

Abstract
Endopeptidase 3.4.24.16 belongs to the zinc-containing metalloprotease family and likely participates in the physiological inactivation of neurotensin.The peptidase displays distinct features in pure primary cultured neurons and astrocytes. Neuronal maturation leads to a decrease in the proportion of endopeptidase 3.4.24.16-bearing neurons and to a concomitant increase in endopeptidase 3.4.24.16 activity and mRNA content. By contrast, there is no change with time in endopeptidase 3.4.24.16 activity or content in astrocytes. Primary cultured neurons exhibit both soluble and membrane-associated endopeptidase 3.4.24.16 activity. The latter behaves as an ectopeptidase on intact plated neurons and resists treatments with 0.2% digitonin and Na2CO3. Further evidence for an association of the enzyme with plasma membranes was provided by cryoprotection experiments and electron microscopic analysis. The membrane-associated form of endopeptidase 3.4.24.16 increased during neuronal differentiation and appears to be mainly responsible for the overall augmentation of endopeptidase 3.4.24.16 activity observed during neuronal maturation. Unlike neurons, astrocytes only contain soluble endopeptidase 3.4.24.16. Astrocytes secrete the enzyme through monensin, brefeldin A, and forskolin-independent mechanisms. This indicates that endopeptidase 3.4.24.16 is not released by classical regulated or constitutive secreting processes. However, secretion is blocked at 4°C and by 8 bromo cAMP and is enhanced at 42°C, two properties reminiscent of that of other secreted proteins lacking a classical signal peptide. By contrast, neurons appear unable to secrete endopeptidase 3.4.24.16.
Keywords: neurons, astrocytes, endopeptidase 3.4.24.16, degradation, secretion, neuropeptides
Endopeptidase 3.4.24.16 is a 75 kDa monomer peptidase that was previously purified from murine tissues (Checler et al., 1986b; Barelli et al., 1988, 1993) and from the brain of various other species (Yoshikawa et al., 1988; Millican et al., 1991; Dahms and Mentlein, 1992; Vincent et al., 1996). Recently, the rat brain endopeptidase 3.4.24.16 was cloned (Dauch et al., 1995) and definitely ascribed to the zinc-containing metalloprotease family according to the consensus sequence of its active site. This enzyme was shown to ubiquitously participate in the catabolism of the neuropeptide neurotensin in vitro in various tissues and cell cultures from central and peripheral origin, giving rise to the biologically inert catabolites neurotensin 1–10 and 11–13 (Checler et al., 1988). Furthermore, we demonstrated that Pro-Ile, a specific dipeptide inhibitor of endopeptidase 3.4.24.16 (Dauch et al., 1991b), drastically protected neurotensin from degradation in a vascularly perfused isolated dog ileum in vivo (Barelli et al., 1994). Finally, we recently established by means of phosphorus-containing peptide inhibitors targeting endopeptidase 3.4.24.16 (Barelli et al., 1992) that these phosphonamide agents enhanced the naloxone-insensitive neurotensin-induced analgesia after intracerebroventricular administration in mice (Vincent et al., 1995). Altogether, these data strongly suggest that endopeptidase 3.4.24.16 is involved in the physiological regulation of neurotensin concentration in both the CNS and the gastrointestinal tract.
We previously established that pure cultured neurons from mouse cerebral hemisphere avidly degrade neurotensin (Checler et al., 1986a;Vincent et al., 1994) and that a restricted population of these cells express neurotensin receptors. Interestingly, 90% of these neurotensin receptor-bearing cells were also found to be immunopositive for endopeptidase 3.4.24.16 (Chabry et al., 1990). This strong correlation between the neurotensin binding and inactivating entities further supports a role of the enzyme in neurotensin inactivation and indicates that pure cultured neurons constitute a relevant model to study endopeptidase 3.4.24.16 cell biology. Unlike neurons, cultured astrocytes exhibit little, if any, high-affinity neurotensin binding, although these cells also exhibit an endopeptidase 3.4.24.16 activity that contributes to the rapid breakdown of neurotensin (Mentlein and Dahms, 1994; Vincent et al., 1994). Pure cultured neurons and astrocytes offer the possibility to examine the behavior of endopeptidase 3.4.24.16 during cell differentiation, subcellular distribution, and secretion processes. We demonstrate here that neuronal and astrocytic endopeptidase 3.4.24.16 exhibit distinct biological properties and likely play complementary roles in cerebral neurotensin catabolism.
MATERIALS AND METHODS
Materials.7-Methoxycoumarin-3-carboxylyl-prolyl-leucyl-glycyl-prolyl-d-lysine-dinitrophenyl (QFS) was from Novabiochem (Meudon, France). Prolyl-isoleucine, cytosine arabinofuranoside, polylysine, digitonin, and 8-bromo-cAMP were purchased from Sigma (St. Louis, MO). Neurotensin was obtained from Neosystem (Strasbourg, France). HAM F12 medium was from Life Technologies. Fetal calf serum was from Boehringer Mannheim. Cpp-Ala-Ala-Tyr-pAb was kindly provided by Drs S. Wilk and M. Orlowski (Mount Sinai School of Medicine, New York, NY). Phosphodiepryl 03 (N-(phenylethylphosphonyl)glycyl-prolyl-hexanoic acid) was synthesized and generously given by Dr V. Dive (CEN Saclay, Gif/Yvette).
Cell cultures. Primary cultures of neurons and astrocytes were prepared from the cerebral hemispheres of 14-d-old mouse embryos as described previously (Chabry et al., 1990; Vincent et al., 1994). Briefly, cells were mechanically dissociated with a pipet in a HAM F12 medium supplemented with 10% fetal calf serum and 0.6% glucose. Dissociated cells were then plated at a density of 3.106 cells in 35 mm plastic tissue culture dishes precoated with polylysine (10 μg/ml) and grown in a humidified atmosphere of 5% CO2/95% air. For neuron cultures, at the beginning of the third day and every 2 d thereafter, cytosine arabinofuranoside (5 μm) was added in the medium during 24 hr to prevent glial cell proliferation. We previously established that these neuronal cultures were virtually devoid of astrocytic cells (<5%) as shown by the lack of glial fibrillary acidic protein (GFAP) immunoreactivity (Vincent et al., 1994). Astrocytes were obtained from neuronal cultures that were not treated with cytosine arabinofuranoside but were maintained in 10% calf serum to prevent oligodendrocyte proliferation. We established that >95% of the cells prepared in this fashion exhibit GFAP, but not galactocerebroside or F4/80 immunoreactivities, indicating that they were virtually devoid of oligodendrocytes and microglia.
Endopeptidase 3.4.24.16 immunolabeling of primary cultured cells.Four-day-old plated neurons and 15-d-old plated astrocytes were immunolabeled for endopeptidase 3.4.24.16 as described (Chabry et al., 1990) using the IgG-purified fraction of a monospecific rabbit polyclonal antiserum directed toward the rat brain purified enzyme (Checler et al., 1989). Briefly, cells were rinsed twice in 50 mm Tris-HCl containing 140 mm NaCl (buffer A) and fixed with glutaraldehyde 3.5% for 1 hr at 4°C. Cryoprotection was performed by incubation with a 0.12 m phosphate (PO4) buffer containing 30% sucrose. Cells were then frozen for 15 sec at −40°C in methyl-2 butane. To block nonspecific labeling, cells were preincubated for 1 hr in buffer A containing 1% BSA (buffer B) and then incubated overnight at 4°C with a 1:250 dilution (in buffer B) of the immune or preimmune IgG fractions. Finally, plated cells were exposed for 90 min to a 1/200 dilution of goat anti-rabbit IgG coupled to peroxidase, and rinsed twice with buffer A before initiating the reaction with 3–3′diaminobenzidine (DAB). Cells were then dehydrated with graded ethanol, counterstained for 30 sec with cresyl violet, coverslipped with glycerol, and examined with a Leitz Aristoplan microscope.
