

Abstract
Tissue inhibitors of metalloproteinases (TIMPs) are natural inhibitors of matrix metalloproteinases (MMPs), enzymes that contribute to cancer and many inflammatory and degenerative diseases. The TIMP N-terminal domain binds and inhibits an MMP catalytic domain, but the role of the TIMP C-terminal domain in MMP inhibition is poorly understood. Here, we employed yeast surface display for directed evolution of full-length human TIMP-1 to develop MMP-3–targeting ultrabinders. By simultaneously incorporating diversity into both domains, we identified TIMP-1 variants that were up to 10-fold improved in binding MMP-3 compared with WT TIMP-1, with inhibition constants (Ki) in the low picomolar range. Analysis of individual and paired mutations from the selected TIMP-1 variants revealed cooperative effects between distant residues located on the N- and C-terminal TIMP domains, positioned on opposite sides of the interaction interface with MMP-3. Crystal structures of MMP-3 complexes with TIMP-1 variants revealed conformational changes in TIMP-1 near the cooperative mutation sites. Affinity was strengthened by cinching of a reciprocal “tyrosine clasp” formed between the N-terminal domain of TIMP-1 and proximal MMP-3 interface and by changes in secondary structure within the TIMP-1 C-terminal domain that stabilize interdomain interactions and improve complementarity to MMP-3. Our protein engineering and structural studies provide critical insight into the cooperative function of TIMP domains and the significance of peripheral TIMP epitopes in MMP recognition. Our findings suggest new strategies to engineer TIMP proteins for therapeutic applications, and our directed evolution approach may also enable exploration of functional domain interactions in other protein systems.
Keywords: matrix metalloproteinase (MMP), tissue inhibitor of metalloproteinase (TIMP), protease inhibitor, directed evolution, protein engineering, protein domain, protein-protein interaction, crystal structure, protein structure, metalloprotease, yeast surface display
Introduction
The majority of eukaryotic proteins consist of at least two domains, and these multidomain proteins can perform elaborate tasks, often employing a binding or active site located at the domain interface (1). The separate domains of a multidomain protein fold independently, and yet the interplay between protein domains has an important role in protein structure, stability, and function (2). Efforts to understand the cooperative impact of individual protein domains on function of multidomain proteins can provide deeper insight into natural protein function and regulation and can also inform new strategies for rational design and engineering of multidomain proteins (3).
Tissue inhibitors of metalloproteinases (TIMPs),2 a family of four proteins in vertebrates, are endogenous inhibitors of matrix metalloproteinases (MMPs) that regulate MMP function and activity (4, 5). TIMPs comprise two domains that pack side by side. The N-terminal domain is widely recognized as the primary inhibitory domain that blocks MMP enzymatic activity by binding at the active site and interacting with the catalytic zinc (5–7). The TIMP C-terminal domain in some cases regulates MMP activation through interactions with noncatalytic MMP hemopexin domains (8) or stimulates MMP-independent cell signaling by binding to membrane receptors (9, 10). Although the TIMP C-terminal domain may also contribute to the binding interface with an MMP catalytic domain, conferring a degree of binding affinity (11, 12), the significance of this interaction and mechanism by which it contributes to MMP recognition have been largely overlooked. Here, we sought to probe the role of the TIMP C-terminal domain and of cooperativity between TIMP domains in MMP recognition.
Directed evolution of proteins is commonly used to improve or alter protein function toward desired properties but, importantly, can also be used to uncover insights relating sequence to function (13–15). Yeast surface display (YSD) is a directed evolution platform for engineering protein binders, including antibodies (16, 17) or other scaffolds (18), and benefits from eukaryotic secretion machinery that provides quality control for displaying full-length, stable, and properly folded proteins (17, 19), making it a suitable platform for engineering multidomain proteins. YSD has typically been applied to evolve single protein domains for altered binding characteristics, as in the recent engineering of the TIMP-2 N-terminal domain for selectivity toward MMP-9 and -14 (20–22). We hypothesized that by simultaneously applying directed evolution to both domains of a full-length TIMP on the yeast surface, we might probe the roles of each individual domain as well as any cooperative dynamic in MMP recognition; by evolving the intact multidomain protein holistically, we could optimize functional domain interplay.
Here, by creating a YSD library incorporating diversity into both domains of TIMP-1 and analyzing mutations of variants selected for improved binding to an already extremely high-affinity natural target, the MMP-3 catalytic domain (MMP-3cd), we aimed to evaluate the potential contributions of each TIMP domain to MMP affinity. The results of our analyses identify an unsuspected role for the TIMP-1 C-terminal domain in evolving MMP-3 affinity, mediated through synergistic effects between distant sites on the opposing TIMP domains. Protein structures of TIMP-1 mutants cocrystallized with MMP-3cd provide insights into the domain cooperativity of TIMP-1 variants with improved MMP-3cd binding. Our results have important implications for development of therapeutic TIMPs with optimized MMP-binding characteristics, while our conceptual approach may be more widely applicable to the application of directed evolution as a tool to probe domain interplay in other multidomain proteins.
Results
Screening of full-length TIMP-1 surface-displayed library identifies variants with improved binding to MMP-3
To explore the roles of each TIMP-1 domain in modulating MMP binding and inhibition, we first developed a platform for YSD of full-length human TIMP-1. The free, exposed N terminus of TIMP-1 is required for MMP binding and inhibition (6, 23); accordingly, we designed a fusion construct connecting the C terminus of TIMP-1 to the N terminus of yeast cell wall protein Aga2p, with secretion directed by the yeast α-factor signal sequence (Fig. 1A). Display of TIMP-1 bearing the correctly processed, mature N terminus was optimized by deletion of Glu-Ala residues from the signal sequence, which otherwise were inefficiently removed by yeast STE13 protease (Fig. 1). The optimized construct was N-terminally processed by yeast protease Kex2, resulting in efficient display of a functional TIMP-1 fusion that demonstrated robust MMP binding, as detected using biotinylated MMP-3cd (Figs. 1C and 2, A and D). MMP-3cd was selected as a model for the TIMP-1/MMP interaction as it offers a well-studied, well-behaved minimal MMP catalytic domain, and its interaction with TIMP-1 has been structurally characterized (6). We also evaluated the potential benefit of codon optimization of the human TIMP-1 gene for yeast but found minimal impact on efficiency of display (Fig. 1D).
Figure 1.
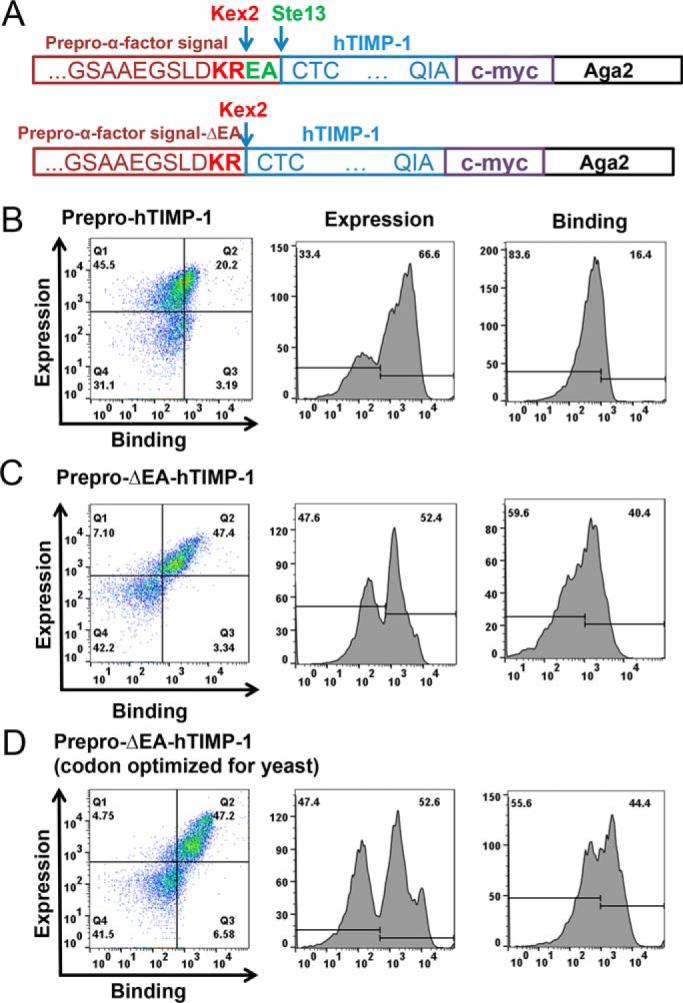
TIMP-1 yeast display and MMP-3cd binding optimization. A, TIMP-1 yeast surface display constructs in pCHA vector. Top construct (prepro-hTIMP-1), prepro-α-factor yeast signal with Kex2 (KR) and Ste13 (EA) cleavage sites fused to the mature N terminus of TIMP-1 followed by c-myc epitope tag fused to the N terminus of Aga2. Bottom construct (prepro-ΔEA-hTIMP-1), prepro-α-factor yeast signal with Kex2 (KR) cleavage site fused to the mature N terminus of TIMP-1 followed by c-myc epitope tag fused to the N terminus of Aga2. B–D, flow cytometry results for MMP-3cd binding to TIMP-1 constructs displayed on the yeast surface. Left panels, dual scatter plot representing MMP-3cd binding on the x axis and TIMP-1 expression (c-myc binding) on the y axis; Q2 represents the cell population dually labeled by biotinylated MMP-3cd and anti-c-myc binding. Middle panels, histograms of TIMP-1 expression (detected by anti-c-myc). Right panels, histograms of MMP-3cd binding. B, yeast display of human TIMP-1 (hTIMP-1) with Kex2 and Ste13 cleavage sites (prepro-hTIMP-1) shows high expression but only modest MMP-3cd binding, suggesting inefficient processing by Ste13. C, yeast display of human TIMP-1 with Kex2 cleavage site only (prepro-ΔEA-hTIMP-1) shows high expression and MMP-3dc binding, indicating more efficient processing of the mature N terminus of TIMP-1. D, yeast display of human TIMP-1, codon-optimized for yeast, with Kex2 cleavage site only shows expression and MMP-3cd binding similar to the noncodon-optimized construct.
