

Abstract
Cancer tissues harbor thousands of mutations, and a given oncogene may be mutated at hundreds of sites, yet only a few of these mutations have been functionally tested. Here, we describe an unbiased platform for the functional characterization of thousands of variants of a single receptor tyrosine kinase (RTK) gene in a single assay. Our in vitro screen for activating mutations (iSCREAM) platform enabled rapid analysis of mutations conferring gain-of-function RTK activity promoting clonal growth. The screening strategy included a somatic model of cancer evolution and utilized a library of 7,216 randomly mutated epidermal growth factor receptor (EGFR) single-nucleotide variants that were tested in murine lymphoid Ba/F3 cells. These cells depend on exogenous interleukin-3 (IL-3) for growth, but this dependence can be compensated by ectopic EGFR overexpression, enabling selection for gain-of-function EGFR mutants. Analysis of the enriched mutants revealed EGFR A702V, a novel activating variant that structurally stabilized the EGFR kinase dimer interface and conferred sensitivity to kinase inhibition by afatinib. As proof of concept for our approach, we recapitulated clinical observations and identified the EGFR L858R as the major enriched EGFR variant. Altogether, iSCREAM enabled robust enrichment of 21 variants from a total of 7,216 EGFR mutations. These findings indicate the power of this screening platform for unbiased identification of activating RTK variants that are enriched under selection pressure in a model of cancer heterogeneity and evolution.
Keywords: epidermal growth factor receptor (EGFR), mutagenesis in vitro, cancer biology, tyrosine-protein kinase (tyrosine kinase), directed evolution, cancer, oncogene, clonal selection, driver mutations, passenger mutations, somatic evolution, tyrosine kinase inhibitors, ERBB, targeted therapies, tyrosine kinase inhibitors
Introduction
There are thousands of mutations in cancer tissues (1, 2), whereas the current list of clinically validated actionable variants only includes a handful of genetic markers (3). Most of the somatic mutations in cancer are expected to be inconsequential passenger mutations that reflect the general genetic instability of the tumors (4). Discovery of the currently known driver mutations has been facilitated, in part, by their accumulation in mutational hot spots within their respective genes. Such examples include the somatic missense mutations at BRAF Val-600, KRAS Gly-12, and EGFR7 Leu-858 (3). However, a great majority of mutations in cancer tissues are observed outside these hot spots (5), and very little direct information is currently available about their functional relevance. The functional characterization of the infrequent mutations, even at the biochemical level, has been technically challenging, given the large number of possible variants. There is a clear clinical need for better understanding of the functional significance of these mutations, both for the identification of markers to predict drug responses and for the discovery of novel actionable targets (6). This need has only become more prominent in the era of large clinical cancer tissue-sequencing efforts and the expansion of the arsenal of available targeted drugs.
EGFR is a member of the ERBB subfamily of receptor tyrosine kinases (RTKs) and a well-characterized oncogene. There are currently 12 tyrosine kinase inhibitors (TKIs) or mAbs targeting EGFR (either as the sole target or among other RTKs) that have been approved by the Food and Drug Administration for clinical use. When used to treat non-small-cell lung cancer, the presence of activating mutations at exons 19 (deletions) or 21 (substitution L858R) in the tumor tissue has been shown to be critical for clinical response to TKIs, such as erlotinib or gefitinib (7). However, over 1,000 different nonsynonymous variants have been reported along the 1,210 amino acid EGFR primary sequence (Fig. 1), whereas functional validation has only been carried out for a fraction of these. Here, we set out to systematically address the activating potential of thousands of random EGFR single-nucleotide variants using an in vitro model of somatic evolution. We provide evidence indicating that few of the clinically rare variants promote significant growth advantage in an EGFR activity–dependent cell model.
Figure 1.

Cancer-associated EGFR mutations. The y axis shows the number of EGFR mutations reported in different cancer types in the COSMIC database (version 86) (http://cancer.sanger.ac.uk/cosmic).8 Please note that the y axis is shown in log10 scale. The total number of EGFR mutations in the database was 21,339. The x axis shows the position of the mutated residue in the EGFR primary sequence. Mutation hot spots, such as L858R and exon 19 deletions, are indicated with labels.
Results
Expression library of random EGFR mutations
To identify activating EGFR mutations from a pool of random mutations, a library of randomly mutated variants was generated by error-prone PCR using the WT human EGFR cDNA as a template (Fig. 2). The mutation frequency was optimized by altering the amount of template DNA used for error-prone PCR and was adjusted to 2.67 mutations per cDNA molecule (Fig. S1), dictating an average of 1.86 amino acid changes per EGFR molecule when translated. After PCR, the mutant EGFR inserts were incorporated into a retroviral mammalian expression vector to create a cDNA expression library.
Figure 2.

iSCREAM pipeline for identification of driver mutations. An expression library including random EGFR mutations was generated by amplifying WT human EGFR cDNA using error-prone PCR and cloning the amplicons into a retroviral mammalian expression vector. The library was subsequently transduced into IL-3–dependent Ba/F3 cells that can survive IL-3 withdrawal if expressing an activated EGFR variant. Upon IL-3 deprivation, clones of Ba/F3 cells expressing WT EGFR or passenger mutations die, whereas clones expressing activating EGFR mutants survive and proliferate. By analyzing the EGFR insert in the surviving cell population as well as in the original library used for infection using targeted next-generation sequencing, it can be determined which mutations are enriched along with the clonal expansion and thus functionally driving the growth.
Cell model dependent on active EGFR signaling
Murine lymphoid Ba/F3 cells are a well-established model for studying activating mutations in tyrosine kinases (8) (Fig. S2). These cells are dependent on exogenous interleukin-3 (IL-3) for survival and growth, but the IL-3 dependence can be compensated by ectopic overexpression of a ligand-stimulated or constitutively active nonreceptor or receptor tyrosine kinase, such as EGFR (9). We hypothesized that the Ba/F3 system could be used as a platform to discriminate activating EGFR mutations from passenger mutations in a massively parallel manner based on the ability of driver mutations to promote IL-3 independence. To this end, the retroviral EGFR mutation library was transduced into Ba/F3 cells, and after selection of stable expression in the presence of puromycin for 48 h, IL-3 was depleted from the growth medium to select for mutations that enabled survival and provided growth advantage (Fig. 2).
Clonal selection of cells expressing 7,216 unique EGFR variants
After culturing the infected cells for 2 weeks in the absence of IL-3, the surviving cell pool was harvested, and the human EGFR cDNA inserts were sequenced from PCR-amplified genomic DNA by ultra-high-depth (>100,000×) targeted next-generation sequencing on an Illumina MiSeq platform (Fig. 2). In addition, the original mutant cDNA library used for the retroviral infection was subjected to sequencing with the same strategy (Fig. S3).
Of the 10,900 theoretically possible unique single-nucleotide variants (SNVs) of EGFR, the analysis contained 9,588 (88.0% of all possible SNVs) after filtering for read depth (>1,000×) and strand bias (<10×) and excluding variants not mapping to the EGFR exons in the reference genome. Of the theoretical maximum of 8,485 nonsynonymous EGFR SNVs, the analysis covered 7,216 (85.0% of all possible nonsynonymous SNVs) that were present in both samples (Fig. S4). On average, each amino acid in the EGFR primary sequence was mutated to 5.63 other amino acids, and 1,159 of the 1,210 residues (95.8%) were mutated to at least to one other amino acid.
The mutations included in the analysis were almost exclusively SNVs, as only 11 insertions or deletions of 2–3 nucleotides in length were present in the library. The distribution of different types of SNVs was also analyzed. At the nucleotide level, the library was found to comprise all possible transitions and transversions and was not biased favoring any particular nucleotide substitutions (Fig. S5A). At the amino acid level, the library included all amino acid changes that it is possible to create by single-point mutations (Fig. S5B).
