

Abstract
Orphanin FQ (OFQ) has recently been reported to be an endogenous ligand for the opioid-like LC132 receptor. The effect of OFQ on high voltage-gated calcium channels (VGCCs) was examined in freshly dissociated rat pyramidal neurons using the whole-cell configuration of the patch-clamp technique. High-threshold Ba2+ currents were reversibly inhibited by OFQ. The depression of the currents was associated with a slowed rate of activation and a change in the activation I–V relationship at step potentials higher than +30 mV. In concentration–response experiments, a mean (±SEM) pEC50 value of 7.0 ± 0.07 and a Hill coefficient of 1.5 ± 0.08 (n = 5) were obtained. The near-maximum inhibition of the Ba2+ currents by OFQ (1 μm) amounted to 31 ± 2.2% of control (n = 15). Opioid receptors could not account for the effects of OFQ on VGCCs, because naloxone, a broad spectrum μ-, δ-, and κ-receptor antagonist, did not reduce the effectiveness of OFQ. When GTP-γ-S was included in the pipette, the depression of the currents by OFQ was irreversible, whereas currents from neurons preincubated with pertussis toxin were not inhibited by OFQ, consistent with the involvement of a PTX-sensitive G-protein. When selective blockers of VGCCs were used, it was demonstrated that all subtypes of VGCCs were affected by OFQ. In conclusion, the effect of OFQ on VGCCs expressed in hippocampal CA3 and CA1 neurons may play an important role in the regulation of hippocampal cell excitability and neurotransmitter release.
Keywords: calcium channel blockers, calcium channel drug effects, opioid drug effects, G-protein, patch clamp, nifedipine, ω-conotoxin-GVIA, ω-agatoxin-IVA, GTP-γ-S, orphanin FQ, nociceptin, opioid receptor, neuromodulation
The family of the G-protein-coupled opioid receptors comprising the μ-, δ-, and κ-receptors (Evans et al., 1992; Kieffer et al., 1992; Meng et al., 1993; Thompson et al., 1993) was recently extended by a novel member (Bunzow et al., 1994; Chen et al., 1994; Fukuda et al., 1994; Mollereau et al., 1994; Wang et al., 1994; Wick et al., 1994; Lachowicz et al., 1995). This receptor, termed among other things LC132 by Bunzow et al. (1994), was cloned by homology-based screening and shares a sequence homology of 64% within the transmembrane regions with the other family members. In situ hybridization analysis revealed that LC132 mRNA is highly expressed in several brain areas, including the cerebral cortex, hippocampus, thalamus, hypothalamus, and the dorsal horn of the spinal cord. When transiently expressed in COS-7 cells, however, the LC132 receptor did not bind any of the typical opioid receptor ligands.
Because of its similarity to the opioid receptor family, it was expected that the LC132 receptor, like the three other opioid receptor subtypes, would be negatively coupled to adenylyl cyclase. In the search for a natural ligand for the LC132 receptor, inhibition of forskolin-stimulated cAMP production in LC132-transfected cells by fractionated extracts from mammalian brain was monitored (Meunier et al., 1995; Reinscheid et al., 1995). A heptadecapeptide was purified that inhibited cAMP production and bound selectively the LC132 receptor in a saturable manner and with nanomolar affinity. Opioid agonists and antagonists did not interact with the effect induced by the peptide, which therefore was assumed to be a natural ligand for the LC132 receptor. This peptide was named orphanin FQ (OFQ) (Reinscheid et al., 1995) or nociceptin (Meunier et al., 1995).
In contrast to the analgesic effect after opioid receptor activation, intracerebroventricular injection of OFQ did not induce analgesia but produced hyperalgesia in the tail-flick test and decreased locomotor activity only at higher doses (Reinscheid et al., 1995). To date, no cellular physiological consequences of LC132 activation in brain neurons have been described. Many receptors that negatively regulate cAMP formation couple in neurons to the inhibition of voltage-gated calcium channels (VGCCs) and the activation of G-protein-activated inwardly rectifying K+ channels (for reviews, see Anwyl, 1991; Hille, 1992, 1994). Therefore, we reasoned that activation of LC132 receptors by OFQ might mediate similar effects. Because LC132 is expressed in hippocampal pyramidal neurons, we examined the effect of OFQ on VGCCs in freshly dissociated CA3 and CA1 neurons. Furthermore, because the diversity of VGCCs is believed to lead to the wide variety of physiological functions controlled by these channels, we identified which types of VGCCs were modulated by OFQ.
A preliminary account of some of these results has been published in abstract form (Knoflach et al., 1996).
MATERIALS AND METHODS
Cell preparation. Pyramidal neurons from the hippocampus of 8- to 11-d-old Sprague Dawley rats were obtained according to methods described by Swartz and Bean (1992). Briefly, slices (450 μm) were cut with a vibratome in an ice-cold solution that contained (in mm): 125 NaCl, 2.5 KCl, 2 CaCl2, 1 MgCl2, 1.25 NaH2PO4, 26 NaHCO3, and 25 d-glucose, bubbled with oxycarbon (95% O2/5% CO2), pH 7.4, and were subsequently incubated at 20°C in the same solution. When neurons were needed for electrophysiological experiments, the CA1 or CA3 regions of three slices were dissected out and treated for 10 min at 37°C with a solution containing (in mm): 82 Na2SO4, 30 K2SO4, 3 MgCl2, 2 HEPES, and 3 mg/ml protease XXIII, pH 7.4 with NaOH. Neurons were isolated by gentle trituration with a Pasteur pipette in a solution containing 1 mg/ml bovine serum albumin and 1 mg/ml trypsin inhibitor and plated on poly-l-ornithine-coated glass coverslips.
