneration in the dentate gyrus of each of the animals examined (data not shown).
Predominance of neuropathology in the hippocampus of the transgenic mice
Because the preponderant expression of the dystrophin brain promoter used in the transgenic constructs is in hippocampus and cortex (Gorecki et al., 1992), we inspected pyramidal cells in the neocortex for pathology and found increased numbers of secondary lysosomal inclusions in the transgenics relative to the control SJL/J mice. These inclusions were structurally similar to those seen in the hippocampal formation (data not shown). At present, we have not examined systematically the pyramidal neurons in the frontal cortices of these animals. We only rarely observed secondary lysosomal inclusions in the Purkinje cells of the same animals, and we found little evidence of neuronal degeneration in the cerebral cortex or cerebellum of these transgenic mice (data not shown).
DISCUSSION
We have shown that aged transgenic mice expressing the C-terminal 100 amino acids of APP in the brain develop neuropathology strikingly similar in many respects to that of AD. Our ultrastructural analyses revealed abnormal degeneration of neurons, neuronal processes, and synapses in the hippocampal formation of these aged transgenic mice. As in AD, this neurodegeneration was accompanied by characteristic cytoskeletal changes. In the hippocampus, deteriorating granule and pyramidal cell dendrites contained disorganized microtubules, and degenerating axons featured increased numbers of neurofilaments, accumulations resembling small secondary lysosomes, and aberrant whorls of membrane. Additional AD-like pathology in these mice included widespread abnormal cytoplasmic lysosomal inclusions, thickened basement membranes associated with blood vessels, and an abundance of debris-laden microglia in the vicinity of large blood vessels.
These pathological features augment the array of neuronal abnormalities similar to those found in AD brain that we described previously in these APP-C100 transgenic mice when they were 4.5 months of age (Kammesheidt et al., 1992). These abnormalities included cell body and neuropil accumulation of Aβ immunoreactivity in the brain, aberrant aggregation of C-terminal epitopes of APP in enlarged intracellular organelles that are similar to the fused lysosomes we had described in AD hippocampus (Benowitz et al., 1989), and the presence of thioflavin S-positive material suggestive of amyloid in the cerebrovasculature. Additional pathological effects of in vivo expression of APP-C100 have been reported in mice transplanted with neuronal cells expressing APP-C100 (Neve et al., 1992; Fukuchi et al., 1994). Most notably, severe cortical atrophy mirrored the cortical shrinkage seen in AD (De la Monte, 1989), and deposition of Aβ immunoreactivity was detected in blood vessel walls and neuropil surrounding the site of the transplant (Fukuchi et al., 1994).
The pathological characteristics of the APP-C100 transgenic mice are remarkably consistent among lines. For example, we noted in our earlier report (Kammesheidt et al., 1992) that intracellular Aβ immunoreactivity was detected in nine of nine different APP-C100 transgenic lines shown to express the transgene, although it spread to the neuropil only in the three lines showing highest expression of the transgene. Mice from one of these lines, line seven, were aged and are described in the present report. Of 25 line-seven transgenic mice >18 months of age examined, all 25 had varying degrees of the degeneration we describe in the hippocampal formation of three representative mice. Only in 3 of 22 nontransgenic control littermates >18 months of age did we detect numbers of degenerating pyramidal or granule cells in excess of that which is typically observed in aged C57BL/6J and SJL/J mice.
We created additional C100 transgenic mice in which the hydrophilic Flag sequence was fused to the N terminus of C100. All of the 14.5-month-old mice in five of these lines that we examined manifested neuropathology that was at least as severe as that seen in the 2-year-old C100 transgenic mice. We expected that the Flag-APP-C100 transgenic mice would show neurodegeneration at an earlier age than the APP-C100 transgenic mice, because earlier in vitro studies had indicated that Flag-APP-C100 was more neurotoxic than APP-C100 (R. Neve, unpublished observations), although pharmacological studies indicated that its specific binding to a receptor was not altered (M. Kozlowski, R. Neve, unpublished observations). Its enhanced toxicity may be attributable to the fact that Flag-APP-C100, with its hydrophilic Flag tag, is less prone to aggregate than C100 (R. Neve, unpublished observations). Our expectations were borne out; however, we cannot rule out the possibility that transgene expression was simply more robust in the Flag-C100 mice than in the APP-C100 transgenic mice. At present, we have not analyzed systematically Flag-APP-C100 mice that are <12 months of age, thus, we cannot comment on the age of onset of neurodegeneration in these lines of transgenic mice.
