

Abstract
The mechanisms by which neurons die after cerebral ischemia and related conditions in vivo are unclear, but they are thought to involve voltage-dependent Na+ channels, glutamate receptors, and nitric oxide (NO) formation because selective inhibition of each provides neuroprotection. It is not known precisely what their roles are, nor whether they interact within a single cascade or in parallel pathways. These questions were investigated using anin vitro primary cell culture model in which striatal neurons undergo a gradual and delayed neurodegeneration after a brief (5 min) challenge with the glutamate receptor agonist NMDA. Unexpectedly, NO was generated continuously by the cultures for up to 16 hr after the NMDA exposure. Neuronal death followed the same general time course except that its start was delayed by ∼4 hr. Application of the NO synthase inhibitor nitroarginine after, but not during, the NMDA exposure inhibited NO formation and protected against delayed neuronal death. Blockade of NMDA receptors or of voltage-sensitive Na+ channels [with tetrodotoxin (TTX)] during the postexposure period also inhibited both NO formation and cell death. The NMDA exposure resulted in a selective accumulation of glutamate in the culture medium during the period preceding cell death. This glutamate release could be inhibited by NMDA antagonism or by TTX, but not by nitroarginine. These data suggest that Na+ channels, glutamate receptors, and NO operate interdependently and sequentially to cause neurodegeneration. At the core of the mechanism is a vicious cycle in which NMDA receptor stimulation causes activation of TTX-sensitive Na+ channels, leading to glutamate release and further NMDA receptor stimulation. The output of the cycle is an enduring production of NO from neuronal sources, and this is responsible for delayed neuronal death. The same neurons, however, could be induced to undergo more rapid NMDA receptor-dependent death that required neither TTX-sensitive Na+ channels nor NO.
Keywords: striatum, glutamate, excitotoxicity, NMDA receptors, sodium channels, nitric oxide
Central neurons are highly vulnerable to periods of ischemia and related insults that other cell types are able to withstand. The reasons for this are poorly understood but studies inin vivo models of cerebral ischemia have identified at least three important participants: glutamate receptors, nitric oxide (NO), and voltage-dependent Na+ channels.
Glutamate has been implicated in the neuronal death after both focal and transient global ischemia because, in experimental animals, glutamate receptor blockade has been found to be neuroprotective (Choi, 1990; Meldrum and Garthwaite, 1990). One consequence of the raised intracellular Ca2+ associated with glutamate receptor activation and other excitatory stimuli is the enzymatic production of NO from the amino acid l-arginine (Garthwaite and Boulton, 1995). In other tissues, endogenously produced NO has been identified as a cytotoxic factor that is important for immunological defense against tumor cells and invading pathogens but that, in excess, can injure normal cells (Moncada et al., 1991; Gross and Wolin, 1995).
The participation of NO in neurodegenerative phenomena remains controversial and confusing. Some laboratories using brain slice or primary tissue culture models of glutamate neurotoxicity have reported that NO is involved (Izumi et al., 1992; Dawson et al., 1993), whereas others, using seemingly similar methods, have convincingly shown that NO plays no obvious role (Demerlé-Pallardy et al., 1991; Hewett et al., 1993; Garthwaite and Garthwaite, 1994). NO synthase inhibitors tested in animal models of cerebral ischemia have also yielded conflicting results, possibly because of differing dosing regimes. Repeated administration of low doses of inhibitor that minimize compromising effects on cerebral blood flow through loss of endothelial NO synthase activity, however, appears to provide substantial protection (Nowicki et al., 1991; Iadecola et al., 1994), and the demonstration that mice whose neuronal NO synthase (nNOS) gene has been deleted are resistant to focal and transient global ischemia (Huang et al., 1994; Panahian et al., 1996) lends credence to the idea that the NO pathway is of pathological significance in vivo. Even when the participation of NO is indicated, it is not known precisely what its role is. NO, or peroxynitrite (formed by a chemical reaction between NO and superoxide anions), could participate directly in cell killing through various mechanisms (Beckman et al., 1990; Gross and Wolin, 1995). Alternatively, NO (or peroxynitrite) could cause the release of glutamate (Meffert et al., 1994; Montague et al., 1994) and/or inhibit glutamate uptake (Pogun et al., 1994) and thereby raise the extracellular glutamate concentration and promote damage through other (possibly NO-independent) mechanisms.
The third important contribution to the pathogenesis of neuronal loss in animal models of focal and global ischemia is made by voltage-sensitive Na+ channels because a variety of inhibitors of Na+ channel function confer at least as much neuroprotection as glutamate receptor blockers or appropriate doses of NOS inhibitors (Taylor and Meldrum, 1995). There is, as yet, no satisfactory explanation for their efficacy. Na+ channel inhibitors have been observed to reduce “pathological” glutamate release, such as occurs during ischemia (Graham et al., 1993; Smith et al., 1993), but whether this is a cause or an effect of their neuroprotective properties has not been investigated. It is unclear, therefore, whether the Na+ channel-dependent pathway is mechanistically related to the glutamate pathway (or the NO pathway) or whether it is part of an independent process.
In the present work, we have used primary cultures of rat striatum, a brain area known to be highly vulnerable to focal and global cerebral ischemia, in an attempt to understand the roles of, and inter-relationships among, glutamate, NO, and Na+channels in the neurodegenerative cascade.
MATERIALS AND METHODS
Primary cell cultures. Primary mixed neuronal–glial cell cultures were prepared from fetal rat brains [embryonic day 18 (E18)] as described previously (Strijbos and Rothwell, 1995) with minor modifications. Briefly, striata (strictly speaking, striata plus globus pallida) were dissected aseptically in ice-cold Ca2+/Mg2+-free HBSS (Life Technologies, Grand Island, NY) containing 10 mmHEPES, 100 U/ml penicillin, and 100 μg/ml streptomycin and were mechanically dissociated by trituration through fire-polished glass pipettes. The cell suspension was diluted in minimal essential medium (MEM; Life Technologies) containingl-alanine-l-glutamine (Glutamax, Life Technologies), 10% heat-inactivated fetal calf serum, 10% horse serum, 100 U/ml penicillin, and 100 μg/ml streptomycin, 1 mm sodium pyruvate, 25 mmglucose, seeded in Primaria tissue culture plates (Falcon) at a density of 7.5 × 105 trypan blue-excluding cells/ml and cultured in a humidified 5% CO2 incubator at 37°C. When non-neuronal cells reached confluency (after 3–6 d), their proliferation was halted by addition of 10 μm cytosine-d-arabinoside (Sigma, Paisley, UK) for 3 d. Thereafter, cultures were maintained in MEM lacking fetal calf serum. Cultures were allowed to mature for 2–3 weeks before being used for experiments. During this period, neurons developed extensive neuritic networks and formed functional synapses (data not shown).
Neurotoxicity. Neuronal cultures were washed twice with prewarmed (37°C) Mg2+-free Dulbecco’s PBS (DPBS) supplemented with 15 mm glucose (Life Technologies). Subsequently, they were exposed to DPBS (37°C), in the absence or presence of 100 μm NMDA and 100 μmd-serine, for various periods of time, typically 5 min. Thereafter, cells were washed twice with prewarmed DPBS and allowed to recover for various periods of time, usually 24 hr, in MEM lacking serum, and the extent of neurodegeneration was then assessed (see below). The role of various putative mediators of NMDA-induced neurotoxicity was investigated by addition of selective inhibitors either during the NMDA exposure period or during the recovery period after washout of NMDA, as indicated.