Electron microscopy. Electron microscopic localization of endopeptidase 3.4.24.16 immunoreactivity was performed in the midbrain tegmentum of the adult rat. Briefly, adult male Sprague–Dawley rats were perfused transaortically with 4% paraformaldehyde and 0.2% glutaraldehyde in 0.1 m (PO4) buffer. The brains were removed, the midbrain blocked, and the blocks immersed in the same fixative for 1 hr. Sections, 35 μm thick, were cut on a Vibratome (Lancer) and collected in 0.1 m phosphate buffer. The peroxidase-antiperoxidase immunocytochemical procedure used for the visualization of endopeptidase 3.4.24.16 immunoreactivity was identical to that described previously (Woulfe et al., 1992). Visualization of bound peroxidase was achieved by reaction with a solution of 0.1 m TBS containing 0.05% DAB and 0.01% H2O2. After reaction with the DAB–H2O2 solution, sections were post-fixed for 1 hr in 2% OsO4, dehydrated in graded ethanol, and embedded in Epon between plastic coverslips. Ultrathin sections were collected from the surface of each Epon block, deposited onto copper grids, counterstained or not with Reynold’s lead citrate, and examined with a Jeol 100 CX electron microscope.
Preparation of cell subcellular fractions. Primary cultured neurons and astrocytes were washed twice with PBS− buffer containing (in mm) 140 NaCl, 8.5 Na2HPO4, 2.7 KCl, 1.5 KH2PO4), pH 7.4, scraped in 5 mm Tris-HCl, pH 7.5, and homogenized with a syringe. Homogenates were centrifuged (4°C, 35 min, 150,000 × g), and supernatants (soluble fraction) were collected. The pellets, referred to as membrane-associated fractions, were resuspended in the same volume of Tris-HCl, pH 7.5. For extraction experiments with Na2CO3, pellets were resuspended in Tris-HCl, pH 7.5, containing 0.1 mNa2CO3, pH 7.5, and maintained at 4°C for 3 hr. Samples were then centrifuged as above, and residual pellets were homogenized in 5 mmTris-HCl, pH 7.5, and submitted to SDS-PAGE and Western blot analysis.
Effect of digitonin on intact neurons and astrocytes.Four-day-old neurons and 15-d-old astrocytes were resuspended in PBS− buffer, pH 7.4, centrifuged for 1 min at 5000 × g, and resuspended in PBS− or in PBS−containing 0.2% digitonin to permeabilize cells without solubilizing membrane proteins. Samples were then maintained at 4°C for 3 hr and centrifuged (4°C, 35 min, 150,000 × g). Supernatants were removed, pellets were resuspended in the same volume of PBS−, and both fractions were analyzed for endopeptidase 3.4.24.16 immunoreactivity by Western blot.
HPLC analysis of neurotensin degradation by plated cells.Four-day-old plated neurons or 15-d-old plated astrocytes were incubated for 2 and 3 hr, respectively, at 37°C, with 10 nmol (10 μm) neurotensin in PBS−containing 1% glucose in the absence or in the presence of 10 mm Pro-Ile or phosphodiepryl 03 [N-(phenylethylphosphonyl)-glycyl-prolyl-l-aminohexanoic acid, 100 nm]. Supernatants were taken out, acidified with 100 μl of 1 m HCl, and the equivalent of 1 nmol of neurotensin was submitted to HPLC analysis. Samples were applied to a reverse phase C18lichrosorb column (Merck, Darmstadt, Germany). Elutions were performed with the trifluoroacetic acid/triethylamine system as described previously (Checler et al., 1988). Absorbance was monitored at 230 nm with a detector setting of 0.05 full scale.
Fluorimetric analysis of QFS degradation. Plated neurons or astrocytes were incubated for 0–240 min at 37°C with QFS (Mcc-Pro-Leu-Gly-Pro-d-Lys-Dnp, 50 nmol, 50 μm) in a final volume of 1 ml of PBS-1% glucose, pH 7.4, in the absence or in the presence of Pro-Ile (10 mm). Cpp-Ala-Ala-Tyr-pAb (0.5 μm) was added to all incubations to prevent the contribution of endopeptidase 3.4.24.15 to QFS-hydrolysing activity. At the end of the incubations, 100 μl of supernatants (5 nmol of QFS) were taken out, acidified with 2 ml of 80 mmsodium formate, pH 3.7, and the activity was fluorimetrically recorded at λex 345 nm and λem405 nm as described previously (Dauch et al., 1991a). Studies on QFS hydrolysis by various cell fractions were performed with 10–20 μg of proteins in a final volume of 100 μl in the conditions described above.
Secretion of endopeptidase 3.4.24.16. For secretion experiments, primary cultured neurons and astrocytes were grown in 35 mm dishes in HAM F12 medium containing 10% FCS during 4 and 15 d, respectively. The medium was removed and the cells were gently washed twice with PBS− buffer. Cells were then incubated at 4°C, 37°C, or 42°C with 1 ml of PBS− or PBS+(PBS− with 1.8 mmCaCl2) buffer containing 1% glucose, in the absence or in the presence of various drugs. At appropriate time intervals, media (referred to as secreted fractions) were collected, and remaining cells (referred to as cellular fractions) were washed twice with PBS−, scraped with a rubber policeman in 1 ml of 5 mm Tris-HCl, pH 7.5, and homogenized with a syringe. Lactate dehydrogenase was assayed as described previously (Kostka et al., 1987) in all secreted fractions, whereas endopeptidase 3.4.24.16 activity was measured in both fractions using QFS as described above. At the end of the secretion procedure, cell viability was checked by trypan blue exclusion.
SDS-PAGE and Western blot analysis. Samples (5–20 μg of proteins) were dried and resuspended in 30 μl of sodium phosphate buffer, pH 7.5, containing 2% SDS and 5% β-mercaptoethanol. Aliquots were then boiled and electrophoresed in 8% acrylamide gels according to the procedure of Laemmli (1970). Proteins were blotted on nitrocellulose according to a procedure described previously (Towbin et al., 1979). Endopeptidase 24.16 was immunolabeled with the IgG-purified fraction of a monospecific polyclonal antiserum developed against the rat brain enzyme (Checler et al., 1989). Hybridization with antibodies and revelation of the IgG-peptidase complex were performed as described previously (Checler et al., 1989), except that the goat anti-rabbit IgG coupled to peroxidase was used at a 1/1000 dilution.
Northern blot analysis. Total mRNAs were isolated from primary cultures of neurons and astrocytes on CsCl gradients as described (Chirgwin et al., 1979). Twenty micrograms of each RNA sample were electrophoresed on a 1% agarose/2.2 mformaldehyde gel. The location of 28S and 18S ribosomal RNA was determined by staining with ethidium bromide. mRNAs were then transferred onto a nylon membrane, crosslinked with ultraviolet light, and hybridized with the PCR fragment (1390 pb) derived from the endopeptidase 3.4.24.16 λ7a clone, obtained as described previously (Dauch et al., 1995), and labeled with 32P by random-priming, Promega) and with a 32P glyceraldehyde-phosphate dehydrogenase (GAPDH) probe (internal standard for cheeking the amount of loaded RNA). Hybridization was performed overnight at 42°C in 50% formamide, 5× SSC, 10% dextran sulfate, 2× Denhardt’s solution, and 0.1 mg/ml heat-denatured calf thymus DNA. The nitrocellulose was washed four times at 65°C in 2× SSC, 0.1% SDS. Autoradiography was performed at −70°C for 3 d.