Electrophysiological experiments. The whole-cell configuration of the patch-clamp technique was used to perform electrophysiological experiments (Hamill et al., 1981). Pipette resistances ranged from 2 to 3 MΩ when filled with a solution containing (in mm): 117 TEACl, 9 HEPES, 9 EGTA, 4.5 MgCl2, 1 GTP, 4 ATP, and 14 phosphocreatine, pH 7.2 with TEAOH, osmolarity 330 mOsm/l. The neurons were perfused with a standard saline solution that contained (in mm): 140 NaCl, 5 KCl, 2 CaCl2, 1 MgCl2, 10 HEPES, 10 d-glucose, pH 7.4 with NaOH, osmolarity adjusted to 340 mOsm/l with sucrose. Whole-cell currents were amplified with an Axopatch 200A amplifier (Axon Instruments, Foster City, CA), filtered at 5 kHz, and digitized at 10 kHz with a Digidata 1200A acquisition board (Axon Instruments) for subsequent storage on a Gateway 2000 P4D-66 personal computer. The data acquisition and analysis were performed with the pClamp 6.0 software package (Axon Instruments). To evoke Ba2+ currents, 50 msec voltage steps to 0 mV from a holding potential of −80 mV were generated while the neuron was perfused with a solution containing (in mm): 25 BaCl2, 145 TEACl, 10 HEPES, 0.1 EGTA, and 10 d-glucose, pH 7.4 with TEAOH, osmolarity adjusted to 340 mOsm/l with sucrose. Leakage currents were subtracted using the P/4 protocol. For drug-testing experiments, Ba2+ currents were generated periodically while the substances were applied locally to the neuron by fast perfusion from a double-barreled pipette assembly (Johnson and Ascher, 1987). The rate of solution exchange of the application system was assessed by measuring the decrease in Na+ current through AMPA/kainate receptors induced by exchanging the normal neuron perfusion solution with a solution containing a 10× lower NaCl concentration. In these experiments, performed on cultured cortical neurons, the rate of solution exchange was consistently ∼30 msec. We anticipated that the solution exchange rate around the freshly isolated neurons, which lacked extensive dendrites, would be considerably faster than this. In concentration–effect experiments, the peak amplitudes of the current inhibition (I) obtained from each concentration of agonist were fitted separately for each neuron using the logistic equation I = Imax/(1+(EC50/[agonist])^Hill), where Imax is the maximum current obtained from the fit, EC50 is the concentration of agonist eliciting a half-maximum effect, and Hill is the Hill coefficient.
OFQ was obtained from Research Genetics (Huntsville, AL), GTP and ATP from Boehringer Mannheim (Mannheim, Germany), ω-conotoxin-GVIA and naloxone from RBI (Natick, MA), and ω-agatoxin-IVA from the Peptide Institute (Osaka, Japan). All other chemicals were obtained from Sigma (St. Louis, MO)
RESULTS
Ba2+ currents through VGCCs
To record Ba2+ currents through VGCCs, 50 msec step potentials to 0 mV were elicited from a holding potential of −80 mV (Fig. 1A). The mean (±SEM) maximum current amplitude recorded from 20 acutely dissociated hippocampal CA3 neurons amounted to 1985 ± 173 pA. When the holding potential was changed to −100 mV and a step potential of −50 mV was used, a low-voltage-activated current was sometimes observed (not shown). The amplitudes of the low-voltage-activated currents, when present, were at least 10-fold smaller than the amplitudes of high-voltage-activated currents and thus were negligible. Hence, the experiments described here were carried out on the high-voltage-activated currents. After the establishment of the whole-cell configuration, an increase in the current amplitude >1–3 min was sometimes observed. To avoid a misleading interpretation of the effect of OFQ, the experiments were started only when the currents had reached a constant amplitude, which was sometimes followed by a variable but linear rundown.
Fig. 1.

OFQ inhibition of high-voltage-gated Ca2+ channel currents. A, Whole-cell currents were evoked in the absence (1, 3) and presence (2) of OFQ (300 nm) according to the protocol shown on top of the superimposed traces. B, Time course of the effect of orphanin FQ (OFQ). The ligand was applied for 45 sec while currents were evoked by 50 msec step potentials to 0 mV from a holding potential of −80 mV at 5 sec intervals.
OFQ inhibition of Ba2+ currents
When OFQ (300 nm) was applied to a neuron, Ba2+ currents evoked by 50 msec step potentials to 0 mV from a holding potential of −80 mV were depressed in a reversible manner (Fig. 1A). In addition, in the presence of OFQ, the rise time of the Ba2+ currents was slowed, and the amplitude of the tail currents was also reduced (Fig.1A). The time course of the effect of OFQ on Ba2+ currents was evaluated by generating step potentials to 0 mV every 5 sec (Fig. 1B). In all neurons tested, reversal of the effect of OFQ was complete or in the same order of magnitude as the “rundown” obtained under control conditions.
Voltage dependence of the inhibition of Ba2+ currents by OFQ
To assess the voltage dependence of the effect of OFQ,I–V relationships in the absence (Fig.2A) and presence (Fig.2B) of OFQ (100 nm) were generated. Depolarizing voltage steps in the range from −45 mV to +30 mV elicited Ba2+ currents whose magnitude depended on the voltage step amplitude (Fig. 2A,B). I–Vrelationships were obtained by plotting the maximum current amplitude as a function of the voltage step potential. As shown in Figure2C, the maximum currents were reached, in both the absence and presence of OFQ, at a voltage step of 0 mV. OFQ also had the greatest inhibitory effect on peak current amplitude at this potential; however, when steady-state activation I–Vrelationships were generated by plotting the current amplitudes evoked 100 msec after the beginning of the test pulse as a function of the voltage step potential, a voltage-independent inhibitory effect of OFQ was observed. When the inhibitions by OFQ of Ba2+ currents evoked by depolarizing pulses to 0 and 25 mV were compared, a significant difference in the mean OFQ effect was found when the currents were measured 10 msec after the beginning of the test pulse (44.5 ± 2.7% inhibition at 0 mV and 27.8 ± 1.8% at 25 mV;p < 0.005, two-tailed t test;n = 4), whereas no significant difference was found when the currents were measured after 100 msec (28.3 ± 5.6% inhibition at 0 mV and 25.6%±7.1% at 25 mV; p > 0.7, two-tailed t test; same neurons). The voltage-independent inhibitory effect of OFQ was not relieved by large depolarizing prepulses, which removed the slowing effect of OFQ (not shown).