In separate experiments, cultures were exposed to lipopolysaccharide (LPS; S. typhosa 0901, Difco, Detroit, MI) at a concentration of 500 ng/ml for various periods of time. When the effects of inhibitors were tested, these were applied simultaneously with, and for the duration of, the LPS application. These experiments were performed under serum-free conditions. Preliminary experiments revealed that the cultures tolerated serum-free conditions for at least 4 d (data not shown).
Assessment of neurodegeneration. Neuronal death was quantified by measuring dehydrogenase activity retained in the cultured cells, at various time points after exposure to the neurotoxic stimulus (typically 24 hr) using the 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide (MTT) assay (Mosmann, 1983). The assay is based on the ability of living cells to convert dissolved MTT into insoluble formazan. Briefly, 100 μl of MTT solution (5 mg/ml) was added for 3 hr to cultures kept in a humidified 5% CO2 incubator at 37°C. Thereafter, the medium was removed and, after solubilizing the formazan (0.04 N HCl in absolute isopropanolol), the optical density was measured (540 nm). Data are expressed as percent viability relative to sham-treated sister cultures.
To check the reliability of the MTT assay as an index of neuronal death, the following control experiments were performed. Phase-contrast photomicrographs were taken of cultures at various time points after treatment with neurotoxin, and the extent of visible neuronal death was compared with MTT results from identically treated sister wells. It was observed that the reductions in culture dehydrogenase activity after neurotoxin treatment, obtained with the MTT assay, correlated well with the loss of neuronal cells observed using phase-contrast microscopy. However, because some dead neurons fragmented and detached from the substratum, phase-contrast microscopy of the attached neurons tended to underestimate the total extent of cell death. To measure the contribution made by neurons to the total measured dehydrogenase enzyme activity, cultures were treated with 300 μmNMDA for 24 hr. This procedure destroys all neurons without affecting the viability of glial cells (Weiss et al., 1993; Strijbos and Rothwell, 1995). After this treatment, 75–80% of dehydrogenase activity was lost, suggesting that the residual 20–25% was attributable to the activity of non-neuronal cells. Finally, the MTT results obtained after neurotoxin treatment correlated well with trypan blue (0.4%) exclusion assays performed on identically treated sister wells, although, as with phase-contrast microscopy, trypan blue staining will tend to underestimate total cell death. These tests confirm previous reports that the readout from the MTT assay correlates well with neuronal cell death (Dorman et al., 1993).
Nitrite plus nitrate measurement. To monitor NO production, the levels of nitrite plus nitrate (NOx) in the tissue culture medium were measured using chemiluminescence, as described previously (Palmer et al., 1987). Briefly, cultures were treated as described above, and samples of culture medium (10 μl) were taken at various intervals after treatment with neurotoxins. Samples, which were kept frozen until assayed, were subjected to nitrate reductase treatment and then injected into a reaction vessel containing sodium iodide (1.5%) in glacial acetic acid under reflux. Under these conditions, nitrite is converted back into NO, which is removed on a stream of nitrogen gas and mixed with ozone. The resulting chemiluminescence was measured with a photomultiplier and quantified using a nitrite standard curve.
Immunocytochemistry. To visualize NOS-containing neurons, cultures were fixed in 4% paraformaldehyde for 20 min, after which they were incubated with an antiserum raised against rat neuronal NOS (1:1000 dilution, kindly donated by Dr. H. H. H. W. Schmidt, Würzburg, Germany). NOS was then visualized using a standard peroxidase/antiperoxidase–diaminobenzidine technique. Cultures were counterstained with hematoxylin/eosin. To quantify the NOS-containing neurons, the numbers of labeled cell bodies were counted in four randomly selected fields in each of 12 cultures, and they were expressed as a percentage of the total population of neuron-like cells.
Amino acid release. To monitor the release of amino acids into the incubation medium, samples (100 μl) were analyzed using a high-performance Locarte amino acid analyzer withdl-homocysteic acid as an internal standard and a lithium buffer system. The amino acid analyzer had a limit of detection of 1–3 pmol/sample. Amino acids from the deproteinated samples were detected fluorometrically as the o-phthalaldehyde derivatives (Lee et al., 1979; Leach et al., 1986).
Statistical analysis. Data are expressed as mean ± SEM and were analyzed for significance using one-way ANOVA.
RESULTS
Kinetics of NMDA-triggered neurodegeneration and NO formation
A brief (5 min) exposure of striatal cultures to 100 μm NMDA caused virtually complete neuronal destruction when assessed 24 hr later, without affecting non-neuronal cells. In contrast to sham-treated sister cultures (Fig.1A), disruption of neuronal cell bodies and the neuritic network was clearly evident using phase-contrast microscopy (Fig. 1B), and this was accompanied by somatic uptake of trypan blue dye (Fig.1C,D) and an ∼75% reduction in cell culture dehydrogenase activity, as indicated by MTT assay; this corresponds to the loss of most of the neuronal population, the residue being the contribution made by non-neuronal cells (see Materials and Methods).
Fig. 1.
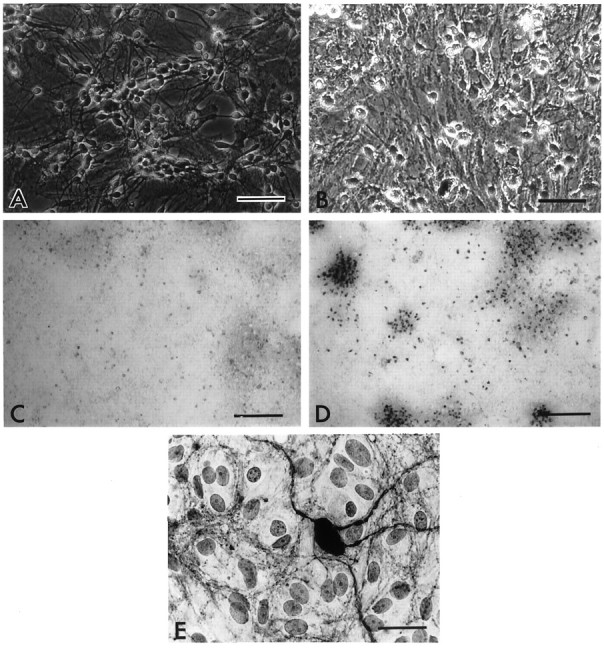
Morphology of NMDA-triggered neuronal death.A, B, Phase-contrast photomicrographs of primary cultures 24 hr after sham treatment (A) or 5 min exposure to 100 μm NMDA (B). Note the phase-bright neuronal somata and dense neuritic network under control conditions (A) and total disruption of these after NMDA treatment (B). Scale bar, 100 μm. C,D, Trypan blue uptake by primary cultures 24 hr after a sham treatment (C) or 5 min exposure to 100 μm NMDA (D). The dye is strongly taken up only by injured neurons, and the confluent glial layer retains its ability to exclude the dye after NMDA exposure. Scale bar, 500 μm. E, Bright-field photomicrograph of an NOS-containing neuron and neurites in a striatal primary culture, revealed by immunocytochemistry. Scale bar, 10 μm.
The 24 hr time point is used frequently as an endpoint for unambiguously determining the extent of cell death. Few attempts have been made to determine the intervening kinetics. The dehydrogenase assay indicated that neurodegeneration occurred in a slow, sigmoidal manner after 5 min exposure to NMDA (Fig.2A). Thus, over the initial 4 hr after washout, no significant neuronal death was observed (confirmed using the trypan blue exclusion test and phase-contrast microscopy; data not shown). Thereafter, increasing loss of viability was evident, culminating in maximal cell loss after 16 hr (again confirmed using trypan blue and phase-contrast microscopy). Approximately 9 hr were required for half-maximal neurodegeneration.