Frequency of activating EGFR mutations
To identify enriched EGFR variants promoting IL-3–independent growth, sequencing data of the EGFR inserts from the cells surviving IL-3 depletion were compared with the original EGFR cDNA library. Of the 7,216 mutations, only 21 (0.29%) were found to be significantly enriched in the surviving Ba/F3 cell pool (q < 0.0001) after correcting for multiple-hypothesis testing using the Benjamini–Hochberg false discovery rate (10) (Fig. 3 and Fig. S6A). Of the 21 mutations, only 2 (0.03% of the 7,216) were present at an allele frequency of ≥2% in the surviving cells (Fig. 3 and Fig. S6B). Interestingly, both of these variants altered the clinically actionable Leu-858 residue of EGFR (11, 12), producing L858R and L858M. Cumulatively, the two Leu-858 variants were enriched by 125-fold and constituted 7.48% of all alleles in the surviving cells (Fig. 3). The scatter plot shown in Fig. 3 is also available in HTML format (Fig. S7), allowing interactive analysis of all mutations, their -fold change values, and allele frequencies in the surviving cells.
Figure 3.

Clonal selection of activating EGFR mutations. The scatter blot demonstrates the enrichment of few specific EGFR mutations in Ba/F3 cells engineered to express 7,216 EGFR variants and to grow in an EGFR activity–dependent manner. The y axis indicates the -fold change of the variant frequency after growing the cells for 2 weeks when compared with the variant frequency in the original mutation library used to transduce the cells. The x axis indicates the position of the mutated residue on the EGFR primary sequence. The size of the dot as well as the intensity of its red color indicate the relative variant frequency of the mutation in the growing cell pool at the 2-week time point. The dashed horizontal line indicates the -fold change level (24.5) for statistically significant difference from the normal distribution after log2 transformation (q < 0.0001).
EGFR mutants with autonomous growth-promoting potential
The experimental pipeline was validated by identifying the Leu-858 residue as the major mutational target. However, the chosen random mutagenesis strategy predicted that there should be on average 2.67 SNVs (corresponding to an average of 1.86 amino acid changes) per EGFR insert (Fig. S1), making it likely that part of the detected mutations co-occurred with other mutations in the same cDNA. Thus, to control for the nonactivating mutations co-evolving in cis with the functional driver mutations, single mutations were selected, cloned into a retroviral vector, and expressed in Ba/F3 cells. Of the six tested variants that were enriched in the surviving cells (shown in boldface type in Fig. 3), three were found to induce IL-3–independent growth in the Ba/F3 cells after a refractory period of 7–14 days (Fig. 4, A and B and Fig. S2). Once established, the same three mutants readily promoted growth upon IL-3 withdrawal (Fig. 4C). As expected, the L858R mutant was one of those supporting growth, as was T790M, also previously reported to enhance EGFR activity (13). As a novel finding, the EGFR mutation A702V also stimulated Ba/F3 cell survival and growth in a ligand-independent manner (Fig. 4 and Fig. S2). Consistent with the model set up to identify only activating mutations, Ba/F3 cells expressing WT EGFR or a variant, D956V, not enriched during the screen, did not survive IL-3 deprivation (Fig. 4, B and C and Fig. S2).
Figure 4.
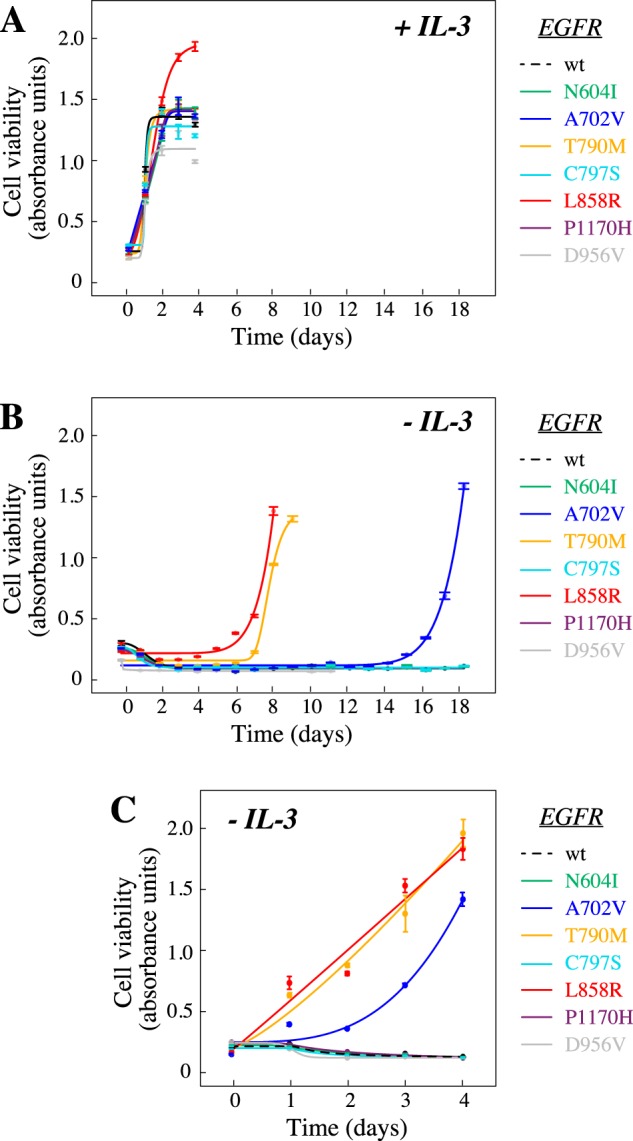
EGFR mutations L858R, T790M, and A702V promote ligand-independent growth. Ba/F3 cells were infected with lentiviral vectors encoding the indicated EGFR variants and cultured in the presence (A) or absence (B and C) of exogenous IL-3. Cell viability was measured by MTT analyses. A, all cell lines were viable in the presence of IL-3. B, after initial withdrawal of IL-3, cells expressing activating mutations emerged as IL-3–independent after a refractory phase of 7–14 days. C, after initial establishment of IL-3 independence, cells harboring activating mutants readily proliferated in the absence of IL-3. Error bars, S.D.
Increased phosphorylation and expression of the activating mutations
To address the phosphorylation status of the three activating EGFR variants in cells maintained in the absence of IL-3, phospho-Western analyses were carried out. As expected, all three variants demonstrated basal IL-3–independent EGFR autophosphorylation unlike WT EGFR expressed in cells maintained in the presence of IL-3 (Fig. 5A). Western blot analysis indicated that the activating variants also seemed to be expressed at greater levels in the absence of IL-3 when compared with WT EGFR (Fig. 5A), whereas all Ba/F3 clones expressed a roughly similar amount of EGFR variants in the presence of IL-3 (Fig. 5B). The establishment of IL-3 independence was, however, not associated with significant differences in the expression of ERBB3 or MET, two receptors with the potential to relay EGFR signaling (14, 15) (Fig. 5A).
Figure 5.

Induction of EGFR expression upon EGFR addiction. A, a Western blot analysis of the indicated cell lines cultured in the presence or absence of IL-3. Two independent clones of cells expressing the A702V variant were analyzed (lanes 4 and 5). B, Western analyses of the indicated Ba/F3 cell lines maintained in the presence of IL-3 and stimulated or not with 10 ng/ml EGF for 10 min. C and D, flow cytometry analyses of the expression of EGFR in the indicated cell lines. The three activating EGFR mutants were analyzed both after maintaining them in the presence (C) or absence (D) of IL-3. E, real-time RT-PCR analysis of EGFR expression in the Ba/F3 cell lines expressing the indicated EGFR variants after maintaining them in the presence or absence of IL-3. Three data points and their means (horizontal lines) are indicated.
The enhanced expression of the activating EGFR variants in cells maintained in the absence versus presence of IL-3 was recapitulated when the expression was measured at the protein level by flow cytometry (Fig. 5, C and D) or at the mRNA level by real-time RT-PCR (Fig. 5E). Taken together, these observations indicate that establishment of IL-3–independent cells with autophosphorylated EGFR at their surfaces is associated with increased EGFR expression, either as a result of induced expression of the transcript or positive selection of cells with the greatest initial EGFR expression.