Fig. 2.

Current–voltage relationship of the OFQ inhibition. Superimposed traces of high-voltage-gated Ca2+channel currents are shown in the absence (A) and presence (B) of 100 nm OFQ. The currents were elicited by 100 msec voltage steps in the range of −45 to +30 mV from a holding potential of −60 mV at 3 sec intervals. The current–voltage relationships shown in C were obtained by plotting the peak current amplitudes in the absence (open circle) and presence (filled circles) of orphanin FQ (OFQ; 100 nm) as a function of the test pulse potential.
Concentration–effect experiments
The concentration–effect relationship for OFQ inhibition of VGCCs was evaluated by applying increasing concentrations of OFQ to the neurons while evoking Ba2+ currents with 0 mV step potentials from a holding potential of −80 mV every 3 sec. The superimposed whole-cell current traces in Figure3A were obtained in the presence and absence of OFQ, at the indicated concentrations. OFQ was always applied until a maximum effect was reached. The application of higher concentrations of OFQ resulted not only in a higher degree but also in a faster onset of inhibition of the currents (Fig. 3B). When the onset of the effect of OFQ was compared at two different concentrations by fitting single exponential curves to the time course of inhibition, the values obtained were significantly different, amounting to 15 ± 1.3 sec (n = 10) and 4.5 ± 0.55 sec (n = 8) at 100 nm and 1 μm OFQ, respectively (p < 0.001, two-tailed t test). The recoveries from inhibition at the same concentrations, however, were not significantly different and yielded values of 33 ± 6.7 sec (n = 10) and 31 ± 3.0 sec (n = 8) at 100 nm and 1 μm OFQ, respectively (p > 0.7, two-tailed t test). The apparent KD values calculated from the mean inhibition onset and recovery values amounted to 78 nm at 100 nm OFQ and to 173 nm at 1 μmOFQ. The peak amplitudes (I) of the current inhibition obtained from each concentration of OFQ were fitted separately for each neuron using the logistic equation (see Materials and Methods). The results from five neurons yielded a pEC50value of 7.0 ± 0.07 and a Hill coefficient of 1.5 ± 0.08. The concentration–response curve in Figure 3C is the fit through the mean normalized current values of these neurons and yields a pEC50 value of 7.00 and a Hill coefficient of 1.4. The near-maximum inhibition of the Ba2+ currents by OFQ (1 μm) measured in 15 neurons amounted to 31 ± 2.2% of control.
Fig. 3.
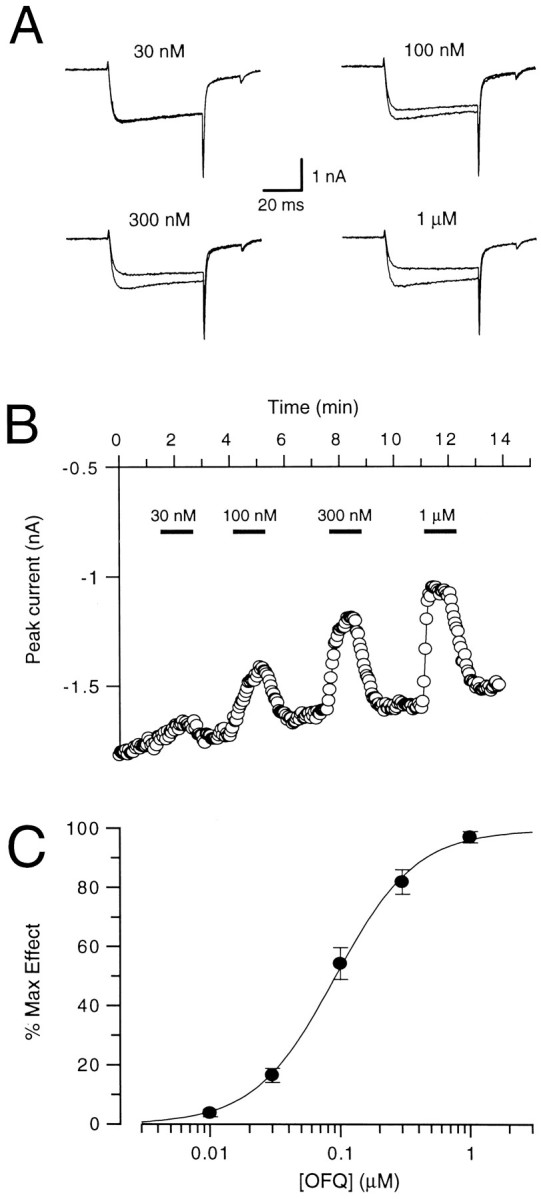
Concentration-dependence of the effect of OFQ on high-voltage-gated Ca2+ channel currents. A, Whole-cell currents evoked by 50 msec voltage steps to 0 mV from a holding potential of −80 mV. The traces of the currents obtained in the absence and presence of the indicated concentrations of OFQ are shown superimposed. B, Time course of the effect of OFQ. The ligand was applied in increasing concentrations for 1 min at 3 min intervals while whole-cell currents were elicited at 3 sec intervals.C, OFQ concentration–response curve. The values of the inhibition of the Ca2+ channel peak current amplitudes obtained with different concentrations of OFQ were fitted individually for each neuron and normalized to the maximum of the fit (100%). The normalized values are plotted as a function of the orphanin FQ (OFQ) concentration. The points represent the mean ± SEM of four neurons, and the sigmoidal curve represents a fit through these points.