Fig. 2.

Kinetics and pharmacology of NMDA-triggered neurodegeneration and NOx formation.A, Kinetics of NMDA-triggered neurodegeneration. Cultures were exposed to NMDA (100 μm, 5 min) and allowed to recover for various periods of time before culture viability (filled circles) and NOxaccumulation (open circles) were measured.B, C, Pharmacology of NMDA-triggered neurodegeneration and NOx accumulation. Cultures were exposed to NMDA (100 μm, 5 min) in the presence or absence of nitroarginine (NO2Arg, 100 μm), and allowed to recover for 24 hr in control medium or medium containing AP5 (100 μm), dizocilpine (10 μm), or nitroarginine (100 μm) before culture viability (B) and NOx accumulation (C) were measured.D, E, Delayed neuronal rescue. Cultures were exposed to NMDA (100 μm, 5 min) and allowed to recover for various periods of time before addition of either nitroarginine (100 μm, D) or dizocilpine (10 μm, E). Culture viability was assessed after 24 hr. All values are mean ± SEM of two to three separate experiments (with at least 3 culture wells per treatment per experiment). *p < 0.01, **p < 0.001 from control groups.
In vivo, the rat striatum and cerebral cortex contain only a small population of NOS-containing neurons; their numbers do not appear to have been quantified accurately, but they have been stated to represent ∼1–2% of the total (Vincent and Johansson, 1983). Because one purpose of the experiments was to evaluate the role of NO in the neurodegenerative process, the content of NOS neurons in the striatal cultures was estimated by immunocytochemistry, using antibodies selective for the neuronal isoform of the enzyme. nNOS-positive somata were represented sparsely (336 ± 21 per 1.2 cm2 culture dish or 0.2 ± 0.3% of total neuron-like cells; n = 12), but a dense network of stained neurites was evident throughout the cultures (Fig.1E).
To assay NO production by the cultures, measurements were made of the accumulation of the stable products, nitrite and nitrate (NOx), in the medium. The levels generated during the NMDA exposure were (and would be expected to be) below the detection limit of the assay, but previous cGMP measurements have shown that NO formation occurs during this phase (Marin et al., 1992). Significant formation of NOx was detected within the first hour after exposure to NMDA. More surprisingly, NOx continued to accumulate in an approximately linear manner for several hours thereafter before slowing down (Fig.2A). The corresponding rate of NO production between 1 and 6 hr after the exposure (assuming 1 NOx per NO) was 33 pmol · mg protein−1 · min−1.
Pharmacology of NMDA-triggered neurodegeneration and NO production
Neuronal death produced by 5 min exposure to NMDA (100 μm) was prevented by the noncompetitive NMDA antagonist dizocilpine (10 μm) or the competitive antagonistd-2-amino-5-phosphonopentanoate (AP5; 100 μm), when added either just before the NMDA exposure (not shown) or immediately after NMDA washout (Fig.2B).
To test for the participation of NO, the cultures were treated with the NOS inhibitor nitroarginine (100 μm). If the inhibitor was added immediately after NMDA washout, a substantial degree of neuroprotection, quantitatively similar to that produced by NMDA antagonists, was observed (Fig. 2B). However, nitroarginine failed to be neuroprotective if it was applied solely during the NMDA exposure (Fig. 2B). Because inhibition of NOS by nitroarginine is expected to be sustained well beyond its washout from the bathing medium, because of slow dissociation from the enzyme (Klatt et al., 1994), these findings suggested that late NO formation, rather than that occurring during or shortly after the exposure, was associated with the neurodegeneration.
To investigate the time window over which NMDA receptor- and NO-dependent mechanisms operate, dizocilpine (10 μm) or nitroarginine (100 μm) was applied at various intervals after washout of NMDA. Significant protection was observed when addition of either antagonist was delayed for up to 4 hr after the NMDA exposure, but by 6 hr the effects of both were lost (Fig.2D,E).
Measurements of NOx accumulation (Fig.2C) revealed a connection between the protection afforded by NMDA antagonism and by NOS inhibition. Thus, at the concentrations giving neuroprotection, the NMDA antagonists AP5 and dizocilpine both inhibited the build-up of NOx in the medium to an extent identical to that produced by nitroarginine. This shows that continuing NMDA receptor activation was responsible for the NO formation and indicates that this, rather than any other concomitant effect of NMDA receptor activity, is a sufficient explanation for the neuronal death.
iNOS and neurodegeneration
Ischemic injury can lead to the expression of the inducible form of NOS (iNOS) (Iadecola et al., 1995), and the observed time course of NO generation, based on NOx measurement, and the kinetics of cell death are not dissimilar to those found in other tissues exposed to iNOS inducers such as LPS or cytokines (Gross and Wolin, 1995). The possibility arose, therefore, that the neurodegeneration triggered by brief NMDA treatment could be effected by iNOS expression which, conceivably (e.g., through activation of gene transcription), could depend on NMDA receptor activity (Gross and Wolin, 1995).
This possibility can be tested pharmacologically, because iNOS and nNOS are differentially sensitive to nitroarginine and the iNOS-selective compound aminoguanidine (Wolff and Lubeskie, 1995). For the positive control, the cultures were treated continuously with LPS to elicit expression of iNOS (Galea et al., 1992; Simmons and Murphy, 1992). This resulted in a biphasic accumulation of NOx in the medium, the first (slower) phase lasting for 48 hr (corresponding to ∼2.5 pmol · mg protein−1 · min−1) and a second (steeper) phase (12 pmol · mg protein−1 · min−1) persisting for at least another 48 hr (Fig.3A). Major neuronal loss accompanied the second phase such that, after an exposure to LPS of 96 hr, the total culture viability was reduced to ∼50% (Fig.3A). There was a close correlation between the loss of viability after different exposure periods and the accumulation of NOx in the medium (Fig. 3B), consistent with the cell death being attributable to NO.
Fig. 3.

iNOS and neurodegeneration. A, Kinetics of LPS-induced neurodegeneration. Cultures were exposed to LPS (500 ng/ml) for various periods of time before culture viability (filled circles) and NOxaccumulation (open circles) were assessed. B, Correlation between LPS-induced reductions in culture viability and NOx accumulation. The data are taken fromA (correlation coefficient = 0.992). C,D, Pharmacology of LPS- and NMDA-induced neurodegeneration and NOx accumulation. Cultures were exposed to either NMDA (100 μm, 5 min) or LPS (500 ng/ml, 96 hr), and the effects of various concentrations of aminoguanidine (AG) or nitroarginine (NO2Arg), applied for 24 hr after NMDA washout or during 96 hr of LPS exposure, on culture viability (C) and NOx accumulation (D) measured. Note the differential inhibition of NMDA and LPS effects by nitroarginine and aminoguanidine, respectively. All values are mean ± SEM of three separate experiments (with at least 3 culture wells per treatment per experiment).