Structural changes in EGFR A702V
As the A702V mutation had not previously been shown to promote EGFR activity, its putative effect on the structure of EGFR was explored. Residue Ala-702 is present at the dimerization interface of the EGFR asymmetric kinase domain homodimer (Fig. 6). The valine side chain was modeled at position 702 in the X-ray structure of an EGFR active kinase asymmetric dimer and indicated that the A702V change in the juxtamembrane segment B (16) of the receiver kinase at the asymmetric kinase dimer interface would likely strengthen a hydrophobic interaction between valine 702 and isoleucine at position 941 of the C-lobe of the activator kinase. Molecular dynamics simulations show that over 100 ns, the A702V mutant (average RMSD (root mean square deviation over main-chain atoms) of 2.0 Å) fluctuates somewhat less than the WT (average RMSD of 2.7 Å) asymmetric dimer (Fig. S8A); similarly, the N-terminal sequence of the structures, juxtamembrane segment B (14) containing sequence position 702, fluctuates less over 100 ns for the mutant (average RMSD of 0.7 Å) than for the WT (average RMSD of 1.1 Å) (Fig. S8B). The relative free energies of binding based on molecular mechanics energy calculations are consistent with a modest increase in stability in the A702V mutant (ΔG = −76 kcal/mol) over the WT (ΔG = −61 kcal/mol) asymmetric dimer structure (Fig. S8C). Given its central interfacial location, it is not surprising that the I941R mutation abrogates the ability of the C-terminal lobe of the activator EGFR kinase domain to dimerize with the receiver kinase and to promote autophosphorylation, as has been shown previously (17).
Figure 6.
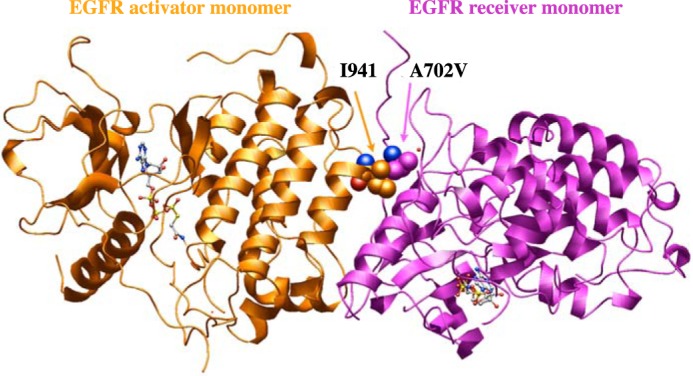
Effect of EGFR A702V on kinase interface. Modeled A702V (receiver kinase) interaction with Ile-941 (activator kinase) at the asymmetric dimer interface in the 2.6 Å resolution EGFR X-ray structure with bound ATP peptide analogue (PDB code 2GS6). A structural water (small red sphere) is nearby. Carbon atoms for the Ile-941 and A702V are shown as orange and magenta spheres, respectively; the red and blue spheres, where seen, correspond to main-chain carbonyl oxygen and amino groups, respectively.
Sensitivity of EGFR mutants to EGFR inhibitors
The growth promoted by the three activating mutations in the absence of IL-3 depended on EGFR expression and kinase activity, as the anti-EGFR cetuximab as well as the EGFR TKI afatinib efficiently reduced survival (Fig. 7A). Interestingly, the EGFR TKI erlotinib was clearly less potent in suppressing the growth of cells expressing EGFR A702V, as compared with cells expressing EGFR L858R. Cells expressing EGFR T790M were completely resistant to erlotinib (Fig. 7A), as expected given its well-demonstrated role as a resistance mutation for first-generation EGFR TKIs (18). Control cells expressing WT EGFR or EGFP (vector control) maintained in the presence of IL-3 were resistant to EGFR inhibition (Fig. 7A and Fig. S9).
Figure 7.

Variable drug sensitivity of activating EGFR mutations. A, drug response curves of Ba/F3 cells expressing the indicated EGFR variants or EGFP (vector control). Cells were cultured for 3 days in the presence of the indicated concentrations of cetuximab, erlotinib, or afatinib. Cells expressing EGFR variants were cultured in the absence of IL-3, and vector control cells were cultured in the presence of IL-3. B, Western blotting analyses of EGFR phosphorylation at Tyr-1110 in Ba/F3 cells expressing the indicated EGFR variants after treating the cells for 3 h with the indicated concentrations of erlotinib or afatinib. Error bars, S.D.
Consistent with the cell viability data, phosphorylation of EGFR at Tyr-1110 of the L858R variant was readily blocked by both erlotinib and afatinib, whereas EGFR A702V was relatively resistant to erlotinib as compared with afatinib (Fig. 7B). Again consistent with previous findings (13), EGFR T790M was resistant to erlotinib but sensitive to high concentrations of afatinib (Fig. 7B).
Compared with the WT active asymmetric kinase domain dimer (e.g. Protein Data Bank (PDB) code 2GS6), the larger, branched hydrophobic side chain of the A702V mutation in the receiver kinase would be in a location to increase hydrophobic interactions with the large branched hydrophobic Ile-941 of the activator kinase (Fig. 6), enhancing kinase–kinase interactions and thus prolonging the activating state of the kinase. In a 100-ns molecular dynamics simulation, the hydrophobic cluster among A702V, Ile-941, and Ala-767 (from receiver kinase) was strictly maintained.
The EGFR–afatinib complex structure (PDB code 4G5J) superposes closely with the receiver kinase domain (RMSD of 0.685 Å over 277 equivalent Cα atoms), which is close to that obtained for the superposition of two structures of the active EGFR domain (RMSD of 0.57 Å over 304 Cα atom pairs for 2GS6 superposed with 2GS2). Afatinib, designed with a reactive acrylamide moiety, inhibits the EGFR kinase via a covalent bond to cysteine in the vicinity of the catalytic residues (19) and may require an active kinase for covalent bond formation to occur as well as for optimal kinase recognition. Indeed, afatinib binds to the active monomer (PDB code 4G5J (19)) as well as to the receiver kinase domain with DFG-in but not to the activator kinase with DFG-out of the asymmetric dimer (PDB code 4G5P (19)). Hence, any process that stabilizes the active asymmetric kinase dimer form longer would be expected to support inhibition by afatinib, as seen in Fig. 7.
In contrast, erlotinib is a noncovalent inhibitor, but crystal structures show that erlotinib recognizes both active (PDB code 1M17) and inactive (PDB code 4HJO) conformations in isolated kinase domains, superposing with an RMSD of 1.33 Å over 177 matched Cα atoms. In the transition from the inactive to the active conformation, the αC helix of the kinase pivots; while the helix C terminus remains in position, the N terminus moves by more than 9 Å (measured between Cα atoms of Ala-755), and Glu-762 of the helix forms a stabilizing salt bridge with Lys-745 of a β-strand. In the ligand complexes, afatinib and erlotinib are located toward the interior face of the αC helix, whereas juxtamembrane segment B crosses orthogonally at the opposite side of the helix and places Ala-702 directly at the C terminus. Thus, Ala-702 is placed adjacent to Ala-767 at the fixed pivot point for movement of the helix critical for kinase activation. Consequently, whereas the helix in the active and inactive structures does not alter its pivot-point position adjacent to Ala-702, in transitioning between the active and inactive conformations, the bulkier valine side chain at residue 702 would likely affect the helix pivot point and could then influence the helix positioning and possible surrounding structures. In contrast to the inactive kinase complex, erlotinib is located close to the αC helix in the active complex; thus, it is plausible that the A702V mutation influences the shape of the binding site itself and interactions with ligands and may explain in part the reduction in inhibition by erlotinib with the A702V replacement.
Discussion
Here, we provide evidence based on an in vitro functional genetics screen that despite thousands of mutations identified in cancer patients, a relatively small number of EGFR variants have the potential to provide growth advantage under selection pressure. In total, 7,216 unique nonsynonymous EGFR SNVs were analyzed in a massively parallel manner based on their ability to promote IL-3–independent growth in the Ba/F3 cell model. Interestingly, only 21 of the 7,216 (0.38%), were significantly (q < 0.0001) enriched during 2 weeks of IL-3 deprivation.