Lack of effect of naloxone
To demonstrate that the effects of OFQ were mediated by the LC132 receptor and not the related opioid receptors, the effect of the broad spectrum opioid receptor antagonist (−)-naloxone on OFQ responses was examined. The inhibition of Ba2+ currents by OFQ (300 nm) measured in the absence and presence of naloxone (10 μm) was not significantly different (24 ± 5.1%,n = 3, and 23 ± 4.6%, n = 3, respectively) (Fig. 4). Thus, the depression of the VGCCs by OFQ is not mediated by any of the μ-, δ-, and κ-opioid receptors.
Fig. 4.

Lack of effect of naloxone on the OFQ modulation of high-voltage-gated Ca2+ channel currents. Orphanin FQ (OFQ; 300 nm) was applied alone or in the presence of the broad spectrum opioid receptor antagonist naloxone (10 μm) for the time indicated by the bars. The currents were elicited by 50 msec voltage steps to 0 mV from a holding potential of −80 mV at 3 sec intervals. In addition to the rapid coapplication with OFQ, naloxone was present in the bath 1 min before and during the test.
G-protein involvement
To confirm that the inhibitory effect of OFQ on VGCCs was mediated via a G-protein pathway, the effect of GTP-γ-S was examined. In neurons recorded with a patch pipette containing 1 mm GTP, the inhibition of the voltage-gated calcium channel currents by OFQ (300 nm) was readily reversible and reproducible, as shown by two consecutive OFQ applications in Figure5A. When the recording pipette contained the nonhydrolyzable analog GTP-γ-S (1 mm) as a replacement for GTP, the maximum Ba2+ current amplitudes were lower (1392 ± 46 pA, n = 3, vs 1985 ± 173 pA,n = 20), and OFQ (300 nm) was without an effect on the currents (not shown). When 100 μm GTP-γ-S was used, however, the depression of the currents by OFQ (300 nm) became irreversible (Fig. 5B). Furthermore, a second application of OFQ remained without an effect (Fig.5B). This suggests that a G-protein is involved in the coupling between the LC132 receptor and the VGCCs. The degree of inhibition of the Ba2+ currents by OFQ (300 nm) in the presence of GTP-γ-S amounted to 34 ± 2.9% of control (n = 3).
Fig. 5.

G-protein involvement in the effect of OFQ on VGCCs. Time courses of the effect of orphanin FQ (OFQ) are shown in the absence (A) and the presence of 100 μm GTP-γ-S (B) in the pipette. Whole-cell currents were elicited by 50 msec voltage steps to 0 mV from a holding potential of −80 mV at 3 sec intervals. In contrast to the reversible effect of OFQ shown in A, the effect of OFQ on the presence of GTP-γ-S in the pipette (B) is irreversible, and a subsequent application of OFQ (same concentration) remained without an effect.
To examine further the involvement of specific G-proteins in the effect of OFQ on VGCCs, neurons were treated with pertussis toxin, an inhibitor of the Gα0/αi G-protein family. For this purpose, neurons were kept for 14–16 hr at 37°C in a culture medium that contained 200 ng/ml pertussis toxin. CA1 pyramidal neurons were used in these experiments, because they displayed a higher rate of survival after the incubation than CA3 pyramidal neurons did. In neurons treated with pertussis toxin, no inhibition of the VGCC currents elicited by step potentials to 0 mV from a holding potential of −80 mV by OFQ (300 nm) was observed (2.0 ± 0.9%; n = 6). In untreated neurons, however, OFQ (same concentration) inhibited the peak current by 22.8 ± 6.3% (n = 4). One neuron was insensitive to OFQ.
Pharmacological distinction of calcium channel subtypes
We used pharmacological means to assess which of the various VGCC types expressed in CA3 pyramidal neurons were being affected by OFQ. When nifedipine (10 μm), an L-type calcium channel inhibitor, was applied to the investigated neurons, the maximum amplitudes of Ba2+ currents induced by a step depolarization to 0 mV were reversibly reduced by 16 ± 1.4% (n = 8) (Fig. 6A). The effect of OFQ on L-type calcium channels was tested by applying OFQ (1 μm) to neurons in the absence and presence of nifedipine (10 μm). The lower degree of inhibition of the Ba2+ current by OFQ obtained when the L-type calcium channels were blocked by nifedipine demonstrates that OFQ is able to modulate L-type calcium channels (Fig. 6A). In seven neurons, the inhibition of the L-type calcium channel currents by OFQ (1 μm), obtained by comparing the maximum amplitude of the currents in the absence and presence of nifedipine and OFQ, amounted to 31 ± 9.2% (Fig. 7B). To demonstrate further this involvement of L-type calcium channels, the ability of OFQ to reduce the slow component of tail currents induced by the dihydropyridine agonist Bay K 8644 was tested. As represented in Figure 6B, the Ba2+ current obtained by a depolarizing step to 0 mV was enhanced by Bay K 8644 (1 μm). Furthermore, the tail current generated in the presence of Bay K 8644 by a voltage step back from 0 mV to −40 mV inactivated with a slower time constant. OFQ inhibited the current generated during the test pulse as well as the maximum tail current amplitude and the slow component of the tail current (Fig.6B). Similar results were obtained with six other neurons, whereas in two neurons OFQ had no effect on the slow phase of the tail current.
