

Abstract
Vitamin D3 is terminally bioactivated in the kidney to 1α,25-dihydroxyvitamin D3 (1,25(OH)2D3) via cytochrome P450 family 27 subfamily B member 1 (CYP27B1), whose gene is regulated by parathyroid hormone (PTH), fibroblast growth factor 23 (FGF23), and 1,25(OH)2D3. Our recent genomic studies in the mouse have revealed a complex kidney-specific enhancer module within the introns of adjacent methyltransferase-like 1 (Mettl1) and Mettl21b that mediate basal and PTH-induced expression of Cyp27b1 and FGF23- and 1,25(OH)2D3-mediated repression. Gross deletion of these segments in mice has severe effects on Cyp27b1 regulation and skeletal phenotype but does not affect Cyp27b1 expression in nonrenal target cells (NRTCs). Here, we report a bimodal activity in the Mettl1 intronic enhancer with components responsible for PTH-mediated Cyp27b1 induction and 1,25(OH)2D3-mediated repression and additional activities, including FGF23 repression, within the Mettl21b enhancers. Deletion of both submodules eliminated basal Cyp27b1 expression and regulation in the kidney, leading to systemic and skeletal phenotypes similar to those of Cyp27b1-null mice. However, basal expression and lipopolysaccharide-induced regulation of Cyp27b1 in NRTCs was unperturbed. Importantly, dietary normalization of calcium, phosphate, PTH, and FGF23 rescued the skeletal phenotype of this mutant mouse, creating an ideal in vivo model to study nonrenal 1,25(OH)2D3 production in health and disease. Finally, we confirmed a conserved chromatin landscape in human kidney that is similar to that in mouse. These findings define a finely balanced homeostatic mechanism involving PTH and FGF23 together with protection from 1,25(OH)2D3 toxicity that is responsible for both adaptive vitamin D metabolism and mineral regulation.
Keywords: cytochrome P450; CRISPR/Cas; ChIP-sequencing (ChIP-Seq); vitamin D; gene regulation; parathyroid hormone; fibroblast growth factor 23 (FGF23); 1,25-dihydroxyvitamin D3; 1-α-hydroxylase; Cyp24a1; Cyp27b1; Cyp27b1-KO; Cyp24a1; parathyroid hormone (PTH)
Introduction
Vitamin D3 undergoes its final bioactivation step in the kidney to hormonal 1α,25-dihydroxyvitamin D3 (1,25(OH)2D3)2 through the actions of the renal P450 enzyme CYP27B1 (1). The hormone's endocrine concentration in the blood is dynamically regulated, however, not only via its synthesis but also through its initial degradation to 1,24,25(OH)3D3 by CYP24A1 (2). This latter renal enzyme is responsible for further metabolic clearance that leads ultimately to calcitroic acid. Of central importance, the expression of CYP27B1 in the kidney is governed by numerous factors, although those of major homeostatic significance include induction by the calcium-regulating hormone PTH (3), suppression by the phosphaturic hormone FGF23 (4), and feedback suppression by 1,25(OH)2D3 (5), all of which link adaptive vitamin D metabolism to the maintenance of mineral homeostasis. Uniquely, renal expression of Cyp24a1 is also regulated by these same hormones, although in a reciprocal manner wherein PTH suppresses whereas both FGF23 and 1,25(OH)2D3 induce expression of this gene (6–9). This regulatory paradigm in the kidney highlights the critical importance of the coordinated regulation of both Cyp27b1 and Cyp24a1 expression for the tight control of circulating 1,25(OH)2D3 and thus orchestration of normal extracellular mineral homeostasis.
Interestingly, recent studies of CYP27B1 expression have revealed that this gene is also expressed at relatively low levels in a wide variety of nonrenal cell types, including skin, bone cells, colonic epithelial cells, and many others as well, an arbitrary collection of functionally diverse cells that we have termed non-renal target cells (NRTCs) (10, 11). Whereas the potential role of Cyp27b1 in these cells remains uncertain, it is nonetheless clear that the Cyp27b1 gene in these tissues is not generally regulated by PTH, FGF23, or 1,25(OH)2D3 as in the kidney but rather by other factors that include pro-inflammatory modulators, such as lipopolysaccharides (LPS), tumor necrosis factor α, and interleukin-1; TCR activation in T cells; and likely additional factors that can exhibit unique regulatory activities in specific NRTCs (12). Presumably, this enables the biological actions of locally produced 1,25(OH)2D3 to contribute to the regulation of functions unique to individual cell types while simultaneously insulating local hormonal regulation from the dynamic changes in PTH, FGF23, and/or 1,25(OH)2D3 that are associated with mineral homeostasis (13). Neither the mechanisms nor the overall biological impact of locally produced 1,25(OH)2D3 is understood, however, particularly in the context of normal levels of residual endocrine 1,25(OH)2D3, where it is likely derived in healthy animals exclusively from the kidney. Despite these uncertainties, the regulated expression of Cyp27b1 and its corresponding 1,25(OH)2D3 turnover partner Cyp24a1, which is also expressed in these nonrenal cell types at very low levels, is widely believed to be central to the highly diverse biological activities that appear to be influenced by the vitamin D system in vivo.
Despite multiple studies beginning in the 1970s, the molecular mechanisms through which PTH, 1,25(OH)2D3, and later FGF23 regulate the expression of Cyp27b1 at the genomic level appear to have resisted virtually all research efforts. This has been due, at least in part, to the lack of in vitro kidney cell models that express appreciable levels of Cyp27b1 or more importantly exhibit regulation by the above three hormones at appropriate levels seen in vivo. Because of this, we turned our attention to the mouse as an in vivo model to understand the mechanisms underlying renal Cyp27b1 expression and regulation. Guided by ChIP-Seq–mediated identification of genetic and epigenetic elements at the Cyp27b1 gene locus in the mouse kidney coupled with the deletion of key genomic regions facilitated by CRISPR/Cas9 gene editing methods in vivo, we discovered a complex, multicomponent endocrine regulatory module located in the introns of the adjacent Mettl1 and Mettl21b genes whose overall modulatory activities were enabled by an open chromatin structure that was unique to the kidney while absent in all nonrenal tissues we examined (13). This module was composed of two independent intronic submodules; both control basal expression of Cyp27b1, whereas one mediates induction by PTH and the other mediates its suppression by FGF23 and 1,25(OH)2D3. Loss of PTH sensitivity led to an aberrant systemic as well as a profound skeletal phenotype approaching that of the Cyp27b1-null mouse, whereas loss of FGF23 and 1,25(OH)2D3 suppression alone led to only minor phenotypic changes restricted to the circulation. The biological absence of this module in the genome of NRTCs coincided with the inability of PTH, FGF23, and 1,25(OH)2D3 to regulate the expression of Cyp27b1 in these tissues. However, sensitivity to LPS induction was retained, indicating that the regulatory module controlling this function was located outside that which modulates renal Cyp27b1 expression.
In our current studies, we further define the enhancer regions that are responsible for Cyp27b1 regulation in the kidney. We then confirm the overall essentiality of the two submodules for controlling the production of endocrine 1,25(OH)2D3 by deleting both regulatory segments from the mouse genome. Accordingly, we show that almost all basal expression of Cyp27b1 as well as its regulatory control by PTH, FGF23, and 1,25(OH)2D3 are lost in this mutant mouse, which leads to a striking reduction in the circulating level of 1,25(OH)2D3 and compensatory and informative changes in additional key vitamin D metabolites as well. This loss of hormone results in severe hypocalcemia and hypophosphatemia, with phenotypic hormonal and skeletal features comparable with those of the Cyp27b1-null mouse. Importantly, the features of this mutant strain as well as those of the previous PTH-insensitive Cyp27b1 mouse (M1-IKO mouse) can be fully rescued via a diet high in calcium (Ca) and phosphate (P), which rapidly normalizes blood Ca and P levels and eventually restores appropriate levels of PTH and FGF23 but eliminates all 1,25(OH)2D3 (14). This therefore provides a novel and useful mouse model for the independent study of the local production of 1,25(OH)2D3 and its biological effects in NRTCs and on disease progression. Finally, we show using ChIP-Seq analysis that the genetic and epigenetic landscape at the CYP27B1 gene locus in human kidney supports a conserved regulatory arrangement similar to the mouse. In summary, this investigation advances our understanding of the genomic mechanisms through which Cyp27b1 and Cyp24a1 are regulated in mouse and perhaps human kidney in vivo; these studies also provide models for the subsequent assessment of the exquisite molecular mechanisms through which Cyp27b1 is regulated in the kidney and for the study of local production of 1,25(OH)2D3 in nonrenal tissues.
Results
Defining the roles of distinct regulatory segments within the Mettl21b intron
As summarized in Fig. 1A, our previous ChIP-Seq studies of the Cyp27b1 gene locus revealed the existence of two regulatory submodules, each of which displayed unique functional, kidney-specific profiles when deleted in the mouse (13). In the present studies, we therefore created several additional deletions within each submodule and a complete module deletion, all of which are summarized in Fig. 1B using a CRISPR/Cas9 approach, and evaluated basal and hormonal response in each of these as well. We began with a dissection of the Mettl21b intronic submodule, given the larger size of the original intronic deletion created in the M21-IKO mouse and because of its apparent ability to control basal Cyp27b1 expression as well as its capacity to mediate suppression of Cyp27b1 expression in the kidney by both FGF23 and 1,25(OH)2D3. Accordingly, we created two separate deletions within the Mettl21b intron as illustrated in Fig. 2A; the first removed the most proximal two elements, including highly repetitive, nonconserved genomic segments within, and the second removed the single most distal element, resulting in mouse strains termed M21-IKOP (more proximal to Cyp27b1) and M21-IKOD (distal), respectively. As seen in Fig. 2B, the overall systemic phenotypes of these two mutant mouse strains were unremarkable, with the M21-IKOD strain alone exhibiting slightly elevated levels of PTH and slightly depressed levels of FGF23 that were similar to the M21-IKO strain; like this latter strain, vitamin D metabolite levels were also altered in the M21-IKOD mouse, with highly elevated 25(OH)D3 and 24,25(OH)2D3 but reduced 1,24,25(OH)3D3 levels (Fig. 2C). To highlight the metabolic difference, the ratio of 25(OH)D3/24,25(OH)2D3 rose from 2 in WT mice to 3.8 in M21-IKOD, which implies less catabolism of 25(OH)D3 together with the higher absolute concentration of 25(OH)D3. Both mutant mice also displayed normal skeletal features similar to the M21-IKO mouse as well (Fig. 2D).
Figure 1.

A, schematic summary of our previous study (13) with the Cyp27b1 genomic locus and ChIP-Seq data to demonstrate control of Cyp27b1 either positively (green) by PTH through the M1 enhancer or negatively (red) by FGF23 via the M21 enhancer or by 1,25(OH)2D3 (1,25D3) via both M1 and M21 (red). B, deletions within the Cyp27b1 gene locus that were created and/or examined in the present study. Black bar, deleted sequence.
Figure 2.

M21-IKO deletions reveal cooperative FGF23 response and differential basal activity. A, schematic depiction of the M21-IKOP (more proximal to Cyp27b1, CRISPR guides G3 to G2) and M21-IKOD (distal, G1 to G3) deletions compared with the total M21-IKO (G1 to G2). Image truncated from Fig. 1A. B, table of serum concentrations of calcium (mg/dl), phosphate (mm), PTH (pg/ml), and FGF23 (pg/ml). C, table of serum concentrations of 25(OH)D3, 24,25(OH)2D3, 1,25(OH)2D3, and 1,24,25(OH)2D3 with n values indicated in parentheses for M21-IKO, M21-IKOP, M21-IKOD, and WT littermate controls. One-way ANOVA with multiple-comparison Tukey post-test: *, p < 0.05 KO versus WT. D, BMD measurements in femur for male (blue) and female (red) in WT (solid bars, n = 6) and M21-IKO (n = 6), M21-IKOP (n = 6), and M21-IKOD (striped, n = 6) mice. *, p < 0.05 paired t test: KO versus WT. Shown is gene expression in the kidney for Cyp27b1 (E), Cyp24a1 (F), and Vdr (G) after treatment in 8–9-week-old M21-IKO, M21-IKOP, and M21-IKOD mice compared with WT littermates with ethanol/PBS vehicle (Veh, gray, n = 6), 10 ng/g bw 1,25(OH)2D3 (1,25D3, black, n = 6) for 6 h, or 50 ng/g bw FGF23 for 3 h (FGF23, green, n = 6). Data are displayed as -fold change (WT vehicle set to 1) from relative quantitation (RQ, mean ± S.E. (error bars)) compared with Gapdh. *, p < 0.05 paired t test: treatment versus vehicle. #, p < 0.05 paired t test: KO vehicle versus WT vehicle.