The analysis covered 88.4% (854 of 966) of unique nonsynonymous SNVs reported in human cancer samples in the COSMIC database (Fig. 1). Collectively, a search of four databases (COSMIC, cBioPortal, GENIE, and CCLE) indicated that 19 of the 21 identified putatively activating mutations were at residues that are mutated in clinical samples, either with an identical missense or nonsense mutation (10 of 21) or with a different amino acid substitution at the same residue (an additional 9 of 21) (Table 1).
Table 1.
EGFR mutations in clinical databases
The 21 activating mutations enriched above the threshold in Fig. 3 are listed together with clinical information available from human samples in the databases COSMIT (v87; http://www.cancer.sanger.ac.uk)8 (48), cBioPortal (http://www.cbioportal.org)8 (49, 50), and GENIE (http://www.genie.cbioportal.org)8 (49–51). Cell lines harboring the indicated mutations according-to CCLE (http://www.portals.broadinstitute.org/ccle)8 (52) database are also listed. Both cases with identical missense or nonsense mutation to the 21 mutations discovered in the screen and cases with a different amino acid substitution at the same residue are listed. Abbreviations for cancer subtypes are as follows: CESC, cervical squamous cell carcinoma; CHOL, cholangiocarcinoma; COAD, colon adenocarcinoma; GBM, glioblastoma multiforme; LGG, lower-grade glioma; LUAD, lung adenocarcinoma; MEL, melanoma; MESO, mesothelioma; NSCLC, non-small cell lung cancer; OV, ovarian serous carcinoma; PRAD, prostate adenocarcinoma; SCC, squamous cell carcinoma; SARC, sarcoma; SKCM, melanoma; STAD, stomach adenocarcinoma; NOS, not specified.

As the mutation strategy adopted for the in vitro analysis involved generation of, on average, 2.7 mutations per cDNA molecule, passenger mutations could evolve alongside the drivers, making the number of true identified driver mutations even smaller than 21. Indeed, cloning and independent expression of six selected mutation hits indicated that only three (50%) were capable of supporting growth. Moreover, none of the mutations, with the exception of the nonsense mutation L833* depicted to truncate the receptor kinase domain, was enriched to a greater extent than L858R, the best-characterized activating EGFR mutation.
In addition to recapitulating the clinical observation about the unique significance of L858R as a product of an EGFR SNV, the distributions of the mutations along the structural domains of EGFR were roughly similar in our analysis and in the mutation maps of clinical sequencing efforts (compare Figs. 1 and 3). Few hits were observed in the extracellular domain, whereas the mostly targeted region was the kinase domain.
In addition to the EGFR L858R mutation, the T790M mutation, previously shown to both promote growth and provide clinical resistance to first generation EGFR TKIs (13), was among the validated findings. As a novel finding, our analysis identified a previously uncharacterized activating EGFR mutation, A702V. Structural analyses indicated that the A702V mutation in the receiver kinase domain likely strengthens the hydrophobic interaction with the activator kinase stabilizing the active kinase dimer. Drug sensitivity assays with different EGFR-targeting therapeutics demonstrated selective sensitivity of Ba/F3 cells expressing EGFR A702V to the second-generation EGFR TKI afatinib, in addition to the EGFR antibody cetuximab. Interestingly, the somatic A702V mutation has also been reported in a clinical case of non-small-cell lung cancer, in a patient with partial response to a protocol including erlotinib (20). These observations suggest that the EGFR A702V mutation may have predictive value as a novel clinical target for EGFR inhibitors.
The reason that other EGFR variants, such as G719A, S768I, or L861Q, previously detected in cancer tissues and proposed to modulate EGFR activity or sensitivity to EGFR inhibitors (21–23), were present but not enriched in the Ba/F3 cell–based screen is currently not known. However, given the pace and the timing of the emergence of IL-3–independent Ba/F3 clones expressing the different EGFR variants (Fig. 4B), it is conceivable that the more active variants, such as L858R, outcompete weaker oncogenes during the selection. This hypothesis was supported by findings suggesting that the EGFR variants demonstrating greater autophosphorylation (Fig. 5A) also emerged with a shorter refractory period upon IL-3 withdrawal (Fig. 4B). Whereas this phenomenon may be considered a limitation for the analysis in poorly detecting some weaker oncogenes, the circumstances are not irrelevant for the evolution of mutations in living cancer tissues. Indeed, also in cancer, the strong oncogenes are expected to outcompete weaker ones (24), and, for example, L858R and T790M are more frequently observed than G719A, S768I, and L861Q (Fig. 1 and Table 1). It is also noteworthy that the uncommon EGFR mutations have been shown to commonly co-occur with other EGFR mutations and that their clinical significance is variable and clearly less well-elucidated, as compared with L858R and T790M (22, 23).
Using the IL-3–dependent lymphoid Ba/F3 cells for the functional selection step and drug sensitivity analyses introduces a cell background that cannot be considered representative of the molecular context of human cancer cells. However, the characteristic rapid pace of cell division, combined with the absolute dependence on exogenous IL-3 that can be compensated by endogenous expression of an active kinase, make the Ba/F3 cells ideal for the functional selection of activating variants. Moreover, the fact that the Ba/F3 cells have been widely used for addressing the functional activation of several receptor as well as nonreceptor tyrosine kinases (8) indicates that the same Ba/F3-based pipeline could be used for analyzing a number of kinases in addition to EGFR. Although effective in identifying activating SNVs, it is, however, important to keep in mind that our model was not designed to screen for short deletions, that, for example, in the case of EGFR have also been shown to have predictive value in the clinic (11, 12). Given the findings that the different EGFR variants also differently promoted their own expression (or selection of a subpopulation of high expressors) (Fig. 5), further development of the pipeline should also involve comprehensive expression analysis of each individual oncogene.
Taken together, our findings indicate that an in vitro clonal selection model with randomly mutated variants can be successfully used to identify gain-of-function mutations, given that a cell line addicted to the signaling of the gene of interest is available. In addition, we provide evidence supporting a conclusion that, despite thousands of somatic mutations reported in the EGFR gene, only a handful (less than 1%) are expected to function as active drivers.
Experimental procedures
Cloning of constructs
WT human EGFR cDNA was amplified from EGFR WT, a gift from Matthew Meyerson (Addgene plasmid 11011) (25), using the primers 5′-GGGGACAAGTTTGTACAAAAAAGCAGGCTTCACCATGCGACCCTCCGGGACGG-3′ and 5′-GGGGACCACTTTGTACAAGAAAGCTGGGTTTCATGCTCCAATAAATTCACTGCTTTGTG-3′ to generate a 3,697-bp amplicon containing EGFR cDNA flanked by attB1 and attB2 gateway recombination sites. The amplicon was cloned into the pDONR221 vector (Tol2Kit) (26) using BP clonase II mix (Invitrogen) to generate pDONR221-EGFR. A retroviral mammalian expression construct was made by a LR-Gateway recombination reaction between the pDONR221-EGFR and pBABEpuro-gateway, a gift from Matthew Meyerson (Addgene plasmid 51070) (27), using LR clonase II mix (Invitrogen).
To create plasmids expressing individual EGFR variants, pDONR221-EGFR was mutated to generate the indicated point mutations using the primers listed in Fig. S10. These mutations were subsequently cloned to pBABEpuro-gateway by LR-Gateway recombination to create the expression constructs.