Fig. 6.

OFQ inhibits the L-type of VGCCs.A, The time course of the effect of orphanin FQ (OFQ; 1 μm) is shown in the absence and presence of nifedipine applied for the times indicated by thebars. The whole-cell currents were evoked at 5 sec intervals by 50 msec voltage steps to 0 mV from a holding potential of −80 mV. B, Superimposed current traces obtained by the protocol shown on top in the absence and presence of Bay K 8644 (Bay K; 1 μm) and the combined presence of Bay K 8644 and OFQ. Bay K 8644 introduced a slow component to the tail currents that was inhibited by OFQ.
Fig. 7.

A, OFQ inhibits the N-type of VGCCs. Orphanin FQ (OFQ; 1 μm) was applied alone or in the presence of ω-conotoxin-GVIA (1 μm) for the time indicated by the bars. The currents were elicited by 50 msec voltage steps to 0 mV from a holding potential of −80 mV at 3 sec intervals. In addition to the rapid coapplication with OFQ, ω-conotoxin-GVIA was present in the bath before the test and until a maximum block was achieved.B, OFQ inhibits all types of VGCCs expressed in CA3 pyramidal neurons. The total height of the bars(open and hatched) represents the mean (± SEM) contribution of the indicated calcium channel type to the total evoked calcium channel current. The hatched barsrepresent the mean (± SEM) inhibition by OFQ of the corresponding calcium channel type.
The sensitivity of the N-type calcium channels to OFQ was determined by comparing the OFQ-mediated inhibition of depolarization-induced Ba2+ currents in the absence and presence of the selective N-type Ca2+ channel blocker ω-conotoxin-GVIA. In these experiments, L-type calcium channels were blocked by nifedipine (10 μm). When ω-conotoxin-GVIA (1 μm) was applied to neurons, the depolarization-evoked currents were reduced in an irreversible manner by 25 ± 2.8% (n = 8) (Fig. 7A). The inhibition of N-type calcium channels by OFQ (1 μm) was more pronounced than that of L-type calcium channels and amounted to 57 ± 6.1% (n = 8) (Fig.7B).
The sensitivity of P/Q-type calcium channels to OFQ was assessed by using a concentration of ω-agatoxin-IVA that blocks these channel types. With 200 nm ω-agatoxin-IVA, the depolarization-elicited Ba2+ currents were inhibited by 31 ± 3.3% of control (n = 8), whereas L-type calcium channels were blocked by 10 μm nifedipine and N-type calcium channels by 1 μm ω-conotoxin-GVIA. OFQ (1 μm) also acted on P/Q-type calcium channels (44 ± 2.7% inhibition of control, n = 7) (Fig.7B).
When all of the L-, N-, and P/Q-type calcium channels were blocked by co-applying nifedipine (10 μm), ω-conotoxin-GVIA (1 μm), and ω-agatoxin-IVA (200 nm), the remaining depolarization-induced Ba2+ current amplitude amounted to 28 ± 4.3% of the total current (n = 8). This remaining current was also sensitive to OFQ. In seven neurons, the inhibition by OFQ (1 μm) amounted to 29 ± 2.3% of the remaining current (Fig. 7B). In these experiments, a rundown of the effect of OFQ is unlikely to have occurred, because in control experiments comprising 10 successive applications of OFQ (100 nm) generated every 3 min, the degree of inhibition of currents by OFQ was comparable throughout (not shown). Accordingly, in the OFQ responses of Figures 4 and 5, no noticeable rundown occurred after two or three successive applications of OFQ, respectively.
DISCUSSION
In the present study, we have shown that OFQ, a natural ligand for the opioid-like receptor LC132, inhibits VGCCs expressed in rat hippocampal pyramidal neurons via a G-protein-mediated pathway. Other members of the opioid receptor family also modulate VGCCs in neurons. The activation of the μ-opioid receptor inhibited VGCCs in rat sensory neurons (Schroeder et al., 1991). A κ-opioid receptor agonist, dynorphin A, reduced the magnitude of inward calcium channel currents in mouse dorsal root ganglion neurons (Macdonald and Werz, 1986). [Met5]-enkephalin also reduced calcium channel currents in submucosal neurons acutely dissociated from guinea pigs, an effect mediated via the δ-opioid receptor (Surprenant et al., 1990). Because the LC132 receptor shares a high sequence homology with the other members of the opioid receptor family (Bunzow et al., 1994), it was expected that activation of the LC132 receptor would trigger similar intracellular signaling mechanisms. The finding that OFQ inhibits high VGCCs corroborates this hypothesis. The experiments performed with the broad spectrum μ-, δ-, and κ-opioid receptor antagonist (−)-naloxone demonstrate that the effects of OFQ were not mediated by any of the μ-, δ-, and κ-opioid receptors but presumably were mediated by the LC132 receptor. The concentration of (−)-naloxone used in the present study (10 μm) is far above those required to displace the μ-, δ-, and κ-opioid receptor-selective radioligands (Ki values of 2.3, 17, and 0.93 nm, respectively) (Raynor et al., 1994) and therefore would be high enough to antagonize all of the μ-, δ-, and κ-opioid receptors. Moreover, in a study in rat dorsal root ganglion neurons, naloxone reduced the inhibition of VGCC currents produced by PLO17 (1 μm) by 91.5% at a 100-fold lower concentration (Moises et al., 1994b).