In line with predictions, LPS-induced loss of viability and NOx accumulation (measured after 96 hr) were both potently inhibited by aminoguanidine, although the IC50 for NOx accumulation (1 μm) was about an order of magnitude less than for neuroprotection. In contrast, nitroarginine was only a weak inhibitor of LPS-induced NOx accumulation (IC50 ∼ 300 μm) and cell death (IC50 > 300 μm; Fig. 3C,3D). When tested against NMDA-triggered neurodegeneration, the absolute and relative potencies of the two antagonists were reversed (Fig.3C,D), indicating that whereas NO generation from iNOS is capable of causing neuronal death, that triggered by NMDA depends primarily on nNOS.
NMDA-triggered amino acid release
A likely explanation for persistent NMDA receptor activation and associated NO production after washout of NMDA itself is that an endogenous receptor agonist, such as glutamate, was being continuously released into the extracellular fluid. The concentrations of glutamate and other amino acids (aspartate, alanine, glutamine, glycine, taurine and hypotaurine) in the bathing medium were thus measured. With sham treatment (only NMDA was omitted), no amino acids were detectable 2 hr later (not shown). When the cultures were exposed for 5 min to NMDA, however, significant concentrations of glutamate and aspartate, but not of the other amino acids, were present after the same interval (2 hr; Fig. 4A). The glutamate concentration (∼1.5 μm) was ∼10-fold higher than that of aspartate. Thereafter, glutamate remained at high levels (peaking at >2.5 μm) for the rest of the 24 hr period. In sham-treated control cultures, by contrast, the concentration attained after 24 hr was only 130 ± 21 nm. The concentration of aspartate in NMDA-treated cultures climbed steadily over the same period but never exceeded 0.5 μm. The other amino acids reached detectable concentrations only 6–8 hr after the NMDA exposure.
Fig. 4.

NMDA-triggered amino acid release. A, Amino acid release. Cultures were exposed to NMDA (100 μm, 5 min) and allowed to recover for 24 hr, during which samples were taken at various intervals for measurement of amino acids. Note that release of glutamate and aspartate occurs much earlier than that of the other amino acids. B, Effects of AP5, TTX, and nitroarginine on glutamate release 2 hr after NMDA washout. Cultures were exposed to NMDA (100 μm, 5 min) and allowed to recover for 2 hr in culture medium containing AP5 (100 μm), TTX (1 μm), or nitroarginine (NO2Arg, 100 μm), after which culture medium was analyzed for glutamate (filled bars) and NOx accumulation (cross-hatched bars).C, Effect of TTX on NMDA-triggered neurodegeneration. Cultures were exposed to NMDA (100 μm, 5 min) and allowed to recover in TTX-containing (1 μm) culture medium for 24 hr, after which culture viability was assessed. All values are mean ± SEM of four separate experiments (with at least 3 culture wells per treatment per experiment). *p < 0.001 difference from control groups;+p < 0.05 difference from NMDA-treated group.
The concentrations of glutamate found in the medium in the aftermath of the initial NMDA insult (∼2 μm) are sufficient to activate NMDA receptors and elicit NO formation (Garthwaite, 1985; Garthwaite et al., 1988). Moreover, the rise in glutamate levels occurred in advance of cell death. These results implicate secondary release of glutamate from endogenous sources in the delayed neurotoxicity elicited by brief NMDA exposure. To investigate whether NO or NMDA receptors were responsible for the release of glutamate, we determined the effects of nitroarginine ord-AP5, added immediately after the NMDA exposure, on the measured glutamate release 2 hr later. In control experiments, NMDA caused the appearance of ∼2.5 μmglutamate. When NMDA-treated cultures were allowed to recover ind-AP5-containing medium, glutamate accumulation was significantly reduced to ∼0.5 μm (Fig.4B). Using the same protocol, however, nitroarginine (100 μm) did not significantly affect glutamate levels. Thus, glutamate release was dependent on NMDA receptors and not NO.
Role of Na+ channels
Most experiments were done with the classical Na+ channel inhibitor tetrodotoxin (TTX; 1 μm). When TTX was applied immediately after NMDA (5 min) washout, a significant degree of neuroprotection (∼60%) was observed (Fig. 4C). At the same time, TTX inhibited significantly the accumulation of NOx in the medium (Fig. 4B) and the release of glutamate (Fig.4B). To examine the possibility that other Na+ channel inhibitors shown to be neuroprotective in animal models of cerebral ischemia work in a similar manner, the compound 619C89 (100 μm) (Graham et al., 1993; Smith et al., 1993) was tested. It was found to inhibit neurodegeneration to an extent not significantly different from that produced by TTX. The values for culture viability in these experiments were: 21 ± 6% (NMDA treatment alone); 55 ± 5% (NMDA followed by 619C89); and 61 ± 8% (NMDA followed by TTX).
Neurodegeneration independent of NO- and TTX-sensitive Na+channels
In view of the many reports that glutamate neurotoxicity is independent of NO (Demerlé-Pallardy et al., 1991; Hewett et al., 1993; Garthwaite and Garthwaite, 1994) and unaffected by blockade of Na+ channels with TTX (Garthwaite and Garthwaite, 1986; Choi, 1987; Rothman et al., 1987), we sought to determine whether the same cultures in which the NO-dependent mechanism normally prevailed could undergo an alternative degenerative route. The ferocity of the initial NMDA exposure seemed a likely determinant and, therefore, we investigated the effect of different exposure periods to NMDA (100 μm) on the ability of nitroarginine, added after NMDA washout, to be protective (Fig.5A). Exposure of cultures to NMDA for 1 min followed by a 24 hr recovery period resulted in near complete neurodestruction as assessed by MTT assay and phase-contrast microscopy. As with 5 min exposure, this neurodegeneration could be substantially prevented by dizocilpine or nitroarginine when added immediately after NMDA washout. With longer exposures (up to 40 min), nitroarginine was progressively less effective such that, at the longest NMDA exposure tested, the compound had lost almost all of its neuroprotectivity (Fig. 5A). This was not because NO generation taking place during the exposure period was able to substitute for the usual delayed NO production, because nitroarginine still failed to render protection if it was present during the exposure (Fig. 5A); neither was it attributable to a failure of NO formation (e.g., because of an early death of the NOS neurons), because measurements made after 24 hr showed that comparable NOx accumulation occurred irrespective of the duration of the exposure (Fig. 5B); nor could it be attributed to a reduced potency or efficacy of nitroarginine (e.g., because of an increase in competing freel-arginine levels) because, when applied immediately after the varying NMDA exposure periods, the inhibitor always blocked NOx formation (Fig.5B).
Fig. 5.