Figure 2.

Screening a library of TIMP-1 mutants for MMP-3 binding. A, schematic diagram illustrates how a library of TIMP-1 mutants (blue/green) was displayed on the yeast surface. TIMP-1 expression was measured using fluorescent conjugated c-myc antibody (red star), and MMP-3 binding was measured using biotinylated MMP-3cd (orange) and fluorescent conjugated streptavidin (purple star). TIMP-1 variants with improved MMP-3cd binding were screened using FACS. B, library diversity was focused in 17 residues of TIMP-1 (loops in red), located in both the N-terminal (blue) and C-terminal (green) domains, that interact with bound MMP-3cd (orange) in PDB structure 1UEA. Targeted residues are located in the AB-loop, C-connector, and EF-loop of the N-terminal domain, and the GH-loop and MTL of the C-terminal domain. C, the WT TIMP-1 N- and C-terminal domain sequences are shown, colored in blue and green, respectively. Segments that interact with MMP-3cd in crystal structure 1UEA are annotated in black text above the sequence, including the N terminus and AB-loop, C-connector, EF-loop, GH-loop, and MTL. TIMP-1 residues diversified in the targeted library are highlighted in red and underlined. D, flow cytometry scatter plots of dually labeled yeast cells show lower MMP-3cd binding signal (x axis) for naïve library (center panel) relative to WT TIMP-1 (left panel); the population after three rounds of FACS sorting (right panel) shows greatly increased MMP-3 binding signal. P1, the diagonal sort gate, represents a population of yeast cells with a high ratio of MMP-3cd binding relative to TIMP-1 expression.
We next generated a targeted library of TIMP-1 mutants, incorporating diversity into MMP contact zones located on both domains of TIMP-1, including 8 residues within the N-terminal domain and 9 residues within the C-terminal domain (Fig. 2, B and C). The TIMP-1 mutant library was constructed aiming to achieve on average three to four mutations per variant through incorporation of degenerate codons and yielded a library size of ∼5 × 106 independent TIMP-1 variants in yeast upon transformation. The YSD TIMP-1 library was screened using fluorescence-activated cell sorting (FACS) by incrementally decreasing the concentration of biotinylated MMP-3cd in successive rounds of sorting, selecting for clones with enhanced affinity toward MMP-3cd (Fig. 2, A and D). After several rounds of FACS to enrich the pool for variants with highest MMP-3 binding relative to TIMP-1 expression, the enriched pool showed significant enhancement in MMP-3 binding compared with both the naïve library and WT TIMP-1 (Fig. 2D). The further enriched TIMP-1 variants isolated after six rounds of FACS screening, representing a total of four unique sequences, showed up to 10-fold binding improvement compared with WT TIMP-1 (Fig. 3, A–C).
Figure 3.

TIMP-1 variants with improved MMP-3 binding. A, flow cytometry scatter plots of dually labeled yeast cells are shown for four yeast-displayed TIMP-1 variants with improved MMP-3cd–binding activity; WT TIMP-1 is shown for reference on the left. The x axis (APC channel) represents biotinylated MMP-3cd binding (250 nm); the y axis (FITC channel) represents TIMP-1 expression. B, median fluorescence MMP-3cd binding signal, corrected for background and normalized to TIMP-1 expression, is plotted for each yeast-displayed TIMP-1 variant stained with 250 nm biotinylated MMP-3cd. Flow cytometry binding experiments were repeated at least twice; plotted values represent average ± S.D. (error bars). C, mutations found in TIMP-1 variants with improved MMP-3 binding are shown, along with their locations in the five targeted MMP-3–interacting loops of TIMP-1 (AB-loop, C-connector (C-conn), EF-loop, GH-loop, and MTL). *, p ≤ 0.05; **, p ≤ 0.01; ***, p ≤ 0.001.
Analysis of TIMP-1 single and double mutants reveals cooperative impact of N- and C-terminal domain mutations on MMP-3 binding
The high-affinity MMP-3-binding TIMP-1 variants obtained from library screening contained four to five mutations per gene; notably, all sequences contained mutations in both the N-terminal and C-terminal domains (Fig. 3C). To identify the mutations directly responsible for affinity improvements, we next generated single-mutant constructs representing the most prevalent mutations and assessed MMP-3cd binding on the yeast surface. Mutations L34G located in the AB-loop and T98D located in the EF-loop of the TIMP-1 N-terminal domain each conferred significant improvement (Fig. 4A), and yet the binding enhancements of these single mutants were modest by comparison with the composite mutants selected from the library (Fig. 3B). Interestingly, the L34G mutation co-occurred repeatedly in combination with Gly-154 mutations in the multiple-turn loop (MTL) of the C-terminal domain located ∼30 Å away. To evaluate the potential functional interaction of these distant residues, we next generated and evaluated several TIMP-1 double-mutant constructs. We found that although TIMP-1 single mutations affecting Gly-154 in isolation had no significant impact on MMP-3 binding (Fig. 4A), several substitutions at this position (Ala, His, and Lys) had a powerful synergistic effect when combined with the L34G mutation (Fig. 4, B–D). In each case examined, the L34G/G154X double mutants showed binding improvements indistinguishable from those of the corresponding library clones, suggesting that mutations at these two sites were sufficient to confer the full functional improvements of selected clones C1, C9, and C14. By contrast, TIMP-1 double mutants combining G154A/H with T98D did not demonstrate similar augmentation of MMP-3 binding affinity above that of the T98D single mutant (Fig. 4, E and F), suggesting that Gly-154 mutations modify MMP affinity in a context-dependent fashion rather than conferring an independent and additive effect.
Figure 4.
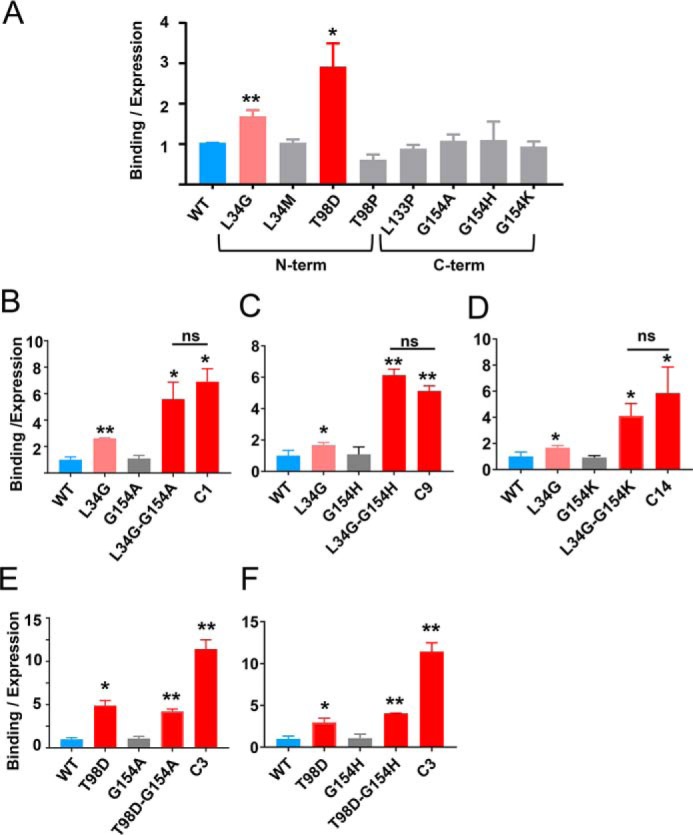
Dissecting functionally important mutations of TIMP-1 variants. A, among tested mutations found in the library-selected TIMP-1 variants with improved MMP-3cd–binding affinity, only L34G and T98D show functional enhancement as single mutations. B–D, Gly-154 mutations to Ala, His, or Lys cooperatively enhance MMP-3cd binding when combined with L34G. B, although not function-enhancing as a single mutation, G154A combined with L34G increases MMP-3cd binding nearly to the level of the composite library-selected variant C1 (L34G/L133P/L152C/G154A). C, likewise, G154H has no significant functional effect as a single mutation but cooperatively enhances MMP-3cd binding in combination with L34G, up to the level of the composite library-selected variant C9 (L34G/S68P/L133H/G154H). D, similarly, G154K offers functional benefit only in combination with L34G where it cooperatively enhances MMP-3cd binding nearly to the level of the composite library-selected variant C14 (L34G/T98P/L133N/S134M/G154K). E, by contrast, the G154A mutation does not confer further enhancement of MMP-3cd binding when combined with the T98D mutation. F, the G154H mutation likewise does not significantly improve MMP-3cd binding in the double T98D/G154H variant. Graphs show MMP-3cd binding to TIMP-1 expression ratio, based on median fluorescence corrected to background signal, for yeast-displayed TIMP-1 mutants stained with 250 nm biotinylated MMP-3cd. Flow cytometry binding experiments were repeated at least twice; plotted values represent average ± S.D. (error bars). *, p ≤ 0.05; **, p ≤ 0.01; ns, not significant.