Many transmitters modulate VGCCs by changing the time course of activation and the voltage dependence of the currents (Bean, 1989;Carbone and Swandulla, 1989; Anwyl, 1991; Luebke and Dunlap, 1994). OFQ also followed this commonly occurring mechanism of modulation, because the time course of activation of calcium channel currents was slower in the presence of OFQ, and a maximum effect on peak current amplitude was measured at a depolarizing step of 0 mV. When the effect of OFQ on the calcium channel currents was measured at steady-state (100 msec), however, a voltage-independent effect remained, similar to previous findings on the inhibitory actions of γ-aminobutyric acid and norepinephrine on N-type calcium channels (Luebke and Dunlap, 1994). Also, in agreement with this latter study, large depolarizing prepulses left the voltage-independent inhibitory action of OFQ unchanged, although they suppressed the slowing effect of OFQ.
OFQ potently and reversibly inhibited the voltage-gated calcium channel currents, with a pEC50 value of 7.0. This value is in agreement with the KD values calculated from the mean onset and recovery of the inhibition of the currents by OFQ (78 nm at 100 nm and 173 nm at 1 μm). In LC132-transfected cells, OFQ inhibited forskolin-stimulated cAMP accumulation with IC50 values of 1.0 nm (Reinscheid et al., 1995) and 0.9 nm(Meunier et al., 1995). The 100-fold difference in potency found in the present study might be attributable to a much lower receptor density in neurons as compared with that in transfected cells, which often show levels of expression several orders of magnitude above physiological levels.
Two lines of evidence for a G-protein-linked inhibition of voltage-gated calcium currents by OFQ are presented here. First, when a low concentration of GTP-γ-S (100 μm) was present in the patch pipette, the inhibition of the currents by OFQ became irreversible. A second application of OFQ had no effect, demonstrating that the G-protein coupling system was presumably activated permanently by OFQ in the presence of GTP-γ-S. This finding is in line with the common observed effect of GTP-γ-S on transmitter modulation of VGCCs (Anwyl, 1991). When a high concentration of the nonhydrolyzable GTP analog GTP-γ-S was present in the patch pipette (1 mm), OFQ was ineffective on calcium channel currents. The observation that the current amplitudes were lower when GTP-γ-S (1 mm) was included in the pipette also indicates, presumably, that a direct inhibition of the currents by GTP-γ-S occurred. This finding is consistent with the decrease of calcium current in hippocampal neurons after activation of caged GTP-γ-S by ultraviolet light (Frank et al., 1996); however, in contrast to baclofen, which remained effective on GTP-γ-S-inhibited currents (Frank et al., 1996), OFQ in the present study was ineffective in the presence of high concentrations of GTP-γ-S. The second evidence for a G-protein-linked effect of OFQ arises from the fact that calcium channel currents recorded from CA1 pyramidal neurons treated with pertussis toxin were not inhibited by OFQ. This suggests an involvement of a Gi or Gotype of G-protein (Hille, 1994). The finding that LC132 receptors couple, in neurons, to VGCCs via a pertussis toxin-sensitive G-protein correlates with studies on the modulation of VGCCs by opioid receptor ligands (Surprenant et al., 1990; Moises et al., 1994a; Nomura et al., 1994; Rhim and Miller, 1994).
In whole-cell recordings, the N-type channel is the most common calcium channel inhibited by transmitters (Anwyl, 1991). OFQ inhibited the ω-conotoxin-GVIA-sensitive component of the calcium current by 57%. This value is in close agreement with the inhibition of the ω-conotoxin-GVIA-sensitive N-type calcium channels by the μ-opioid agonist PLO17 found in rat sensory neurons (55%) (Rusin and Moises, 1995). The finding that OFQ inhibits the L-type channel is of particular interest, because in rat sensory neurons μ-opioid receptor activation did not modulate the L-type channel (Rusin and Moises, 1995), although in GH3 cells transfected with rat μ-opioid receptor cDNA, a coupling of the μ-opioid receptors to L-type channels was reported (Piros et al., 1995). The low percentage of L-type calcium channels found in rat sensory neurons (15% of the total current) cannot account for the lack of effect of the μ-opioid agonist PLO17 on L-type calcium channels, because in the present study, a similarly low contribution of L-type calcium channels to the total high-voltage-activated current was found (16%). In a study performed on rat CA3 hippocampal neurons, a higher percentage of L-type calcium channels than that found here was reported (36%) (Mintz et al., 1992), whereas in CA1 hippocampal neurons an L-type percentage of 38% and 19% were found by Ishibashi et al. (1995) and Mintz et al. (1992), respectively. The variation of L-type channel contribution to the total calcium channel current could be attributable to the different dihydropyridines used: nifedipine in the present study and nimodipine or nitrendipine in the other two studies. A heterogeneity in calcium channel expression, however, cannot be excluded.
The effect of OFQ on the ω-agatoxin-IVA-sensitive calcium channels, presumably P/Q-types, is of importance, because in addition to N-type calcium channels, these channels have also been implicated in controlling neurotransmitter release (Horne and Kemp, 1991; Luebke et al., 1993; Takahashi and Momiyama, 1993; Wheeler et al., 1994).
In conclusion, the G-protein-mediated effect of OFQ on all of the different types of VGCCs expressed in hippocampal CA3 neurons may play an important role in the regulation of hippocampal cell excitability, synaptic transmission, and plasticity.
Footnotes
Correspondence should be addressed to Dr. John A. Kemp, F. Hoffmann-La Roche Ltd., Pharma Division, Preclinical Research, CH-4070 Basel, Switzerland.