Interestingly, whereas deletion of the single segment in the M21-IKOD mouse revealed a Cyp27b1 RNA expression phenotype similar to that of the M21-IKO mouse (reduced basal activity and suppression by FGF23 and 1,25(OH)2D3 treatment) as seen in Fig. 2E, collective independent deletion of the two more proximal segments in the M21-IKOP strain showed a functional difference; whereas sensitivity to the two hormones as seen in the M21-IKO strain was recapitulated, the residual basal expression of Cyp27b1 remained intact only in M21-IKOP mice. In both strains, however, FGF23 suppression of Cyp27b1 expression remained present unlike in the M21-IKO mouse. These results suggest that whereas control of basal expression was restricted to the more distal epigenetic component, suppression by FGF23 was perhaps mediated via all three regions. Interestingly, Cyp24a1 expression in the M21-IKO mouse kidney was also suppressed homeostatically in the M21-IKOD mouse (Fig. 2F). This reduction correlated with the decrease in basal Cyp27b1 expression seen in M21-IKOD mice, apparently leading to elevated levels of 25(OH)D3 and 24,25(OH)2D3 that were also observed in Fig. 2C. Vdr expression levels were modestly up-regulated in response to both 1,25(OH)2D3 and FGF23 (Fig. 2G) (13). As with the M21-IKO mouse, these deletions (M21-IKOP and M21-IKOD) had no effect on Cyp27b1 expression or LPS induction in any of the NRTCs examined, including the thyroparathyroid (TPTG) tissue (data not shown) (13). As there is little known about the methyltransferase function of Mettl1 (15) and Mettl21b may have an interaction with eukaryotic elongation factor 1α (eEF1A) (16), we also created a deletion mutation of the Mettl21b gene itself with two different frameshift mutations in exon 1, which resulted in the absence of expression of mature, full-length Mettl21b. These animals were completely WT in their systemic parameters, size, weight, skeletal composition, and kidney expression of Cyp27b1, Cyp24a1, and Vdr (data not shown).
FGF23 suppression is restricted to the Mettl21b intronic enhancer, whereas 1,25(OH)2D3 suppression spans both kidney M1 and M21 enhancers
Given the possibility that Cyp27b1 suppression might be mediated via several redundant activities within the complex M21 submodule, we returned to the M21-IKO mouse and examined the ability of these two hormones to suppress Cyp27b1 expression in a more thorough fashion as a function of time following FGF23 and 1,25(OH)2D3 treatment. As seen in Fig. 3A, the results confirm as previously suggested that although basal Cyp27b1 expression is reduced in this mouse, the gene is fully refractory to transient suppression by FGF23. Cyp24a1 (Figs. 2 and 3B) as well as the Vdr (Fig. 3C) were also down-regulated by FGF23 via a direct response, as demonstrated previously (13). In contrast, however, temporal 1,25(OH)2D3 suppression was still present as was seen in the control M1-IKO mouse (Fig. 3D). In both cases, however, Cyp27b1 suppression was less robust than that apparent in the WT controls. These data indicate that whereas FGF23 activity is restricted to the M21 intronic submodule, 1,25(OH)2D3 suppression spans both the submodules in M21 and the M1, as demonstrated clearly in the M1/M21 double deletion mouse (discussed in detail below).
Figure 3.
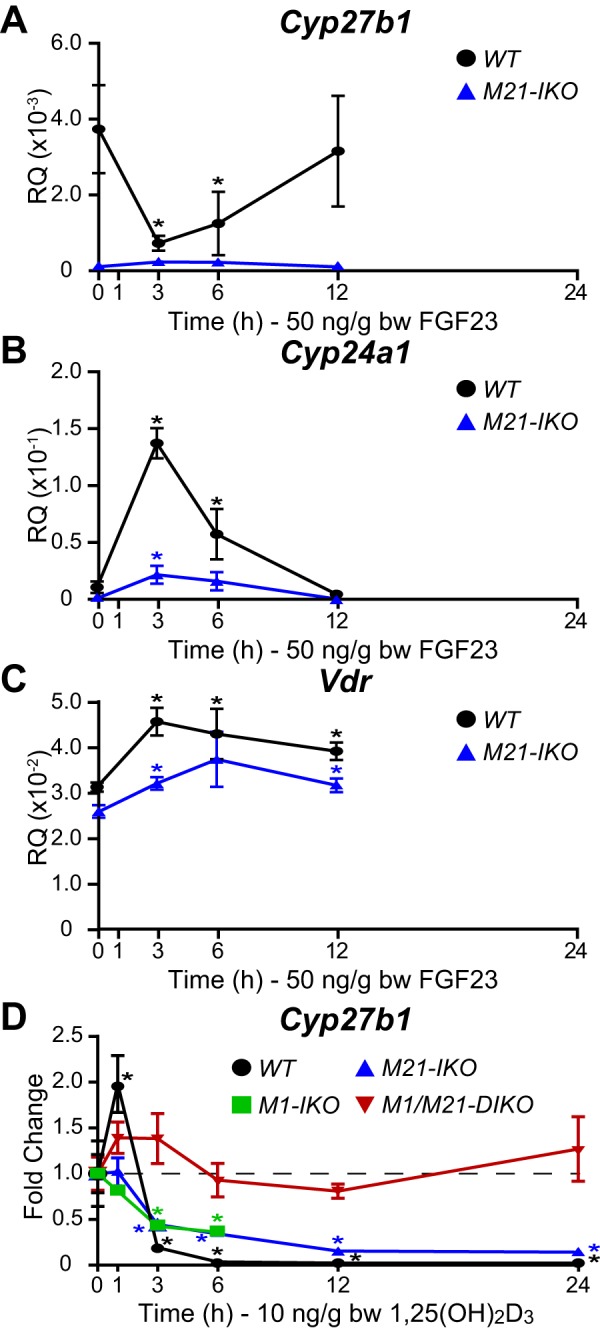
Time course of Cyp27b1 repression in response to FGF23 and 1,25(OH)2D3 in M21-IKO mice. A–C, gene expression in the kidney for Cyp27b1 (A), Cyp24a1 (B), and Vdr (C) in M21-IKO and WT littermate mice treated with 50 ng/g bw FGF23 for 0, 1, 3, 6, or 12 h. Data are displayed as relative quantitation (RQ, mean ± S.E. (error bars)) compared with Gapdh. n = 6 for each time point. *, p < 0.05 paired t test: time point versus vehicle. D, Cyp27b1 gene expression in WT, M1-IKO, M21-IKO, and M1/M21-DIKO (double deletion) mice with 10 ng/g bw 1,25(OH)2D3 for 0, 1, 3, 6, 12, and 24 h. Data are displayed as -fold change (vehicle set to 1) from relative quantitation (mean ± S.E.) compared with Gapdh. n = 6 for each time point. *, p < 0.05 paired t test: time point versus vehicle.
Bimodal activities of the single regulatory segment located in the Mettl1 intron
Our initial studies identified an epigenetically active open chromatin region within a Mettl1 intron that binds both the VDR and CREB and that mediates up-regulation of Cyp27b1 expression by PTH exclusively in the kidney (13). As a result, deletion of this small region of ∼300 bp, as documented in the M1-IKO mouse, strikingly reduced basal Cyp27b1 expression, caused resistance to PTH, and culminated in a phenotype that approached that of the Cyp27b1-null mouse. Based upon this observation, as well as the results documented in Fig. 3 relative to 1,25(OH)2D3 suppression, we created two additional mutant mouse strains in an attempt to separate these two functions genomically. As illustrated in Fig. 4A, the first mutant contained a deletion of the more proximal ∼100 bp of the regulatory segment defined originally in the M1-IKO mouse (termed the M1-IKOP mouse), and the second contained a deletion of the more distal ∼200 bp (termed the M1-IKOD mouse). The DNA sequences of this entire region indicated an enrichment of multiple VDRE and CREB motifs (Fig. 4B) (17–19). Interestingly, the systemic phenotypes of each of these two mutant strains were much less striking than that seen originally in the M1-IKO mouse. Thus, neither exhibited hypocalcemia or hypophosphatemia, and whereas PTH and FGF23 levels tended strongly toward those of WT littermates, neither were as extreme as those seen in the M1-IKO mouse (Fig. 4C). Vitamin D metabolite levels were also distinctly different, relative not only to WT and M1-IKO mice but also to each other. With respect to 1,25(OH)2D3 levels, both were higher than that seen in the M1-IKO mouse and approached WT levels (Fig. 4D). The near normal nature of the systemic features of Ca and P in the M1-IKOD and M1-IKOP mice relative to the M1-IKO mouse was likely responsible for the normal bone mineral densities (BMDs) seen in both males and females of the two strains (Fig. 4E). Based upon these observations, we isolated RNA from the kidneys of these mutant mice following treatment with a single injection of either vehicle, PTH, FGF23, or 1,25(OH)2D3 and assessed transcript levels for Cyp27b1, Cyp24a1, and Vdr relative to WT and M1-IKO mice. As can be seen in Fig. 4F, whereas basal expression levels of Cyp27b1 in vehicle-treated M1-IKOP mice were strikingly reduced relative to WT mice and comparable with that seen in the M1-IKO mouse, basal expression of Cyp27b1 and protein in the M1-IKOD mice was statistically elevated above both M1-IKOP and WT mice as well (Fig. 4, F and I). These findings were supported by the coordinated suppression of Cyp24a1 expression in the M1-IKO and M1-IKOP mice but not in the M1-IKOD mouse, where Cyp24a1 is up-regulated relative to WT controls (Fig. 4G). Vdr RNA and VDR protein levels in both the M1-IKOP and M1-IKOD mice resembled those of WT mice (Fig. 4, H and I). In contrast, the suppressive effect of FGF23 and 1,25(OH)2D3 on Cyp27b1 expression was retained in each of the mouse strains examined despite the striking differences in basal expression seen in each strain (Fig. 4F).
Figure 4.

Deletions within the M1-IKO region reveal bimodal activity separating PTH induction and 1,25(OH)2D3 repression. A, schematic of M1-IKO, M1-IKOP, and M1-IKOD deletions compared with ChIP-Seq peak data from Meyer et al. (13), truncated from Fig. 1A. B, sequence and location depiction of CRISPR RNA guide targets, M1-IKO deletions, and putative VDR and CREB DNA response elements. C, table of serum calcium (mg/dl), phosphate (mm), PTH (pg/ml), and FGF23 (pg/ml) concentrations. D, table of serum 25(OH)D3, 24,25(OH)2D3, 1,25(OH)2D3, and 1,24,25(OH)2D3 concentrations with n values indicated in parentheses for M1-IKO, M1-IKOP, M1-IKOD, and WT littermates (pooled WT values same as in Fig. 2). One-way ANOVA with multiple-comparison Tukey post-test: *, p < 0.05 KO versus WT. E, BMD measurements in femur and body weights (g) for male (blue) and female (red) in WT (solid bars, n = 6) and M1-IKO (n = 6), M1-IKOP (n = 6), and M1-IKOD (striped, n = 6) mice. *, p < 0.05 paired t test: KO versus WT. Gene expression in the kidney for Cyp27b1 (F), Cyp24a1 (G), and Vdr (H) after treatment in 8–9-week-old M1-IKO, M1-IKOP, and M1-IKOD mice compared with WT littermates with ethanol/PBS vehicle (Veh, gray, n = 6), 230 ng/g bw PTH for 1 h (PTH, blue, n = 6), 10 ng/g bw 1,25(OH)2D3 (1,25D3, black, n = 6) for 6 h, or 50 ng/g bw FGF23 for 3 h (FGF23, green, n = 6). Data are displayed as -fold change (WT vehicle set to 1) from relative quantitation (RQ, mean ± S.E. (error bars)) compared with Gapdh. *, p < 0.05 paired t test: treatment versus vehicle. #, p < 0.05 paired t test: KO vehicle versus WT vehicle. I, Western blot analysis of CYP27B1 protein expression from mitochondrial extracts compared with loading control VDAC1/PORIN protein expression (left). VDR protein expression compared with β-tubulin protein from whole-cell extract (right).