To extend our findings and quantitatively evaluate protease-inhibitory activity of TIMP-1 variants in solution, we next cloned, expressed, and purified soluble forms of several of the mutant TIMP-1 proteins using a human cell expression system. We then assessed inhibition of MMP-3cd activity by soluble WT TIMP-1 and TIMP-1 variants L34G, L34G/G154A, and C1 using an enzyme inhibition assay that measured the reduced rate of cleavage of a fluorometric substrate in the presence of increasing concentrations of the inhibitor. Data were fitted to Morrison's tight binding equation (Equation 1) to determine equilibrium inhibition constants (Ki) (Fig. 5 and Table 1). Importantly, these solution studies corroborated the YSD findings; variant C1 showed >5-fold improvement compared with WT TIMP-1, an effect fully recapitulated by theTIMP-1-L34G/G154A variant (Table 1). Thus, we find consistent evidence from both solution studies and YSD demonstrating that TIMP-1 C-terminal domain mutations can act cooperatively with N-terminal domain mutations to modulate MMP binding and inhibition.
Figure 5.

Ki determination using Morrison fits of inhibition assays. Equilibrium inhibition constants (Ki) of purified soluble TIMP-1 and variants toward MMP-3cd were measured by the reduction in cleavage rates of a fluorogenic substrate in the presence of increasing concentration of the inhibitors. Data were plotted as initial velocities versus TIMP variant concentration and fitted by multiple regression to Morrison's tight binding inhibition equation as shown. The Ki value determined for each TIMP-1 variant is indicated on the plot and in Table 1. A, WT TIMP-1. B, TIMP-1-L34G. C, TIMP-1-L34G/G154A. D, TIMP-1-C1 (L34G/L133P/L151C/G154A).
Table 1.
Ki values for TIMP-1 variants
| TIMP-1 mutant | Ki |
|---|---|
| pm | |
| WT | 93.4 ± 8.1 |
| L34G | 33.5 ± 4.2 |
| L34G/G154A | 16.7 ± 1.5 |
| C1 | 17.8 ± 1.8 |
Crystal structures of TIMP-1 variants in complex with MMP-3 show conformational changes that stabilize interdomain interactions and MMP-3 binding
To gain structural insights into the effect of amino acid substitutions on the binding mechanism of TIMP-1 with MMP-3cd, we cocrystallized the TIMP-1-L34G and TIMP-1-C1 (L34G/L151C/L133P/G154A) mutants in complex with MMP-3cd and solved the crystal structures of the complexes. The TIMP-1-L34G/MMP-3cd and TIMP-1-C1/MMP-3cd structures were solved by molecular replacement and refined against diffraction data extending to resolutions of 2.37 and 2.67 Å, respectively. Data collection and refinement statistics are summarized in Table 2. Each complex shows the expected protein architecture of TIMP-1 and MMP-3cd as described previously for the complex with WT TIMP-1 (PDB code 1UEA) (6); however, notable deviations are seen in the vicinity of the mutated residues that can account for the observed improvements in binding and inhibition.
Table 2.
X-ray crystallographic data collection and refinement statistics
r.m.s.d., root mean square deviation.
| Crystal data | ||
| Structure name | TIMP-1-L34G/MMP-3cd | TIMP-1-C1/MMP-3cd |
| PDB code | 6MAV | 6N9D |
| Data collection | ||
| Resolution range (Å) | 52.59–2.37 (2.46–2.37) | 43.99–2.67 (2.77–2.67) |
| Space group | P 65 2 2 | P 65 2 2 |
| a, b, c (Å) | 69.70, 69.70, 321.33 | 70.03, 70.03, 319.51 |
| α, β, γ (°) | 90, 90, 120 | 90, 90, 120 |
| Rmerge | 0.066 (0.791) | 0.247 (1.838) |
| Rmeas | 0.069 (1.180) | 0.253 (1.882) |
| Rpim | 0.028 (0.328) | 0.055 (0.402) |
| CC1/2 | 0.999 (0.933) | 0.982 (0.668) |
| Multiplicity | 12.1 (12.7) | 20.4 (21.1) |
| Completeness (%) | 99.9 (100.0) | 99.5 (89.3) |
| Mean I/σ(I) | 19.1 (3.1) | 18.8 (2.2) |
| Refinement | ||
| Unique reflections used in refinement | 19,911 | 14,162 |
| Rwork/Rfree | 0.217/0.298 | 0.184/0.258 |
| Number of non-hydrogen atoms | 2,681 | 2,675 |
| Macromolecules | 2,643 | 2,624 |
| Ligands | 5 | 5 |
| Solvent | 33 | 46 |
| Protein residues | 335 | 336 |
| r.m.s.d. bonds (Å) | 0.010 | 0.009 |
| r.m.s.d. angles (°) | 1.35 | 1.30 |
| Ramachandran favored (%) | 91 | 94 |
| Ramachandran outliers (%) | 1.2 | 0.3 |
| Average B-factor | 77.60 | 55.10 |
| Macromolecules | 77.70 | 55.20 |
| Ligands | 69.80 | 51.60 |
| Solvent | 71.30 | 49.20 |
A striking feature observed in both mutant structures, proximal to the L34G mutation at the enzyme/inhibitor interface, is a reciprocal “clasp” formed by Tyr-35 of TIMP-1 and Tyr-153 of MMP-3 (Fig. 6, A and B). The two Tyr rings, one from each protein, stretch across the interface to form H-bonds with the partner protein, TIMP-1 Tyr-35 with the MMP-3 Phe-154 backbone carbonyl and MMP-3 Tyr-155 with the TIMP-1 Gly-34 backbone carbonyl. Additionally, the two Tyr aromatic rings form hydrophobic and edge-to-face π interactions bridging the interface. Formation of this reciprocal tyrosine clasp is facilitated by substantial backbone displacements in the TIMP-1-L34G AB-loop and the MMP-3 S-loop compared with the WT TIMP-1/MMP-3cd complex (Fig. 6C). In the WT TIMP-1 structure, bulkier Leu-34 pushes these two loops apart, preventing Tyr-35 of TIMP-1 from H-bonding to the Phe-154 carbonyl, whereas in the L34G mutant structure the distance between oxygens is shortened from 3.5 to 3 Å. The H-bond between MMP-3 Tyr-155 and TIMP-1 Gly-34 is already present in the WT TIMP-1 complex with a distance of 2.6 Å between oxygens; however, the shift of the MMP-3 S-loop in the mutant complexes enables this short H-bond to be cinched even tighter in the TIMP-1 mutant complexes (Fig. 6).
Figure 6.

Crystal structure of TIMP-1-L34G mutant bound to MMP-3cd. A, cartoon representation of the TIMP-1-L34G/MMP-3cd complex crystal structure is shown (right); TIMP-1-L34G is in blue (N-terminal domain) and green (C-terminal domain) with the mutated residue in yellow, and MMP-3cd is in orange with the catalytic zinc ion shown as a gray sphere. The inset panel (left) shows interactions of TIMP-1-L34G Tyr-35 and MMP-3cd Tyr-153, which form a reciprocal tyrosine clasp at the binding interface. B, stick representation of the protein environment surrounding the reciprocal tyrosine clasp with 2Fo − Fc map contoured at 1.5σ. C, superposition of the mutant TIMP-1-L34G/MMP-3cd crystal structure (colored as above) with the WT TIMP-1/MMP-3cd structure (PDB code 1UEA; shown in pale blue/pale orange) highlights the conformational changes in the AB-loop of TIMP-1-L34G, which result in the cinching of the reciprocal tyrosine clasp.