REFERENCES
- 1.Anwyl R. Modulation of vertebrate neuronal calcium channels by transmitters. Brain Res Brain Res Rev. 1991;16:265–281. doi: 10.1016/0165-0173(91)90010-6. [DOI] [PubMed] [Google Scholar]
- 2.Bean BP. Neurotransmitter inhibition of neuronal calcium currents by changes in channel voltage dependence. Nature. 1989;340:153–156. doi: 10.1038/340153a0. [DOI] [PubMed] [Google Scholar]
- 3.Bunzow JR, Saez C, Mortrud M, Bouvier C, Williams JT, Low M, Grandy DK. Molecular cloning and tissue distribution of a putative member of the rat opioid receptor gene family that is not a μ, δ or κ opioid receptor type. FEBS Lett. 1994;347:284–288. doi: 10.1016/0014-5793(94)00561-3. [DOI] [PubMed] [Google Scholar]
- 4.Carbone E, Swandulla D. Neuronal calcium channels: kinetics, blockade and modulation. Prog Biophys Mol Biol. 1989;54:31–58. doi: 10.1016/0079-6107(89)90008-4. [DOI] [PubMed] [Google Scholar]
- 5.Chen Y, Fan Y, Liu J, Mestek A, Tian M, Kozak CA, Yu L. Molecular cloning, tissue distribution and chromosomal localization of a novel member of the opioid receptor gene family. FEBS Lett. 1994;347:279–283. doi: 10.1016/0014-5793(94)00560-5. [DOI] [PubMed] [Google Scholar]
- 6.Evans CJ, Keith DE, Jr, Morrison H, Magendzo K, Edwards RH. Cloning of a δ opioid receptor by functional expression. Science. 1992;258:1952–1955. doi: 10.1126/science.1335167. [DOI] [PubMed] [Google Scholar]
- 7.Frank C, Engert F, Tokutomi N, Lux HD. Different effects of baclofen and GTPγS on voltage-activated Ca2+ currents in rat hippocampal neurons in vitro. Eur J Pharmacol. 1996;295:87–92. doi: 10.1016/0014-2999(95)00621-4. [DOI] [PubMed] [Google Scholar]
- 8.Fukuda K, Kato S, Mori K, Nishi M, Takeshima H, Iwabe N, Miyata T, Houtani T, Sugimoto T. cDNA cloning and regional distribution of a novel member of the opioid receptor family. FEBS Lett. 1994;343:42–46. doi: 10.1016/0014-5793(94)80603-9. [DOI] [PubMed] [Google Scholar]
- 9.Hamill OP, Marty A, Neher E, Sakmann B, Sigworth FJ. Improved patch-clamp techniques for high-resolution current recording from cells and cell-free membrane patches. Pflüegers Arch. 1981;391:85–100. doi: 10.1007/BF00656997. [DOI] [PubMed] [Google Scholar]
- 10.Hille B. G protein-coupled mechanisms and nervous signaling. Neuron. 1992;9:187–195. doi: 10.1016/0896-6273(92)90158-a. [DOI] [PubMed] [Google Scholar]
- 11.Hille B. Modulation of ion-channel function by G protein-coupled receptors. Trends Neurosci. 1994;17:531–536. doi: 10.1016/0166-2236(94)90157-0. [DOI] [PubMed] [Google Scholar]
- 12.Horne AL, Kemp JA. The effect of ω-conotoxin GVIA on synaptic transmission within the nucleus accumbens and hippocampus of the rat in vitro. Br J Pharmacol. 1991;103:1733–1739. doi: 10.1111/j.1476-5381.1991.tb09855.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ishibashi H, Rhee JS, Akaike N. Regional differences of high voltage-activated Ca2+ channels in rat CNS neurones. NeuroReport. 1995;6:1621–1624. doi: 10.1097/00001756-199508000-00008. [DOI] [PubMed] [Google Scholar]
- 14.Johnson JW, Ascher P. Glycine potentiates the NMDA response in cultured mouse brain neurons. Nature. 1987;325:529–531. doi: 10.1038/325529a0. [DOI] [PubMed] [Google Scholar]
- 15.Kieffer BL, Befort K, Gaveriaux Ruff C, Hirth CG. The δ-opioid receptor: isolation of a cDNA by expression cloning and pharmacological characterization. Proc Natl Acad Sci USA. 1992;89:12048–12052. doi: 10.1073/pnas.89.24.12048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Knoflach F, Reinscheid RK, Civelli O, Kemp JA. Orphanin FQ inhibition of voltage-gated calcium channels in rat hippocampal CA3 neurones in vitro. J Physiol (Lond) 1996;494:55. doi: 10.1523/JNEUROSCI.16-21-06657.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lachowicz JE, Shen Y, Monsma FJ, Jr, Sibley DR. Molecular cloning of a novel G protein-coupled receptor related to the opiate receptor family. J Neurochem. 1995;64:34–40. doi: 10.1046/j.1471-4159.1995.64010034.x. [DOI] [PubMed] [Google Scholar]
- 18.Luebke JI, Dunlap K. Sensory neuron N-type calcium currents are inhibited by both voltage-dependent and -independent mechanisms. Pflügers Arch. 1994;428:499–507. doi: 10.1007/BF00374571. [DOI] [PubMed] [Google Scholar]
- 19.Luebke JI, Dunlap K, Turner TJ. Multiple calcium channel types control glutamatergic synaptic transmission in the hippocampus. Neuron. 1993;11:895–902. doi: 10.1016/0896-6273(93)90119-c. [DOI] [PubMed] [Google Scholar]
- 20.Macdonald RL, Werz MA. Dynorphin A decreases voltage-dependent calcium conductance of mouse dorsal root ganglion neurones. J Physiol (Lond) 1986;377:237–249. doi: 10.1113/jphysiol.1986.sp016184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Meng F, Xie GX, Thompson RC, Mansour A, Goldstein A, Watson SJ, Akil H. Cloning and pharmacological characterization of a rat κ opioid receptor. Proc Natl Acad Sci USA. 1993;90:9954–9958. doi: 10.1073/pnas.90.21.9954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Meunier JC, Mollereau C, Toll L, Suaudeau C, Moisand C, Alvinerie P, Butour JL, Guillemot JC, Ferrara P, Monsarrat B, Mazarguil H, Vassart G, Parmentier M, Costentin J. Isolation and structure of the endogenous agonist of opioid receptor-like ORL1 receptor. Nature. 1995;377:532–535. doi: 10.1038/377532a0. [DOI] [PubMed] [Google Scholar]