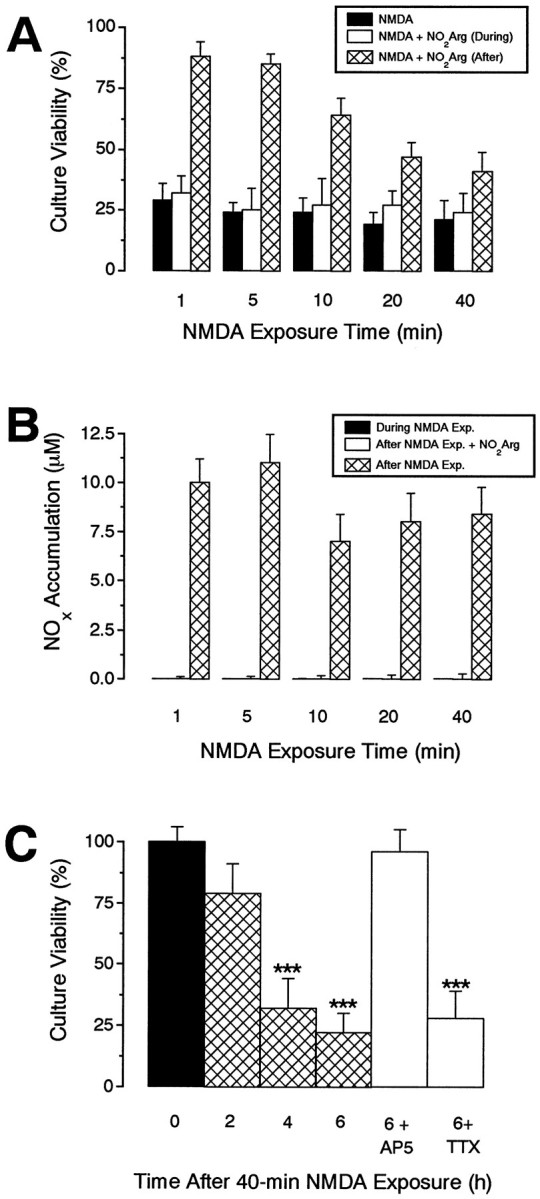
NMDA-triggered neurodegeneration can be independent of NOS and TTX-sensitive sodium channels. A,B, Effects of prolonged NMDA exposure on culture viability and NOx accumulation. Cultures were exposed to NMDA (100 μm) for various periods in the presence or absence of nitroarginine (NO2Arg, 100 μm) and were then allowed to recover for 24 hr in the presence or absence of nitroarginine (100 μm) before culture viability (A) and NOx accumulation (B) were assessed. Note that nitroarginine becomes progressively less effective in providing neuroprotection with increasing NMDA exposure times (A), whereas it efficiently inhibits NOx accumulation irrespectively (B).C, Kinetics and pharmacology of NO-independent NMDA-triggered neurodegeneration. Cultures were exposed to NMDA (100 μm, 40 min), and culture viability was assessed at 0, 2, 4, and 6 hr later. Other cultures were exposed to NMDA (100 μm, 40 min) and recovered for 6 hr in culture medium containing AP5 (100 μm) or TTX (1 μm). Note that the kinetics of neurodegeneration is faster than is observed after a brief (5 min) NMDA exposure (Fig. 2A) and that it is insensitive to TTX. All values are mean ± SEM of two to three separate experiments (with at least 3 culture wells per treatment per experiment). *p < 0.001 from control groups (time = 0 hr).
An explanation for NO-independent neurodegeneration is that the neurons died through other routes before the NO-dependent mechanism could become operational. To test this idea, we charted the progress of cell death after prolonged (40 min) NMDA exposure. Major loss of viability was observed by 4 hr, but not by 2 hr, after administration (Fig.5C). In addition, even though the NMDA antagonist AP5 (added immediately after NMDA washout) was able to prevent the neuronal loss under these conditions, Na+ channel blockade with TTX could not (Fig. 5C).
DISCUSSION
Despite proliferous publications over several years on glutamate-induced neurodegeneration in vitro, and on ischemic brain damage in vivo, little understanding of the underlying mechanisms has emerged. Our evidence obtained using a primary culture model shows that key phenomenological properties of the pathogenesis of neuronal death in vivo, namely, a dependence on Na+ channels, NMDA receptors, and nNOS, can be incorporated into a mechanistic sequence that explicitly defines the roles of these proteins and that implicitly identifies several specific questions that can now be subjected to direct experimental tests. Although the experiments were carried out using cultures of striatum, some of the features of the pathological mechanism we have elucidated have been noted previously in studies of other brain areas, including cerebral cortex and hippocampus (Dawson et al., 1993; Vigé et al., 1993; Dubinsky et al., 1995), indicating that our results may be more broadly applicable. The scheme suggested by our findings is shown in Figure 6, the main points of which are as follows.
Fig. 6.

Hypothetical scheme accounting for NMDA-triggered neurodegeneration. With brief (5 min) exposure (thick arrows), the neurons die through enduring NO formation, which is the product of a self-perpetuating cycle involving Na+-channel activity, which causes glutamate release and then further stimulation of NMDA receptors. The neuronal cell death resulting from this pathway manifests a prolonged (3–4 hr) delay, and it takes ∼16 hr to become maximal. NO generated from iNOS can also kill the neurons. A possible mechanism for NO-induced cell death is through peroxynitrite (ONOO−) formation after reaction with superoxide anions (·O−2). The sites of action of the inhibitors used experimentally (identified inparentheses) are indicated. When the initial stimulation of NMDA receptors is more prolonged (e.g., 40 min), the neurons die more quickly (within 4–6 hr) and in a mechanistically distinct manner (thin arrows) that requires glutamate release (now independent of TTX-sensitive Na+ channels) and NMDA receptor stimulation, but the associated effector mechanism is NO-independent (despite abundant NO generation). This mechanism is presumed to be Ca2+-dependent, and it may involve activation of catabolic enzymes, such as phospholipases, proteases, and nucleases. The broken lines signify alternative routes through which Na+ channels and glutamate, in principle, could mediate neuronal death, although these were not visible in the experimental paradigms examined.AMPAR, AMPA receptors; mGluR, metabotropic glutamate receptors.
Degeneration is delayed
The neurodegeneration taking place after transient exposure of the striatal cultures to NMDA has a delayed onset and is slow, requiring 16 hr for completion. The time course is very much more gradual than in several other reported experiments, including those on brain slices of cerebellum or hippocampus (Hajos et al., 1986; Garthwaite and Garthwaite, 1989) or on cortical cultures (Maulucci-Gedde and Choi, 1987). In these same models, no protection from glutamate or NMDA-induced neurodegeneration was afforded by either NOS inhibition or TTX (Garthwaite and Garthwaite, 1986, 1994; Choi, 1987; Hewett et al., 1993) and, therefore, it is possible that the neurons succumbed in a manner similar to when, in the present experiments, the cultures were given longer initial NMDA exposures. On the other hand, time courses of neuronal loss resembling those we observed after brief NMDA exposure have been noted previously (Dawson et al., 1993; Dubinsky et al., 1995) and, in the one study in which they were tested, NOS inhibitors were cytoprotective (Dawson et al., 1993). In apparent conflict with our findings, however, it was found that NOS inhibitors added solely for the duration of the initial NMDA challenge were effective. Possibly, the more limited washing procedure that seems to have been used in that study left sufficient residual compound to sustain NOS inhibition during the postexposure period.
Degeneration is caused by secondary glutamate release
The initial exposure to NMDA (lasting 1–40 min) is not destructive in itself; it merely acts as a trigger for a sustained, self-perpetuating cycle that is fueled by the release of glutamate from endogenous sources. The concentration of glutamate that appears in the bulk medium after the NMDA exposure (∼2 μm) is sufficient to activate NMDA receptors (Patneau and Mayer, 1990) and, even in the presence of extracellular Mg2+ (which imposes a voltage-dependent block of NMDA receptor channels), raise cytosolic Ca2+ levels (Burgard and Hablitz, 1995), and stimulate NO formation (Garthwaite, 1985; Garthwaite et al., 1988). It has also been shown to be a toxic concentration to cultured neurons when the numbers of underlying astrocytes, which remove extracellular glutamate by cellular uptake, are kept low (Rosenberg and Aizenman, 1989). In addition, a reduction by TTX in the measured concentration to ∼0.5 μm (which is too low to stimulate appreciable NO formation through NMDA receptors) was associated with a marked inhibition of NOxaccumulation. Thus, the concentration of glutamate in the medium is likely to be a reasonable reflection of the concentration directly bathing the neurons. The bulk concentration of released aspartate was lower (112 nm at 2 hr after NMDA exposure). In view of this and the 10-fold lower potency of aspartate at NMDA receptors compared with that of glutamate (Garthwaite, 1985; Patneau and Mayer, 1990), it is unlikely that aspartate contributed significant biological effects. The other amino acids measured only reached detectable levels in the medium after 6–8 hr, when cell death was already taking place, suggesting that, unlike with glutamate, they appeared as a result of nonspecific cellular leakage.