The TIMP-1-C1/MMP-3cd crystal structure reflects near-identical conformational changes in the TIMP-1 AB-loop and MMP-3cd S-loop, resulting from the L34G mutation, with consequent cinching of the reciprocal tyrosine clasp. In addition, mutations in the C-terminal domain of TIMP-1 produce significant conformational changes in this domain, the effect of which is to strengthen interactions both with the TIMP-1 N-terminal domain and with MMP-3cd. TIMP-1-C1 mutation G154A results in adoption of an α-helical conformation by residues 154–157 of the multiple-turn loop at the interface between the TIMP-1 N- and C-terminal domains (Fig. 7, A, D, and E). Glycine, the native TIMP-1 residue at this position, is known to destabilize α-helical secondary structures (24), and each of the mutations of this residue identified in our library screen, to Ala, His, and Lys, are predicted to confer greater propensity for α-helix formation (25, 26), explaining the surprising array of varied substitutions selected at this position. This alteration in secondary structure allows the two domains of TIMP-1 to pack together more tightly and introduces a new H-bond stabilizing the interdomain interaction (Fig. 7D). Additional conformational alterations in the TIMP-1-C1 structure result in closer contact with MMP-3cd, including hydrophobic contacts of TIMP-1-C1 Leu-152 with MMP-3 Thr-215, Tyr-220, and Leu-222 side chains (Fig. 7B) and a new intradomain H-bond between TIMP-1 residues Gln-150 and Pro-133 (Fig. 7C). In sum, the stabilizing effects of the mutations on TIMP-1 C-terminal domain secondary structure and on interdomain interactions serve to integrate the domains of this multidomain protein, producing functional cooperativity, which in turn improves MMP-3cd binding.
Figure 7.

Crystal structure of TIMP-1-C1 variant bound to MMP3cd. A, cartoon representation of the TIMP-1-C1/MMP-3cd complex crystal structure is shown using the same color scheme described for Fig. 6. Altered inter- and intramolecular interactions attributable to TIMP-1-C1 mutations are highlighted in B–D. B, a hydrophobic cluster is formed at the intermolecular interface between TIMP-1 Leu-152 and MMP-3cd residues Thr-215, Tyr-220, and Leu-222. C, a new H-bond within the TIMP-1-C1 C-terminal domain is formed between the carbonyl of Pro-133 and the side chain of Gln-150. D, as a consequence of the G154A mutation, residues 154–157 adopt an α-helical conformation, and a new interdomain H-bond is formed between the side chain of Ser-155 in the C-terminal domain and the carbonyl of Pro-6 in the N-terminal domain. E, superposition of the TIMP-1-C1/MMP-3cd crystal structure (colored as above) with the WT TIMP-1/MMP-3cd structure (PDB code 1UEA; shown in pale blue/pale orange) highlights conformational changes in the multiple-turn loop of TIMP-1-C1, which facilitate stabilizing interdomain interactions within TIMP-1 as well as favorable hydrophobic interactions at the interface with MMP-3cd. F, stick representation of the protein environment surrounding the hydrophobic cluster at the intermolecular interface with 2Fo − Fc map contoured at 1.0σ.
Discussion
Despite the common belief that the N-terminal domain of a TIMP is solely responsible for MMP binding and inhibition, here we have found that the C-terminal domain of TIMP-1 and the interplay between the two domains play a significant role in MMP-3 binding. We further have shown that not only the MMP-contacting interface but also the interactions between TIMP domains can be optimized in an integral fashion to achieve desired binding characteristics. Prior studies seeking to define and reshape the protease-inhibitory function of TIMPs have focused on the N-terminal domain due to the early finding that the isolated N-TIMP domain is independently capable of MMP inhibition with only modest loss of affinity compared with full-length TIMPs (7, 12). Site-directed mutagenesis experiments identified several key amino acid residues, mutation of which can shift the inhibition specificity of N-TIMPs (27–35). More recently, directed evolution efforts have employed phage display or YSD screening of N-TIMP-2 libraries to more broadly scan the sequence determinants of binding specificity at the N-TIMP/MMP interface (20–22, 36). These studies have shown TIMPs to be malleable scaffolds, tolerant of mutation and relatively easy to reengineer for an altered spectrum of protease-inhibitory activity. However, prior work has overlooked the potential functional role of the TIMP C-terminal domain, which can contribute up to a third of the contact surface area at the TIMP/MMP interface (11). Furthermore, the more complex tertiary structures of multidomain proteins are often accompanied by more complex functional capabilities (1) where cooperation between domains renders an intact protein as more than the sum of its parts. Here, by applying evolutionary pressure to a library of full-length TIMP-1 variants, diversified in both domains, we have uncovered such functional cooperativity between TIMP domains.
Our structural analyses of TIMP-1 variants lend insight into how TIMP mutations at the periphery of the interface with an MMP, in both the N- and C-terminal domains, can stabilize binding interactions. Within the N-terminal domain AB-loop, we identified the L34G mutation, which facilitates tighter cinching of a reciprocal tyrosine clasp involving Tyr-35 of TIMP-1 and Tyr-153 of MMP-3. Our structures demonstrate shortening of one intermolecular H-bond and formation of a new additional H-bond. H-bonds have long been appreciated as major contributors to protein–protein binding affinity and specificity (37, 38), where the strength of H-bonds generally correlates well with interatomic distance between the proton donor and acceptor (39). Tyrosine residues in particular are highly represented in protein/protein interaction hot spots (40, 41), likely due to the ability to form aromatic π interactions as well as H-bonds, both of which are evidenced in our structures. Within the C-terminal domain, we identified the G154A mutation, responsible for conformational changes in the multiple-turn loop that impact both the binding interface with MMP-3cd and the interface between the TIMP domains. The interface with MMP-3cd gains new hydrophobic stabilizing contacts, while the interdomain interface is stabilized by the defined α-helical secondary structure adopted by TIMP residues 154–157 and by a new interdomain H-bond. Interestingly, TIMP-1 Gly-154, a residue with low helical propensity, was found mutated to Ala, Lys, and His, residues with higher helical propensities (25, 26), in three of four isolated clones with enhanced MMP-3cd–binding affinity. Given that we observed a function-enhancing role for these substitutions only in combination with the L34G mutation, it may be that a G154X mutation alone is insufficient to fully stabilize the helical turn but requires additional structural stability conferred by the reciprocal tyrosine clasp on the opposite side of the intermolecular interface.
Because MMPs represent high-value but challenging therapeutic targets, our results suggest new avenues for engineering MMP-targeted therapeutic proteins. Many human MMPs contribute to pathology of cancer, fibrosis, arthritis, cardiovascular, pulmonary, and other diseases when improperly expressed (23, 42, 43), but clinical trials of broad-spectrum MMP inhibitors in cancer and arthritis proved disappointing (44, 45). Engineered designer TIMPs have been viewed as a possible pathway to achieve greater selectivity and efficacy (23, 46, 47), and recent campaigns to develop such selectivity have employed YSD platforms for directed evolution of N-TIMP-2 aided by computational library design (20–22). Our present findings suggest that future TIMP engineering efforts may benefit from employing a full-length TIMP scaffold and, further, that maximum advantage of the multidomain scaffold may be best achieved through simultaneous optimization to capture cooperative mutations across domains.
Finally, the utility of our approach for identifying and engineering domain cooperativity may extend beyond the TIMP family and have relevance for other systems. Prior studies have demonstrated how connectivity and cooperativity of multiple domains contribute to protein folding, stability, and function (2, 48, 49). Dynamic interactions between individual domains orchestrate folding pathways, determining kinetic and equilibrium properties of cooperative folding (49). Favorable interactions between domains can dictate the stability of individual domain structures (48), and thus interdomain interactions have been critical in the natural evolution of function in multidomain proteins. Directed evolution can recapitulate this natural evolution of multidomain functionality, e.g. when a low-affinity peptide-binding PDZ domain was linked to an unrelated, functionally inert fibronectin type III domain (FN3), optimization of FN3 led to a highly selective domain-interdependent “affinity clamp” (1). To date, however, the ability to pinpoint specific sequence and structural determinants of domain cooperativity has been limited. Here, directed evolution simultaneously targeting residues within both domains of TIMP-1 enabled us to search for and identify pairs of residues responsible for interdomain cooperativity in TIMP-1 function. This strategy may be more broadly useful to detect domain cooperativity, probing the structure–function paradigm, and to engineer enhanced synergistic function into other multidomain proteins for varied practical applications.
Experimental procedures
Strains and plasmids
The yeast Saccharomyces cerevisiae strain EBY100 (MATa AGA1::GAL1-AGA1::URA3 ura3–52 trp1 leu2-Δ200 his3-Δ200 pep4::HIS3 prb11.6R can1 GAL) was used for yeast surface display of human TIMP-1 protein and its variants. Yeast display plasmids for displaying WT TIMP-1 and TIMP-1 mutants at the N terminus of Aga2p protein on the yeast surface were derived from the pCHA backbone, pCHA-VRC01 vector (50) purchased from Addgene.