The data presented here comprise compelling evidence to support the hypothesis that APP-C100 is critically involved in the etiology of AD. Synaptic loss correlates more strongly with the degree of cognitive impairment in AD than does amyloid deposition (DeKosky and Scheff, 1990; Terry et al., 1990). Interestingly, the Flag-APP-C100 mice evince, by the age of 1 year, pronounced behavioral deficits relative to controls, and the behavioral impairment correlates well with the extent of hippocampal neurodegeneration quantified in the mouse brains (J. Berger-Sweeney et al., unpublished observations). It has been proposed that synaptic–axonal damage precedes and plays an important causative role in the genesis of plaques in AD (Masliah et al., 1993,1994). Synaptic and axonal–dendritic degeneration of unusual magnitude has been demonstrated unequivocally at the ultrastructural level in all APP-C100 transgenic mice >18 months of age and in all Flag-APP-C100 mice >12 months of age that we examined.
Is the neurodegenerative activity a function of the holo-APP-C100 fragment or of Aβ that may be generated by proteolytic cleavage of C100? We showed previously (Yankner et al., 1989) that the neurotoxicity of C100 in vitro can be removed from the cell culture medium by preabsorption of the medium with an antibody to either the N terminus or the C terminus of APP-C100. These data led us to suggest that the intact fragment exerts the neurotoxic effects, although they do not rule out the possibility that the Aβ peptide may comprise the precise pathogenic domain within the C100 fragment. Furthermore, in vivo models for the action of Aβ in the brain have produced phenotypes very different from those described for the APP-C100 models, although strain differences may have accounted for some of the differences. Transplantation of APP-C100-producing neuronal cells into mouse brain caused cortical atrophy (Neve et al., 1992;Fukuchi et al., 1994), the appearance of the Alz-50 antigen (Neve et al., 1992), and deposition of Aβ (Fukuchi et al., 1994) in the vicinity of the transplant. In contrast, transplantation of human neurons that secrete Aβ into rodent brain did not cause detectable lesions (Mantione et al., 1995). Further, expression of Aβ in the brains of transgenic mice failed to elicit an AD phenotype in one case (Wirak et al., 1991; Jucker et al., 1992) and caused gliosis, apoptosis, and a curious appearance of neurons with unstained perinuclear cytoplasm in another (LaFerla et al., 1995). Gross overexpression (≥10-fold higher than endogenous levels of mouse APP) of human APP containing the V717F mutation in transgenic mice (Games et al., 1995) resulted in β-amyloid deposition. There were preliminary indications, at the light microscope level, of decreased synaptophysin and MAP2 immunoreactivity in the molecular layer of the hippocampal dentate gyrus of the mice. These abnormalities may have been the result of overexpression of APP or of the expression of the APP mutation. Overexpression of normal APP in vitro can lead to the accumulation of APP-C100-like C-terminal fragments and to the death of the neuronal cells in which these fragments build up (Fukuchi et al., 1992; Yoshikawa et al., 1992). The APP V717F mice were not, however, examined for the presence of APP-C100.
The APP-C100 and Flag-APP-C100 transgenic mice thus recreate many aspects of the synaptic damage, neuronal death, disruption of the neuronal cytoskeleton, and lysosomal abnormalities that are seen in the brains of individuals with AD. The phenotype of these animals leads us to suggest that APP-C100 is a critical component of the molecular mechanism of AD neurodegeneration.
Footnotes
This research was supported by grants HD19932 (M.L.O.G.) and AG12954 (R.L.N.) from National Institutes of Health and by Janssen Research Foundation. We thank Drs. Amaea Walker, Frederick Boyce, and Ralph Nixon for helpful discussions. We appreciate generous gifts of antibodies from Drs. S. Gandy (369A), B. Cummings (E1-42), and D. Selkoe (C4). Critical reading of this manuscript by Drs. Frederick Boyce and Daniel Alkon is greatly appreciated.
Correspondence should be addressed to Dr. Rachael Neve, 202 MRC, McLean Hospital, 115 Mill Street, Belmont, MA 02178.