Roles of the NMDA receptor
The pivotal mechanism by which the released glutamate brings about cell death is through the NMDA receptor, which performs three important functions. First, it is responsible for further glutamate release, because NMDA receptor blockade reduced the concentration of glutamate in the medium in the period preceding the usual onset of neuronal cell death by 75%. The relevance of this finding is likely to extend to the intact brain because, in rat striatum in vivo, NMDA receptor activation has been shown to evoke delayed glutamate release (Dijk et al., 1995) and ischemia-induced glutamate release is inhibited by NMDA antagonists (Ghribi et al., 1994). Second, NMDA receptor activity represents the dominant mechanism by which NO is generated in the pathological setting, even though several alternative pathways, in principle, could be operative, including non-NMDA receptor activation and nonspecific cellular depolarization (Garthwaite and Garthwaite, 1987; Southam et al., 1991). Third, when a longer initial NMDA challenge is imposed, the NMDA receptor is able to mediate a more fulminant, NO-independent type of neurodegeneration. The reasons why the neurons follow this different route will have to await experimental tests, but a possibility is that a prolonged initial exposure to NMDA, at a concentration (100 μm) giving near-maximal stimulation of NMDA receptors, causes a greater net intracellular accumulation of Ca2+ than would be achieved during the same period of time by a 5 min exposure to NMDA followed by glutamate at concentrations (low micromolar) that are submaximal for NMDA receptors. Different effector mechanisms, such as enzymes activated by higher Ca2+ concentrations than the calmodulin-dependent NOS (Choi, 1990; Meldrum and Garthwaite, 1990), may then be engaged and lead to more rapid cell death.
Finally, of course, it is brief NMDA receptor activation that acts as the trigger for the sequence of events that culminates in neuronal degeneration. The critical mechanisms taking place during NMDA exposure that set the process in motion remain to be studied. One possibility is that the potential for generating the fatal NMDA receptor–glutamate release cycle is preexisting, but covert, and it simply requires sufficient NMDA receptor stimulation to make it operational. Alternatively, the initial exposure might cause long-term changes in one or more key components (e.g., NMDA receptors, Na+ channels, or NOS) that are required for the cycle or its output (NO) to become pathologically significant.
Na+ channels are required for glutamate release
Voltage-dependent Na+ channels represent a major component of the cycle linking NMDA receptor activity to glutamate release. This finding provides a tangible explanation for the neuroprotective effects of Na+ channel inhibitors in cerebral ischemia in vivo and is consistent with results showing that these agents diminish the associated release of glutamate (Graham et al., 1993; Smith et al., 1993; Taylor and Meldrum, 1995). NMDA-induced glutamate release in striatum in vivo has also been found to depend critically on voltage-sensitive Na+ channels (Dijk et al., 1995). The precise link between Na+ channels and extracellular glutamate accumulation, however, remains to be investigated. Several possibilities exist, including release by Na+-dependent action potentials and reversed glutamate uptake after intracellular Na+ loading (Szatkowski and Attwell, 1994), perhaps as a result of prolonged influx of the ion through noninactivating Na+ channels. The source of the glutamate, however, remains to be determined, and a contribution from non-neuronal cells cannot be excluded.
With the more severe initial NMDA insult giving rise to more rapid degeneration, secondary glutamate release is probably still required because NMDA receptor blockade after the exposure was still protective. Moreover, judging by the rate of NO generation, glutamate release was still taking place but, given the lack of neuroprotection by TTX in this paradigm, other mechanisms (or TTX-resistant Na+ channels) are presumably responsible.
NO as the effector
So far, with respect to neurodegeneration the most important output of the NMDA receptor/Na+ channel/glutamate release cycle is a sustained neuronal formation of NO. This is indicated by the fact that nNOS inhibition prevented most of the cell death without significantly affecting glutamate release. Consistent with NO being the downstream effector, iNOS expression led to neuronal death, albeit more slowly (perhaps connected with the lower rate of NO formation compared with that occurring post-NMDA) and exogenously added NO-donating compounds can replicate NMDA-triggered neuronal death (Dawson et al., 1993) (our unpublished observations). Understanding how NO causes cell death will require further investigation, but peroxynitrite formation (Beckman et al., 1990; Gross and Wolin, 1995) is an appealing possibility.
Window for neuroprotection
The mechanism permits an appreciable window of opportunity for cytoprotection, amounting to ∼4 hr. This is similar to the window observed with NMDA antagonists in vivo after focal ischemia (Bielenberg and Beck, 1991; Hatfield et al., 1992) or local injection of NMDA receptor agonists (Bakker and Foster, 1991), but it is less than would be predicted from the time course of the neurodegeneration itself, suggesting that beyond a certain point the neurons are destined to die through processes that no longer require NO. The identification of these processes will be important because they could allow intervention later in the neurodegenerative cascade than can be achieved by existing putative stroke therapies.
Footnotes
This study was supported by the Medical Research Council (UK).
Correspondence should be addressed to Professor J. Garthwaite, The Cruciform Project, University College London, 140 Tottenham Court Road, London W1P 9LN, UK.
Present addresses are as follows. Dr. Strijbos: The Cruciform Project, The Rayne Institute, University College London, 5 University Street, London WC1 6JQ, UK. Dr. Leach:Chemical and Life Sciences, University of Greenwich, Wellington Street, London SE18 6PF, UK. Prof. Garthwaite: The Cruciform Project, University College London, 140 Tottenham Court Road, London W1P 9LN, UK.