Yeast surface display of TIMP-1
pCHA-TIMP-1 plasmids (the yeast display vectors) were transformed into the yeast strain EBY100 by electroporation using a Bio-Rad Gene Pulser and 2-mm electroporation cuvettes. The yeast cells were grown in minimal SD-CAA medium, pH 6 (20 g/liter dextrose, 6.7 g/liter yeast nitrogen base lacking amino acids, 5.4 g/liter Na2HPO4, 8.6 g/liter NaH2PO4·H2O, and 5 g/liter Bacto casamino acids). After 16 h growth at 30 °C, cells were pelleted at 3000 × g and resuspended in SGR-CAA medium (20 g/liter galactose, 6.7 g/liter yeast nitrogen base lacking amino acids, 5.4 g/liter Na2HPO4, 8.6 g/liter NaH2PO4·H2O, 5 g/liter Bacto casamino acids, and 20 g/liter raffinose) for induction. Cells were grown in SGR-CAA medium at 30 °C for 16 h before harvesting. The yeast cells displaying TIMP-1 variants were collected at an OD600 of 0.2 and washed with cold PBSA buffer (8 g/liter NaCl, 0.2 g/liter KCl, 1.44 g/liter Na2HPO4, 0.24 g/liter KH2PO4, pH 7.4, and 1% BSA). The cells were first incubated with 100–250 nm biotinylated MMP-3cd and mouse anti-c-myc 9e10 (Sigma) for 1 h on ice. The cells were then washed and resuspended in 100 μl of cold PBSA buffer containing fluorescein-conjugated goat anti-mouse antibody (Thermo Fisher) and streptavidin-Alexa Fluor 647 (each at 1:100 dilution) and incubated on ice for 30 min. After harvesting and washing with cold PBSA buffer, the cells were centrifuged and resuspended in 750 μl of PBSA buffer. Flow cytometry data were then collected using an Attune NxT flow cytometer from at least 10,000 cell events per sample and analyzed using FlowJo software (FlowJo, LLC).
Generating the targeted library of TIMP-1 mutants
A library of human TIMP-1 gene variants, targeting for mutation 17 residues in five loops shown to interact with MMP catalytic domains in crystal structures (Fig. 2C), was purchased from Invitrogen GeneArt Gene Synthesis. The library of mutants was constructed by assembling gene blocks of synthetic oligonucleotides with 13% chance of NNS degenerate codon incorporation at each position targeted for mutation (where N = any nucleotide and S = G or C), to achieve on average three to four mutations per variant. The yeast display TIMP-1 mutant library was generated by PCR amplification of the TIMP-1 gene from the targeted TIMP-1 library using primers with 50-bp overhangs containing homology to sequence upstream and downstream of the TIMP-1 gene in the pCHA-TIMP-1 yeast display vector. PCR products of TIMP-1 gene variants were gel-purified and concentrated using Pellet Paint® Co-Precipitant (EMD Millipore) following the manufacturer's protocol. The pCHA-TIMP-1 vector was double-digested using BsrGI and BamHI restriction enzymes, and the pCHA digested vector (1 μg) and purified PCR product (5 μg) of the TIMP-1 mutant library were mixed and electrotransformed into yeast cells as described previously (51). Five separate electrotransformations were performed and combined. The yeast cells were then resuspended in cold YPD medium (20 g/liter glucose, 20 g/liter peptone, and 10 g/liter yeast extract) and grown at 30 °C with shaking for 1 h. The library size was estimated by plating yeast transformants in serial dilution and found to contain about 5 × 106 independent variants. The yeast cells containing the library of TIMP-1 mutants were pelleted and resuspended in 50 ml of SD-CAA medium, pH 4.5 (same as SD-CAA medium described above, but phosphate components were substituted with 13.7 g/liter sodium citrate dehydrate and 8.4 g/liter citric acid anhydrous) and grown for 16 h at 30 °C with shaking.
Screening the yeast-displayed TIMP-1 library using FACS
Prior to each round of sorting, yeast cells were grown and induced; counted (OD600 of 1 = 107 cells/ml); incubated with biotinylated MMP-3cd followed by anti-c-myc, streptavidin, and secondary antibody; washed; and suspended in PBSA buffer as described above under “Yeast surface display of TIMP-1.” Samples were maintained on ice and covered from light until loading onto a flow cytometer for analysis (BD Accuri) or library sorting (BD Aria II). Sorted cells were recovered in SD-CAA, pH 4.5, containing 1% penicillin-streptavidin and incubated at 30 °C overnight. Yeast surface protein expression was then induced by culturing cells in SGR-CAA medium at 30 °C overnight. The initial library (∼5 × 107 yeast) was subjected to two rounds of screening (staining, sorting, recovery, and regrowth) using a concentration of 100 nm biotinylated MMP-3cd. Screening was continued through four additional rounds, for a total of six rounds, under equilibrium sorting conditions employing incrementally decreasing MMP-3cd concentration from 50 to 5 nm.
DNA sequencing
Following rounds of sorting, recovered yeast were cultured on SD-CAA plates, and plasmid DNA was extracted from the isolated individual yeast clones using the ZymoprepTM yeast plasmid miniprep II kit (ZymoResearch). TIMP-1 mutant plasmids extracted from yeast were transformed into Escherichia coli cells and plated on LB-Amp (100 μg/ml ampicillin). The pCHA-TIMP-1 variant plasmid was then extracted and purified from the bacteria using the Qiagen Miniprep kit according to the manufacturer's protocol (Qiagen) and sequenced (Eurofins Scientific) with primers up- and downstream of the TIMP-1 gene. Sequences were analyzed using SnapGene software (SnapGene, GSL Biotech, LLC).
Soluble TIMP expression and purification
The pTT-TIMP-1 vector, a generous gift from Dr. B. Toussaint (Grenoble, France), was used for expression of full-length human TIMP-1 (52). TIMP-1 mutants were amplified from the corresponding yeast display vector (pCHA-TIMP-1) using PCR and inserted between HindIII and BamHI restriction sites of the pTT-TIMP-1 vector. WT TIMP-1 and TIMP-1 variants were expressed in HEK293-FreeStyle cells (Thermo Fisher Scientific) in Gibco® FreeStyleTM 293 Expression Medium (Thermo Fisher Scientific) on a shaker incubator at 37 °C and 8% CO2. The HEK293-FreeStyle cells were grown to a cell density of 1–1.5 × 106 in 300-ml culture, transfected with 300 μg of pTT-TIMP-1 plasmid using 0.9 ml of 1 mg/ml polyethylenimine (PEI) transfection reagent, and incubated on the shaker incubator for 72 h before harvesting the cells and collecting the supernatant media. For purification, the clarified medium was concentrated, dialyzed into buffer A (50 mm HEPES, pH 6.8, and 25 mm NaCl), and purified over an SP-Sepharose column using a linear gradient of buffer B (50 mm HEPES, pH 6.8, and 0.5 m NaCl). Fractions containing the WT or mutant TIMP-1 were assessed by silver-stained SDS-PAGE. To prepare homogenous purified proteins for crystallization, TIMP-1 variants were then enzymatically deglycosylated with peptide:N-glycosidase F (New England Biolabs). Deglycosylation of TIMP-1 protein was confirmed by SDS-PAGE followed by purification by size-exclusion chromatography using a Superdex-75 column (GE Healthcare) that was equilibrated and eluted with 50 mm HEPES, pH 6.8, containing 150 mm NaCl. The highly pure deglycosylated TIMP-1 fractions identified by silver-stained SDS-PAGE were combined and concentrated using Amicon Ultra-15 centrifugal filter units with a molecular weight cutoff of 10,000.
MMP expression, purification, and biotinylation
ProMMP-3cd was expressed from E. coli using a pET3a construct featuring human proMMP-3 lacking the C-terminal hemopexin domain, a generous gift of H. Nagase (53). ProMMP-3cd was refolded, purified, and activated as described previously (53). Briefly, recombinant protein was extracted from inclusion bodies in a solution containing 8 m urea, 20 mm Tris-HCl, pH 8.6, 20 mm DTT, and 50 μm ZnCl2. The protein was purified on Q-Sepharose equilibrated with 8 m urea, 20 mm Tris-HCl, pH 8.6, and 50 μm ZnCl2 and eluted using a linear gradient of NaCl to 0.5 m. Fractions containing proMMP-3cd were combined, diluted to an A280 of 0.3, and refolded by stepwise dialysis with 50 mm Tris-HCl, pH 7.5, 10 mm CaCl2, and 150 mm NaCl. Subsequently, purified proMMP-3cd was activated overnight in the presence of the organomercurial compound 4-aminophenyl mercuric acetate (1 mm at 37 °C). Concentrations of active MMP-3cd were determined by titration against a TIMP-1 reference stock as described previously (54). MMP-3cd was biotinylated using the EZ-Link NHS-PEG4 biotinylation kit (Thermo Scientific) according to the manual with addition of biotin in a 1:10 molar ratio (protein:biotin) and incubated at room temperature for 30 min. The biotinylated MMP-3cd was purified using Zeba spin desalting columns (Thermo Scientific) and tested for degree of biotinylation using the 4′-hydroxyazobenzene-2-carboxylic acid assay as described in the kit protocol.