Generation of expression library of random EGFR mutants
The expression library for human EGFR mutants was generated by error-prone PCR using the GeneMorph II random mutagenesis kit (catalogue no. 200550, Agilent Technologies). Using pDONR221-EGFR as the template and the primer pair 5′-TTGATGCCTGGCAGTTCCCTA-3′ (which binds 78 bp upstream of the attL1 site at the 5′-end of the EGFR insert) and 5′-ATCTTGTGCAATGTAACATCAGAGATT-3′ (which binds 80 bp downstream of the attL2 site at the 3′-end of the EGFR insert), 4,043-bp amplicons were produced containing randomly mutated EGFR cDNAs flanked by the attL recombination sites. Following the instructions in the GeneMorph II random mutagenesis kit manual, 10 cycles of amplification were performed. The PCR product was gel-purified (NucleoSpin Gel and PCR Clean-up kit; Macherey Nagel) and cloned into a pBABEpuro-gateway vector with LR clonase II. ccdB-sensitive Escherichia coli (NovaBlue) were transformed with the LR reaction product to amplify the random mutation library plasmid.
Cell culture and generation of stable lines
To generate stable Ba/F3 lines expressing the EGFR library or single EGFR mutants, amphotropic Phoenix HEK293 cells were transfected with the respective pBABEpuro-gateway plasmid or the empty vector control using Fugene 6 transfection reagent (Promega) for production of retroviruses. Ba/F3 cells were transduced with the retroviral supernatants, and stable cell populations were selected in the presence of 2 μg/ml puromycin (Gibco) for 48 h. Ba/F3 cells were maintained in RPMI 1640 (Lonza), containing 1 mm l-glutamine (Lonza), 50 units/ml penicillin-streptomycin (Lonza), 10% FBS (Biowest), 5% conditioned medium from WEHI cells (as a source of IL-3), and 1 μg/ml puromycin. To select for EGFR mutations promoting growth in the absence of IL-3, 2.5 × 107 Ba/F3 cells stably transduced with the EGFR random mutant library were washed twice with 20 ml of PBS and maintained in a volume of 40 ml of RPMI 1640, containing 1 mm l-glutamine, 50 units/ml penicillin-streptomycin, 10% FBS, and 1 μg/ml puromycin.
A549 and NCI-H661 human lung cancer cells (ATCC) were maintained in Dulbecco's modified Eagle's medium (Lonza), containing 1 mm l-glutamine, 50 units/ml penicillin-streptomycin, and 10% FBS.
Next-generation sequencing
Genomic DNA (100 ng) extracted from the Ba/F3 cells (NucleoSpin Tissue, Macherey Nagel) as well as the original plasmid library (5 ng) used for transduction were PCR-amplified using primers 5′-GGGGACAAGTTTGTACAAAAAAGCAGGCTTCACCATGCGACCCTCCGGGACGG-3′ and 5′-GGGGACCACTTTGTACAAGAAAGCTGGGTTTCATGCTCCAATAAATTCACTGCTTTGTG-3′ and a 1:1 mixture of Phusion (Thermo) and Velocity (Bioline) high-fidelity DNA polymerases. The Nextera XT DNA Sample Preparation Kit (Illumina) was used to prepare sequencing libraries from these amplicons, which were sequenced on MiSeq with 150-bp paired-end sequencing, producing 6 million reads/sample. Filtering of low-quality reads and trimming of Nextera XT adapter sequences from the reads was performed with trimmomatic (version 0.36) (28) using the parameters recommended in the user guide for paired end sequencing (http://www.usadellab.org/cms/?page=trimmomatic).8 The trimmed reads were aligned to “hg19” human reference genome using BWA-MEM (version 0.7.15) (29). The produced Sequence Alignment/Map (SAM) files were converted to Binary Alignment Map (BAM) files, sorted, and indexed using samtools (version 1.3.1) (30). Variant calling was carried out using samtools by setting the maximum depth to 300,000 to accommodate all of the reads aligned to the reference genome. The NexteraXT libraries were not generated using a strand-specific protocol, and therefore, to identify potential sequencing artifacts, the result from bamreadcount (https://github.com/genome/bam-readcount)8 was used to calculate strand bias (ratio of the number of forward reads to the number of reverse reads aligning to a particular locus) for each detected variant in the samples. Variants with strand bias less than 0.1 and larger than 10 (i.e. reads aligning at a locus with one orientation (5′–3′) being 10 times more abundant than the other orientation) were filtered out. Annovar (version 2017-07-17 01:17:05-0400) (31) was used for functional annotation of the variants. Data analysis was carried out using R (version 3.4.1) (32), and plots were made using “ggplot2” (version 2.2.1) and “plotly” (version 4.7.1) R packages (33, 34). The source code used to analyze the data produced in this study can be accessed at https://gitlab.utu.fi/deecha/iSCREAM.EGFR.8
Statistical analyses
To identify the EGFR mutations that were enriched during the clonal expansion of Ba/F3 cells under IL-3 depletion, -fold changes were calculated by comparing the variant allele frequencies of the mutations in the surviving cells with that in the original EGFR mutant expression library. The approach was named in vitro screen for activating mutations (iSCREAM). Using all of the observed -fold changes, a normal distribution was fitted (μ = 0.093 ± 0.011, σ = 0.967 ± 0.008) to log2-transformed -fold change values using the “fitdistrplus” package in R (35). Then p values were calculated for all of the mutations, and correction for multiple testing was performed using the Benjamini–Hochberg method (10). Mutations with q < 0.0001 were considered significantly enriched, which corresponds to a false discovery rate of 0.01%.
Cell proliferation assay
Ba/F3 cells expressing WT EGFR or selected EGFR mutants were seeded at a density of 20,000 cells/well in 96-well plates. Cell viability was assessed using the MTT assay (Promega) in the presence or absence of 5% conditioned medium from WEHI cells as a source of IL-3. The growth curves indicating mean ± S.D. were plotted using the “ggplot2” R package (33). Sigmoidal curves were fitted using the “drc” (version 3.0-1) package in R (36).
Western blot analysis
Cells were harvested and lysed in lysis buffer (10 mm Tris-HCl, pH 7.4, 1% Triton X-100, 150 mm NaCl, 1 mm EDTA, 10 mm NaF), supplemented with 2 mm phenylmethylsulfonyl fluoride, 10 mg/ml aprotinin, 10 mg/ml leupeptin, 10 mm Na4P2O7, and 1 mm Na3VO4. Lysates were analyzed for EGFR phosphorylation using the phospho-EGFR–specific antibody (catalogue no. 2220, Cell Signaling Technology) recognizing phosphorylated Tyr-1110. Anti-EGFR (sc-03 (Santa Cruz Biotechnology, Inc.) and catalogue no. 4267 (Cell Signaling Technology)), anti-ERBB3 (catalogue no. 4754, Cell Signaling Technology), and anti-MET (catalogue no. 8198, Cell Signaling Technology) were used to analyze total EGFR, ERBB3, and MET expression, respectively, and anti-β-actin (A5441; Sigma Aldrich) as a loading control. HRP-conjugated anti-rabbit (catalogue no. A16104, Invitrogen), HRP-conjugated anti-mouse (catalogue no. sc-2005, Santa Cruz Biotechnology), IRDye 800CW anti-rabbit (catalogue no. 925-32213, LI-COR), and IRDye 680RD anti-mouse (catalogue no. 925-68072, LI-COR) were used as secondary antibodies. The blots were imaged using chemiluminescence (WesternBright ECL HRP, Advansta) on the ImageQuant LAS-4000 imaging system (Fujifilm) or using near-IR fluorescence on the Odyssey CLx imaging system (LI-COR).
To address the effect of EGFR inhibitors on EGFR phosphorylation, cells were seeded at a density of 3 million cells/well in 12-well plates and treated with erlotinib (10–1000 nm; Santa Cruz Biotechnology) or afatinib (1–100 nm; Santa Cruz Biotechnology) for 3 h.
Flow cytometry analysis of EGFR expression
Ba/F3 clones were washed with azide-free PBS and stained with eBioscience Fixable Viability Dye eFluor 780 (catalogue no. 65-0865, Thermo Fisher Scientific) according to the manufacturer's protocol. The cells were fixed with 4% paraformaldehyde and permeabilized with ice-cold methanol. Fixed cells were incubated with anti-EGFR (1:100; catalogue no. 4267, Cell Signaling Technologies) followed by incubation with Alexa Fluor 488–conjugated anti-rabbit (1:200; catalogue no. A-11034, Thermo Fisher Scientific). An LSR Fortessa flow cytometer was used with the BD FACSDiva software (version 8.0.1). The data were analyzed using the FlowJo software (version 10.5.3).