Protein concentration. Protein concentrations were determined by the Bradford method according to the manufacturer’s recommendations with white egg lysosyme as the standard.
RESULTS
It has been clearly established that neurons in primary cultures can enter a differentiation program (Yavin and Yavin, 1974). Morphologically, these cells are spherical in shape at a nondifferentiated stage and progressively display extended processes that ultimately give rise to a typical network of interconnecting fibers after 24 hr to 4 d in culture. These same features were observed in our primary neuronal cultures 4 d after plating (Fig.1). The cells were consistently GFAP-immunonegative, indicating a virtual lack of astrocytic growth (not shown). By contrast, a subpopulation of cultured neurons showed positive immunostaining for the IgG-purified fraction of an antiserum specific to the rat brain endopeptidase 3.4.24.16 (Fig. 1). The proportion of endopeptidase 3.4.24.16-immunopositive neurons varied with the degree of cell differentiation, decreasing from almost 100% 2 hr after plating to a plateau value of 40% between 4 and 8 d (Fig.2A). Parallel measurement of the Pro-Ile-sensitive QFS-hydrolysing activity that can be ascribed to endopeptidase 3.4.24.16 (Dauch et al., 1991a) showed a marked increase in total endopeptidase 3.4.24.16 activity during neuronal differentiation (Fig. 2A). Northern blot analysis also showed an apparent increase in the 5 kb endopeptidase 3.4.24.16 mRNA during the same period (Fig. 3), and ethidium bromide labeling revealed similar amounts of both 18S and 28S ribosomal mRNAs (not shown). Overall, this suggests that the level of enzyme expression is under the control of transcriptional events that are modulated during differentiation, although a difference in the stability of the mRNA taking place during maturation cannot be definitely excluded.
Fig. 1.

Immunolabeling of endopeptidase 3.4.24.16 in primary cultures of neurons and astrocytes. Neurons and astrocytes were cultured for 4 and 15 d, respectively, in the conditions described in Materials and Methods. After fixation and cryoprotection, cells were incubated overnight at 4°C with the IgG-purified fractions of the immune (top panels) or preimmune (bottompanels) rabbit antiserum developed against rat brain endopeptidase 3.4.24.16. After exposure to a goat anti-rabbit IgG coupled to peroxidase, endopeptidase 3.4.24.16-bearing cells were revealed with diaminobenzidine (brown cells) as described, and immunonegative cells still reacted with cresyl violet (blue cells). Photographs were taken with Kodacolor 100 film at 200× magnification.
Fig. 2.

Endopeptidase 3.4.24.16 activity in whole homogenate of primary cultured neurons and astrocytes: effect of time in culture. Neurons and astrocytes were primary-cultured as described in Materials and Methods. At the indicated times, dishes were rinsed twice with PBS−, cells were scraped in 5 mm Tris-HCl, pH 7.5, and homogenized with a syringe. Endopeptidase 3.4.24.16 activity (black bars) was monitored in neuron (A) and astrocyte (B) homogenates (50 and 25 μl, respectively) by means of the QFS assay and quantified as described in Materials and Methods. 1 unit = 1 nmol of QFS hydrolysed/hr/25 or 50 μl. Values are the mean ± SEM of six independent determinations performed with six different cultures. The white bars in A indicate the percent of neurons expressing endopeptidase 3.4.24.16 during neuronalin vitro differentiation and were taken from Chabry et al. (1990).
Fig. 3.

Northern blot analysis of endopeptidase 3.4.24.16 mRNA during in vitro neuronal differentiation. Total mRNAs (20 μg) were isolated as described in Materials and Methods from neurons cultured for the indicated times and from 15-d-old-plated astrocytes. RNAs were electrophoresed, blotted on a nitrocellulose sheet, and hybridized with the32P-labeled PCR fragment derived from the λ7a clone encoding rat brain endopeptidase 3.4.24.16 and the GAPDH probe (see Materials and Methods). RNA molecular weights are indicated on the left. Autoradiograms (top) were analyzed by densitometry. Bars indicate the mean of two independent quantifications performed with two distinct cell cultures and correspond to the 5 kb E:3.4.24.16 versus the GAPDH mRNA density recovered in the same sample. Ratio values did not differ by >0.005.
Unlike in neurons, astrocytes in culture for 15 d displayed homogenous endopeptidase 3.4.24.16 immunostaining (Fig. 1). Such a uniform distribution of astrocytic endopeptidase 3.4.24.16 was observed at all times in culture (not shown). Furthermore, the total activity of the enzyme did not vary with the time in culture (Fig.2B).
Subcellular fractionation indicated that the main endopeptidase 3.4.24.16 activity was present in the 150,000 × gsupernatant of whole neuron and astrocyte homogenates (Fig.4A). However, 10–20% of the activity was consistently recovered in the membrane-associated pellet after high-speed centrifugation (Fig. 4A). The dual distribution of the enzyme activity in the soluble and membrane-associated compartments was corroborated by the immunodetection of endopeptidase 3.4.24.16 in both fractions (Fig.4A, bottom). To examine whether the enzyme activity was inside vesicular organelles or loosely bound to membranes, we examined the effect of digitonin (at a concentration that permeabilizes cells and intracellular vesicles without altering membrane integrity) (Sambamurti et al., 1992) and Na2 CO3 (which depletes the membrane from loosely associated proteins). Neuronal membrane-associated endopeptidase 3.4.24.16 resisted treatments with digitonin (Fig. 4B) and Na2CO3 (Fig. 4C) indicating, at a biochemical level, that the enzyme was neither inside vesicles nor weakly absorbed to the membrane, but that it was truly membrane-associated. By contrast, these treatments drastically lowered or fully abolished both the immunolabeling observed in astrocytic membranes (Fig. 4B,C) and the enzymatic activity (not shown), indicating that no fraction of astrocytic endopeptidase 3.4.24.16 was genuinely membrane-associated.
Fig. 4.

Effect of digitonin and Na2CO3 on endopeptidase 3.4.24.16 immunoreactivity in neurons and astrocytes. A, Homogenates, soluble and membrane-associated fractions of 4-d-old neurons and 15-d-old astrocytes, were prepared as described in Materials and Methods and assayed for their Pro-Ile-sensitive QFS-hydrolysing activity (1 unit = 1 nmol of QFS hydrolysed/hr). Values represent the mean ± SEM of six determinations performed with independent cultures. Bottom panel corresponding to Western blot analysis of proteins (10 μg) in soluble and membrane-associated fractions of neurons and astrocytes shows a single immunoreactive band around 75 kDa. B, Cells were prepared as described in Materials and Methods and incubated in the absence (Control) or in the presence of 0.2% Digitonin. Aliquots were then centrifuged (4°C, 35 min, 150,000 × g), and 5–15 μg of protein of supernatants (S) and pellets (P) were electrophoresed and analyzed by Western blot. C, Membrane-associated fractions of primary cultured neurons and astrocytes (5 and 50 μg of protein, respectively) were treated with 0.1 mNa2CO3 and centrifuged, and resulting pellets (P) were analyzed by Western blot.
The membrane-associated form of neuronal endopeptidase 3.4.24.16 activity was only marginal at early stages of differentiation, but increased drastically during maturation (Fig.5A). The soluble fraction of the enzyme also increased during the same period, but proportionally less, so that the net result was an augmentation of the membrane-associated versus soluble enzyme (Fig. 5B). This increased recovery of the enzyme in the membrane-associated fraction was corroborated by an enhanced immunoreactivity detected by Western blot in neuronal cultures (Fig. 5C).