Active TIMP-1 concentration determination by titration
Concentrations of purified TIMP-1 variants were determined by titration against a reference stock of MMP-3cd previously titrated against WT TIMP-1, essentially as described previously (54). Briefly, MMP-3cd (200 nm) was preincubated for 1 h at 25 °C with a range of substoichiometric concentrations of the TIMP-1 variant. MMP/TIMP mixtures were then assayed for residual proteolytic activity using colorimetric thiopeptolide substrate Ac-Pro-Leu-Gly-[2-mercapto-4-methylpentanoyl]-Leu-Gly-OC2H5 (Enzo Life Sciences). MMP/TIMP mixtures were diluted 40-fold into a reaction cuvette containing the thiopeptolide substrate at a final concentration 100 μm in 50 mm HEPES, pH 6.0, 10 mm CaCl2, 0.05% Brij-35, and 1 mm 5,5′-dithiobis(2-nitrobenzoic acid). Reactions were followed for 10 min at 37 °C on a Varian spectrophotometer (Thermo Scientific), and linear initial rates were measured as an increase in the absorbance at 410 nm (ϵ410 = 13,600 m−1 cm−1). Data were fitted using linear regression analyses and extrapolated to the stoichiometric equivalence point in Prism 7 (GraphPad Software, San Diego, CA).
TIMP-1/MMP-3 inhibition studies
Equilibrium inhibition constants (Ki) of WT TIMP-1 and TIMP-1 variants toward MMP-3 were measured using a method appropriate for tight binding inhibition, similar to a method described previously (21). MMP-3cd (0.24 nm) was incubated with 0.04-2.5 nm WT TIMP-1 or TIMP-1 variant in TCNB buffer (50 mm Tris, pH 7.5, 100 mm NaCl, 10 mm CaCl2, and 0.05% Brij) for 1 h at 37 °C. Thereafter, the fluorogenic substrate Mca-Pro-Leu-Gly-Leu-Dpa-Ala-Arg-NH2 (where Mca is (7-methoxycoumarin-4-yl)acetyl and Dpa is N-3-(2,4-dinitrophenyl)-l-2,3-diaminopropionyl) (Anaspec, CA) was added to the reaction at a final concentration of 10 μm, and fluorescence was monitored with 340/30 excitation and 400/30 emission filters using a Synergy HT plate reader (BioTek, Winooski, VT) at 37 °C. Fluorescence readings were recorded every minute for 120 min, and enzymatic rates were determined from the slope of the linear portion of the fluorescence signal. To determine Ki, data were plotted as initial velocities versus TIMP-1 or variant concentration and fitted by multiple regression to Morrison's tight binding inhibition equation (55) (Equation 1) where Vt is enzyme velocity in the presence of inhibitor, V0 is enzyme velocity in the absence of inhibitor, [E] is enzyme concentration, [I] is inhibitor concentration, [S] is substrate concentration, Km is the Michaelis–Menten constant, and Kiapp is an apparent inhibition constant given by Equation 2. Data were fitted using Prism 7 (GraphPad Software) (Fig. 5). Reported inhibition constants are average values obtained from two independent experiments, each with duplicate samples. Calculations were performed using a Km value of 11.23 μm for MMP-3cd as determined from three Michaelis–Menten kinetic experiments in our laboratory.
| (Eq. 1) |
| (Eq. 2) |
Crystallization and X-ray diffraction
TIMP-1 variants and MMP-3cd protein were mixed at a 1:1 molar ratio, and TIMP-1 variant/MMP-3cd protein complexes were concentrated to 2.8–4.8 mg/ml prior to crystallization screening. Crystallization was conducted using the hanging-drop method with the protein solution mixed 1:1 (v/v) with a reservoir solution. TIMP-1-L34G/MMP-3cd protein crystals were grown in condition 92 of the Anatrace Top96 crystallization kit (Anatrace) (0.1 m ammonium acetate, 0.1 m Bis-Tris HCl, pH 5.5, and 17% (w/v) PEG 10,000). TIMP-1-C1/MMP-3cd protein crystals were grown in condition 41 of the Anatrace Top96 crystallization kit (Anatrace) (0.2 m sodium acetate, 0.1 m sodium cacodylate HCl, pH 6.5, and 18% (w/v) PEG 8000). TIMP-1 variant/MMP-3cd protein crystals appeared over 48 h and were grown over a few weeks. Crystals were flash-cooled in liquid nitrogen using cryoprotectant buffer matched to the crystallization reservoir solution and additionally containing 30% dextrose. Single-wavelength (1.0 Å) native X-ray diffraction data were collected at 100 K at Advanced Light Source beamline 8.2.2, Lawrence Berkeley National Laboratory. TIMP-1 variant complex structures were each solved from single crystals that diffracted to 2.37 Å (for TIMP-1-L34G) and 2.67 Å resolution (for TIMP-1-C1). The X-ray data were processed with xia2 (56) using DIALS (57) for indexing, refinement, and integration with POINTLESS (58) and AIMLESS (59) for scaling and merging. Rfree flags were assigned to a random 5% of reflections, and this test set was maintained throughout all subsequent stages of structure solution and refinement.
Structure determination and refinement
X-ray crystal structures of the protein complexes of MMP-3cd/TIMP-1 variants were solved using CCP4 software by molecular replacement using the program Molrep (60). The previously solved structure of human MMP-3cd/WT TIMP-1 (PDB code 1UEA) without the corresponding Zn and Ca ions was used as a search model. Following molecular replacement, Phenix.refine was used for sequential automated refinements (61) alternating with manual adjustments in Coot (62). Because of the flexibility of MMP-3cd and TIMP-1 protein segments and the possibility that they might demonstrate local trajectories of motion partially decoupled from the larger protein domains, we employed TLS refinement using the automatic TLS group finding tool in the Phenix software for refinement of both structures. Several residues in the TIMP-1 variant chains showed only weak and indistinct density and were left unmodeled in the final models, including residues Gly-52, Asp-53, Ala-54, and Ala-55 in both structures and additionally Leu-51 in the TIMP1-L34G/MMP-3cd structure. The final stage of restrained refinement included water molecules with peaks greater than 1σ and within acceptable H-bonding distances from neighboring protein atoms. The coordinates and structure factors for the TIMP-1-L34G/MMP-3cd and TIMP-1-C1/MMP-3cd structures have been submitted to the Worldwide Protein Data Bank under the accession codes 6MAV and 6N9D, respectively. Structure figures were generated using PyMOL (Schrödinger, LLC).
Author contributions
M. R.-S., G. P. D., D. C. R., and E. S. R. conceptualization; M. R.-S. and B. S. data curation; M. R.-S. and K. A. G. formal analysis; M. R.-S. and E. S. R. validation; M. R.-S., K. A. G., B. S., and E. S. R. investigation; M. R.-S. and E. S. R. visualization; M. R.-S. and B. S. methodology; M. R.-S. and E. S. R. writing-original draft; M. R.-S. and E. S. R. writing-review and editing; B. S. resources; G. P. D., D. C. R., and E. S. R. funding acquisition; E. S. R. supervision; E. S. R. project administration.
Acknowledgments
We thank Dr. Laura Lewis-Tuffin at the cell sorting facility at Mayo Clinic, Florida for help with FACS of TIMP-1 libraries; Rachel Henin and Alexandra Hockla for MMP-3 protein expression, purification, and biotinylation; and Matt Coban for advice with crystallographic data processing. We also thank Dr. Niv Papo for helpful discussions. Diffraction data were measured at beamline 8.2.2 of the Berkeley Center for Structural Biology, Advanced Light Source (ALS), Lawrence Berkeley National Laboratory. The Berkeley Center for Structural Biology is supported in part by the Howard Hughes Medical Institute. The Advanced Light Source is a Department of Energy Office of Science User Facility under Contract DE-AC02-05CH11231. The ALS-ENABLE beamlines are supported in part by the National Institutes of Health, National Institute of General Medical Sciences, Grant P30 GM124169.
This work was supported by National Institutes of Health Grant R21 CA205471 (to E. S. R.) and United States Department of Defense Grant W81XWH-16-2-0030 (to G. P. D. and D. C. R.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
The atomic coordinates and structure factors (codes 6MAV and 6N9D) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- TIMP
- tissue inhibitor of metalloproteinases
- MMP
- matrix metalloproteinase
- YSD
- yeast surface display
- MMP-3cd
- MMP-3 catalytic domain
- MTL
- multiple-turn loop
- FN3
- fibronectin type III domain
- TLS
- translation/libration/screw
- PDB
- Protein Data Bank
- Bis-Tris
- 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol.