As documented in Fig. 4F, however, the most interesting observation was that whereas PTH induction was fully lost in the M1-IKO mouse as described previously, the deletion created in the M1-IKOP mouse did not lead to a complete loss of PTH response but rather limited the induction of Cyp27b1 to ∼3-fold. These results suggest the likelihood of significant complexity within the DNA sequence(s) that serves to mediate PTH control of Cyp27b1, perhaps at the deletion boundary, although the ability of the M1-IKOP mouse to mount a WT response to PTH implies that the proximal region is not involved in PTH regulation. The consequence of this limited PTH response is profound, however, as it precludes both the strong rise in PTH levels as well as the striking skeletal phenotype that is eventually seen in the M1-IKO mouse.
Finally, increased basal expression of Cyp27b1 in the M1-IKOD mouse indicates that the removal of the more proximal ∼200 bp leads to a 3-fold de-repression of Cyp27b1 expression (Fig. 4F). Thus, it is possible that this region retains DNA sequence elements that mediate Cyp27b1 suppression by 1,25(OH)2D3 and that the loss of these sequences alleviates the repressive actions of 1,25(OH)2D3 in this M1 intronic region. Despite this, however, as in the intron of Mettl21b, none of the deletions utilized to evaluate the components in the introns of Mettl1 altered the basal or LPS-inducible activity of Cyp27b1 in NRTCs (data not shown). Interestingly, however, Cyp27b1 expression in the TPTG of the M1-IKOP mouse mirrored that of the M1-IKO mouse with a lowered baseline (13), whereas the M1-IKOD was unchanged (data not shown). The establishment of these genomic/functional correlates will enable further studies designed to elucidate the molecular basis for the actions of PTH, FGF23, 1,25(OH)2D3, and perhaps other hormones and to define the transcription factors and upstream signaling pathways responsible for mediating these systemic hormones' actions at the level of the Cyp27b1 gene.
Calcitonin induces secondary renal Cyp27b1 expression via an up-regulation of serum PTH
Calcitonin (CT) represents an historical inducer of renal Cyp27b1 and represents a potential candidate for Cyp27b1 up-regulation by the proximal segment in the Mettl21b intron (20). To confirm that CT induces Cyp27b1 in the kidney, we injected increasing concentrations of salmon CT (20–2000 ng/g) into WT mice, assessed the temporal response of Cyp27b1 expression to the hormone, and noted a striking up-regulation of the target gene (Fig. 5A). Systemic measurements of PTH as seen in Fig. 5B, however, revealed that this treatment caused a surprising and dramatic up-regulation of this hormone, suggesting that the positive effects of CT on Cyp27b1 expression might be secondary to PTH up-regulation. To test this hypothesis, we treated WT, M1-IKO, and M21-IKO mice with 20 ng/g CT and measured Cyp27b1 expression as a function of time (Fig. 5, C and D). As can be seen in Fig. 5C, whereas CT strongly induced Cyp27b1 in the kidney of WT mice and the M21-IKO mice, this induction was fully abrogated in M1-IKO mice, where Cyp27b1 is refractory to exogenous PTH. Accordingly, this experiment demonstrates an unexpected finding that strong up-regulation of Cyp27b1 by CT is indeed secondary to the hormone's ability to induce PTH, making it unlikely that the unknown factor active in the single distal segment of the Mettl21b intron is CT.
Figure 5.
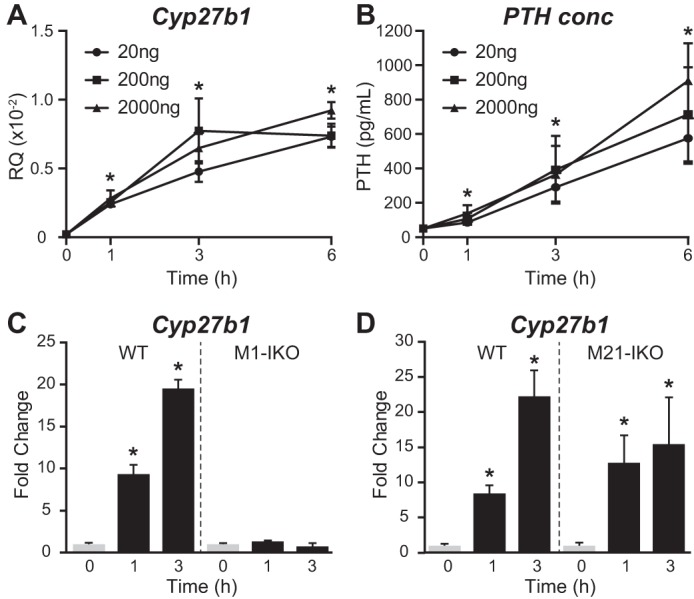
CT treatment rapidly induces serum PTH levels and Cyp27b1 expression. Cyp27b1 expression in the kidney (A) and serum PTH levels (B) in WT mice treated with 20, 200, or 2000 ng/g bw CT for 0, 1, 3, or 6 h. Data are displayed as relative quantitation (RQ, mean ± S.E. (error bars)) compared with Gapdh or pg/ml. n = 6 for each time point. *, p < 0.05 paired t test: time point versus vehicle. C and D, kidney Cyp27b1 expression in M1-IKO (C) or M21-IKO (D) mice compared with WT treated with 20 ng/g bw CT for 0, 1, or 3 h. Data are displayed as -fold change (vehicle set to 1) from relative quantitation (mean ± S.E.) compared with Gapdh. n = 6 for each time point. *, p < 0.05 paired t test: treatment versus vehicle.
Creation of a unique kidney-specific Cyp27b1 enhancer deletion mouse model
Having established loss of function correlates with the specific structural components within the introns of Mettl21b and Mettl1 genes, we next tested the hypothesis that these two submodules comprise a comprehensive regulatory module that mediates full expression and hormonal regulation of Cyp27b1 in mouse kidney and is thereby fully responsible for the circulating levels of endocrine 1,25(OH)2D3. We utilized a sequential CRISPR/Cas9 approach to delete both the Mettl1 and Mettl21b intronic regulatory segments simultaneously, as illustrated in Fig. 1, and then assessed the resulting systemic and skeletal consequences of this dual intronic deletion (termed the M1/M21-DIKO mouse) on both basal and hormone-regulated expression of renal Cyp27b1. As can be seen in Fig. 6A, deletion of both regulatory components resulted in a complex systemic and skeletal phenotype that was more extreme than that seen in the M1-IKO mouse (13) and that paralleled that of the Cyp27b1-null mouse, which produces undetectable levels of 1,25(OH)2D3 (21, 22). Indeed, the M1/M21-DIKO mouse was extremely hypocalcemic and hypophosphatemic, coincident with highly elevated PTH levels and with FGF23 levels well below those seen in the Cyp27b1-null mouse. As expected, these systemic features were accompanied by low BMDs in these mice (at 8 weeks of age in both sexes), indicative of a highly aberrant skeleton (Fig. 6B).
Figure 6.

Phenotype of the M1 and M21 double intronic deletion (M1/M21-DIKO) mouse is similar to the Cyp27b1-null mouse. A, table of serum calcium (mg/dl), phosphate (mm), PTH (pg/ml), and FGF23 (pg/ml) concentrations with n values indicated in parentheses for C27KO, M1-IKO, M21-IKO, M1/M21-DIKO, and WT littermates (pooled WT values same as in Fig. 2). One-way ANOVA with multiple-comparison Tukey post-test: *, p < 0.05 KO versus WT. B, BMD measurements in femur for males (blue) and females (red) in WT (solid bars, n = 6) and M1-IKO (n = 6), M21-IKO (n = 6), and M1/M21-DIKO (striped, n = 6) mice. *, p < 0.05 paired t test: KO versus WT. C–E, gene expression in the kidney for Cyp27b1 (C), Cyp24a1 (D), Vdr (E) after treatment in 8–9-week-old M1-IKO, M21-IKO, and M1/M21-DIKO mice compared with WT littermates with ethanol/PBS vehicle (Veh, gray, n = 6), 230 ng/g bw PTH for 1 h (PTH, blue, n = 6), 10 ng/g bw 1,25(OH)2D3 (1,25D3, black, n = 6) for 6 h, or 50 ng/g bw FGF23 for 3 h (FGF23, green, n = 6). Data are displayed as -fold change (WT vehicle set to 1) from relative quantitation (RQ, mean ± S.E. (error bars)) compared with Gapdh. *, p < 0.05 paired t test: treatment versus vehicle. #, p < 0.05 paired t test: KO vehicle versus WT vehicle. F, table of serum 25(OH)D3, 24,25(OH)2D3, 1,25(OH)2D3, and 1,24,25(OH)3D3 concentrations with n values indicated in parentheses as in A (pooled WT values same as in Fig. 2). G, gene expression in the duodenum for Calb1, S100g, Trpv6, and Atp2b1 of M1/M21-DIKO mice compared with WT littermates treated with 10 ng/g bw 1,25(OH)2D3 for 0, 1, 3, 6, 12, and 24 h. Data are displayed as relative quantitation (mean ± S.E.) compared with Gapdh or pg/ml. *, p < 0.05 paired t test: time point versus vehicle. H, Cyp27b1 expression in kidney, skin, and calvaria in M1/M21-DIKO (DIKO) mice treated with 10 μg/g LPS for 6 h. Data are displayed as relative quantitation (mean ± S.E.) compared with Gapdh. *, p < 0.05 paired t test: treatment versus vehicle. #, p < 0.05 paired t test: KO vehicle versus WT vehicle.
Of equal importance, as seen in Fig. 6C, RT-qPCR analysis of isolated kidney RNA from the M1/M21-DIKO mouse revealed that basal Cyp27b1 expression was reduced to a level even lower than that seen in the M1-IKO mouse, exhibiting a 99.7% reduction relative to WT mice in this experiment. Moreover, renal expression of Cyp27b1, as anticipated, was fully refractory to exogenous treatment with a single time point injection of PTH, FGF23, or 1,25(OH)2D3, as seen in Fig. 6C. Renal Cyp27b1 expression in the M1/M21-DIKO mouse was also refractory to suppression by 1,25(OH)2D3 as a function of time, as documented in Fig. 3D, fully supporting our earlier contention that 1,25(OH)2D3 suppression was mediated through both submodules. Not surprisingly, examination of renal levels of Cyp24a1 expression as in Fig. 6D also revealed a striking down-regulation of the latter gene that was even more pronounced than that seen in either the M1-IKO or the M21-IKO mice; direct response of Cyp24a1 to PTH suppression and to FGF23 induction was difficult to assess given the exceptionally low baselines, although 1,25(OH)2D3 induction remained intact. Vdr levels were also basally reduced as well compared with WT controls (Fig. 6E).
Unexpectedly, despite the low levels of renal Cyp27b1 expression and the striking systemic and skeletal phenotype of this mouse, blood levels of 1,25(OH)2D3, although lower that than seen in the M1-IKO mouse, were measurable, as seen in Fig. 6F. We hypothesize that circulating 1,25(OH)2D3 levels in both of these mouse strains are inappropriately high relative to their aberrant phenotypes, likely due to the striking suppression of renal Cyp24a1 expression, which restricts the turnover of what little 1,25(OH)2D3 is produced by highly reduced renal Cyp27b1 expression. That Cyp24a1 expression is functionally low is also supported by the absence of 24,25(OH)2D3 and 1,24,25(OH)3D3 and a consequential elevation in the levels of 25(OH)D3 substrate that is present in M1/M21-DIKO, M1-IKO, and Cyp27b1-null mice. The inability of these levels of 1,25(OH)2D3 to direct adequate uptake of calcium from the gut is equally supported by the trend toward a reduction in the expression of intestinal S100g, Trpv6, and Atp2b1, genes known to be involved in calcium absorption from the gut, although loss of Trpv6 expression appears to dominate (Fig. 6G) (23, 24).
Finally, it is important to note that basal expression of Cyp27b1 in the M1/M21-DIKO mouse is again uninterrupted in NRTCs, such as bone and skin, as was observed in both M1-IKO and M21-IKO mice (13) and fully inducible by the inflammatory modulator LPS (Fig. 6H). As a control, Il-6 expression was strongly induced in response to LPS injection (data not shown) (13). This supports our hypothesis that the regulation of Cyp27b1 in nonrenal cells is likely mediated by a control region that is distinct from that of the kidney-specific module. The absence of any change in NRTC Cyp27b1 also reinforces the idea that residual 1,25(OH)2D3 in the blood is not derived from an increase in 1,25(OH)2D3 production from NRTCs, including the parathyroid gland, although these alternative sources are always possible. Thus, we have created a truly unique “knockout” of Cyp27b1 expression selectively in the kidney by removing the key noncoding enhancers that represent the primary determinants of Cyp27b1 expression in this tissue, with a small residual level of 1,25(OH)2D3 remaining as a result of the dramatic suppression of Cyp24a1 expression and activity.