Fig. 5.

Soluble and membrane-associated endopeptidase 3.4.24.16 activity during differentiation of primary cultured neurons.A, Primary cultured neurons were scraped at the indicated differentiation times in 5 mm Tris-HCl, pH 7.5, and subcellular fractions were prepared as described in Materials and Methods. Both soluble (white bars) and membrane-associated (black bars) fractions were tested for their QFS-hydrolysing activities as described in Materials and Methods.B illustrates the ratio between QFS-hydrolysing activity in membrane-associated versus soluble fractions according to differentiation time. C, Five micrograms of membrane-associated fractions taken at 1, 3, 5, and 7 d of culture were dried, submitted to an 8% acrylamide gel, and analyzed by Western blot with the anti-E 3.4.24.16 IgG fraction as described in Materials and Methods.
To determine whether this membrane-associated form of endopeptidase 3.4.24.16 behaved as an ectopeptidase, i.e., with its catalytic site exposed to the extracellular medium, we submitted neurotensin and QFS to degradation by intact plated neurons. Figure6B indicates that exposure of the peptide to these intact cells resulted in the production of neurotensin 1–10, the formation of which was significantly reduced (p < 0.0001) by previous treatment of the cells with the endopeptidase 3.4.24.16-specific dipeptide inhibitor Pro-Ile (Dauch et al., 1991b) and by phosphodiepryl 03 (Fig.6D) (Barelli et al., 1992). Furthermore, plated neurons efficiently cleaved QFS in a Pro-Ile-sensitive manner (Fig.6E). By contrast, plated astrocytes also generated neurotensin 1–10 (Fig. 6A), but the formation of this catabolic product was insensitive to the endopeptidase 3.4.24.16 inhibitors (Fig. 6C). Accordingly, plated astrocytes displayed very little, if any, Pro-Ile-sensitive QFS-hydrolysing activity (Fig. 6E).
Fig. 6.
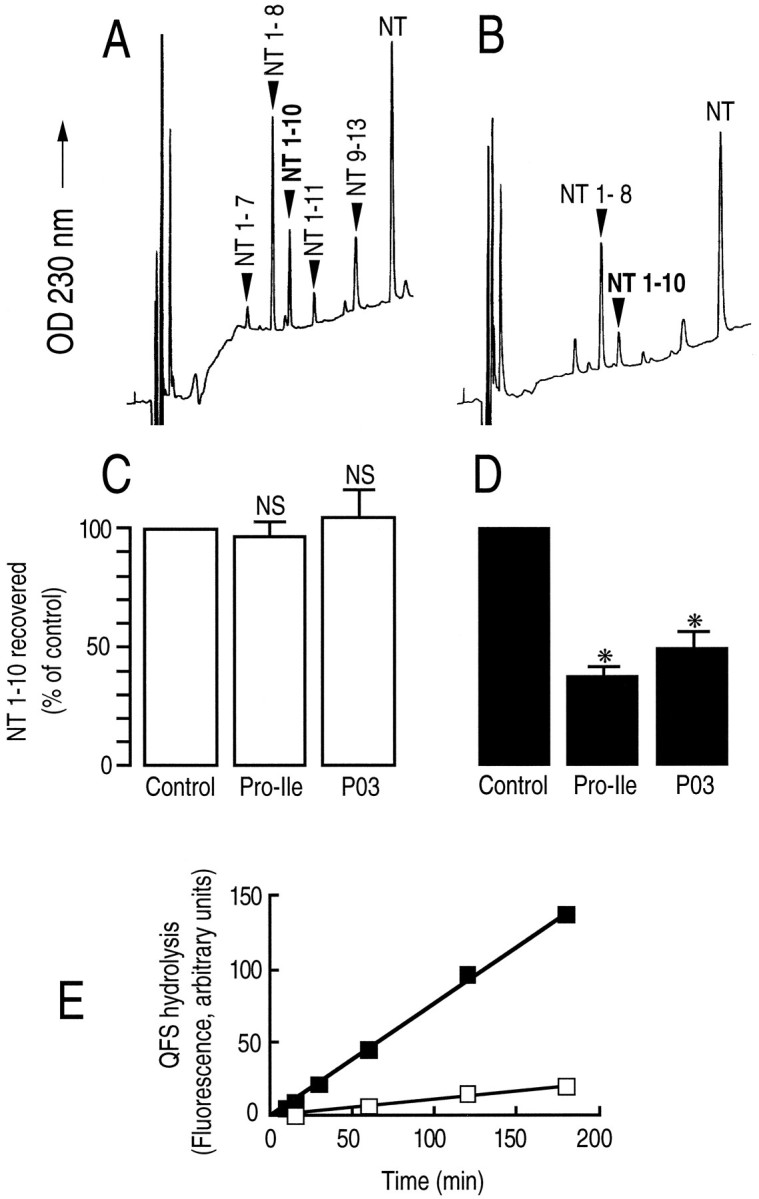
Endopeptidase 3.4.24.16 activity on plated neurons. Neurotensin (10 nmol, 10 μm) was incubated with 15-d-old cultured astrocytes (A) and with 4-d-old cultured neurons (B) for 2 and 3 hr, respectively, as described in Materials and Methods, in the absence (A, B) or in the presence (C, D) of Pro-Ile (10 mm) or phosphodiepryl 03 (100 nm). One hundred microliters of acidified medium were submitted to HPLC analysis. Arrows indicate the elution time of synthetic neurotensin fragments. Bars (C, D) represent the NT (1–10) recovered and are expressed as the percent of NT (1–10) recovered in the absence of inhibitor (Control). Values are the mean ± SEM of three to nine independent determinations. *p < 0.0001. NS, Nonstatistically significant. E, The Pro-Ile-sensitive QFS-hydrolysing activity detectable on plated neurons (▪) or astrocytes (□) was monitored as described in Materials and Methods.
Having established that the neuronal membrane-associated form of endopeptidase 3.4.24.16 increased during differentiation (Fig. 5), we examined the ability of intact plated neurons to cleave QFS at various stages of differentiation. As can be seen in Figure 7, the ectoenzyme form of endopeptidase 3.4.24.16 was not detectable 2 hr after plating, but gradually increased thereafter, clearly confirming that endopeptidase 3.4.24.16 undergoes a targeting to a membrane-associated compartment during neuronal maturation.
Fig. 7.

QFS hydrolysis by plated neurons during in vitro differentiation. Neuronal cultures corresponding to 2 hr (▴), 1 d (○), 2 d (▪), or 4 d (•) of differentiation were incubated for the indicated times with QFS (50 nmol, 50 μm) in 1 ml of PBS-1% glucose, pH 7.4, as described in Materials and Methods, in the absence or in the presence of Pro-Ile (10 mm). At the end of the incubations, 100 μl of supernatants were removed and acidified, and endopeptidase 3.4.24.16 activity was fluorimetrically monitored as described in Materials and Methods. Curves represent kinetics of the Pro-Ile-sensitive QFS-hydrolysing activity and correspond to the mean of three independent determinations.
To definitely establish that neuronal endopeptidase 3.4.24.16 genuinely exists in a membrane-associated form, we examined the effect of cryoprotection of neurons and astrocytes on the immunolabeling of endopeptidase 3.4.24.16. In the absence of cryoprotection, one would expect to lose the immunolabel attributable to intracellular, but not to membrane-associated, endopeptidase 3.4.24.16 (Rosene and Rhodes, 1990). Figure 8 indicates that 4-d-plated neurons retained their immunopositivity in the absence of cryoprotection, confirming that a population of the cells indeed expressed a membrane-bound form of endopeptidase 3.4.24.16. By contrast, in the absence of cryoprotection, astrocytic cultures were totally devoid of immunoreactivity (Fig. 8) indicating that, unlike neurons, astrocytes contain only a soluble form of endopeptidase 3.4.24.16 in agreement with our biochemical analysis.