- 23.Mintz IM, Adams ME, Bean BP. P-type calcium channels in rat central and peripheral neurons. Neuron. 1992;9:85–95. doi: 10.1016/0896-6273(92)90223-z. [DOI] [PubMed] [Google Scholar]
- 24.Moises HC, Rusin KI, Macdonald RL. μ-Opioid receptor-mediated reduction of neuronal calcium current occurs via a G(o)-type GTP-binding protein. J Neurosci. 1994a;14:3842–3851. doi: 10.1523/JNEUROSCI.14-06-03842.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Moises HC, Rusin KI, Macdonald RL. μ- and κ-opioid receptors selectively reduce the same transient components of high-threshold calcium current in rat dorsal root ganglion sensory neurons. J Neurosci. 1994b;14:5903–5916. doi: 10.1523/JNEUROSCI.14-10-05903.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mollereau C, Parmentier M, Mailleux P, Butour JL, Moisand C, Chalon P, Caput D, Vassart G, Meunier JC. ORL1, a novel member of the opioid receptor family: cloning, functional expression and localization. FEBS Lett. 1994;341:33–38. doi: 10.1016/0014-5793(94)80235-1. [DOI] [PubMed] [Google Scholar]
- 27.Nomura K, Reuveny E, Narahashi T. Opioid inhibition and desensitization of calcium channel currents in rat dorsal root ganglion neurons. J Pharmacol Exp Ther. 1994;270:466–474. [PubMed] [Google Scholar]
- 28.Piros ET, Prather PL, Loh HH, Law PY, Evans CJ, Hales TG. Ca2+ channel and adenylyl cyclase modulation by cloned μ-opioid receptors in GH3 cells. Mol Pharmacol. 1995;47:1041–1049. [PubMed] [Google Scholar]
- 29.Raynor K, Kong H, Chen Y, Yasuda K, Yu L, Bell GI, Reisine T. Pharmacological characterization of the cloned κ-, δ-, and μ-opioid receptors. Mol Pharmacol. 1994;45:330–334. [PubMed] [Google Scholar]
- 30.Reinscheid RK, Nothacker H-P, Bourson A, Ardati A, Henningsen RA, Bunzow JR, Grandy DK, Langen H, Monsma FJ, Jr, Civelli O. Orphanin FQ: a neuropeptide that activates an opioid-like G protein-coupled receptor. Science. 1995;270:792–794. doi: 10.1126/science.270.5237.792. [DOI] [PubMed] [Google Scholar]
- 31.Rhim H, Miller RJ. Opioid receptors modulate diverse types of calcium channels in the nucleus tractus solitarius of the rat. J Neurosci. 1994;14:7608–7615. doi: 10.1523/JNEUROSCI.14-12-07608.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rusin KI, Moises HC. μ-Opioid receptor activation reduces multiple components of high-threshold calcium current in rat sensory neurons. J Neurosci. 1995;15:4315–4327. doi: 10.1523/JNEUROSCI.15-06-04315.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schroeder JE, Fischbach PS, Zheng D, McCleskey EW. Activation of μ opioid receptors inhibits transient high- and low-threshold Ca2+ currents, but spares a sustained current. Neuron. 1991;6:13–20. doi: 10.1016/0896-6273(91)90117-i. [DOI] [PubMed] [Google Scholar]
- 34.Surprenant A, Shen KZ, North RA, Tatsumi H. Inhibition of calcium currents by noradrenaline, somatostatin and opioids in guinea-pig submucosal neurones. J Physiol (Lond) 1990;431:585–608. doi: 10.1113/jphysiol.1990.sp018349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Swartz KJ, Bean BP. Inhibition of calcium channels in rat CA3 pyramidal neurons by a metabotropic glutamate receptor. J Neurosci. 1992;12:4358–4371. doi: 10.1523/JNEUROSCI.12-11-04358.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Takahashi T, Momiyama A. Different types of calcium channels mediate central synaptic transmission. Nature. 1993;366:156–158. doi: 10.1038/366156a0. [DOI] [PubMed] [Google Scholar]
- 37.Thompson RC, Mansour A, Akil H, Watson SJ. Cloning and pharmacological characterization of a rat μ opioid receptor. Neuron. 1993;11:903–913. doi: 10.1016/0896-6273(93)90120-g. [DOI] [PubMed] [Google Scholar]
- 38.Wang JB, Johnson PS, Imai Y, Persico AM, Ozenberger BA, Eppler CM, Uhl GR. cDNA cloning of an orphan opiate receptor gene family member and its splice variant. FEBS Lett. 1994;348:75–79. doi: 10.1016/0014-5793(94)00557-5. [DOI] [PubMed] [Google Scholar]
- 39.Wheeler DB, Randall A, Tsien RW. Roles of N-type and Q-type Ca2+ channels in supporting hippocampal synaptic transmission. Science. 1994;264:107–111. doi: 10.1126/science.7832825. [DOI] [PubMed] [Google Scholar]
- 40.Wick MJ, Minnerath SR, Lin X, Elde R, Law PY, Loh HH. Isolation of a novel cDNA encoding a putative membrane receptor with high homology to the cloned μ, δ, and κ opioid receptors. Brain Res Mol Brain Res. 1994;27:37–44. doi: 10.1016/0169-328x(94)90181-3. [DOI] [PubMed] [Google Scholar]