Establishing animal models for the study of homeostasis and vitamin D metabolism in NRTCs
The studies presented thus far provide an initial genomic framework with which to develop further molecular and homeostatic insights into the mechanisms through which PTH, FGF23, and 1,25(OH)2D3 regulate renal Cyp27b1 expression and blood levels of 1,25(OH)2D3. Perhaps equally important, however, is the apparent creation of an in vivo animal model as described by the M1/M21-DIKO mouse above, wherein systemic 1,25(OH)2D3 production from the endocrine kidney has been largely and uniquely curtailed without altering the potential for 1,25(OH)2D3 production in nonrenal tissues. This model is therefore distinct from that of the global Cyp27b1-null mouse. It thus provides us with the opportunity to both explore homeostatic response mechanisms through renal Cyp27b1 expression and study the local production of 1,25(OH)2D3 and its effects on the biological activities of NRTCs in vivo in the absence and presence of exogenously added 1,25(OH)2D3. It also provides us with the ability to assess the impact of exogenous vitamin D and its metabolites, particularly that of 25(OH)D3, on NRTC production of 1,25(OH)2D3, an assessment of considerable clinical importance. We reasoned, however, that normalizing key parameters of mineral homeostasis in this mouse using a high-Ca and -P rescue diet would be highly advantageous to minimize the complexity of the highly aberrant metabolic and structural phenotype, a manipulation that has been used extensively to normalize both systemic and skeletal parameters that could present potential confounding in vivo parameters. To approach this, we first assessed the time course of response to the rescue diet as compared with the normal Ca and P control diet in Cyp27b1-null mice in the absence of 1,25(OH)2D3, with treatment beginning immediately prior to weaning, because a reported conflict exists in the literature with regard to the time course of normalization of mineral, hormones, and the renal expression of Cyp27b1 and Cyp24a1 (9, 25). In our experience, although Ca and P levels are rapidly corrected upon exposure to the diet, as might be expected, both normalization of hormones and restoration of WT Cyp27b1 and Cyp24a1 renal expression levels are strikingly retarded. As can be seen in Fig. 7A, whereas Ca and P levels are fully restored at 8 weeks of age, normalization of PTH and FGF23 levels and the concomitant correction of renal Cyp27b1, Cyp24a1, and Vdr expression do not occur until 16 weeks (Fig. 7, B and C) (note that whereas mutant Cyp27b1 transcripts produce a defective CYP27B1 protein that cannot make 1,25(OH)2D3, the normal regulation of the mutant transcript is retained). Based upon this time course, we then conducted a similar rescue study with M1-IKO and M1/M21-DIKO using Cyp27b1-null and WT mice as controls. As can be seen in Fig. 8A, whereas the normal Ca and P control diet resulted in hypocalcemia, hypophosphatemia, hyperparathyroidism, and reduced FGF23 levels in all three mutants compared with WT mice, treatment with the rescue diet from weaning to 16 weeks raised Ca and P levels in all cases to those approaching WT mice and reduced PTH and raised FGF23 levels, although at this time point, PTH levels were still slightly above normal and FGF23 levels were well above normal. The overshoot in FGF23 levels is likely responsible for the lower than normal P levels seen due to phosphate diuresis from the kidney (4). As a result of the general normalization of these systemic parameters in the M1/M21-DIKO mouse on rescue diet, both body weights and BMDs were increased, and reduced femur lengths were restored as well (Fig. 8, B and C). Similar effects were seen in the M1-IKO and the Cyp27b1-null mouse (data not shown).
Figure 7.
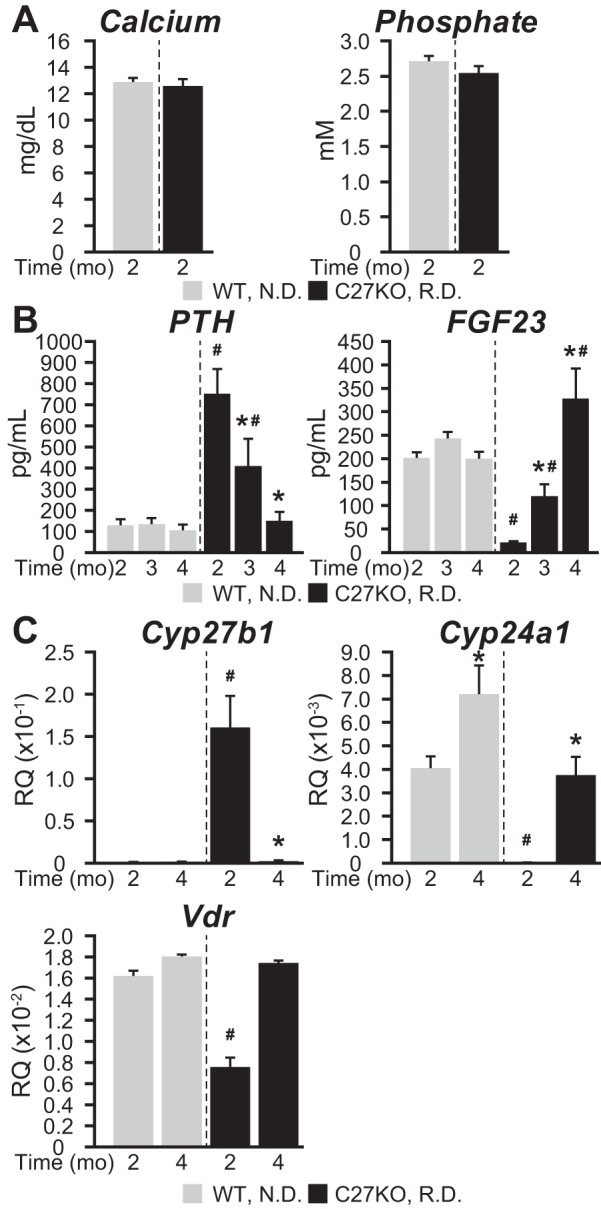
Serum PTH and FGF23 levels in Cyp27b1-null mice are normalized after 4 months of dietary rescue. A, serum calcium (mg/dl) and phosphate (mm) concentrations after 2 months of rescue diet (RD, 20% lactose, 2% Ca, 1.25% P) versus normal control diet (ND, 0.6% Ca, 0.4% P). B, serum PTH (pg/ml), and FGF23 (pg/ml) concentrations after 2, 3, and 4 months of RD versus ND. C, Cyp27b1, Cyp24a1, Vdr expression levels after 2 and 4 months of RD versus ND. Data are displayed as relative quantitation (RQ, mean ± S.E. (error bars)) compared with Gapdh. n = 4 for WT groups, n = 6 for KO groups. *, p < 0.05 paired t test: treatment versus vehicle. #, p < 0.05 paired t test: KO time point versus WT time point.
Figure 8.

Dietary rescue of the M1/M21-DIKO mouse. A, table of serum calcium (mg/dl), phosphate (mm), PTH (pg/ml), and FGF23 (pg/ml) concentrations with n values indicated in parentheses for C27KO, M1-IKO, M21-IKO, and M1/M21-DIKO fed control diet (control, 0.6% Ca, 0.4% P) or rescue diet (rescue, 20% lactose, 2% Ca, 1.25% P) for 4 months versus WT littermates (pooled WT values same as in Fig. 2). One-way ANOVA with multiple-comparison Tukey post-test: *, p < 0.05 control or rescue versus WT. B, femur length from WT mice fed control diet (control), M1/M21-DIKO mice on control diet (control) or rescue diet (rescue) for 4 months (n = 2, male top, female bottom). C, BMD measurements in femur and body weights (g) for WT (gray, n = 6, control diet) and M1/M21-DIKO (n = 6) mice fed control diet (blue) or rescue diet (black) for 4 months. *, p < 0.05 paired t test: KO versus WT. D, gene expression for Cyp27b1, Cyp24a1, and Vdr in the kidney of WT (gray), M1-IKO, and M1/M21-DIKO mice after control diet (blue) or rescue diet (black). Data are displayed as relative quantitation (RQ, mean ± S.E. (error bars)) compared with Gapdh. *, p < 0.05 paired t test: KO versus WT control diet. #, p < 0.05 paired t test: KO rescue diet versus WT control diet. E, table of serum 25(OH)D3, 24,25(OH)2D3, 1,25(OH)2D3, and 1,24,25(OH)2D3 concentrations with n values indicated in parentheses as in A (pooled WT values same as in Fig. 2). F, gene expression for Cyp27b1, Cyp24a1, and Vdr in the skin of WT (gray), M1-IKO, and M1/M21-DIKO mice after control diet (blue) or rescue diet (black) as in D.
Based upon these phenotypic features of the rescued M1-IKO and M1/M21-DIKO mice, we isolated RNA and assessed the levels of Cyp27b1, Cyp24a1, and Vdr transcripts in the kidneys of these mutant mice. As expected (see Fig. 2E), basal levels of Cyp27b1 were considerably lower in both the M1-IKO and the M1/M21-DIKO mice (Fig. 8D). The anticipated Cyp27b1 responses to rescue diet-induced changes in PTH and FGF23 were also observed in these mice. Thus, Cyp27b1 levels were lower in rescued M1-IKO mice, which retained Cyp27b1 sensitivity to FGF23 but not PTH, but were unaffected in rescued M1/M21-DIKO mice wherein Cyp27b1 expression remained insensitive to any corrective changes in both PTH and FGF23. As anticipated, however, rescue diet–induced changes in these two hormones homeostatically up-regulated Cyp24a1 expression in the M1-IKO mouse and particularly in the M1/M21-DIKO mice. Vdr expression levels were also raised in both strains. These data support our hypothesis that the reduced expression of Cyp24a1 in the kidney is critical to the inappropriately high levels of circulating 1,25(OH)2D3 seen in both of these mice, a view substantiated by further assessment of circulating metabolites of vitamin D. Accordingly, the additional measurement of vitamin D metabolites in both M1-IKO and M2/M21-DIKO mice, as seen in Fig. 8E, revealed a striking rescue diet–induced increase in 24,25(OH)2D3, a concomitant decrease in 25(OH)D3 substrate, a suppression of circulating 1,25(OH)2D3 to below detectable levels of the assay, and a concomitant rise in 1,24,25(OH)3D3 indicative of high rates of renal catabolism of 1,25(OH)2D3.
Again, as seen in Fig. 8F, no effect of the rescue diet was seen on the expression of Cyp27b1, Cyp24a1, or Vdr in any of the NRTC tissues examined (skin analyses are displayed in Fig. 8F). Aside from strongly supporting the role of low Cyp24a1 in the maintenance of inappropriately high 1,25(OH)2D3 in parallel with the strikingly aberrant mineral homeostatic phenotype observed, these results support the idea that diet-rescued M1/M21-DIKO mice exhibit normal levels of Ca and P, near normal PTH and FGF23, and generally normal circulating levels of key vitamin D metabolites while at the same time exhibiting undetectable levels of circulating endocrine 1,25(OH)2D3. Thus, it seems likely that this mouse model will be useful not only in evaluating the molecular basis for mineral homeostasis but also for assessing the potential roles and mechanisms associated with NRTC Cyp27b1 expression and the enzyme's sensitivity to circulating levels of exogenously added 25(OH)D3. Finally, given the critical changes to both PTH and FGF23 in the rescued mouse, we then administered a dose of PTH and confirmed that Cyp27b1 expression was indeed insensitive to PTH induction even in the absence of high PTH and low FGF23 (data not shown).