REFERENCES
- 1.Bakker MHM, Foster AC. An investigation of the mechanisms of delayed neurodegeneration caused by direct injection of quinolinate into the rat striatum in vivo . Neuroscience. 1991;42:387–395. doi: 10.1016/0306-4522(91)90383-y. [DOI] [PubMed] [Google Scholar]
- 2.Beckman JS, Beckman TW, Chen J, Marshall PA, Freeman BA. Apparent hydroxyl radical production by peroxynitrite: implications for endothelial injury from nitric oxide and superoxide. Proc Natl Acad Sci USA. 1990;87:1620–1624. doi: 10.1073/pnas.87.4.1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bielenberg GW, Beck T. The effects of dizocilpine (MK-801), phencyclidine and nomodipine on infarct size 48 hr after middle artery occlusion in the rat. Brain Res. 1991;552:338–342. doi: 10.1016/0006-8993(91)90101-z. [DOI] [PubMed] [Google Scholar]
- 4.Burgard EC, Hablitz JJ. N -methyl-d-aspartate receptor-mediated calcium accumulation in neocortical neurons. Neuroscience. 1995;69:351–362. doi: 10.1016/0306-4522(95)00273-l. [DOI] [PubMed] [Google Scholar]
- 5.Choi DW. Ionic dependence of glutamate neurotoxicity. J Neurosci. 1987;7:369–379. doi: 10.1523/JNEUROSCI.07-02-00369.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Choi DW. Cerebral hypoxia: some new approaches and unanswered questions. J Neurosci. 1990;10:2493–2501. doi: 10.1523/JNEUROSCI.10-08-02493.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dawson VL, Dawson TM, Uhl GR, Snyder SH. Mechanisms of nitric oxide-mediated neurotoxicity in primary brain cultures. J Neurosci. 1993;13:2651–2661. doi: 10.1523/JNEUROSCI.13-06-02651.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Demerlé-Pallardy C, Lonchampt M-O, Chabrier P-E, Braquet P. Absence of implication ofl-arginine/nitric oxide pathway on neuronal cell injury induced by l-glutamate or hypoxia. Biochem Biophys Res Commun. 1991;181:456–464. doi: 10.1016/s0006-291x(05)81441-x. [DOI] [PubMed] [Google Scholar]
- 9.Dijk SN, Francis PT, Stratmann GC, Bowen DM. NMDA-induced glutamate and aspartate release from rat cortical pyramidal neurones: evidence for modulation by a 5-HT1A antagonist. Br J Pharmacol. 1995;115:1169–1174. doi: 10.1111/j.1476-5381.1995.tb15020.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dorman DC, Bolon B, Morgan KT. The toxic effects of formate in dissociated primary neural cell cultures. Toxicol Appl Pharmacol. 1993;122:265–272. doi: 10.1006/taap.1993.1195. [DOI] [PubMed] [Google Scholar]
- 11.Dubinsky JM, Kristal BS, Elizondo-Fournier M. On the probabilistic nature of excitotoxic neuronal death in hippocampal neurons. Neuropharmacology. 1995;34:701–711. doi: 10.1016/0028-3908(95)00041-4. [DOI] [PubMed] [Google Scholar]
- 12.Galea E, Feinstein DL, Reis DJ. Induction of calcium-independent nitric oxide synthase activity in primary rat glial cultures. Proc Natl Acad Sci USA. 1992;89:10945–10949. doi: 10.1073/pnas.89.22.10945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Garthwaite G, Garthwaite J. In vitro neurotoxicity of excitatory amino acid analogues during cerebellar development. Neuroscience. 1986;17:755–767. doi: 10.1016/0306-4522(86)90043-6. [DOI] [PubMed] [Google Scholar]
- 14.Garthwaite G, Garthwaite J. Neurotoxicity of excitatory amino acid receptor agonists in young rat hippocampal slices. J Neurosci Methods. 1989;29:33–42. doi: 10.1016/0165-0270(89)90106-4. [DOI] [PubMed] [Google Scholar]
- 15.Garthwaite G, Garthwaite J. Nitric oxide does not mediate acute glutamate neurotoxicity, not is it neuroprotective, in rat brain slices. Neuropharmacology. 1994;33:1431–1438. doi: 10.1016/0028-3908(94)90046-9. [DOI] [PubMed] [Google Scholar]
- 16.Garthwaite J. Cellular uptake disguises action ofl-glutamate on N -methyl-d-aspartate receptors. Br J Pharmacol. 1985;85:297–307. doi: 10.1111/j.1476-5381.1985.tb08860.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Garthwaite J, Boulton CL. Nitric oxide signaling in the central nervous system. Annu Rev Physiol. 1995;57:683–706. doi: 10.1146/annurev.ph.57.030195.003343. [DOI] [PubMed] [Google Scholar]
- 18.Garthwaite J, Garthwaite G. Cellular origins of cyclic GMP responses to excitatory amino acid receptor agonists in rat cerebellum in vitro . J Neurochem. 1987;48:29–39. doi: 10.1111/j.1471-4159.1987.tb13123.x. [DOI] [PubMed] [Google Scholar]
- 19.Garthwaite J, Charles SL, Chess-Williams R. Endothelium-derived relaxing factor release on activation of NMDA receptors suggests role as intercellular messenger in the brain. Nature. 1988;336:385–388. doi: 10.1038/336385a0. [DOI] [PubMed] [Google Scholar]
- 20.Ghribi O, Callebert J, Plotkine M, Boulu RG. Competitive NMDA receptor blockers reduce striatal glutamate accumulation in ischaemia. NeuroReport. 1994;5:1253–1255. doi: 10.1097/00001756-199406020-00024. [DOI] [PubMed] [Google Scholar]
- 21.Graham SH, Chen J, Sharp FR, Simon RP. Limiting ischemic injury by inhibition of excitatory amino acid release. J Cereb Blood Flow Metab. 1993;13:88–97. doi: 10.1038/jcbfm.1993.11. [DOI] [PubMed] [Google Scholar]
- 22.Gross SS, Wolin MS. Nitric oxide: pathophysiological mechanisms. Annu Rev Physiol. 1995;57:737–769. doi: 10.1146/annurev.ph.57.030195.003513. [DOI] [PubMed] [Google Scholar]
- 23.Hajos F, Garthwaite G, Garthwaite J. Reversible and irreversible neuronal damage caused by excitatory amino acid analogues in rat cerebellar slices. Neuroscience. 1986;18:417–436. doi: 10.1016/0306-4522(86)90163-6. [DOI] [PubMed] [Google Scholar]
- 24.Hatfield RH, Gill R, Brazell C. The dose-response relationship and therapeutic window for dizocilpine (MK-801) in a rat focal ischaemia model. Eur J Pharmacol. 1992;216:1–7. doi: 10.1016/0014-2999(92)90201-e. [DOI] [PubMed] [Google Scholar]
- 25.Hewett SJ, Corbett JA, McDaniel ML, Choi DW. Inhibition of nitric oxide formation does not protect murine cortical cell cultures from N -methyl-d-aspartate neurotoxicity. Brain Res. 1993;625:337–341. doi: 10.1016/0006-8993(93)91078-7. [DOI] [PubMed] [Google Scholar]
- 26.Huang Z, Huang PL, Panahian N, Dalkara T, Fishman MC, Moskowitz MA. Effects of cerebral ischemia in mice deficient in neuronal nitric oxide synthase. Science. 1994;265:1883–1885. doi: 10.1126/science.7522345. [DOI] [PubMed] [Google Scholar]
- 27.Iadecola C, Pelligrino DA, Moskowitz MA, Lassen NA. Nitric oxide synthase inhibition and cerebrovascular regulation. J Cereb Blood Flow Metab. 1994;14:175–192. doi: 10.1038/jcbfm.1994.25. [DOI] [PubMed] [Google Scholar]
- 28.Iadecola C, Xu X, Zhang F, El-Fakahany EE, Ross ME. Marked induction of calcium-independent nitric oxide synthase activity after focal ischemia. J Cereb Blood Flow Metab. 1995;15:52–59. doi: 10.1038/jcbfm.1995.6. [DOI] [PubMed] [Google Scholar]