REFERENCES
- 1.Benowitz LI, Rodriguez W, Paskevich P, Mufson EJ, Schenk D, Neve RL. The amyloid precursor protein is concentrated in neuronal lysosomes in normal and Alzheimer disease subjects. Exp Neurol. 1989;106:237–250. doi: 10.1016/0014-4886(89)90156-8. [DOI] [PubMed] [Google Scholar]
- 2.Buxbaum JD, Gandy SE, Cicchetti P, Ehrlich ME, Czernik AJ, Fracasso RP, Ramabhadran TV, Unterbeck AJ, Greengard P. Processing of Alzheimer β/A4 amyloid precursor protein: modulation by agents that regulate protein phosphorylation. Proc Natl Acad Sci USA. 1990;87:6003–6006. doi: 10.1073/pnas.87.15.6003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Carpenter AF, Carpenter PW, Markesbery WR. Morphometric analysis of microglia in Alzheimer’s disease. J Neuropathol Exp Neurol. 1993;52:601–608. doi: 10.1097/00005072-199311000-00007. [DOI] [PubMed] [Google Scholar]
- 4.Cataldo AM, Hamilton DJ, Nixon RA. Lysosomal abnormalities in degenerating neurons link neuronal compromise to senile plaque development in Alzheimer disease. Brain Res. 1994;640:68–80. doi: 10.1016/0006-8993(94)91858-9. [DOI] [PubMed] [Google Scholar]
- 5.Cummings BJ, Su JH, Geddes JW, Van Nostrand WE, Wagner SL, Cunningham DD, Cotman CW. Aggregation of the amyloid precursor protein within degenerating neurons and dystrophic neurites in Alzheimer’s disease. Neuroscience. 1992;48:763–777. doi: 10.1016/0306-4522(92)90265-4. [DOI] [PubMed] [Google Scholar]
- 6.DeKosky ST, Scheff SW. Synapse loss in frontal cortex biopsies in Alzheimer’s disease: correlation with cognitive severity. Ann Neurol. 1990;27:457–464. doi: 10.1002/ana.410270502. [DOI] [PubMed] [Google Scholar]
- 7.De la Monte SM. Quantitation of cerebral atrophy in preclinical and end-stage Alzheimer’s disease. Ann Neurol. 1989;5:450–459. doi: 10.1002/ana.410250506. [DOI] [PubMed] [Google Scholar]
- 8.Fukuchi K, Kamino K, Deeb SS, Smith AC, Dang T, Martin GM. Overexpression of amyloid precursor protein alters its normal processing and is associated with neurotoxicity. Biochem Biophys Res Commun. 1992;182:165–173. doi: 10.1016/s0006-291x(05)80126-3. [DOI] [PubMed] [Google Scholar]
- 9.Fukuchi K, Sopher B, Furlong CE, Smith AC, Dang NT, Martin GM. Selective neurotoxicity of COOH-terminal fragments of the β-amyloid precursor protein. Neurosci Lett. 1993;154:145–148. doi: 10.1016/0304-3940(93)90192-n. [DOI] [PubMed] [Google Scholar]
- 10.Fukuchi K, Kunkel DD, Schwartzkroin PA, Kamino K, Ogburn CE, Furlong CE, Martin GM. Overexpression of a C-terminal portion of the β-amyloid precursor protein in mouse brains by transplantation of transformed neuronal cells. Exp Neurol. 1994;127:253–264. doi: 10.1006/exnr.1994.1101. [DOI] [PubMed] [Google Scholar]
- 11.Games D, Adams D, Alessandrini R, Barbour R, Berthelette P, Blackwell C, Carr T, Clemens J, Donaldson T, Gillespie F, Guido T, Hagopian S, Johnson-Wood K, Khan K, Lee M, Leibowitz P, Lieberburg I, Little S, Masliah E, McConlogue L, Montoya-Zavala Mucke L, Paganini L, Penniman E, Power M, Schenk D, Seubert P, Snyder B, Soriano F, Tan H, Vitale J, Wadsworth S, Wolozin B, Zhao J. Alzheimer-type neuropathology in transgenic mice overexpressing V717F β-amyloid precursor protein. Nature. 1995;373:523–527. doi: 10.1038/373523a0. [DOI] [PubMed] [Google Scholar]
- 12.Gorecki DC, Monaco AP, Derry JM, Walker AP, Barnard EA, Barnard PJ. Expression of four alternative dystrophin transcripts in brain regions regulated by different promoters. Hum Mol Genet. 1992;1:505–510. doi: 10.1093/hmg/1.7.505. [DOI] [PubMed] [Google Scholar]
- 13.Hamos JE, DeGennaro LJ, Drachman DA. Synaptic loss in Alzheimer’s disease and other dementias. Neurology. 1989;39:355–361. doi: 10.1212/wnl.39.3.355. [DOI] [PubMed] [Google Scholar]