Fig. 8.

Effect of cryoprotection on endopeptidase 3.4.24.16 immunoreactivity in primary cultured neurons and astrocytes. Four-day-old plated neurons and 15-d-old plated astrocytes were fixed with glutaraldehyde, cryoprotected (+) or not (−) with NaK2 buffer containing 30% sucrose, and processed for immunochemical detection of endopeptidase 3.4.24.16 as described in Materials and Methods. Photographs were taken with Kodacolor 100 film at 200× magnification.
In keeping with the preceding observations, electron microscopic analysis of endopeptidase 3.4.24.16 immunoreactivity in the midbrain tegmentum of the adult rat brain revealed striking differences in the subcellular distribution of the immunolabeling between neurons and astrocytes (Fig. 9). In nerve cell bodies (Fig.9a) and dendrites (Fig. 9c–e), the immunoreactivity was mainly concentrated over restricted portions of the plasma membrane. Dense peroxidase deposits were also associated with intracellular organelles clustered beneath the immunoreactive membrane segments (Fig. 9c–e, arrows). By contrast, in astrocytes, the immunolabel was diffusely distributed throughout the cytoplasm of both cell bodies (not shown) and distal processes (Fig. 9a,b), with no obvious predilection for either the plasma membrane or intracellular organelles. Incubation with preimmune serum, omission of the primary antiserum, or previous incubation of immune serum with purified endopeptidase 3.4.24.16 totally abolished both neuronal and glial immunoreactivity. Taken together, these data indicate that the differences between the subcellular distribution of neuronal and glial forms of the enzyme observed in cell culture also occur in vivo.
Fig. 9.
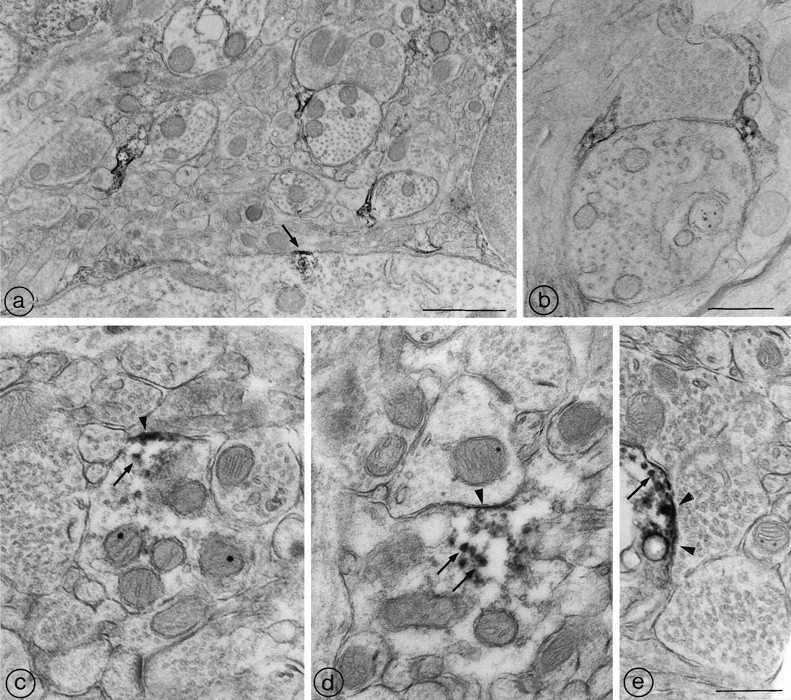
Ultrastructural distribution of endopeptidase 3.4.24.16 in the midbrain tegmentum of the adult rat. a, Three densely immunoreactive astrocytic leaflets are visible in this field of the midbrain tegmentum. Each of them is apposed to a cross-sectioned dendritic profile. At the bottom of the field, a neuronal soma exhibits a discrete patch of endopeptidase 3.4.24.16 immunoreactivity. The immunoreactivity is restricted to a short segment of the plasma membrane (arrow) and underlying cytoplasm. Scale bar, 1 μm. b, Two immunoreactive astrocytic leaflets seal off a synaptic junction between an axon terminal and an unlabeled dendritic shaft. Note that the reaction product pervades the entire glial cytoplasm. Scale bar, 0.5 μm.c, d, e, Endopeptidase 3.4.24.16-immunoreactive dendrites. In all three of these labeled dendritic shafts, the reaction product is concentrated along restricted zones of the plasmalemma (arrowheads), as well as within microtubules and/or vesicular organelles (arrows). Whereas the subplasmalemmal labeling in c and d is clearly extrasynaptic, the reaction product in e may be masking a postsynaptic specialization. Scale bar, 0.5 μm.
Our previous electron microscopic study revealed the occurrence, within the midbrain tegmentum, of endocytic invaginations at the level of appositions between endopeptidase 3.4.24.16-immunoreactive neurons and astrocytes, suggesting a possible translocation of the enzyme between these two elements (Woulfe et al., 1992). This finding prompted us to investigate the possibility of a secretion of endopeptidase 3.4.24.16 by either astrocytes or neurons. In astrocyte cultures, our experiments revealed a time-dependent secretion of endopeptidase 3.4.24.16 that reached by 10 hr a plateau value corresponding to ∼40% of the activity initially present in whole cells (Fig.10A). Parallel experiments indicated that <2% of total lactate dehydrogenase activity was present in the medium after 1 hr and that this value did not change during the kinetics of secretion (not shown), ruling out the possibility of nonspecific leaking of enzymatic material in the extracellular medium. By contrast, virtually no endopeptidase 3.4.24.16 activity was recovered from the supernatant of cultured neurons (Fig.10A). As can be seen in Figure 10 (B,C), the increase in endopeptidase 3.4.24.16 activity (Fig. 10B) or immunoreactivity (Fig. 10C) in astrocyte supernatant was accompanied by a decrease of both parameters in the corresponding cell extracts. Altogether, the sum of secreted and cell-extracted activities corresponded to the activity initially present in whole-cell homogenates. Furthermore, the secretion process was not greatly affected by previous treatment of astrocytes by cycloheximide (Table1). Altogether, this indicates that the turnover of the peptidase in the astrocytes was markedly slow. Treating the cells with 8 bromo cAMP or lowering the temperature drastically reduced or fully blocked the secretion of endopeptidase 3.4.24.16 from cultured astrocytes, whereas increasing the temperature accelerated the secretion process (Table 1). Forskolin did not influence the secretion of endopeptidase 3.4.24.16 by astrocytes (Table 1). Furthermore, several agents known to affect secretory processes such as ionomycin, TPA, monensin, brefeldin A, and chloroquin did not influence the secretion of endopeptidase 3.4.24.16 by astrocytes (Table 1).
Fig. 10.

Secretion of endopeptidase 3.4.24.16 by cultured astrocytes. A, Time course of endopeptidase 3.4.24.16 recovery in the medium of 15-d-old cultured astrocytes (•) and 4-d-old cultured neurons (□). Incubations were performed in PBS+/1% glucose, pH 7.4. At the indicated times, medium was taken off and enzymatic activity was fluorimetrically measured with QFS as detailed in Materials and Methods. Values are expressed as the percentage of whole-cell activity detectable att = 0 and are the mean ± SEM of four independent experiments. B illustrates the medium (white bars) and cellular (black bars) endopeptidase 3.4.24.16 activity in astrocytes at each time of the secretion kinetics. Values are mean ± SEM of three independent experiments. C, Western blot analysis of endopeptidase 3.4.24.16 protein content in media and cells during astrocyte-secretion experiments. Five micrograms of proteins were loaded onto an 8% SDS-PAGE and immunoblotted as described in Materials and Methods.