The genetic and epigenetic landscape of CYP27B1 in the human kidney is similar to that in mouse
Our studies have defined a complex module that mediates the selective basal and hormone-regulated expression of Cyp27b1 in the mouse. Interestingly, genes in this chromosomal location are syntenic in the human genome, although they are located in reverse on the opposite strand. Thus, potential regulatory active regions could be present in humans in a fashion similar to those in the mouse (13). Support for this idea has emerged with the identification of SNPs in regions of METTL1 and METTL21b (EEF1AKMT3) that link the severity of autoimmune diseases purported to be sensitive to 1,25(OH)2D3, such as multiple sclerosis, to Cyp27b1 expression (26–28). Unfortunately, none of the available renal proximal tubule cell lines of human origin, including the popular RPTEC cell line, exhibit informative genetic and epigenetic profiles across the CYP27B1 gene locus that would facilitate our understanding of renal CYP27B1 regulation (data not shown). We therefore obtained several donated cadaver kidneys and conducted both RNA and ChIP-Seq analyses of isolated segments of the cortex from these organs. Significant levels of CYP27B1, CYP24A1, and VDR RNA were detected by RT-qPCR analysis of this tissue (CT values of 27, 25, and 27, which equates to RQ values of 0.032 ± 0.003, 0.13 ± 0.012, and 0.030 ± 0.002 (mean ± S.E.), respectively). Based upon this analysis, we then performed ChIP-Seq analysis using antibodies to the VDR, pCREB, H3K4me1, and H3K27ac, transcription factors, and histone modification that we initially utilized to characterize the kidneys of vitamin D–sufficient WT mice. As is documented in Fig. 9, the ChIP-Seq data tracks reveal the presence of both VDR and pCREB at several sites across the CYP27B1 landscape and specifically within the introns of METTL1 and METTL21B, where they were similarly observed within the mouse Cyp27b1 gene locus. These sites aligned in general with H3K4me1 and H3K27ac histone modifications that are known to represent partial signatures of active regulatory enhancers, suggesting in turn that the sites of VDR and CREB binding are indeed localized to regions that may mediate regulation of CYP27B1 by PTH and perhaps 1,25(OH)2D3. It is clear, however, that not all sites are similar to that seen in the mouse, suggesting that there may be differences; the exact location within the METTL1 intron is not identical, and only a single site is present in the large intron of METTL21B. Despite these differences, we speculate that a kidney-specific module analogous to that seen in the mouse may exist in humans and that the regulation of CYP27B1 by PTH, FGF23, and 1,25(OH)2D3 could be very similar.
Figure 9.

ChIP-Seq analysis of the human kidney reveals conserved genomic occupancy relative to the mouse. ChIP-Seq analysis of the mouse kidney (13) (top, as in Fig. 1A) was contrasted with that derived from isolated human kidney cortex. Overlaid ChIP-Seq data tracks for pCREB and VDR from mouse kidney (top) are displayed as either basal or vehicle (yellow, n = 3) or treated for 1 h with PTH or 1,25(OH)2D3 (blue, n = 3) as indicated. Overlapping data (vehicle and treatment) appear as green. Bottom, human kidney ChIP-Seq data for pCREB, VDR, H3K4me1, and H3K27ac are displayed in triplicate (blue, yellow, and pink; overlaps appear as brown). DNase-Seq data from the ENCODE project (normal human embryonic kidney (NHEK), embryonic stem cell line H1, MCF7 breast cancer cells, CD4-positive T cell line) are shown to highlight specificity retained for adult kidney. Regions of interest are highlighted in light gray boxes. The human CYP27B1 locus is in the opposite direction on chromosome 12, and therefore, we reversed the human locus to highlight conservation of the peaks to the mouse. Genomic repeats and SNPs are also displayed aligned to the data at the bottom. Genomic location and scale are indicated (top), and maximum height of tag sequence density for each data track is indicated on the y axis (top left, each track, normalized to input and 107 tags). The direction of transcription is indicated by the arrow, and exons are indicated by boxes.
Discussion
Our previous studies have revealed the presence of a kidney-specific genomic module in the mouse that controls expression of the Cyp27b1 gene responsible for the production of endocrine 1,25(OH)2D3 (13). In these current studies, we further localized the complex genomic components within the two individual submodules that serve to mediate basal as well as homeostatic regulation by PTH, FGF23, and 1,25(OH)2D3. The properties of these control elements are unique as for PTH; others display redundancy as for FGF23 and 1,25(OH)2D3. We also show that the removal of both components of this regulatory module using a sequential CRISPR/Cas9 approach in vivo fully eliminates all basal and hormone-regulated expression of Cyp27b1 in the kidney while leaving the basal expression of this gene intact in NRTCs. This maneuver causes both a striking reduction in the circulating levels of 1,25(OH)2D3 and a severe systemic and skeletal phenotype. Whereas these widespread deficiencies are rescued when these animals are fed a high-Ca and -P diet, homeostatic up-regulation of renal Cyp24a1 reduces the small amounts of residual 1,25(OH)2D3 that are present in this mouse to undetectable levels, thereby imposing normal degradation rates on 1,25(OH)2D3 production, providing the first animal model in which to study the nature of local NRTC-produced 1,25(OH)2D3. Finally, this module appears to be present in a similar, although perhaps not identical, layout in the human kidney.
Gross CRISPR/Cas9-mediated dissection of the intronic Mettl1 and Mettl21b components that comprise the Cyp27b1 regulatory module has revealed considerable functional complexity that is attached to each of the epigenetically defined segments located within these two distinct but independent submodules. Three potential enhancers are located within the more complex Mettl21b intron. The most distal retains an inducible basal function, the deletion of which causes a significant loss of basal Cyp27b1 expression. This component also mediates FGF23 suppression, although this activity is attenuated relative to that of the WT mouse, suggesting additional components. It was therefore not surprising to observe that the removal of the two more proximal segments also attenuated FGF23 suppression, indicating that the action of this hormone was redundant across at least two if not three sites in the Mettl21b intron. These two segments are also located in a region of the genome that retains many repetitive elements and does not appear to be conserved within the human sequence as we previously documented (13). As identified earlier, however, FGF23 suppression of Cyp27b1 was fully lost upon deletion of the entire M21 submodule, indicating that FGF23 activity was entirely limited to this particular component of the renal module. Interestingly, suppression by 1,25(OH)2D3, although attenuated, was retained upon full deletion of the M21 module, suggesting that this hormone also operated within intronic M1. A full time course of response in the M1-IKO mouse confirmed these differences between 1,25(OH)2D3 and FGF23, both of which were fully lost in the M1/M21-DIKO mouse. It is also noteworthy that whereas the activities of 1,25(OH)2D3 cannot be specifically localized to each of the three segments within the M21 intron due to residual activity within M1, the idea that the actions of 1,25(OH)2D3 span both submodules (M1 and M21) is supported by the localization of the VDR at each of these sites within both submodules via ChIP-Seq analysis (13). This interpretation suggests that the potent and sustained suppressive effects of 1,25(OH)2D3 on Cyp27b1 expression are due to its ability to reinforce the suppressive actions of FGF23 within M21 while opposing the inducing actions of PTH within M1. Finally, loss of the basal activity of Cyp27b1 indicates the presence of an inducing function within the Mettl21b intron. Whereas we speculate that this may be due to an unknown systemic factor, it seems clear that this factor is not CT, a peptide hormone known to up-regulate Cyp27b1 (20), because our results suggest that this induction in intact mice is secondary to the up-regulation of PTH. Additional studies will be necessary to better resolve each of these genomic sites of action.
Additional investigation to resolve the actions of PTH in the M1 submodule were informative yet in some respects puzzling. Restricted deletion of the more proximal segment of the M1 region in the M1-IKOP mouse supported our speculation that basal expression and sensitivity to PTH induction were linked, at least at this level of resolution. Nevertheless, the results also suggested that these segments and perhaps the DNA sequence elements themselves are complex, given the fact that whereas basal expression was fully lost, a residual sensitivity (3×) to PTH induction was retained. Surprisingly, however, this very modest response of Cyp27b1 to PTH was of clear significance, because 1,25(OH)2D3 was sufficiently high in these mice to maintain normal blood Ca and P levels and to prevent the largely uncontrolled up-regulation of PTH and full suppression of FGF23 that is seen in the M1-IKO mouse. How this is accomplished in the M1-IKOP mouse is uncertain at present, although it does indicate that the sensitivity of renal Cyp27b1 expression to PTH is exquisite. Surprisingly, Cyp24a1 expression remained suppressed to some degree in this mouse despite lowered PTH and higher FGF23 levels, resulting in above normal levels of 24,25(OH)2D3 and particularly of 1,24,25(OH)3D3. The discordant activity of Cyp24a1 expression at very low RNA levels suggests the potential for a very narrow window of expression that leads to considerable CYP24A1 enzyme activity, although it is worth noting that the 25(OH)D3 substrate is exceptionally high when Cyp24a1 RNA is low, likely accounting for the higher than normal 24,25(OH)2D3 levels that are frequently seen at these low levels of Cyp24a1 expression and the higher than normal ratio of 25(OH)D3/24,25(OH)2D3 that is seen. Interestingly, creation of the M1-IKOD mouse resulted in an unexpected rise in the basal expression of Cyp27b1 concomitant with full retention of Cyp27b1 response to PTH. We speculate that this increase in the basal level may reflect a de-repression of renal Cyp27b1 expression due to loss of M1-mediated 1,25(OH)2D3 suppression, although alternative explanations are possible as well.
Although the above studies revealed many of the individual features of the kidney-specific Cyp27b1 regulatory module, we reasoned that the creation and analysis of a mutant mouse in which both submodules had been removed would be necessary to confirm the predicted boundaries of the entire module and to determine the degree to which the loss of this module could impact both the basal and hormone-regulated expression of Cyp27b1 and, perhaps more importantly, to assess whether this module was indeed kidney-specific. Observations such as this would support the creation of a mouse model unable to express Cyp27b1 exclusively in the kidney, thereby selectively deficient in endocrine-derived 1,25(OH)2D3, while at the same time maintaining full expression of basal and regulated Cyp27b1 in NRTCs. As described under “Results,” simultaneous loss of the two submodules did indeed result in such a phenotype in the M1/M21-DIKO mouse. This loss of expression of renal Cyp27b1 was clearly due to strongly reduced basal activity, likely the result of the additive effects brought about by loss of the unknown basal inducer in the M21 intron and loss of the PTH inducer in the M1 intron. This observation would support the idea that the unknown regulator is not PTH.
The deletion of this complex dual module also resulted in a Cyp27b1 gene fully refractory to transcriptional regulation by PTH, FGF23, and 1,25(OH)2D3. The loss of both basal and hormone regulated expression of Cyp27b1 in the kidney results in a dramatic systemic and skeletal phenotype as described earlier that was entirely reminiscent of the Cyp27b1-null mouse (21, 30). Due to the exceptionally high levels of PTH and to the absence of FGF23, this reduction in Cyp27b1 expression was again accompanied by an important and striking suppression of renal Cyp24a1 expression; high PTH is known to suppress whereas low FGF23 fails to induce Cyp24a1 expression. These changes in Cyp27b1 and Cyp24a1 are consistent with the altered vitamin D metabolite levels measured in the blood of these mice; 24,25(OH)2D3 levels were very low, whereas 1,25(OH)2D3 levels were strikingly deficient. Such findings are similar to those measured in Cyp27b1-null mice with the exception that 1,25(OH)2D3 levels, although undetectable in the latter mouse, are initially only deficient in the M1/M21-DIKO mouse.
These data could support the idea that this residual 1,25(OH)2D3 might be secreted from numerous NRTC sources rather than from the kidney because basal Cyp27b1 expression is retained in those tissues. Although this is possible, we speculate that this blood level of 1,25(OH)2D3 is actually the result of the striking loss of renal Cyp24a1 expression and activity, which eliminates the renal turnover of 1,25(OH)2D3 that accompanies the striking decrease in Cyp27b1 expression. Significantly, this highlights the importance of the dual contribution of both enzymes to the maintenance of circulating 1,25(OH)2D3, which results in an inappropriately high, detectable level in view of the overall detrimental phenotype that is evident in M1/M21-DIKO mice.
Nevertheless, irrespective of this speculation, our results confirm the creation of an important mouse model selectively deficient in Cyp27b1 expression in the kidney, having been uniquely created through the removal of a key regulatory module for this gene whose multiple activities are restricted to the kidney and absent at nonrenal cellular sites of Cyp27b1 expression. The potential utility of such a model is extensive and includes the ability to study homeostatic regulation and selective administration of vitamin D metabolites, such as 25(OH)D3, 1,25(OH)2D3, 24,25(OH)2D3, and 1,24,25(OH)3D3, or analogs of these metabolites. It also includes the potential assessment of the mechanisms through which NRTC production of 1,25(OH)2D3 occurs in the absence of blood metabolites and the impact of substrate levels on this production, among others.