- 29.Izumi Y, Benz AM, Clifford DB, Zorumski CF. Nitric oxide inhibitors attenuate N -methyl-d-aspartate excitotoxicity in rat hippocampal slices. Neurosci Lett. 1992;135:227–230. doi: 10.1016/0304-3940(92)90442-a. [DOI] [PubMed] [Google Scholar]
- 30.Klatt P, Schmidt K, Brunner F, Mayer B. Inhibitors of brain nitric oxide synthase. J Biol Chem. 1994;269:1674–1680. [PubMed] [Google Scholar]
- 31.Leach MJ, Marden CM, Miller AA. Pharmacological studies on lamotrigine, a novel potential antiepileptic drug. II. Neurochemical studies on the mechanism of action. Epilepsia. 1986;27:490–497. doi: 10.1111/j.1528-1157.1986.tb03573.x. [DOI] [PubMed] [Google Scholar]
- 32.Lee HM, Forde MD, Lee MC, Bucher DJ. Fluorometric microbore amino acid analyser: the construction of an inexpensive, highly sensitive instrument using o -phthalaldehyde as a detection reagent. Anal Biochem. 1979;96:298–307. doi: 10.1016/0003-2697(79)90585-2. [DOI] [PubMed] [Google Scholar]
- 33.Marin P, Lafon-Cazal M, Bockaert J. A nitric oxide synthase activity selectively stimulated by NMDA receptors depends on protein kinase C activation in mouse striatal neurons. Eur J Neurosci. 1992;4:425–432. doi: 10.1111/j.1460-9568.1992.tb00892.x. [DOI] [PubMed] [Google Scholar]
- 34.Maulucci-Gedde M, Choi DW. Cortical neurons exposed to glutamate rapidly leak preloaded 51chromium. Exp Neurol. 1987;96:420–429. doi: 10.1016/0014-4886(87)90059-8. [DOI] [PubMed] [Google Scholar]
- 35.Meffert MK, Premack BA, Schulman H. Nitric oxide stimulates Ca2+-independent synaptic vesicle release. Neuron. 1994;12:1235–1244. doi: 10.1016/0896-6273(94)90440-5. [DOI] [PubMed] [Google Scholar]
- 36.Meldrum BS, Garthwaite J. Excitatory amino acid neurotoxicity and neurodegenerative disease. Trends Pharmacol Sci. 1990;11:379–387. doi: 10.1016/0165-6147(90)90184-a. [DOI] [PubMed] [Google Scholar]
- 37.Moncada S, Palmer RMJ, Higgs EA. Nitric oxide: physiology, pathophysiology, and pharmacology. Pharmacol Rev. 1991;43:109–142. [PubMed] [Google Scholar]
- 38.Montague PR, Gancayco CD, Winn MJ, Marchase RB, Friedlander MJ. Role of NO production in NMDA receptor-mediated neurotransmitter release in cerebral cortex. Science. 1994;263:973–977. doi: 10.1126/science.7508638. [DOI] [PubMed] [Google Scholar]
- 39.Mossman T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J Immunol Methods. 1983;65:55–63. doi: 10.1016/0022-1759(83)90303-4. [DOI] [PubMed] [Google Scholar]
- 40.Nowicki JP, Duval D, Poignet H, Scatton B. Nitric oxide mediates neuronal death after focal cerebral ischemia in the mouse. Eur J Pharmacol. 1991;204:339–340. doi: 10.1016/0014-2999(91)90862-k. [DOI] [PubMed] [Google Scholar]
- 41.Palmer RMJ, Ferrige AG, Moncada S. Nitric oxide release accounts for the biological activity of endothelium-derived relaxing factor. Nature. 1987;327:524–526. doi: 10.1038/327524a0. [DOI] [PubMed] [Google Scholar]
- 42.Panahian N, Yoshida T, Huang PL, Hedley-Whyte ET, Dalkara T, Fishman MC, Moskowitz MA. Attenuated hippocampal damage after global ischemia in mice mutant in neuronal nitric oxide synthase. Neuroscience. 1996;72:343–354. doi: 10.1016/0306-4522(95)00563-3. [DOI] [PubMed] [Google Scholar]
- 43.Patneau DK, Mayer ML. Structure-activity relationships for amino acid transmitter candidates acting at N -methyl-d-aspartate and quisqualate receptors. J Neurosci. 1990;10:2385–2399. doi: 10.1523/JNEUROSCI.10-07-02385.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pogun S, Dawson V, Kuhar MJ. Nitric oxide inhibits3H-glutamate transport in synaptosomes. Synapse. 1994;18:21–26. doi: 10.1002/syn.890180104. [DOI] [PubMed] [Google Scholar]
- 45.Rosenberg PA, Aizenman E. Hundred-fold increase in neuronal vulnerability to glutamate toxicity in astrocyte-poor cultures of rat cerebral cortex. Neurosci Lett. 1989;103:162–168. doi: 10.1016/0304-3940(89)90569-7. [DOI] [PubMed] [Google Scholar]
- 46.Rothman SM, Thurston JH, Hauhart RE. Delayed neurotoxicity of excitatory amino acids in vitro . Neuroscience. 1987;22:471–480. doi: 10.1016/0306-4522(87)90347-2. [DOI] [PubMed] [Google Scholar]
- 47.Simmons ML, Murphy S. Induction of nitric oxide synthase in glial cells. J Neurochem. 1992;59:897–905. doi: 10.1111/j.1471-4159.1992.tb08328.x. [DOI] [PubMed] [Google Scholar]
- 48.Smith SE, Lekieffre D, Sowinski P, Meldrum BS. Cerebroprotective effect of BW619C89 after focal or global cerebral ischaemia in the rat. NeuroReport. 1993;4:1339–1342. doi: 10.1097/00001756-199309150-00013. [DOI] [PubMed] [Google Scholar]
- 49.Southam E, East SJ, Garthwaite J. Excitatory amino acid receptors coupled to the nitric oxide:cyclic GMP pathway in rat cerebellum during development. J Neurochem. 1991;56:2072–2081. doi: 10.1111/j.1471-4159.1991.tb03468.x. [DOI] [PubMed] [Google Scholar]
- 50.Strijbos PJLM, Rothwell NJ. Interleukin-1β attenuates excitatory amino acid-induced neurodegeneration in vitro : involvement of nerve growth factor. J Neurosci. 1995;15:3468–3474. doi: 10.1523/JNEUROSCI.15-05-03468.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Szatkowski M, Attwell D. Triggering and execution of neuronal death in brain ischaemia: two phases of glutamate release by different mechanisms. Trends Neurosci. 1994;17:359–365. doi: 10.1016/0166-2236(94)90040-x. [DOI] [PubMed] [Google Scholar]
- 52.Taylor CP, Meldrum BS. Na+channels as targets for neuroprotective drugs. Trends Pharmacol Sci. 1995;16:309–316. doi: 10.1016/s0165-6147(00)89060-4. [DOI] [PubMed] [Google Scholar]
- 53.Vigé X, Carreau A, Scatton B, Nowicki JP. Antagonism by NG- nitro-l-arginine of l-glutamate-induced neurotoxicity in cultured neonatal rat cortical neurons: prolonged application enhances neuroprotective efficacy. Neuroscience. 1993;55:893–901. doi: 10.1016/0306-4522(93)90306-z. [DOI] [PubMed] [Google Scholar]
- 54.Vincent SR, Johansson O. Striatal neurons containing both somatostatin- and avian pancreatic polypeptide (APP)-like immunoreactivities and NADPH-diaphorase activity: a light and electron microscopic study. J Comp Neurol. 1983;217:264–270. doi: 10.1002/cne.902170304. [DOI] [PubMed] [Google Scholar]
- 55.Weiss JH, Hartley DM, Koh J, Choi DW. AMPA receptor activation potentiates zinc neurotoxicity. Neuron. 1993;10:43–49. doi: 10.1016/0896-6273(93)90240-r. [DOI] [PubMed] [Google Scholar]
- 56.Wolff DJ, Lubeskie A. Aminoguanidine is an isoform-selective, mechanism-based inactivator of nitric oxide synthase. Arch Biochem Biophys. 1995;316:290–301. doi: 10.1006/abbi.1995.1040. [DOI] [PubMed] [Google Scholar]