Table 1.
Effects of various drugs on endopeptidase 3.4.24.16 secretion by cultured astrocytes
| Experimental conditions | Secreted E 3.4.24.16 activity (% of control) |
|---|---|
| PBS+(control) | 100 |
| Ionomycin 200 nm | 96 ± 7.5 |
| Forskolin 20 μm | 138 ± 10 |
| TPA 0.1 μg/ml | 136 ± 55 |
| KCl 60 mm | 112 ± 20 |
| 8 Br cAMP 0.5 mm | 28 ± 6 |
| Cycloheximide 100 μm | 73 ± 13 |
| Monensin 10 μm | 88 ± 11.5 |
| Brefeldin A 10 μg/ml | 108 ± 18 |
| Chloroquine 100 μm | 115 ± 22 |
| Colchicine 10 μm | 120 ± 9 |
| 4°C | 3 ± 3 |
| 42°C* | 348 ± 88 |
| PBS− | 38 ± 4.8 |
Fifteen-day-old plated astrocytes were incubated for 6 hr (except *, which was incubated for 1 hr) in the conditions indicated, then secreted endopeptidase 3.4.24.16 activity was measured with QFS as described in Materials and Methods. Values correspond to the QFS-hydrolysing activity recovered in the medium and are expressed as the percent of the activity recovered in the medium in control conditions. Values are the mean ± SEM of three to five independent experiments.
DISCUSSION
Neuronal endopeptidase 3.4.24.16 expression is regulated during the maturation process. At a nondifferentiated stage, most of the neurons expressed the enzyme in a soluble form. During differentiation, the population of immunopositive neurons decreased, whereas the activity of the enzyme concomitantly increased. Overall, this indicates that the neuronal differentiation program leads to the selection of a restricted population of neurons overexpressing endopeptidase 3.4.24.16. Although one cannot exclude the possibility that the appearance of the network of synaptic connections could also contribute to enhanced stability of the enzymatic activity, this increase in the production of the enzyme appears likely to be attributable to the activation of transcriptional events, given the concomitant apparent increase in the 5 kb endopeptidase 3.4.24.16 mRNA. It is striking that Northern blot analysis performed on the whole rat brain tissue with the same labeled probe also revealed two polyA+ mRNA species of 3 and 5 kb, but with increased intensity for the former species (Dauch et al., 1995). One possible explanation of such difference in the relative intensities could be overloaded total mRNAs (present study), causing increased nonspecific background concentrating into an apparent band at 5 kb. However, this seems unlikely because Northern blot analysis performed on identical amounts of total mRNA from whole rat brain also led to prominent 3 kb mRNA labeling (not shown). Whether the distinct pattern observed between the previous study (Dauch et al., 1995) and the present work could be accounted for by species difference, i.e., rat versus mice, remains to be established.
The increase in endopeptidase 3.4.24.16 activity during neuronal maturation coincides with the appearance of a membrane-associated form of endopeptidase 3.4.24.16. This late-occurring form appears genuinely associated with the membrane as it resists treatments with digitonin and Na2CO3 and displays an ectopeptidasic activity in intact plated neurons. The existence of this ectopeptidase form was further confirmed by the resistance of endopeptidase 3.4.24.16 immunoreactivity in the absence of cryoprotection. Unlike neurons, all astrocytes expressed endopeptidase 3.4.24.16 between 15 and 60 d in culture. The activity of the enzyme did not vary during this time period. Accordingly, the membrane-associated form of the enzyme detected in neurons never appeared in astrocytes.
Electron microscopy confirmed that neurons and astrocytes exhibited the same differential subcellular distribution in intact brain as they did in culture. In neurons, endopeptidase 3.4.24.16 was associated mainly with segments of the plasma membrane and intracellular membrane-bound organelles, in keeping with the occurrence of the membrane-associated form of the enzyme demonstrated biochemically. Interestingly, labeled vesicles were usually clustered immediately beneath the hot spots of membrane immunostaining, suggesting that they might correspond to recycling portions of the membrane. Whether the enzyme participates in the intracellular degradation of neurotensin after internalization of ligand-receptor complexes as this occurs in neurons of the substantia nigra and ventral tegmental area remains to be demonstrated. By contrast, in astrocytes, endopeptidase 3.4.24.16 was distributed throughout the cytoplasm of cell bodies and processes in conformity with biochemical results that suggest an exclusive soluble form of the enzyme in this cell type.
It should be noted that Serizawa et al. (1995) reported on the purification and characterization of a rat liver peptidase called oligopeptidase M that displays an N-terminal sequence identical with that deduced from the endopeptidase 3-4-24-16 sequence (Dauch et al., 1996). Biochemical analysis of the subcellular distribution of oligopeptidase M indicated that the activity seems to be associated with the inner mitochondrial membrane fraction (Serizawa et al., 1995). However, our characterization of the endopeptidase 3.4.24.16 at the electron microscopic level never revealed the association of the enzyme with this intracellular organelle.
The recent molecular cloning of rat brain endopeptidase 3.4.24.16 allowed to reveal two mRNA species of 3 and 5 kb, respectively (Dauch et al., 1995). It is not yet clear whether the two mRNAs exhibit variable length in their 3′ noncoding region or whether the high molecular weight mRNA represents an intermediate, immature form of the low molecular weight mRNA. However, the possibility that the two mRNAs correspond to the membrane-associated and soluble forms of endopeptidase 3.4.24.16 may be readily excluded, because we consistently detected a single immunolabeled protein of the expected molecular weight in crude tissues thought to display the two distinct forms of endopeptidase 3.4.24.16 (Checler et al., 1989). Furthermore, both a major soluble form of the enzyme and a minor fraction behaving as the membrane-associated form (and of the same apparent molecular weight) were recovered from COS-7 cells transiently expressing the 2448 bp isolated cDNA encoding endopeptidase 3.4.24.16 (Dauch et al., 1995).
The sequence of endopeptidase 3.4.24.16 did not reveal the presence of a signal peptide that could serve to anchor the enzyme as it is the case for type II intrinsic proteins (for review, see Ehlers and Riordan, 1991). Furthermore, although three putative glycosylation sites exist in the enzyme, our experiments on purified (Checler et al., 1986b) and recombinant endopeptidase 3.4.24.16 (Dauch et al., 1995) clearly showed that the peptidase did not undergo glycosylation. Finally, the enzyme does not belong to the glycosyl phosphatidylinositol-anchored proteins, as shown by our experiments demonstrating that the renal membrane-associated endopeptidase 3.4.24.16 was not released by treatment with phosphatidylinositol-specific phospholipase C (Barelli et al., 1993). Two structural features remain that could account for the anchoring of endopeptidase 3.4.24.16 to the membrane. First, there exists few hydrophobic domains, as deduced from the hydropathic profile of the enzyme (Dauch et al., 1995), that could mediate protein–protein interactions. Second, we detected two stretches of charged residues in the middle of the endopeptidase 3.4.24.16 sequence. Interestingly, a similar cluster of charged residues was found in a proteolytic activity, the N-arginine dibasic convertase (Pierotti et al., 1994), as well as in two other types of proteins: carboxypeptidase E (CPE), where this type of sequence (Fricker et al., 1986) acts as an amphiphilic helix responsible for the attachment of CPE to the membrane (Mitra et al., 1994), and complexins in which comparable stretches of hydrophilic residues have been implicated in their interaction with the SNAP-receptor core complex (McMahon et al., 1995). Altogether, these observations are consistent with the possibility that endopeptidase 3.4.24.16 interacts with proteins able to target the enzyme to the membrane via its hydrophilic sequence. Whether such putative intermediate protein is present in neurons but not in astrocytes remains to be established. The deletion of the stretch of polarized residues by site-directed mutagenesis and subsequent transfection analysis should allow us to assess whether the removal of this sequence abolishes the occurrence of the membrane-associated form of endopeptidase 3.4.24.16 in transfectants.