As with other mouse models carrying mutant genes involved in vitamin D–mediated mineral regulation (e.g. Vdr-null and Cyp27b1-null), we deemed the restoration of the systemic and skeletal features of these mice using a high-Ca and -P rescue diet to be useful in providing a less complex physiologic environment with which to explore the questions posed above. We therefore initiated a dietary rescue study of the mineral phenotype by establishing a time course of normalization of the calcemic, phosphatemic, hormonal, and skeleton phenotype as well as correction of enzymatic expression in the kidneys following treatment using Cyp27b1-null mice and then M1-IKO and M1/M21-DIKO mice. Whereas this rescue diet has been utilized abundantly to “normalize” mutant mouse strains, the time course of full phenotypic rescue diverges in many studies (9). Thus, whereas Ca and P levels are rapidly normalized (<4 weeks after initiation of rescue diet), coordinated restoration of WT levels of PTH and FGF23 and correction of the abnormal expression of renal Cyp27b1 and Cyp24a1 require additional exposure in our studies until the mice are 12–16 weeks of age. This phenomenon occurs even when the rescue diet is made available to the dams and neonates immediately after birth (data not shown). As with Cyp27b1-null mice, M1-IKO and M1/M21-DIKO mice followed a similar temporal trajectory to normalization. It is unclear why PTH and FGF23 levels as well as renal Cyp27b1 and Cyp24a1 expression do not correct more rapidly in concert with blood mineral levels. The absence of 1,25(OH)2D3 does not provide an explanation for this delay, however, as dietary rescue in Vdr-null mice occurs at a similar temporal pace and is not facilitated or altered by existing levels of 1,25(OH)2D3 in both M1-IKO and M1/M21-DIKO mice.
Interestingly, because Cyp27b1 expression displays differential sensitivity to FGF23 and PTH regulation in M1-IKO and M1/M21-DIKO mice, these mutant mice also collectively confirmed an important homeostatically driven principle regarding the key role of renal Cyp24a1 in the regulation of 1,25(OH)2D3. Specifically, whereas both Cyp27b1 was suppressed and Cyp24a1 was raised in the M1-IKO mouse, only Cyp24a1 was raised in the M1/M21-DIKO mouse, as Cyp27b1 expression was fully refractory to regulation by PTH and FGF23. Strikingly, this rise in renal Cyp24a1 expression in both mutant mouse strains resulted in the loss of detectable levels of 1,25(OH)2D3 and a concomitant rise in 1,24,25(OH)3D3, in concert with a related decrease in 25(OH)D3 and a coordinated rise in 24,25(OH)2D3 levels. These effects directly in the M1/M21-DIKO mouse, therefore, clearly highlight the exclusive ability of renal Cyp24a1 to influence not only vitamin D hormone levels but also the profiles of additional vitamin D metabolites. They also support our idea that residual levels of circulating 1,25(OH)2D3 in the M1/M21-DIKO mouse on a normal diet are likely due to the absence of renal 1,25(OH)2D3 degradation rather than to secretion of the hormone from unknown NRTC sources. Regardless, the rescue of the general systemic, skeletal, and renal phenotype of the M1/M21-DIKO mouse by a high-Ca and -P diet, which is fully deficient in endocrine 1,25(OH)2D3 yet near normal with respect to other vitamin D metabolites, together with the mechanistic lessons learned appears to result in an ideal mouse model with which to explore the questions posed in the discussion above.
Our initial analyses of the Cyp27b1 gene locus in the mouse kidney raised the important question of whether the human CYP27B1 locus was similarly arranged and whether its known regulation by PTH, FGF23, and 1,25(OH)2D3 is mediated by similar functional elements. Importantly, our investigation of discarded human kidney tissue conducted by RT-qPCR and ChIP-Seq analyses as above revealed that the CYP27B1 gene locus is indeed similar to that of the mouse. Accordingly, we observed localization of both VDR and pCREB at sites within the introns of METTL1 and METTL21b that also aligned with histone H3K4me1 and H3K27ac enrichment that represents potential signatures of active regulatory enhancers. A single site rather than multiple sites was observed in the intron of METTL21B, however, and additional sites of potential binding for the VDR were also noted. Additional studies will be required to advance our understanding of the human kidney and to assess hormonal regulation, perhaps through the isolation of human proximal tubules in vitro, although as with the mouse, neither these isolates nor human cell lines have thus far been useful in delineating genomic mechanisms. It is unclear at present when the kidney-specific module emerges during development or why it is lost in both mouse and human kidney cell lines. Further investigation of the human kidney and other tissues will be useful, because multiple attempts to correlate the expression of CYP27B1 with human physiology and disease have been reported, and numerous observations indicate that vitamin D and/or 1,25(OH)2D3 may influence autoimmune disease progression as well. Perhaps most interesting currently are the efforts to correlate the presence of SNPs within the CYP27B1 locus in human populations with autoimmune diseases, and indeed many have been identified (26–28, 31–34). Interestingly, variants that seem most likely to influence CYP27B1 expression in humans are found within the vicinity of the renal module that, as documented in this report in the mouse, are responsible exclusively for expression of Cyp27b1 and production of endocrine 1,25(OH)2D3. These variants do not appear to be located directly within the regions we have identified here in the human kidney, however. Because CYP27B1 expression in mouse and human NRTCs is not generally regulated by PTH, FGF23, or 1,25(OH)2D3, it will be interesting to determine whether extrarenal regulation of CYP27B1 by inflammation and other modulators is mediated by genomic sites located outside the kidney module as is evident in the mouse.
In summary, we have identified the functional activity of the multiple regulatory components that are located within the kidney that mediate the kidney-specific regulation of Cyp27b1 in the mouse. We have also identified a variety of homeostatic consequences for other components of the machinery that control not only vitamin D metabolite levels but mediator proteins as well. Importantly, deletion of both submodules results in a mutant mouse that is vitamin D–deficient and that, when rescued by high dietary Ca and P, results in a mouse fully depleted of circulating 1,25(OH)2D3. NRTC expression of Cyp27b1 is unhindered, however, providing a model for the future investigation of the mechanism and roles played through the local production of 1,25(OH)2D3.
Experimental procedures
Reagents
The following reagents were used for in vivo injections. 1α,25(OH)2D3 was obtained from SAFC Global (Madison, WI), PTH (1–84, human) was obtained from Bachem (Torrence, CA) (H-1370.0100), mouse fibroblast growth factor 23 was from R&D Systems (Minneapolis, MN) (FGF23, 2629-FG-025), LPS was from Sigma (L6529), and salmon calcitonin was from Bachem (H-2260.0001, Torrence, CA). Antibodies used for ChIP-Seq analysis of VDR (C-20, sc-1008, lot H1216) were purchased from Santa Cruz Biotechnology, Inc. (Dallas, TX). H3K4me1 (ab8895, lot GR283603-1), H3K27ac (ab4729, lot GR3187597-1), and H3K36me3 (ab9050, lot GR273247-1) were purchased from Abcam (Cambridge, MA). Phosphorylated CREB (Ser-133) (pCREB, 06-519, lot 2762242) and H3K9ac (06-942, lot 2664263) were purchased from Millipore Corp. (Billerica, MA). Traditional genotyping PCR was completed with GoTaq (Promega, Madison, WI), and all real-time qPCR was completed with StepOnePlus using TaqMan for gene expression assays (Applied Biosystems, Foster City, CA). Primers were obtained from IDT (Coralville, IA).
Gene expression
Dissected tissues were frozen immediately in liquid nitrogen and stored at −80 °C. Frozen tissues were homogenized in TRIzol reagent (Life Technologies, Inc.), and RNA was isolated as per the manufacturer's instructions. 1 μg of isolated total RNA was DNase-treated, reverse-transcribed using the High Capacity cDNA kit (Applied Biosystems), and then diluted to 100 μl with RNase/DNase-free water. qPCR was performed using primers specific to a select set of differentially expressed genes by Taqman analyses. TaqMan Gene Expression probes (Applied Biosystems) were used for RT-PCR and are found in Table 1.
Table 1.
TaqMan assays, CRISPR guides, and genotyping primers
| Sequence-based reagents | Sequence/Source | Reference |
|---|---|---|
| TaqMan assays | ||
| Gapdh | Applied Biosystems | 4352339E |
| Cyp27b1 | Applied Biosystems | Mm01165918 |
| Cyp24a1 | Applied Biosystems | Mm00487244 |
| Vdr | Applied Biosystems | Mm00437297 |
| Mettl1 | Applied Biosystems | Mm00487686 |
| Mettl21b | Applied Biosystems | Mm01165909 |
| GAPDH | Applied Biosystems | Hs02758991_g1 |
| CYP27B1 | Applied Biosystems | Hs00168017_m1 |
| CYP24A1 | Applied Biosystems | Hs00167999_m1 |
| VDR | Applied Biosystems | Hs01045840_m1 |
| METTL1 | Applied Biosystems | Hs01096147_g1 |
| METTL21B | Applied Biosystems | Hs00919597_g1 |
| Primers for CRISPR | ||
| M1-IKO guide 1 | GATTAGTTGACCTTTCCTCC-TGG | Meyer et al. (13) |
| M1-IKO guide 2 | CAGGAACTCCAGACCATGAG-AGG | Meyer et al. (13) |
| M1-IKO guide 3 | GTTGTTCTGTCTAGCTCCAG-TGG | This paper |
| M1-IKO guide 4 | GCGTCAGAGATCAGCTCCAG-TGG | This paper |
| M21-IKO guide 1 | CCTACCTTCCCGCTACTGTT-GGG | Meyer et al. (13) |
| M21-IKO guide 2 | CCCTTCCTTAGGGACTTCAT-GGG | Meyer et al. (13) |
| M21-IKO guide 3 | GCATGCAATGGCCCTCCGTT-TGG | This paper |
| Primers for genotyping | ||
| M1-IKO span F | AGTGGAGTTTGCAGACATAGGCT | Meyer et al. (13) |
| M1-IKO span R | TTACCTGTCTATAGGGAAGATG | Meyer et al. (13) |
| M1-IKO internal F | TCTACTCTGGGTCTGTGGCCTT | Meyer et al. (13) |
| M1-IKO internal R | AGCTAGACAGAACAACCGGGG | Meyer et al. (13) |
| M1-IKOP span F | TCTACTCTGGGTCTGTGGCCTT | This paper |
| M1-IKOP span R | ATGTAACAGGAACTCCAGACCAT | This paper |
| M1-IKOP internal F | GGCTCCAGTCCCTCCGTGAGCC | This paper |
| M1-IKOP internal R | GCTGACCCGGCTCACTGACCC | This paper |
| M1-IKOD span F | Same as M1-IKO span F | Meyer et al. (13) |
| M1-IKOD span R | Same as M1-IKO span R | Meyer et al. (13) |
| M1-IKOD internal F | Same as M1-IKO internal F | Meyer et al. (13) |
| M1-IKOD internal R | Same as M1-IKO internal R | Meyer et al. (13) |
| M21-IKO span F | TTCCTCCACTGAGACAAGAGTTA | Meyer et al. (13) |
| M21-IKO span R | CCTTGCTACTTTCCAACAGCCTGCCT | Meyer et al. (13) |
| M21-IKO internal F | ATGTATCCTCTCCCTCCTGAACA | Meyer et al. (13) |
| M21-IKO internal R | CCCCATACAATAGGGTTTCTCTCTG | Meyer et al. (13) |
| M21-IKOP span F | Same as M21-IKO span F | Meyer et al. (13) |
| M21-IKOP span R | TCATTGTCTTTAGCCCTGATCTTAC | This paper |
| M21-IKOP internal F | CCGAGCTAATCTTCAACCCTTCT | This paper |
| M21-IKOP internal R | CTTGTGTTCCCCATTCTTTGCTC | This paper |
| M21-IKOD span F | Same as M21-IKO span F | Meyer et al. (13) |
| M21-IKOD span R | Same as M21-IKO span R | Meyer et al. (13) |
| M21-IKOD internal F | Same as M21-IKO internal F | Meyer et al. (13) |
| M21-IKOD internal R | Same as M21-IKO internal R | Meyer et al. (13) |
ChIP followed by sequencing (ChIP-Seq)
ChIP was performed using antibodies listed under “Reagents.” ChIP was performed as described previously with several modifications (13, 35). Human kidney samples were obtained from the University of Wisconsin Madison Organ Procurement Organization with approval. These kidneys represent an organ donation from a 69-year-old female. In brief, human kidney cortex was dissected from whole kidney tissue, placed into 1.5% formaldehyde cross-linking solution, and then subjected to a ChIP procedure and immunoprecipitation using either a control IgG or the indicated antibodies. Statistical analysis and data processing for ChIP-Seq assays were performed as reported previously (36). The isolated DNA (or input DNA acquired prior to precipitation) was then validated by qPCR and further prepared for ChIP-Seq analysis. ChIP-Seq libraries were prepared as described previously (13, 36). All human data were mapped to the hg19 genome build.