- 14.Ivins KJ, Neve KA, Feller DJ, Fidel SA, Neve RL. Antisense GAP-43 inhibits the evoked release of dopamine from PC12 cells. J Neurochem. 1993;60:626–633. doi: 10.1111/j.1471-4159.1993.tb03194.x. [DOI] [PubMed] [Google Scholar]
- 15.Jucker M, Walker LC, Martin LJ, Kitt CA, Kleinman HK, Ingram DK, Price DL. Age-associated inclusions in normal and transgenic mouse brain. Science. 1992;255:1443–1455. doi: 10.1126/science.1542796. [DOI] [PubMed] [Google Scholar]
- 16.Kalaria RN. The blood-brain barrier and cerebral microcirculation in Alzheimer disease. Cerebrovasc Brain Metab Rev. 1992;4:226–260. [PubMed] [Google Scholar]
- 17.Kammesheidt A, Boyce FM, Spanoyannis AF, Cummings BJ, Ortegon M, Cotman CW, Vaught JL, Neve RL. Amyloid deposition and neuronal pathology in transgenic mice expressing the carboxyterminal fragment of the Alzheimer amyloid precursor in the brain. Proc Natl Acad Sci USA. 1992;89:10857–10861. doi: 10.1073/pnas.89.22.10857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kozlowski MR, Spanoyannis A, Manly SP, Fidel SA, Neve RL. The neurotoxic carboxy-terminal fragment of the Alzheimer amyloid precursor binds to a neuronal cell surface molecule: pH dependence of the neurotoxicity and the binding. J Neurosci. 1992;12:1679–1687. doi: 10.1523/JNEUROSCI.12-05-01679.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.LaFerla FM, Tinkle BT, Bieberich CJ, Haudenschild CC, Jay G. The Alzheimer’s Aβ peptide induces neurodegeneration and apoptic cell death in transgenic mice. Nature Genet. 1995;9:21–30. doi: 10.1038/ng0195-21. [DOI] [PubMed] [Google Scholar]
- 20.Mantione JR, Kleppner SF, Miyazono M, Wertkin AM, Lee VM-Y, Trojanowski JQ. Human neurons that constitutively secrete Aβ do not induce Alzheimer’s disease pathology following transplantation and long-term survival in the rodent brain. Brain Res. 1995;671:333–337. doi: 10.1016/0006-8993(94)01400-c. [DOI] [PubMed] [Google Scholar]
- 21.Masliah E, Mallory M, Deerinck T, DeTeresa R, Lamont S, Miller A, Terry RD, Carragher B, Ellisman M. Re-evaluation of the structural organization of neuritic plaques in Alzheimer’s disease. J Neuropathol Exp Neurol. 1993;52:619–632. doi: 10.1097/00005072-199311000-00009. [DOI] [PubMed] [Google Scholar]
- 22.Masliah E, Honer WG, Mallory M, Voigt M, Kushner P, Hansen L, Terry R. Topographical distribution of synaptic-associated proteins in the neuritic plaques of Alzheimer’s disease hippocampus. Acta Neuropathol (Berl) 1994;87:135–142. doi: 10.1007/BF00296182. [DOI] [PubMed] [Google Scholar]
- 23.Neve RL, Boyce FM. Construction and analysis of transgenic mice expressing amyloidogenic fragments of the Alzheimer amyloid protein precursor. In: Perez-Polo JR, editor. Paradigms of neural injury, methods in neurosciences, Vol 30. Academic; San Diego: 1996. pp. 298–314. [Google Scholar]
- 24.Neve RL, Harris P, Kosik KS, Kurnit DM, Donlon TA. Identification of cDNA clones for the human microtubule-associated protein tau and chromosomal localization of the genes for tau and microtubule-associated protein 2. Mol Brain Res. 1986;1:271–280. doi: 10.1016/0169-328x(86)90033-1. [DOI] [PubMed] [Google Scholar]
- 25.Neve RL, Perrone-Bizzozero NI, Finklestein S, Zwiers H, Bird E, Kurnit DM, Benowitz LI. The neuronal growth-associated protein GAP-43 (B-50, F1): neuronal specificity, developmental regulation and regional expression of the human and rat cDNAs. Mol Brain Res. 1987;2:177–183. doi: 10.1016/s0006-8993(87)80012-4. [DOI] [PubMed] [Google Scholar]