We have demonstrated that primary cultured astrocytes secreted endopeptidase 3.4.24.16. As discussed previously, the sequence of rat brain endopeptidase 3.4.24.16 does not contain the typical signal peptide necessary to trigger classical regulated and constitutive secretory mechanisms. Accordingly, astrocytic endopeptidase 3.4.24.16 secretion was not prevented by treatment with brefeldin A or monensin, two agents known to interfere with endoplasmic reticulum and Golgi transits, respectively. Furthermore, forskolin did not modify the enzyme secretion, indicating that this mechanism was not under the control of cAMP-regulated events.
Several studies recently have postulated the existence of an additional, nonconventional secretory pathway for proteins lacking the canonical signal peptide sequences (for review, see Muesh et al., 1990;Halban and Irminger, 1994). Interleukin-1β is an example of a protein that lacks a signal peptide and is secreted through a temperature-dependent mechanism (Rubartelli et al., 1990). Interleukin-1β secretion is blocked at 4°C and highly increased at 42°C, two properties that are reminiscent of those observed in the case of astrocytic endopeptidase 3.4.24.16 secretion. Similarly, endopeptidase 3.4.22.19, which also lacks a signal peptide, is released from the astrocyte-derived C6 glioma cell line (Ferro et al., 1993). Therefore, endopeptidase 3.4.24.16 is not the only example of proteolytic activity undergoing a secretion process without exhibiting the structural requirements generally associated with classical secretory proteins. The molecular events or putative additional cofactors that prevent this type of secretion from occurring in neurons remain to be determined.
To our knowledge, the present study is the first demonstration of major differences in the properties of a peptidase linked to the nature of the host cell. Whether these differences reflect complementary roles of the enzyme on peptide inactivation remains to be established. In this context, it is interesting to note that, at the level of neurotensinergic pathways, endopeptidase 3.4.24.16-immunopositive astrocytic elements are often directly apposed to endopeptidase 3.4.24.16-immunoreactive nerve cell bodies and/or dendrites (Woulfe et al., 1992). It is therefore tempting to speculate that although the endopeptidase 3.4.24.16 secreted form of astrocytes would act in the extracellular space, thereby restricting diffusion of released neurotensin, the neuronal membrane-associated activity would be responsible for the physiological inactivation of the peptide either in the synaptic cleft, beside neurotensin receptors, or inside early endosomal compartments in which receptor-ligand complexes would have been internalized (Mazella et al., 1991).
Footnotes
This work was supported by the Centre National de la Recherche Scientifique and the Institut National de la Santé et de la Recherche Médicale. We are grateful to Drs. J. Mazella and J. Chabry for advice concerning cell immunolabeling, and to S. Soldera for mRNA preparation. We thank Dr. M. Orlowski (Mount Sinai School of Medicine, New York, NY), who gave us Cpp-Ala-Ala-Tyr-pAb. We are indebted to Dr. V. Dive (Centre Energie Atomique, Saclay, France), who synthesized and provided us with phosphodiepryl 03. We thank J. Kervella for secretarial assistance and F. Aguila for artwork.
Correspondence should be addressed to Frédéric Checler, Institut de Pharmacologie Moleculaire et Cellulaire, CNRS UPR 411, 660 Route des Lucioles, Sophia Antipolis, 06560 Valbonne, France.
REFERENCES
- 1.Barelli H, Vincent JP, Checler F. Peripheral inactivation of neurotensin: isolation and characterization of a metallopeptidase from rat ileum. Eur J Biochem. 1988;175:481–489. doi: 10.1111/j.1432-1033.1988.tb14220.x. [DOI] [PubMed] [Google Scholar]
- 2.Barelli H, Dive V, Yiotakis A, Vincent JP, Checler F. Potent inhibition of endopeptidase 24.16 and endopeptidase 24.15 by the phosphonamide peptide N -(phenylethylphosphonyl)-Gly-l-Pro-l-aminohexanoic acid. Biochem J. 1992;287:621–625. doi: 10.1042/bj2870621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Barelli H, Vincent JP, Checler F. Rat kidney endopeptidase 24.16: purification, physico-chemical characteristics and specificity towards opiates, tachykinins and neurotensin-related peptides. Eur J Biochem. 1993;211:79–90. doi: 10.1111/j.1432-1033.1993.tb19872.x. [DOI] [PubMed] [Google Scholar]
- 4.Barelli H, Fox-Threlkeld JET, Dive V, Daniel EE, Vincent JP, Checler F. Role of endopeptidase 3.4.24.16 in the catabolism of neurotensin, in vivo , in the vascularly perfused dog ileum. Br J Pharmacol. 1994;112:127–132. doi: 10.1111/j.1476-5381.1994.tb13041.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chabry J, Checler F, Vincent JP, Mazella J. Colocalization of neurotensin receptors and of the neurotensin-degrading enzyme endopeptidase 24–16 in primary cultures of neurons. J Neurosci. 1990;10:3916–3921. doi: 10.1523/JNEUROSCI.10-12-03916.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Checler F, Mazella J, Kitabgi P, Vincent JP. High affinity receptor sites and rapid proteolytic inactivation of neurotensin in primary cultured neurons. J Neurochem. 1986a;47:1742–1748. doi: 10.1111/j.1471-4159.1986.tb13083.x. [DOI] [PubMed] [Google Scholar]
- 7.Checler F, Vincent JP, Kitabgi P. Purification and characterization of a novel neurotensin degrading peptidase from rat brain synaptic membranes. J Biol Chem. 1986b;261:11274–11281. [PubMed] [Google Scholar]
- 8.Checler F, Barelli H, Kitabgi P, Vincent JP. Neurotensin metabolism in various tissues from central and peripheral origins. Ubiquitous involvement of a novel neurotensin degrading metalloendopeptidase. Biochimie. 1988;70:75–82. doi: 10.1016/0300-9084(88)90161-7. [DOI] [PubMed] [Google Scholar]
- 9.Checler F, Barelli H, Vincent JP. Tissue distribution of a novel neurotensin-degrading metallopeptidase. An immunological approach using monospecific polyclonal antibodies. Biochem J. 1989;257:549–554. doi: 10.1042/bj2570549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chirgwin JM, Przybyla AE, MacDonald WJ, Rutter RJ. Isolation of biologically active ribonucleic acid from sources enriched in ribonuclease. Biochemistry. 1979;18:5294–5299. doi: 10.1021/bi00591a005. [DOI] [PubMed] [Google Scholar]
- 11.Dahms P, Mentlein R. Purification of the main somatostatin-degrading proteases from rat and pig brains, their action on other neuropeptides, and their identification as endopeptidases 24.15 and 24.16. Eur J Biochem. 1992;208:145–154. doi: 10.1111/j.1432-1033.1992.tb17168.x. [DOI] [PubMed] [Google Scholar]