CRISPR-generated and transgenic mice
The guides used for CRISPR-Cas9–mediated genome editing were optimized for the least number of potential off-target sites and fewest sites within coding exons using the Zhang laboratory CRISPR Design tool and cross-referenced with Liu laboratory CRISPR-DO. The guides (see Table 1) were annealed and cloned into plasmid pX330 or pX458 obtained from the Zhang laboratory via Addgene (Cambridge, MA) as described recently (37). Resulting PCR products were then transcribed in vitro utilizing the T7 MEGAshortscript (Life Technologies) kit (38). The mixture of 50 ng/μl of the produced RNA guides and 40 ng/μl of Cas9 protein in injection buffer (5 mm Trizma base, 5 mm Tris-HCl, 0.1 mm EDTA, pH 7.4) was injected into the pronucleus of 1-day fertilized embryos isolated from hyperovulating female C57BL/6 mice as described previously (39) and implanted into recipient females by the University of Wisconsin Madison Biotechnology Genome Editing and Animal Models Core. For the creation of the M1/M21-DIKO mouse, homozygous M21-IKO knockout females were hyperovulated as described (39) and mated to homozygous M21-IKO knockout males prior to CRISPR microinjection with M1-IKO guides (Table 1). This ensured that the M1 deletion would be allelic with the M21 deletion. The resultant pups were genotyped with spanning primers (Table 1), cloned, and sequenced. The top 10 predicted potential off-target sites were examined by PCR and sequencing analysis.
Animal studies
Genetically modified mice were outbred with C57BL/6 mice (Jackson Laboratory, Bar Harbor, ME) as heterozygotes. Mice were housed in high-density ventilated caging in the Animal Research Facility of the University of Wisconsin (Madison, WI) under 12-h light/dark cycles at 72 °F and 45% humidity. All mice used in this study were maintained on a standard rodent chow diet (5008, Lab Diet, St. Louis, MO), aged 8–9 weeks, and then backcrossed 5 or more generations unless otherwise indicated. For the high-Ca/P rescue diet study, 3-week-old mice were fed either a rescue diet (Envigo TD.96348, 20% lactose, 2% Ca, 1.25% P) or normal control diet (Envigo, TD.97191, 0.6% Ca, 0.4% P) for 2, 3, or 4 months, at which time animals were euthanized, and tissues were collected. All experiments and tissue collections were performed in the procedure rooms in the Research Animal Facility of the University of Wisconsin (Madison, WI). All animal studies were reviewed and approved by the Research Animal Care and Use Committee of the University of Wisconsin (Madison, WI) under protocol A005478. Animals were subjected to intraperitoneal injection of 10 ng/g body weight (bw) 1,25(OH)2D3 (in propylene glycol), 230 ng/g bw PTH (1–84) (in PBS), 50 ng/g bw FGF23 (in PBS + 0.1% BSA), 10 μg/g bw LPS (in PBS), or vehicle (EtOH or PBS). Animals were sacrificed, and tissues were collected 1 h after PTH injection, 3 h after FGF23 injection, and 6 h after 1,25(OH)2D3 and LPS injection. Unless otherwise indicated, all experiments were conducted with equal numbers of males and females (n ≥ 6). Data were reported as mixed, as no differences were found between sexes.
Western blot analysis
Mitochondrial extracts from whole kidneys were prepared with a Potter–Elvehjem homogenization tube using 5 ml of homogenization buffer (250 mm sucrose, 19 mm HEPES, 10 mm KCl, pH 7.4, plus protease inhibitor mixture (Roche Applied Science)). The resulting homogenate was centrifuged (4,000 × g) for 1 min (4 °C). The supernatant was centrifuged (9,000 × g) for 20 min (4 °C), and the resulting pellet was suspended in 1 ml of homogenization buffer and centrifuged (9,000 × g) for 10 min (4 °C). The pellet was solubilized in 500 μl of SDS loading buffer (100 mm Tris-HCl, pH 6.8, 4% SDS, 0.2% bromphenol blue, 20% glycerol, 200 mm DTT). 30 μl of sample was heated to 95 °C for 5 min and subjected to 12% SDS-PAGE. The kidney mitochondrial extracts were subjected to Western blot analysis as described previously (36) using primary antibodies to CYP27B1 (G-20, sc-49644, Santa Cruz Biotechnology, lot B1114, 1:500) and VDAC/porin (ab15895, Abcam, lot GR264581-2, 1:5,000) followed by secondary antibodies donkey α-goat IgG-HRP (sc-2020, Santa Cruz Biotechnology, 1:2,000) and goat α-rabbit IgG-HRP (sc-2004, Santa Cruz Biotechnology, 1:5,000), respectively.
Whole-kidney tissue lysates were prepared using Tissue Lysis Buffer (10 mm Tris-Cl (pH 8), 300 mm KCl, 1 mm EDTA, 2 mm DTT, and protease inhibitor mixture) and protein concentrations were measured using a protein assay (Bio-Rad). 80 μg of lysates was denatured and subjected to 12% SDS-PAGE. Subsequently, whole-kidney lysates were subjected to Western blot analysis as described previously (36) using primary antibodies to VDR (9A7 (40), 1:2,000) and β-tubulin (H-235, sc-9104, Santa Cruz Biotechnology; lot E0913, 1:5,000) followed by secondary antibodies goat α-rat IgG-HRP (sc-2032, Santa Cruz Biotechnology, 1:2,000) and goat α-rabbit IgG-HRP (1:5,000), respectively. Due to the overlapping molecular weights of VDR and β-tubulin, one aliquot of sample was run over two gels and immunoblotted as described above.
Blood chemistry
Cardiac blood was collected at the time of sacrifice. Collected blood was split into serum- or EDTA-treated plasma and incubated at room temperature for 30 min followed by centrifugation at 6,000 rpm for 12 min (twice) to obtain serum or EDTA plasma. Serum calcium and phosphate levels were measured using the QuantiChromTM calcium assay kit (DICA-500, BioAssay Systems, Hayward, CA) and QuantiChromTM phosphate assay kit (DIPI-500, BioAssay Systems). Circulating intact FGF23 and PTH were measured in EDTA plasma via a mouse/rat FGF-23 (intact) ELISA kit (catalog no. 60-6800, Immutopics, San Clemente, CA) and a mouse PTH(1–84) ELISA kit (catalog no. 60-2305, Immutopics), respectively. As experiments were performed in parallel, WT littermates were pooled for increased statistical power.
Quantification of serum vitamin D metabolites
Serum 25(OH)D3, 24,25(OH)2D3, 1,25(OH)2D3, and 1,24,25(OH)3D3 were quantified by LC-MS/MS using previously published methods (9, 41, 42) and as reported recently (13). As experiments were performed in parallel, WT littermates were pooled for increased statistical power.
BMD
At 8–9 weeks of age, BMDs of the CRISPR-generated mice and their WT littermates were measured and analyzed by dual X-ray absorptiometry with a PIXImus densitometer (GE-Lunar Corp., Madison, WI) as described previously (29).
Statistical evaluation and data availability
Data were analyzed using GraphPad Prism version 8 software (GraphPad Software, Inc., La Jolla, CA) and in consultation with the University of Wisconsin Statistics Department. All values are reported as the mean ± S.E., and differences between group means were evaluated using one-way ANOVA, two-way ANOVA, or Student's t test as indicated in the figure legends. All human kidney ChIP-Seq data have been deposited in the Gene Expression Omnibus (GSE129585).
Author contributions
M. B. M. and J. W. P. conceptualization; M. B. M, N. A. B, S. M. L., and M. K. investigation; R. R. R. human tissue procurement; M. B. M. and J. W. P. writing-original draft; N. A. B., M. K., and G. J. writing-review and editing; M. B. M. data curation.
Acknowledgments
We thank members of the Pike Laboratory for contributions during manuscript preparation. We also thank Kathy Krentz and the University of Wisconsin Biotechnology Center–Genome Editing and Animal Models Core for generating the CRISPR/Cas9 enhancer-deleted mice. We acknowledge the Organ Procurement Organization at the University of Wisconsin (Madison, WI) for making organ tissues available. We also thank Waters Corp. (through a collaboration with Queen's University) for generously providing the LC-MS/MS instrumentation used in this study.
This work was supported by the Department of Biochemistry, University of Wisconsin (Madison, WI); University of Wisconsin Carbone Cancer Center Support Grant P30; and NIDDK, National Institutes of Health, Grant R01-DK117475 (to J. W. P.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
All human kidney ChIP-Seq data have been deposited in the Gene Expression Omnibus under accession number GSE129585.
- 1,25(OH)2D3
- 1α,25-dihydroxyvitamin D3
- FGF23
- fibroblast growth factor 23
- PTH
- parathyroid hormone
- TPTG
- thyroparathyroid gland
- NRTC
- non-renal target cell
- CT
- calcitonin
- Ca
- calcium
- P
- phosphate
- CREB
- cAMP-response element–binding protein
- M1-IKO
- Mettl1 intronic knockout
- M21-IKO
- Mettl21b intronic knockout
- M1/M21-DIKO
- Mettl1 and Mettl21b double intronic knockout
- LPS
- lipopolysaccharide(s)
- eEF1A
- eukaryotic elongation factor 1α
- qPCR
- quantitative PCR
- H3K4me1
- monomethylated histone H3 Lys-4
- H3K27ac
- acetylated histone H3 Lys-27
- H3K36me3
- trimethylated histone H3 Lys-36
- H3K9ac
- acetylated histone H3 Lys-9
- bw
- body weight
- HRP
- horseradish peroxidase
- BMD
- bone mineral density
- ANOVA
- analysis of variance.