Real-time RT-PCR
RNA samples were extracted with TRIsure (Bioline) according to the manufacturer's protocol. cDNA was synthesized with the SensiFast cDNA synthesis kit (Bioline). The analysis was carried out using TaqMan Universal Master Mix II (Applied Biosystems) with the following primers and probes: human EGFR forward, 5′-CCA CCT GTG CCA TCC AAA CT-3′ (Pharmacia); human EGFR reverse, 5′-GGC GAT GGA CGG GAT CTT-3′ (Pharmacia); human EGFR probe, 5′-FAM-CCA GGT CTT GAA GGC TGT CCA ACG AAT-TAMRA-3′ (Eurogentech); mouse GAPDH, Universal ProbeLibrary Mouse GAPDH Gene Assay 5046211001 (Sigma). Thermal cycling was performed using the QuantStudio 12K Flex Real-Time PCR System (Thermo Fisher Scientific) for 2 min at 50 °C and 10 min at 95 °C, followed by 40 cycles of 15 s at 95 °C and 1 min at 60 °C. Samples were analyzed in triplicates, and the S.D. of the CT values was <5% of the mean. EGFR mRNA expression was quantified using mouse GAPDH mRNA expression as a reference with the 2−ΔCT method.
Drug sensitivity assay
Ba/F3 cells were seeded at a density of 20,000 cells/well in 96-well plates. The cells were incubated in the presence of 0.0006–10 μm erlotinib (Santa Cruz Biotechnology) or afatinib (Santa Cruz Biotechnology) or 0.0006–10 μg/ml cetuximab (Erbitux, Merck) for 72 h before measuring the number of viable cells with the MTT assay. IL–3-independent clones were cultured in the absence of, and the control Ba/F3 cells were transduced with an empty vector in the presence of, 5% WEHI cell conditioned medium. Sigmoidal dose-response curves were fitted using 4-parameter logistic regression with the “drc” (version 3.0-1) package in R (36). Dose-response curves indicating mean ± S.D. were plotted using “ggplot2” (33).
Structural analyses of EGFR kinase domains
To evaluate the likely consequences of the A702V mutation in human EGFR, the A702V mutation was modeled into the X-ray structures (PDB codes 2GS2 (17) (2.8 Å resolution) and 2GS6 with bound ATP analogue (17) (2.6 Å resolution)) of EGFR using the mutation function of the modeling and visualization program Bodil (37). Symmetry operations were used to generate the asymmetric dimer structures consisting of a receiver and activator kinase domain. The rotamer function in Bodil was used to explore alternative conformations of the valine side chain. Ala-702 is located on the extended juxtamembrane segment B, and in the receiver kinase, this segment extends across the dimer interface with the activator kinase. Visualization with Bodil indicates that the A702V mutation would very likely enhance hydrophobic contacts with Ile-941 of the activator kinase, strengthening the interface of the active asymmetric dimer. Fig. 6 was prepared using Bodil.
Human EGFR domains (2GS2, active, no bound ligand (17); erlotinib-bound, active 1M17 (38) and inactive 4HJO (39); and 4G5J, afatinib-bound (19)) were superposed with the active domain bound to an ATP analogue (2GS6) using Vertaa in Bodil. RMSD values are reported over equivalent Cα atom pairs from the two structures within 3.5 Å of each other after superpositioning. The respective 2.6, 2.75, and 2.8 Å resolution structures of human EGFR domains with bound erlotinib, active (1M17) and inactive (4HJO), and afatinib (4G5J) were obtained from the PDB.
Molecular dynamic simulations and free energy of binding calculations
To understand the dynamic states and differences between the WT (PDB code 2GS2) and A702V mutant EGFR kinase asymmetric dimer structures, molecular dynamics simulations were carried out using the AMBER package (version 18) (40) and the ff14SB force field (41). The A702V mutant structure was generated using chimera (42). To carry out molecular dynamics simulations, both the WT and mutant EGFR structures were solvated with explicit TIP3P water molecules (43) in an octahedral box. The distance between the dimer surface atoms and the edge of the box was set to 10 Å. Following this, 16 sodium ions were added to neutralize both dimer systems. Additionally, 150 mm Na+/Cl− ions were incorporated into the system. Periodic boundary conditions were ensured, and the particle mesh Ewald algorithm (44) was used to treat electrostatic interactions with a distance cutoff of 9 Å. Before conducting the production simulation, 3,000 cycles of energy minimization were carried out while gradually tapering off from the 25 kcal mol−1 Å−2 restraint applied to the solute atoms. The systems were then heated to 300 K during 100 ps with a 10 kcal mol−1 Å−2 restraint on solute atoms. Afterward, a 900-ps equilibration at constant pressure (1 bar) was employed while reducing the restraint gradually to 0.1 kcal mol−1 Å−2. The equilibration was finalized with restraint-free 10-ns simulation. Finally, the production simulation was performed for 100 ns at constant temperature (300 K) and pressure (1 bar) that were maintained using the Berendsen algorithm (45). Trajectories were saved every 10 ps, and the resulting snapshots were further analyzed using the programs CPPTRAJ (46) and VMD (47). These trajectories were also utilized to compute free energy of binding calculations for both the WT and mutant EGFR dimers using the molecular mechanics-generalized Born surface area (MMGBSA) method, implemented in the AMBER program (40).
Author contributions
D. C., K. J. K., I. P., and K. E. conceptualization; D. C., I. P., V. K. O., M. K., M. Z. T., J. P. K., P. A. J., M. S. J., and K. E. formal analysis; D. C. validation; D. C., M. Z. T., M. S. J., and K. E. visualization; D. C., K. J. K., I. P., M. S. J., L. L. E., and K. E. methodology; D. C., K. J. K., I. P., M. S. J., and K. E. writing-original draft; D. C., K. J. K., I. P., J. P. K., P. A. J., M. S. J., L. L. E., and K. E. writing-review and editing; V. K. O., M. K., M. Z. T., and M. S. J. data curation; J. P. K. resources; K. E. supervision; K. E. funding acquisition; K. E. investigation; K. E. project administration.
Supplementary Material
Acknowledgments
We thank Jukka Lehtonen for scientific IT support (Biocenter Finland); Markku Karhunen for sharing statistical expertise; and Minna Santanen, Mika Savisalo, and Maria Tuominen for skillful technical assistance. We acknowledge the CSC-IT Center for Science for computational resources and the Turku Centre for Biotechnology Functional Genomics facility for RT-PCR service.
P. A. Jänne has received consulting fees from AstraZeneca, Boehringer Ingelheim, Pfizer, Merrimack Pharmaceuticals, Roche/Genentech, Chugai Pharmaceuticals, ACEA Biosciences, and Ariad Pharmaceuticals and sponsored research funding from Astellas Pharmaceuticals, AstraZeneca, Daiichi Sankyo, and PUMA and receives post-marketing royalties on DFCI-owned intellectual property on EGFR mutations licensed to Lab Corp. K. Elenius has a research agreement with Boehringer Ingelheim and ownership interest in Abomics, Orion, and Roche.
This article was selected as one of our Editors' Picks.
This article contains Figs. S1–S10.
Please note that the JBC is not responsible for the long-term archiving and maintenance of this site or any other third party hosted site.
- EGFR
- epidermal growth factor receptor
- RTK
- receptor tyrosine kinase
- TKI
- tyrosine kinase inhibitor
- IL
- interleukin
- SNV
- single-nucleotide variant
- RMSD
- root mean square deviation
- PDB
- Protein Data Bank
- iSCREAM
- in vitro screen for activating mutations
- HRP
- horseradish peroxidase
- MTT
- 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide.