References
- 1. Huang J., Koide A., Makabe K., and Koide S. (2008) Design of protein function leaps by directed domain interface evolution. Proc. Natl. Acad. Sci. U.S.A. 105, 6578–6583 10.1073/pnas.0801097105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Vogel C., Bashton M., Kerrison N. D., Chothia C., and Teichmann S. A. (2004) Structure, function and evolution of multidomain proteins. Curr. Opin. Struct. Biol. 14, 208–216 10.1016/j.sbi.2004.03.011 [DOI] [PubMed] [Google Scholar]
- 3. Bagowski C. P., Bruins W., and Te Velthuis A. J. (2010) The nature of protein domain evolution: shaping the interaction network. Curr. Genomics 11, 368–376 10.2174/138920210791616725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Brew K., and Nagase H. (2010) The tissue inhibitors of metalloproteinases (TIMPs): an ancient family with structural and functional diversity. Biochim. Biophys. Acta 1803, 55–71 10.1016/j.bbamcr.2010.01.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Batra J., and Radisky E. S. (2014) Tissue inhibitors of metalloproteinases (TIMPs): inhibition of Zn-dependent metallopeptidases, in Encyclopedia of Inorganic and Bioinorganic Chemistry (Scott R. A., ed) John Wiley and Sons, Chichester, UK [Google Scholar]
- 6. Gomis-Rüth F. X., Maskos K., Betz M., Bergner A., Huber R., Suzuki K., Yoshida N., Nagase H., Brew K., Bourenkov G. P., Bartunik H., and Bode W. (1997) Mechanism of inhibition of the human matrix metalloproteinase stromelysin-1 by TIMP-1. Nature 389, 77–81 10.1038/37995 [DOI] [PubMed] [Google Scholar]
- 7. Murphy G., Houbrechts A., Cockett M. I., Williamson R. A., O'Shea M., and Docherty A. J. (1991) The N-terminal domain of tissue inhibitor of metalloproteinases retains metalloproteinase inhibitory activity. Biochemistry 30, 8097–8102 10.1021/bi00247a001 [DOI] [PubMed] [Google Scholar]
- 8. Morgunova E., Tuuttila A., Bergmann U., and Tryggvason K. (2002) Structural insight into the complex formation of latent matrix metalloproteinase 2 with tissue inhibitor of metalloproteinase 2. Proc. Natl. Acad. Sci. U.S.A. 99, 7414–7419 10.1073/pnas.102185399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Jung K. K., Liu X. W., Chirco R., Fridman R., and Kim H. R. (2006) Identification of CD63 as a tissue inhibitor of metalloproteinase-1 interacting cell surface protein. EMBO J. 25, 3934–3942 10.1038/sj.emboj.7601281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Fernandez C. A., Roy R., Lee S., Yang J., Panigrahy D., Van Vliet K. J., and Moses M. A. (2010) The anti-angiogenic peptide, loop 6, binds insulin-like growth factor-1 receptor. J. Biol. Chem. 285, 41886–41895 10.1074/jbc.M110.166439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Batra J., Soares A. S., Mehner C., and Radisky E. S. (2013) Matrix metalloproteinase-10/TIMP-2 structure and analyses define conserved core interactions and diverse exosite interactions in MMP/TIMP complexes. PLoS One 8, e75836 10.1371/journal.pone.0075836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Huang W., Suzuki K., Nagase H., Arumugam S., Van Doren S. R., and Brew K. (1996) Folding and characterization of the amino-terminal domain of human tissue inhibitor of metalloproteinases-1 (TIMP-1) expressed at high yield in E. coli. FEBS Lett. 384, 155–161 10.1016/0014-5793(96)00304-3 [DOI] [PubMed] [Google Scholar]
- 13. Romero P. A., and Arnold F. H. (2009) Exploring protein fitness landscapes by directed evolution. Nat. Rev. Mol. Cell Biol. 10, 866–876 10.1038/nrm2805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Bonsor D. A., and Sundberg E. J. (2011) Dissecting protein-protein interactions using directed evolution. Biochemistry 50, 2394–2402 10.1021/bi102019c [DOI] [PubMed] [Google Scholar]
- 15. Yuen C. M., and Liu D. R. (2007) Dissecting protein structure and function using directed evolution. Nat. Methods 4, 995–997 10.1038/nmeth1207-995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Boder E. T., and Wittrup K. D. (1997) Yeast surface display for screening combinatorial polypeptide libraries. Nat. Biotechnol. 15, 553–557 10.1038/nbt0697-553 [DOI] [PubMed] [Google Scholar]
- 17. Boder E. T., Raeeszadeh-Sarmazdeh M., and Price J. V. (2012) Engineering antibodies by yeast display. Arch. Biochem. Biophys. 526, 99–106 10.1016/j.abb.2012.03.009 [DOI] [PubMed] [Google Scholar]
- 18. Mei M., Zhou Y., Peng W., Yu C., Ma L., Zhang G., and Yi L. (2017) Application of modified yeast surface display technologies for non-antibody protein engineering. Microbiol. Res. 196, 118–128 10.1016/j.micres.2016.12.002 [DOI] [PubMed] [Google Scholar]
- 19. Raeeszadeh-Sarmazdeh M., Patel N., Cruise S., Owen L., O'Neill H., and Boder E. T. (2019) Identifying stable fragments of Arabidopsis thaliana cellulose synthase subunit 3 by yeast display. Biotechnol. J. 14, e1800353 10.1002/biot.201800353 [DOI] [PubMed] [Google Scholar]
- 20. Arkadash V., Radisky E. S., and Papo N. (2018) Combinatorial engineering of N-TIMP2 variants that selectively inhibit MMP9 and MMP14 function in the cell. Oncotarget 9, 32036–32053 10.18632/oncotarget.25885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Arkadash V., Yosef G., Shirian J., Cohen I., Horev Y., Grossman M., Sagi I., Radisky E. S., Shifman J. M., and Papo N. (2017) Development of high affinity and high specificity inhibitors of matrix metalloproteinase 14 through computational design and directed evolution. J. Biol. Chem. 292, 3481–3495 10.1074/jbc.M116.756718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Shirian J., Arkadash V., Cohen I., Sapir T., Radisky E. S., Papo N., and Shifman J. M. (2018) Converting a broad matrix metalloproteinase family inhibitor into a specific inhibitor of MMP-9 and MMP-14. FEBS Lett. 592, 1122–1134 10.1002/1873-3468.13016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Radisky E. S., Raeeszadeh-Sarmazdeh M., and Radisky D. C. (2017) Therapeutic potential of matrix metalloproteinase inhibition in breast cancer. J. Cell. Biochem. 118, 3531–3548 10.1002/jcb.26185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Chakrabartty A., Schellman J. A., and Baldwin R. L. (1991) Large differences in the helix propensities of alanine and glycine. Nature 351, 586–588 10.1038/351586a0 [DOI] [PubMed] [Google Scholar]
- 25. Blaber M., Zhang X.-J., and Matthews B. W. (1993) Structural basis of amino acid α helix propensity. Science 260, 1637–1640 10.1126/science.8503008 [DOI] [PubMed] [Google Scholar]
- 26. Pace C. N., and Scholtz J. M. (1998) A helix propensity scale based on experimental studies of peptides and proteins. Biophys. J. 75, 422–427 10.1016/S0006-3495(98)77529-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Butler G. S., Hutton M., Wattam B. A., Williamson R. A., Knäuper V., Willenbrock F., and Murphy G. (1999) The specificity of TIMP-2 for matrix metalloproteinases can be modified by single amino acid mutations. J. Biol. Chem. 274, 20391–20396 10.1074/jbc.274.29.20391 [DOI] [PubMed] [Google Scholar]
- 28. Meng Q., Malinovskii V., Huang W., Hu Y., Chung L., Nagase H., Bode W., Maskos K., and Brew K. (1999) Residue 2 of TIMP-1 is a major determinant of affinity and specificity for matrix metalloproteinases but effects of substitutions do not correlate with those of the corresponding P1′ residue of substrate. J. Biol. Chem. 274, 10184–10189 10.1074/jbc.274.15.10184 [DOI] [PubMed] [Google Scholar]
- 29. Williamson R. A., Hutton M., Vogt G., Rapti M., Knäuper V., Carr M. D., and Murphy G. (2001) Tyrosine 36 plays a critical role in the interaction of the AB loop of tissue inhibitor of metalloproteinases-2 with matrix metalloproteinase-14. J. Biol. Chem. 276, 32966–32970 10.1074/jbc.M101843200 [DOI] [PubMed] [Google Scholar]