References
- 1. Haussler M. R., Chandler J. S., Pike J. W., Brumbaugh P. F., Speer D. P., and Pitt M. J. (1980) Physiological importance of vitamin D metabolism. Prog. Biochem. Pharmacol 17, 134–142 [PubMed] [Google Scholar]
- 2. Prosser D. E., and Jones G. (2004) Enzymes involved in the activation and inactivation of vitamin D. Trends Biochem. Sci. 29, 664–673 10.1016/j.tibs.2004.10.005 [DOI] [PubMed] [Google Scholar]
- 3. Brenza H. L., and DeLuca H. F. (2000) Regulation of 25-hydroxyvitamin D3 1a-hydroxylase gene expression by parathyroid hormone and 1,25-dihydroxyvitamin D3. Arch. Biochem. Biophys. 381, 143–152 10.1006/abbi.2000.1970 [DOI] [PubMed] [Google Scholar]
- 4. Saito H., Maeda A., Ohtomo S., Hirata M., Kusano K., Kato S., Ogata E., Segawa H., Miyamoto K., and Fukushima N. (2005) Circulating FGF-23 is regulated by 1α,25-dihydroxyvitamin D3 and phosphorus in vivo. J. Biol. Chem. 280, 2543–2549 10.1074/jbc.M408903200 [DOI] [PubMed] [Google Scholar]
- 5. DeLuca H. F. (1974) Vitamin D: the vitamin and the hormone. Fed. Proc. 33, 2211–2219 [PubMed] [Google Scholar]
- 6. Chandler J. S., Chandler S. K., Pike J. W., and Haussler M. R. (1984) 1,25-Dihydroxyvitamin D3 induces 25-hydroxyvitamin D3–24-hydroxylase in a cultured monkey kidney cell line (LLC-MK2) apparently deficient in the high affinity receptor for the hormone. J. Biol. Chem. 259, 2214–2222 [PubMed] [Google Scholar]
- 7. Zierold C., Darwish H. M., and DeLuca H. F. (1995) Two vitamin D response elements function in the rat 1,25-dihydroxyvitamin D 24-hydroxylase promoter. J. Biol. Chem. 270, 1675–1678 10.1074/jbc.270.4.1675 [DOI] [PubMed] [Google Scholar]
- 8. Quarles L. D. (2012) Skeletal secretion of FGF-23 regulates phosphate and vitamin D metabolism. Nat. Rev. Endocrinol. 8, 276–286 10.1038/nrendo.2011.218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kaufmann M., Lee S. M., Pike J. W., and Jones G. (2015) A high-calcium and phosphate rescue diet and VDR-expressing transgenes normalize serum vitamin D metabolite profiles and renal Cyp27b1 and Cyp24a1 expression in VDR null mice. Endocrinology 156, 4388–4397 10.1210/en.2015-1664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hewison M., Burke F., Evans K. N., Lammas D. A., Sansom D. M., Liu P., Modlin R. L., and Adams J. S. (2007) Extra-renal 25-hydroxyvitamin D3–1α-hydroxylase in human health and disease. J. Steroid Biochem. Mol. Biol. 103, 316–321 10.1016/j.jsbmb.2006.12.078 [DOI] [PubMed] [Google Scholar]
- 11. Bikle D. D., Patzek S., and Wang Y. (2018) Physiologic and pathophysiologic roles of extra renal CYP27b1: case report and review. Bone Rep. 8, 255–267 10.1016/j.bonr.2018.02.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Adams J. S., Rafison B., Witzel S., Reyes R. E., Shieh A., Chun R., Zavala K., Hewison M., and Liu P. T. (2014) Regulation of the extrarenal CYP27B1-hydroxylase. J. Steroid Biochem. Mol. Biol. 144, 22–27 10.1016/j.jsbmb.2013.12.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Meyer M. B., Benkusky N. A., Kaufmann M., Lee S. M., Onal M., Jones G., and Pike J. W. (2017) A kidney-specific genetic control module in mice governs endocrine regulation of the cytochrome P450 gene. J. Biol. Chem. 292, 17541–17558 10.1074/jbc.M117.806901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Dardenne O., Prud'homme J., Hacking S. A., Glorieux F. H., and St-Arnaud R. (2003) Correction of the abnormal mineral ion homeostasis with a high-calcium, high-phosphorus, high-lactose diet rescues the PDDR phenotype of mice deficient for the 25-hydroxyvitamin D-1α-hydroxylase (CYP27B1). Bone 32, 332–340 10.1016/S8756-3282(03)00023-1 [DOI] [PubMed] [Google Scholar]
- 15. Bahr A., Hankeln T., Fiedler T., Hegemann J., and Schmidt E. R. (1999) Molecular analysis of METTL1, a novel human methyltransferase-like gene with a high degree of phylogenetic conservation. Genomics 57, 424–428 10.1006/geno.1999.5780 [DOI] [PubMed] [Google Scholar]
- 16. Malecki J., Aileni V. K., Ho A. Y. Y., Schwarz J., Moen A., Sørensen V., Nilges B. S., Jakobsson M. E., Leidel S. A., and Falnes P. (2017) The novel lysine specific methyltransferase METTL21B affects mRNA translation through inducible and dynamic methylation of Lys-165 in human eukaryotic elongation factor 1α (eEF1A). Nucleic Acids Res. 45, 4370–4389 10.1093/nar/gkx002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kerner S. A., Scott R. A., and Pike J. W. (1989) Sequence elements in the human osteocalcin gene confer basal activation and inducible response to hormonal vitamin D3. Proc. Natl. Acad. Sci. U.S.A. 86, 4455–4459 10.1073/pnas.86.12.4455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ozono K., Liao J., Kerner S. A., Scott R. A., and Pike J. W. (1990) The vitamin D-responsive element in the human osteocalcin gene: association with a nuclear proto-oncogene enhancer. J. Biol. Chem. 265, 21881–21888 [PubMed] [Google Scholar]
- 19. Meyer M. B., Goetsch P. D., and Pike J. W. (2012) VDR/RXR and TCF4/β-catenin cistromes in colonic cells of colorectal tumor origin: impact on c-FOS and c-MYC gene expression. Mol. Endocrinol 26, 37–51 10.1210/me.2011-1109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Shinki T., Ueno Y., DeLuca H. F., and Suda T. (1999) Calcitonin is a major regulator for the expression of renal 25-hydroxyvitamin D3–1α-hydroxylase gene in normocalcemic rats. Proc. Natl. Acad. Sci. U.S.A. 96, 8253–8258 10.1073/pnas.96.14.8253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Panda D. K., Miao D., Tremblay M. L., Sirois J., Farookhi R., Hendy G. N., and Goltzman D. (2001) Targeted ablation of the 25-hydroxyvitamin D 1α-hydroxylase enzyme: evidence for skeletal, reproductive, and immune dysfunction. Proc. Natl. Acad. Sci. U.S.A. 98, 7498–7503 10.1073/pnas.131029498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Panda D. K., Miao D., Bolivar I., Li J., Huo R., Hendy G. N., and Goltzman D. (2004) Inactivation of the 25-hydroxyvitamin D 1α-hydroxylase and vitamin D receptor demonstrates independent and interdependent effects of calcium and vitamin D on skeletal and mineral homeostasis. J. Biol. Chem. 279, 16754–16766 10.1074/jbc.M310271200 [DOI] [PubMed] [Google Scholar]
- 23. Christakos S. (2012) Recent advances in our understanding of 1,25-dihydroxyvitamin D3 regulation of intestinal calcium absorption. Arch. Biochem. Biophys. 523, 73–76 10.1016/j.abb.2011.12.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lee S. M., Riley E. M., Meyer M. B., Benkusky N. A., Plum L. A., DeLuca H. F., and Pike J. W. (2015) 1,25-Dihydroxyvitamin D3 controls a cohort of vitamin D receptor target genes in the proximal intestine that is enriched for calcium-regulating components. J. Biol. Chem. 290, 18199–18215 10.1074/jbc.M115.665794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Li Y. C., Amling M., Pirro A. E., Priemel M., Meuse J., Baron R., Delling G., and Demay M. B. (1998) Normalization of mineral ion homeostasis by dietary means prevents hyperparathyroidism, rickets, and osteomalacia, but not alopecia in vitamin D receptor-ablated mice. Endocrinology 139, 4391–4396 10.1210/endo.139.10.6262 [DOI] [PubMed] [Google Scholar]
- 26. International Multiple Sclerosis Genetics Consortium, Wellcome Trust Case Control Consortium 2, Sawcer S., Hellenthal G., Pirinen M., Spencer C. C., Patsopoulos N. A., Moutsianas L., Dilthey A., Su Z., Freeman C., Hunt S. E., Edkins S., Gray E., Booth D. R., et al. (2011) Genetic risk and a primary role for cell-mediated immune mechanisms in multiple sclerosis. Nature 476, 214–219 10.1038/nature10251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Raychaudhuri S., Remmers E. F., Lee A. T., Hackett R., Guiducci C., Burtt N. P., Gianniny L., Korman B. D., Padyukov L., Kurreeman F. A., Chang M., Catanese J. J., Ding B., Wong S., van der Helm-van Mil A. H., et al. (2008) Common variants at CD40 and other loci confer risk of rheumatoid arthritis. Nat. Genet. 40, 1216–1223 10.1038/ng.233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ramagopalan S. V., Dyment D. A., Cader M. Z., Morrison K. M., Disanto G., Morahan J. M., Berlanga-Taylor A. J., Handel A., De Luca G. C., Sadovnick A. D., Lepage P., Montpetit A., and Ebers G. C. (2011) Rare variants in the CYP27B1 gene are associated with multiple sclerosis. Ann. Neurol 70, 881–886 10.1002/ana.22678 [DOI] [PubMed] [Google Scholar]
- 29. Onal M., St John H. C., Danielson A. L., and Pike J. W. (2016) Deletion of the distal Tnfsf11 RL-D2 enhancer that contributes to PTH-mediated RANKL expression in osteoblast lineage cells results in a high bone mass phenotype in mice. J. Bone Miner. Res. 31, 416–429 10.1002/jbmr.2698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Dardenne O., Prud'homme J., Arabian A., Glorieux F. H., and St-Arnaud R. (2001) Targeted inactivation of the 25-hydroxyvitamin D3-1α-hydroxylase gene (CYP27B1) creates an animal model of pseudovitamin D-deficiency rickets. Endocrinology 142, 3135–3141 10.1210/endo.142.7.8281 [DOI] [PubMed] [Google Scholar]
- 31. Ramagopalan S. V., Heger A., Berlanga A. J., Maugeri N. J., Lincoln M. R., Burrell A., Handunnetthi L., Handel A. E., Disanto G., Orton S. M., Watson C. T., Morahan J. M., Giovannoni G., Ponting C. P., Ebers G. C., and Knight J. C. (2010) A ChIP-seq defined genome-wide map of vitamin D receptor binding: associations with disease and evolution. Genome Res. 20, 1352–1360 10.1101/gr.107920.110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Sundqvist E., Bäärnhielm M., Alfredsson L., Hillert J., Olsson T., and Kockum I. (2010) Confirmation of association between multiple sclerosis and CYP27B1. Eur. J. Hum. Genet. 18, 1349–1352 10.1038/ejhg.2010.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Alcina A., Fedetz M., Fernández O., Saiz A., Izquierdo G., Lucas M., Leyva L., García-León J. A., Abad-Grau M. del M., Alloza I., Antigüedad A., Garcia-Barcina M. J., Vandenbroeck K., Varadé J., de la Hera B., et al. (2013) Identification of a functional variant in the KIF5A-CYP27B1-METTL1-FAM119B locus associated with multiple sclerosis. J. Med. Genet. 50, 25–33 10.1136/jmedgenet-2012-101085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Karaky M., Alcina A., Fedetz M., Barrionuevo C., Potenciano V., Delgado C., Izquierdo G., and Matesanz F. (2016) The multiple sclerosis-associated regulatory variant rs10877013 affects expression of CYP27B1 and VDR under inflammatory or vitamin D stimuli. Mult. Scler. 22, 999–1006 10.1177/1352458515610208 [DOI] [PubMed] [Google Scholar]
- 35. Meyer M. B., Zella L. A., Nerenz R. D., and Pike J. W. (2007) Characterizing early events associated with the activation of target genes by 1,25-dihydroxyvitamin D3 in mouse kidney and intestine in vivo. J. Biol. Chem. 282, 22344–22352 10.1074/jbc.M703475200 [DOI] [PubMed] [Google Scholar]
- 36. Meyer M. B., Benkusky N. A., and Pike J. W. (2014) The RUNX2 cistrome in osteoblasts: characterization, down-regulation following differentiation, and relationship to gene expression. J. Biol. Chem. 289, 16016–16031 10.1074/jbc.M114.552216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Meyer M. B., Benkusky N. A., and Pike J. W. (2015) Selective distal enhancer control of the Mmp13 gene identified through clustered regularly interspaced short palindromic repeat (CRISPR) genomic deletions. J. Biol. Chem. 290, 11093–11107 10.1074/jbc.M115.648394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Wang H., Yang H., Shivalila C. S., Dawlaty M. M., Cheng A. W., Zhang F., and Jaenisch R. (2013) One-step generation of mice carrying mutations in multiple genes by CRISPR/Cas-mediated genome engineering. Cell 153, 910–918 10.1016/j.cell.2013.04.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Meyer M., de Angelis M. H., Wurst W., and Kühn R. (2010) Gene targeting by homologous recombination in mouse zygotes mediated by zinc-finger nucleases. Proc. Natl. Acad. Sci. U.S.A. 107, 15022–15026 10.1073/pnas.1009424107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Pike J. W., Donaldson C. A., Marion S. L., and Haussler M. R. (1982) Development of hybridomas secreting monoclonal antibodies to the chicken intestinal 1α,25-dihydroxyvitamin D3 receptor. Proc. Natl. Acad. Sci. U.S.A. 79, 7719–7723 10.1073/pnas.79.24.7719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kaufmann M., Gallagher J. C., Peacock M., Schlingmann K. P., Konrad M., DeLuca H. F., Sigueiro R., Lopez B., Mourino A., Maestro M., St-Arnaud R., Finkelstein J. S., Cooper D. P., and Jones G. (2014) Clinical utility of simultaneous quantitation of 25-hydroxyvitamin D and 24,25-dihydroxyvitamin D by LC-MS/MS involving derivatization with DMEQ-TAD. J. Clin. Endocrinol. Metab. 99, 2567–2574 10.1210/jc.2013-4388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kaufmann M., Morse N., Molloy B. J., Cooper D. P., Schlingmann K. P., Molin A., Kottler M. L., Gallagher J. C., Armas L., and Jones G. (2017) Improved screening test for idiopathic infantile hypercalcemia confirms residual levels of serum 24,25-(OH)2D3 in affected patients. J. Bone Miner. Res. 32, 1589–1596 10.1002/jbmr.3135 [DOI] [PubMed] [Google Scholar]