References
- 1. Lawrence M. S., Stojanov P., Polak P., Kryukov G. V., Cibulskis K., Sivachenko A., Carter S. L., Stewart C., Mermel C. H., Roberts S. A., Kiezun A., Hammerman P. S., McKenna A., Drier Y., Zou L., et al. (2013) Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature 499, 214–218 10.1038/nature12213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Zehir A., Benayed R., Shah R. H., Syed A., Middha S., Kim H. R., Srinivasan P., Gao J., Chakravarty D., Devlin S. M., Hellmann M. D., Barron D. A., Schram A. M., Hameed M., Dogan S., et al. (2017) Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat. Med. 23, 703–713 10.1038/nm.4333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hyman D. M., Taylor B. S., and Baselga J. (2017) Implementing genome-driven oncology. Cell 168, 584–599 10.1016/j.cell.2016.12.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Vogelstein B., Papadopoulos N., Velculescu V. E., Zhou S., Diaz L. A. Jr., and Kinzler K. W. (2013) Cancer genome landscapes. Science 339, 1546–1558 10.1126/science.1235122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Chang M. T., Asthana S., Gao S. P., Lee B. H., Chapman J. S., Kandoth C., Gao J., Socci N. D., Solit D. B., Olshen A. B., Schultz N., and Taylor B. S. (2016) Identifying recurrent mutations in cancer reveals widespread lineage diversity and mutational specificity. Nat. Biotechnol. 34, 155–163 10.1038/nbt.3391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Dienstmann R., Jang I. S., Bot B., Friend S., and Guinney J. (2015) Database of genomic biomarkers for cancer drugs and clinical targetability in solid tumors. Cancer Discov. 5, 118–123 10.1158/2159-8290.CD-14-1118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Mok T. S., Wu Y. L., Thongprasert S., Yang C.-H., Chu D.-T., Saijo N., Sunpaweravong P., Han B., Margono B., Ichinose Y., Nishiwaki Y., Ohe Y., Yang J. J., Chewaskulyong B., Jiang H., Duffield E. L., et al. (2009) Gefitinib or carboplatin–paclitaxel in pulmonary adenocarcinoma. N. Engl. J. Med. 361, 947–957 10.1056/NEJMoa0810699 [DOI] [PubMed] [Google Scholar]
- 8. Warmuth M., Kim S., Gu X. J., Xia G., and Adrián F. (2007) Ba/F3 cells and their use in kinase drug discovery. Curr. Opin. Oncol. 19, 55–60 10.1097/CCO.0b013e328011a25f [DOI] [PubMed] [Google Scholar]
- 9. Jiang J., Greulich H., Jänne P. A., Sellers W. R., Meyerson M., and Griffin J. D. (2005) Epidermal growth factor-independent transformation of Ba/F3 cells with cancer-derived epidermal growth factor receptor mutants induces gefitinib-sensitive cell cycle progression. Cancer Res. 65, 8968–8974 10.1158/0008-5472.CAN-05-1829 [DOI] [PubMed] [Google Scholar]
- 10. Benjamini Y., and Hochberg Y. (1995) Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B 57, 289–300 10.1111/j.2517-6161.1995.tb02031.x [DOI] [Google Scholar]
- 11. Paez J. G., Jänne P. A., Lee J. C., Tracy S., Greulich H., Gabriel S., Herman P., Kaye F. J., Lindeman N., Boggon T. J., Naoki K., Sasaki H., Fujii Y., Eck M. J., Sellers W. R., et al. (2004) EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science 304, 1497–1500 10.1126/science.1099314 [DOI] [PubMed] [Google Scholar]
- 12. Lynch T. J., Bell D. W., Sordella R., Gurubhagavatula S., Okimoto R. A., Brannigan B. W., Harris P. L., Haserlat S. M., Supko J. G., Haluska F. G., Louis D. N., Christiani D. C., Settleman J., and Haber D. A. (2004) Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N. Engl. J. Med. 350, 2129–2139 10.1056/NEJMoa040938 [DOI] [PubMed] [Google Scholar]
- 13. Regales L., Balak M. N., Gong Y., Politi K., Sawai A., Le C., Koutcher J. A., Solit D. B., Rosen N., Zakowski M. F., and Pao W. (2007) Development of new mouse lung tumor models expressing EGFR T790M mutants associated with clinical resistance to kinase inhibitors. PLoS One 2, e810 10.1371/journal.pone.0000810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Jo M., Stolz D. B., Esplen J. E., Dorko K., Michalopoulos G. K., and Strom S. C. (2000) Cross-talk between epidermal growth factor receptor and c-Met signal pathways in transformed cells. J. Biol. Chem. 275, 8806–8811 10.1074/jbc.275.12.8806 [DOI] [PubMed] [Google Scholar]
- 15. van Lengerich B., Agnew C., Puchner E. M., Huang B., and Jura N. (2017) EGF and NRG induce phosphorylation of HER3/ERBB3 by EGFR using distinct oligomeric mechanisms. Proc. Natl. Acad. Sci. U.S.A. 114, E2836–E2845 10.1073/pnas.1617994114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Jura N., Endres N. F., Engel K., Deindl S., Das R., Lamers M. H., Wemmer D. E., Zhang X., and Kuriyan J. (2009) Mechanism for activation of the EGF receptor catalytic domain by the juxtamembrane segment. Cell 137, 1293–1307 10.1016/j.cell.2009.04.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Zhang X., Gureasko J., Shen K., Cole P. A., and Kuriyan J. (2006) An allosteric mechanism for activation of the kinase domain of epidermal growth factor receptor. Cell 125, 1137–1149 10.1016/j.cell.2006.05.013 [DOI] [PubMed] [Google Scholar]
- 18. Kobayashi S., Boggon T. J., Dayaram T., Jänne P. A., Kocher O., Meyerson M., Johnson B. E., Eck M. J., Tenen D. G., and Halmos B. (2005) EGFR mutation and resistance of non-small-cell lung cancer to gefitinib. N. Engl. J. Med. 352, 786–792 10.1056/NEJMoa044238 [DOI] [PubMed] [Google Scholar]
- 19. Solca F., Dahl G., Zoephel A., Bader G., Sanderson M., Klein C., Kraemer O., Himmelsbach F., Haaksma E., and Adolf G. R. (2012) Target binding properties and cellular activity of afatinib (BIBW 2992), an irreversible ErbB family blocker. J. Pharmacol. Exp. Ther. 343, 342–350 10.1124/jpet.112.197756 [DOI] [PubMed] [Google Scholar]
- 20. Reckamp K. L., Krysan K., Morrow J. D., Milne G. L., Newman R. A., Tucker C., Elashoff R. M., Dubinett S. M., and Figlin R. A. (2006) A phase I trial to determine the optimal biological dose of celecoxib when combined with erlotinib in advanced non-small cell lung cancer. Clin. Cancer Res. 12, 3381–3388 10.1158/1078-0432.CCR-06-0112 [DOI] [PubMed] [Google Scholar]
- 21. Beau-Faller M., Prim N., Ruppert A.-M., Nanni-Metéllus I., Lacave R., Lacroix L., Escande F., Lizard S., Pretet J.-L., Rouquette I., de Crémoux P., Solassol J., de Fraipont F., Bièche I., Cayre A., et al. (2014) Rare EGFR exon 18 and exon 20 mutations in non-small-cell lung cancer on 10 117 patients: a multicentre observational study by the French ERMETIC-IFCT network. Ann. Oncol. 25, 126–131 10.1093/annonc/mdt418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. O'Kane G. M., Bradbury P. A., Feld R., Leighl N. B., Liu G., Pisters K.-M., Kamel-Reid S., Tsao M. S., and Shepherd F. A. (2017) Uncommon EGFR mutations in advanced non-small cell lung cancer. Lung Cancer 109, 137–144 10.1016/j.lungcan.2017.04.016 [DOI] [PubMed] [Google Scholar]
- 23. Galli G., Corrao G., Imbimbo M., Proto C., Signorelli D., Ganzinelli M., Zilembo N., Vitali M., de Braud F., Garassino M. C., and Lo Russo G. (2018) Uncommon mutations in epidermal growth factor receptor and response to first and second generation tyrosine kinase inhibitors: a case series and literature review. Lung Cancer 115, 135–142 10.1016/j.lungcan.2017.12.002 [DOI] [PubMed] [Google Scholar]