- 12.Dauch P, Barelli H, Vincent JP, Checler F. Fluorimetric assay of the neurotensin-degrading metalloendopeptidase, endopeptidase 24–16. Biochem J. 1991a;280:421–426. doi: 10.1042/bj2800421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dauch P, Vincent JP, Checler F. Specific inhibition of endopeptidase 24.16 by dipeptides. Eur J Biochem. 1991b;202:269–276. doi: 10.1111/j.1432-1033.1991.tb16372.x. [DOI] [PubMed] [Google Scholar]
- 14.Dauch P, Vincent JP, Checler F. Molecular cloning and expression of rat brain endopeptidase 3.4.24.16. J Biol Chem. 1995;270:27266–27271. doi: 10.1074/jbc.270.45.27266. [DOI] [PubMed] [Google Scholar]
- 15.Ehlers MRW, Riordan JF. Membrane proteins with soluble counterparts: role of proteolysis in the release of transmembrane proteins. Biochemistry. 1991;30:10065–10074. doi: 10.1021/bi00106a001. [DOI] [PubMed] [Google Scholar]
- 16.Ferro ES, Tambourgi DV, Gobersztejn F, Gomes MD, Sucupira M, Armelin MCS, Kipnis TL, Camargo ACM. Secretion of a neuropeptide-metabolizing enzyme similar to endopeptidase 22.19 by glioma C6 cells. Biochem Biophys Res Commun. 1993;191:275–281. doi: 10.1006/bbrc.1993.1213. [DOI] [PubMed] [Google Scholar]
- 17.Fricker LD, Evans CJ, Esch FS, Herbert E. Cloning and sequence analysis of cDNA for bovine carboxypeptidase E. Nature. 1986;323:461–464. doi: 10.1038/323461a0. [DOI] [PubMed] [Google Scholar]
- 18.Halban PA, Irminger JC. Sorting and processing of secretory proteins. Biochem J. 1994;299:1–18. doi: 10.1042/bj2990001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kostka P, Ahmad S, Berezin I, Kwan CY, Daniel EE. Subcellular fractionation of the longitudinal smooth muscle/myentheric plexus (LSM/MP) of dog ileum: dissociation of the distribution of two plasma membrane marker enzymes. J Neurochem. 1987;49:1124–1132. doi: 10.1111/j.1471-4159.1987.tb10002.x. [DOI] [PubMed] [Google Scholar]
- 20.Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 21.Mazella J, Leonard K, Chabry J, Kitabgi P, Vincent JP, Beaudet A. Binding and internalization of iodinated neurotensin in neuronal cultures from embryonic mouse brain. Brain Res. 1991;564:249–255. doi: 10.1016/0006-8993(91)91460-i. [DOI] [PubMed] [Google Scholar]
- 22.McMahon HT, Missler M, Li C, Südhof TC. Complexins: cytosolic proteins that regulate SNAP receptor function. Cell. 1995;83:111–119. doi: 10.1016/0092-8674(95)90239-2. [DOI] [PubMed] [Google Scholar]
- 23.Mentlein R, Dahms P. Endopeptidases 24.16 and 24.15 are responsible for the degradation of somatostatin, neurotensin and other neuropeptides by cultivated rat cortical astrocytes. J Neurochem. 1994;62:27–36. doi: 10.1046/j.1471-4159.1994.62010027.x. [DOI] [PubMed] [Google Scholar]
- 24.Millican PE, Kenny AJ, Turner AJ. Purification and properties of a neurotensin-degrading endopeptidase from pig brain. Biochem J. 1991;276:583–591. doi: 10.1042/bj2760583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mitra A, Song L, Fricker LD. The C-terminal region of carboxypeptidase E is involved in membrane binding and intracellular routing in AtT-20 cells. J Biol Chem. 1994;269:19876–19881. [PubMed] [Google Scholar]
- 26.Muesch A, Hartmann E, Rohde K, Rubartelli A, Sitia R, Rapoport TA. A novel pathway for secretory proteins? Trends Biochem Sci. 1990;15:86–88. doi: 10.1016/0968-0004(90)90186-f. [DOI] [PubMed] [Google Scholar]
- 27.Pierotti AR, Prat A, Chesneau V, Gaudoux F, Leseney AM, Foulon T, Cohen P. N -arginine dibasic convertase, a metalloendopeptidase as a prototype of a class of processing enzymes. Proc Natl Acad Sci USA. 1994;91:6078–6082. doi: 10.1073/pnas.91.13.6078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rosene DL, Rhodes KJ. Cryoprotection and freezing methods to control ice crystal artifact in frozen sections of fixed and unfixed brain tissue. In: Cohn M, editor. Methods in neurosciences. Quantitative and qualitative microscopy, Vol. 3. Academic; San Diego: 1990. pp. 360–384. [Google Scholar]
- 29.Rubartelli A, Cozzolino F, Talio M, Sitia R. A novel secretory pathway for interleukin-1β, a protein lacking a signal sequence. EMBO J. 1990;9:1503–1510. doi: 10.1002/j.1460-2075.1990.tb08268.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sambamurti K, Shioi J, Anderson JP, Papolla MA, Robakis NK. Evidence for intracellular cleavage of the Alzheimer’s amyloid precursor in PC12 cells. J Neurosci Res. 1992;33:319–329. doi: 10.1002/jnr.490330216. [DOI] [PubMed] [Google Scholar]
- 31.Serizawa A, Dando PM, Barrett AJ. Characterization of a mitochondrial metallopeptidase reveals neurolysin as a homologue of thimet oligopeptidase. J Biol Chem. 1995;270:2092–2098. doi: 10.1074/jbc.270.5.2092. [DOI] [PubMed] [Google Scholar]
- 32.Towbin H, Staehelin T, Gordon J. Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: procedure and some applications. Proc Natl Acad Sci USA. 1979;76:4350–4354. doi: 10.1073/pnas.76.9.4350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Vincent B, Vincent JP, Checler F. Neurotensin and neuromedin N undergo distinct catabolic processes in astrocytes and primary cultured neurons. Eur J Biochem. 1994;221:297–306. doi: 10.1111/j.1432-1033.1994.tb18741.x. [DOI] [PubMed] [Google Scholar]
- 34.Vincent B, Dive V, Yiotakis A, Smadja C, Maldonado R, Vincent JP, Checler F. Phosphorus-containing peptides as mixed inhibitors of endopeptidase 3.4.24.15 and 3.4.24.16: effect on neurotensin degradation in vitro and in vivo . Br J Pharmacol. 1995;115:1053–1063. doi: 10.1111/j.1476-5381.1995.tb15918.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Vincent B, Vincent JP, Checler F. Purification and characterization of human endopeptidase 3.4.24.16. Comparison with the porcine counterpart indicates a unique cleavage site on neurotensin. Brain Res. 1996;709:51–58. doi: 10.1016/0006-8993(95)01260-5. [DOI] [PubMed] [Google Scholar]
- 36.Woulfe J, Checler F, Beaudet A. Light and electron microscopic localization of the neutral metalloendopeptidase EC 3.4.24.16 in the mesencephalon of the rat. Eur J Neurosci. 1992;4:1309–1319. doi: 10.1111/j.1460-9568.1992.tb00156.x. [DOI] [PubMed] [Google Scholar]
- 37.Yavin E, Yavin Z. Attachment and culture of dissociated cells from rat embryos cerebral hemispheres on polylysine-coated surface. J Cell Biol. 1974;62:540–546. doi: 10.1083/jcb.62.2.540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yoshikawa T, Tashiro T, Takahashi K. Specificity of action on neuropeptides of an endopeptidase from the synaptosomal membranes of guinea-pig brain. J Biochem. 1988;104:1007–1010. doi: 10.1093/oxfordjournals.jbchem.a122566. [DOI] [PubMed] [Google Scholar]