- 30. Nagase H., and Brew K. (2003) Designing TIMP (tissue inhibitor of metalloproteinases) variants that are selective metalloproteinase inhibitors. Biochem. Soc. Symp. 70, 201–212 [DOI] [PubMed] [Google Scholar]
- 31. Wei S., Chen Y., Chung L., Nagase H., and Brew K. (2003) Protein engineering of the tissue inhibitor of metalloproteinase 1 (TIMP-1) inhibitory domain. In search of selective matrix metalloproteinase inhibitors. J. Biol. Chem. 278, 9831–9834 10.1074/jbc.M211793200 [DOI] [PubMed] [Google Scholar]
- 32. Lee M. H., Rapti M., and Murphy G. (2003) Unveiling the surface epitopes that render tissue inhibitor of metalloproteinase-1 inactive against membrane type 1-matrix metalloproteinase. J. Biol. Chem. 278, 40224–40230 10.1074/jbc.M305678200 [DOI] [PubMed] [Google Scholar]
- 33. Lee M. H., Rapti M., Knaüper V., and Murphy G. (2004) Threonine 98, the pivotal residue of tissue inhibitor of metalloproteinases (TIMP)-1 in metalloproteinase recognition. J. Biol. Chem. 279, 17562–17569 10.1074/jbc.M312589200 [DOI] [PubMed] [Google Scholar]
- 34. Lee M. H., Rapti M., and Murphy G. (2005) Total conversion of tissue inhibitor of metalloproteinase (TIMP) for specific metalloproteinase targeting: fine-tuning TIMP-4 for optimal inhibition of tumor necrosis factor-α-converting enzyme. J. Biol. Chem. 280, 15967–15975 10.1074/jbc.M500897200 [DOI] [PubMed] [Google Scholar]
- 35. Hamze A. B., Wei S., Bahudhanapati H., Kota S., Acharya K. R., and Brew K. (2007) Constraining specificity in the N-domain of tissue inhibitor of metalloproteinases-1; gelatinase-selective inhibitors. Protein Sci. 16, 1905–1913 10.1110/ps.072978507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Bahudhanapati H., Zhang Y., Sidhu S. S., and Brew K. (2011) Phage display of tissue inhibitor of metalloproteinases-2 (TIMP-2): identification of selective inhibitors of collagenase-1 (metalloproteinase 1 (MMP-1)). J. Biol. Chem. 286, 31761–31770 10.1074/jbc.M111.253328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Fersht A. R. (1987) The hydrogen bond in molecular recognition. Trends Biochem. Sci. 12, 301–304 10.1016/0968-0004(87)90146-0 [DOI] [Google Scholar]
- 38. Lo Conte L., Chothia C., and Janin J. (1999) The atomic structure of protein-protein recognition sites. J. Mol. Biol. 285, 2177–2198 10.1006/jmbi.1998.2439 [DOI] [PubMed] [Google Scholar]
- 39. Jeffrey G. A., and Jeffrey G. A. (1997) An Introduction to Hydrogen Bonding, Oxford University Press, New York [Google Scholar]
- 40. Moreira I. S., Fernandes P. A., and Ramos M. J. (2007) Hot spots—a review of the protein-protein interface determinant amino-acid residues. Proteins 68, 803–812 10.1002/prot.21396 [DOI] [PubMed] [Google Scholar]
- 41. Binz H. K., Amstutz P., Kohl A., Stumpp M. T., Briand C., Forrer P., Grütter M. G., and Plückthun A. (2004) High-affinity binders selected from designed ankyrin repeat protein libraries. Nat. Biotechnol. 22, 575–582 10.1038/nbt962 [DOI] [PubMed] [Google Scholar]
- 42. Kessenbrock K., Plaks V., and Werb Z. (2010) Matrix metalloproteinases: regulators of the tumor microenvironment. Cell 141, 52–67 10.1016/j.cell.2010.03.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Hu J., Van den Steen P. E., Sang Q. X., and Opdenakker G. (2007) Matrix metalloproteinase inhibitors as therapy for inflammatory and vascular diseases. Nat. Rev. Drug Discov. 6, 480–498 10.1038/nrd2308 [DOI] [PubMed] [Google Scholar]
- 44. Coussens L. M., Fingleton B., and Matrisian L. M. (2002) Matrix metalloproteinase inhibitors and cancer: trials and tribulations. Science 295, 2387–2392 10.1126/science.1067100 [DOI] [PubMed] [Google Scholar]
- 45. Turk B. (2006) Targeting proteases: successes, failures and future prospects. Nat. Rev. Drug Discov. 5, 785–799 10.1038/nrd2092 [DOI] [PubMed] [Google Scholar]
- 46. Nagase H., and Brew K. (2002) Engineering of tissue inhibitor of metalloproteinases mutants as potential therapeutics. Arthritis Res. 4, Suppl. 3, S51–S61 10.1186/ar573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Levin M., Udi Y., Solomonov I., and Sagi I. (2017) Next generation matrix metalloproteinase inhibitors—novel strategies bring new prospects. Biochim. Biophys. Acta Mol. Cell Res. 1864, 1927–1939 10.1016/j.bbamcr.2017.06.009 [DOI] [PubMed] [Google Scholar]
- 48. Bhaskara R. M., and Srinivasan N. (2011) Stability of domain structures in multi-domain proteins. Sci. Rep. 1, 40 10.1038/srep00040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Itoh K., and Sasai M. (2008) Cooperativity, connectivity, and folding pathways of multidomain proteins. Proc. Natl. Acad. Sci. U.S.A. 105, 13865–13870 10.1073/pnas.0804512105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Mata-Fink J., Kriegsman B., Yu H. X., Zhu H., Hanson M. C., Irvine D. J., and Wittrup K. D. (2013) Rapid conformational epitope mapping of anti-gp120 antibodies with a designed mutant panel displayed on yeast. J. Mol. Biol. 425, 444–456 10.1016/j.jmb.2012.11.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Chao G., Lau W. L., Hackel B. J., Sazinsky S. L., Lippow S. M., and Wittrup K. D. (2006) Isolating and engineering human antibodies using yeast surface display. Nat. Protoc. 1, 755–768 10.1038/nprot.2006.94 [DOI] [PubMed] [Google Scholar]
- 52. Crombez L., Marques B., Lenormand J. L., Mouz N., Polack B., Trocme C., and Toussaint B. (2005) High level production of secreted proteins: example of the human tissue inhibitor of metalloproteinases 1. Biochem. Biophys. Res. Commun. 337, 908–915 10.1016/j.bbrc.2005.09.136 [DOI] [PubMed] [Google Scholar]
- 53. Suzuki K., Kan C. C., Hung W., Gehring M. R., Brew K., and Nagase H. (1998) Expression of human pro-matrix metalloproteinase 3 that lacks the N-terminal 34 residues in Escherichia coli: autoactivation and interaction with tissue inhibitor of metalloproteinase 1 (TIMP-1). Biol. Chem. 379, 185–191 [DOI] [PubMed] [Google Scholar]
- 54. Batra J., Robinson J., Soares A. S., Fields A. P., Radisky D. C., and Radisky E. S. (2012) Matrix metalloproteinase-10 (MMP-10) interaction with tissue inhibitors of metalloproteinases TIMP-1 and TIMP-2: binding studies and crystal structure. J. Biol. Chem. 287, 15935–15946 10.1074/jbc.M112.341156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Morrison J. F. (1969) Kinetics of the reversible inhibition of enzyme-catalysed reactions by tight-binding inhibitors. Biochim. Biophys. Acta 185, 269–286 10.1016/0005-2744(69)90420-3 [DOI] [PubMed] [Google Scholar]
- 56. Winter G. (2010) xia2: an expert system for macromolecular crystallography data reduction. J. Appl. Crystallogr. 43, 186–190 10.1107/S0021889809045701 [DOI] [Google Scholar]
- 57. Winter G., Waterman D. G., Parkhurst J. M., Brewster A. S., Gildea R. J., Gerstel M., Fuentes-Montero L., Vollmar M., Michels-Clark T., Young I. D., Sauter N. K., and Evans G. (2018) DIALS: implementation and evaluation of a new integration package. Acta Crystallogr. D Struct. Biol. 74, 85–97 10.1107/S2059798317017235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Evans P. (2006) Scaling and assessment of data quality. Acta Crystallogr. D Biol. Crystallogr. 62, 72–82 10.1107/S0907444905036693 [DOI] [PubMed] [Google Scholar]
- 59. Evans P. R., and Murshudov G. N. (2013) How good are my data and what is the resolution? Acta Crystallogr. D Biol. Crystallogr. 69, 1204–1214 10.1107/S0907444913000061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., and Read R. J. (2007) Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 10.1107/S0021889807021206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Afonine P. V., Grosse-Kunstleve R. W., and Adams P. D. (2005) The Phenix refinement framework, in CCP4 Newsletter, Number 42, Summer 2005, Daresbury Laboratory, Daresbury, UK [Google Scholar]
- 62. Emsley P., and Cowtan K. (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 10.1107/S0907444904019158 [DOI] [PubMed] [Google Scholar]