- 26.Neve RL, Kammesheidt A, Hohmann CF. Brain transplants of cells expressing the carboxyterminal fragment of the Alzheimer amyloid protein precursor cause specific neuropathology in vivo. Proc Natl Acad Sci USA. 1992;89:3448–3452. doi: 10.1073/pnas.89.8.3448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Oster-Granite ML, Herndon RM. The development of the cerebellar cortex of the Syrian hamster, Mesocricetus auratus. Foliation, cytoarchitectonic, Golgi, and electron microscopic studies. J Comp Neurol. 1976;169:443–479. doi: 10.1002/cne.901690404. [DOI] [PubMed] [Google Scholar]
- 28.Perlmutter LS, Myers MA, Barrón E. Vascular basement membrane components and the lesions of Alzheimer’s disease: light and electron microscopic analyses. Microsc Res Tech. 1994;28:204–215. doi: 10.1002/jemt.1070280305. [DOI] [PubMed] [Google Scholar]
- 29.Prickett KS, Amberg DC, Hopp TP. A calcium-dependent antibody for identification and purification of recombinant proteins. Biotechniques. 1989;7:580–589. [PubMed] [Google Scholar]
- 30.Quon D, Wang Y, Catalano R, Scardina JM, Murakami K, Cordell B. Formation of β-amyloid protein deposits in brains of transgenic mice. Nature. 1991;352:239–241. doi: 10.1038/352239a0. [DOI] [PubMed] [Google Scholar]
- 31.Selkoe DJ, Podlisney MB, Joachim CL, Vickers EA, Lee G, Fritz LC, Oltersdorf T. β-Amyloid precursor protein of Alzheimer disease occurs as 110- to 135-kilodalton membrane-associated proteins in neural and nonneural tissues. Proc Natl Acad Sci USA. 1988;85:7341–7345. doi: 10.1073/pnas.85.19.7341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sopher BL, Fukuchi K, Smith AC, Leppig KA, Furlong CE, Martin GM. Cytotoxicity mediated by conditional expression of a carboxyl-terminal derivative of the β-amyloid precursor protein. Mol Brain Res. 1994;26:207–217. doi: 10.1016/0169-328x(94)90092-2. [DOI] [PubMed] [Google Scholar]
- 33.Terry RC, Peck A, DeTeresa R, Horoupian DS. Some morphometric aspects of the brain in senile dementia of the Alzheimer type. Ann Neurol. 1981;10:184–192. doi: 10.1002/ana.410100209. [DOI] [PubMed] [Google Scholar]
- 34.Terry RD, Masliah E, Salmon DP, Butters N, DeTeresa R, Hill R, Hansen LA, Katzman R. Physical basis of cognitive alterations in Alzheimer’s disease: synapse loss is the major correlate of cognitive impairment. Ann Neurol. 1990;30:572–580. doi: 10.1002/ana.410300410. [DOI] [PubMed] [Google Scholar]
- 35.Vickers JC, Riederer BM, Marugg RA, Buee-Scherrer V, Buee L, Delacourte A, Morrison JH. Alterations in neurofilament protein immunoreactivity in human hippocampal neurons related to normal aging and Alzheimer’s disease. Neuroscience. 1994;62:1–13. doi: 10.1016/0306-4522(94)90310-7. [DOI] [PubMed] [Google Scholar]
- 36.Wirak DO, Bayney R, Ramabhadran TV, Fracasso RP, Hart JT, Hauer PE, Hsiau P, Pekar SI, Scangos GA, Trapp BD, Unterbeck AJ. Deposits of amyloid β protein in the central nervous system of transgenic mice. Science. 1991;253:323–325. doi: 10.1126/science.1857970. [DOI] [PubMed] [Google Scholar]
- 37.Wolozin BL, Pruchnicki A, Dickson DW, Davies P. A neuronal antigen in the brains of Alzheimer patients. Science. 1986;232:648–650. doi: 10.1126/science.3083509. [DOI] [PubMed] [Google Scholar]
- 38.Yankner BA, Dawes LR, Fisher S, Villa-Komaroff L, Oster-Granite ML, Neve RL. Neurotoxicity of a fragment of the amyloid precursor associated with Alzheimer’s disease. Science. 1989;245:417–420. doi: 10.1126/science.2474201. [DOI] [PubMed] [Google Scholar]
- 39.Yoshikawa K, Aizawa T, Hayashi Y. Degeneration in vitro of post-mitotic neurons overexpressing the Alzheimer amyloid protein precursor. Nature. 1992;359:64–67. doi: 10.1038/359064a0. [DOI] [PubMed] [Google Scholar]