- 24. Martincorena I., Raine K. M., Gerstung M., Dawson K. J., Haase K., Van Loo P., Davies H., Stratton M. R., and Campbell P. J. (2017) Universal patterns of selection in cancer and somatic tissues. Cell 171, 1029–1041.e21 10.1016/j.cell.2017.09.042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Greulich H., Chen T. H., Feng W., Jänne P. A., Alvarez J. V., Zappaterra M., Bulmer S. E., Frank D. A., Hahn W. C., Sellers W. R., and Meyerson M. (2005) Oncogenic transformation by inhibitor-sensitive and -resistant EGFR mutants. PLoS Med. 2, 1167–1176 10.1371/journal.pmed.0020313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kwan K. M., Fujimoto E., Grabher C., Mangum B. D., Hardy M. E., Campbell D. S., Parant J. M., Yost H. J., Kanki J. P., and Chien C.-B. (2007) The Tol2kit: a multisite gateway-based construction kit forTol2 transposon transgenesis constructs. Dev. Dyn. 236, 3088–3099 10.1002/dvdy.21343 [DOI] [PubMed] [Google Scholar]
- 27. Greulich H., Kaplan B., Mertins P., Chen T.-H. T., Tanaka K. E., Yun C.-H. C., Zhang X., Lee S.-H., Cho J., Ambrogio L., Liao R., Imielinski M., Banerji S., Berger A. H., Lawrence M. S., et al. (2012) Functional analysis of receptor tyrosine kinase mutations in lung cancer identifies oncogenic extracellular domain mutations of ERBB2. Proc. Natl. Acad. Sci. U.S.A. 109, 14476–14481 10.1073/pnas.1203201109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Bolger A. M., Lohse M., and Usadel B. (2014) Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120 10.1093/bioinformatics/btu170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Li H. (2013) Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv 1303.3997 [q-bio.GN] [Google Scholar]
- 30. Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R., and 1000 Genome Project Data Processing Subgroup (2009) The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079 10.1093/bioinformatics/btp352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Wang K., Li M., and Hakonarson H. (2010) ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 38, e164 10.1093/nar/gkq603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. RCore Team (2018) R: A Language and Environment for Statistical Computing, R Project for Statistical Computing, Vienna, Austria [Google Scholar]
- 33. Wickham H. (2009) ggplot2: Elegant Graphics for Data Analysis, Springer-Verlag; New York [Google Scholar]
- 34. Sievert C. (2018) plotly for R. https://plotly-book.cpsievert.me
- 35. Delignette-Muller M. L., and Dutang C. (2015) fitdistrplus: an R Package for fitting distributions. J. Stat. Softw. 64, 1–34 10.18637/jss.v064.i04 [DOI] [Google Scholar]
- 36. Ritz C., Baty F., Streibig J. C., and Gerhard D. (2015) Dose-response analysis using R. PLoS One 10, e0146021 10.1371/journal.pone.0146021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Lehtonen J. V., Still D.-J., Rantanen V. V., Ekholm J., Björklund D., Iftikhar Z., Huhtala M., Repo S., Jussila A., Jaakkola J., Pentikäinen O., Nyrönen T., Salminen T., Gyllenberg M., and Johnson M. S. (2004) BODIL: a molecular modeling environment for structure-function analysis and drug design. J. Comput. Aided. Mol. Des. 18, 401–419 10.1007/s10822-004-3752-4 [DOI] [PubMed] [Google Scholar]
- 38. Stamos J., Sliwkowski M. X., and Eigenbrot C. (2002) Structure of the epidermal growth factor receptor kinase domain alone and in complex with a 4-anilinoquinazoline inhibitor. J. Biol. Chem. 277, 46265–46272 10.1074/jbc.M207135200 [DOI] [PubMed] [Google Scholar]
- 39. Park J. H. H., Liu Y., Lemmon M. A. A., and Radhakrishnan R. (2012) Erlotinib binds both inactive and active conformations of the EGFR tyrosine kinase domain. Biochem. J. 448, 417–423 10.1042/BJ20121513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Case D. A., Betz R. M., Cerutti D. S., Cheatham I. I. I., Darden T. E. T. A., Duke R. E., Giese T. J., Gohlke H., Goetz A. W., Homeyer N., Izadi S., Janowski P., Kaus J., Kovalenko A., Lee T. S., et al. (2018) AMBER 2018, University of California, San Francisco, CA [Google Scholar]
- 41. Maier J. A., Martinez C., Kasavajhala K., Wickstrom L., Hauser K. E., and Simmerling C. (2015) ff14SB: improving the accuracy of protein side chain and backbone parameters from ff99SB. J. Chem. Theory Comput. 11, 3696–3713 10.1021/acs.jctc.5b00255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Pettersen E. F., Goddard T. D., Huang C. C., Couch G. S., Greenblatt D. M., Meng E. C., and Ferrin T. E. (2004) UCSF Chimera—a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 10.1002/jcc.20084 [DOI] [PubMed] [Google Scholar]
- 43. Jorgensen W. L., Chandrasekhar J., Madura J. D., Impey R. W., and Klein M. L. (1983) Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 79, 926–935 10.1063/1.445869 [DOI] [Google Scholar]
- 44. Essmann U., Perera L., Berkowitz M. L., Darden T., Lee H., and Pedersen L. G. (1995) A smooth particle mesh Ewald method. J. Chem. Phys. 103, 8577–8593 10.1063/1.470117 [DOI] [Google Scholar]
- 45. Berendsen H. J. C., Postma J. P. M., van Gunsteren W. F., DiNola A., and Haak J. R. (1984) Molecular dynamics with coupling to an external bath. J. Chem. Phys. 81, 3684–3690 10.1063/1.448118 [DOI] [Google Scholar]
- 46. Roe D. R., and Cheatham T. E. 3rd (2013) PTRAJ and CPPTRAJ: software for processing and analysis of molecular dynamics trajectory data. J. Chem. Theory Comput. 9, 3084–3095 10.1021/ct400341p [DOI] [PubMed] [Google Scholar]
- 47. Humphrey W., Dalke A., and Schulten K. (1996) VMD: visual molecular dynamics. J. Mol. Graph. 14, 33–38, 27–28 10.1016/0263-7855(96)00018-5 [DOI] [PubMed] [Google Scholar]
- 48. Forbes S. A., Beare D., Boutselakis H., Bamford S., Bindal N., Tate J., Cole C. G., Ward S., Dawson E., Ponting L., Stefancsik R., Harsha B., Kok C. Y., Jia M., Jubb H., et al. (2017) COSMIC: somatic cancer genetics at high-resolution. Nucleic Acids Res. 45, D777–D783 10.1093/nar/gkw1121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Gao J., Aksoy B. A., Dogrusoz U., Dresdner G., Gross B., Sumer S. O., Sun Y., Jacobsen A., Sinha R., Larsson E., Cerami E., Sander C., and Schultz N. (2013) Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 6, pl1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Cerami E., Gao J., Dogrusoz U., Gross B. E., Sumer S. O., Aksoy B. A., Jacobsen A., Byrne C. J., Heuer M. L., Larsson E., Antipin Y., Reva B., Goldberg A. P., Sander C., and Schultz N. (2012) The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2, 401–404 10.1158/2159-8290.CD-12-0095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. AACR Project GENIE Consortium (2017) AACR project GENIE: powering precision medicine through an international consortium. Cancer Discov. 7, 818–831 10.1158/2159-8290.CD-17-0151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Barretina J., Caponigro G., Stransky N., Venkatesan K., Margolin A. A., Kim S., Wilson C. J., Lehár J., Kryukov G. V., Sonkin D., Reddy A., Liu M., Murray L., Berger M. F., Monahan J. E., et al. (2012) The cancer cell line encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 483, 603–607 10.1038/nature11003 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.