

Abstract
The human complement component, C5a, binds two different seven-transmembrane receptors termed C5aR1 and C5aR2. C5aR1 is a prototypical G-protein–coupled receptor that couples to the Gαi subfamily of heterotrimeric G-proteins and β-arrestins (βarrs) following C5a stimulation. Peptide fragments derived from the C terminus of C5a can still interact with the receptor, albeit with lower affinity, and can act as agonists or antagonists. However, whether such fragments might display ligand bias at C5aR1 remains unexplored. Here, we compare C5a and a modified C-terminal fragment of C5a, C5apep, in terms of G-protein coupling, βarr recruitment, endocytosis, and extracellular signal-regulated kinase 1/2 mitogen-activated protein kinase activation at the human C5aR1. We discover that C5apep acts as a full agonist for Gαi coupling as measured by cAMP response and extracellular signal-regulated kinase 1/2 phosphorylation, but it displays partial agonism for βarr recruitment and receptor endocytosis. Interestingly, C5apep exhibits full-agonist efficacy with respect to inhibiting lipopolysaccharide-induced interleukin-6 secretion in human macrophages, but its ability to induce human neutrophil migration is substantially lower compared with C5a, although both these responses are sensitive to pertussis toxin treatment. Taken together, our data reveal that compared with C5a, C5apep exerts partial efficacy for βarr recruitment, receptor trafficking, and neutrophil migration. Our findings therefore uncover functional bias at C5aR1 and also provide a framework that can potentially be extended to chemokine receptors, which also typically interact with chemokines through a biphasic mechanism.
Keywords: G-protein–coupled receptor (GPCR), arrestin, complement, cell signaling, signal transduction
Introduction
The complement peptide C5a, a potent chemotactic agent and an anaphylatoxin, is one of the most critical activation products of the human complement system (1). C5a is a 74-amino acid-long peptide that is generated upon the enzymatic cleavage of complement component C5 by C5 convertases. Abnormal levels of C5a and subsequent signaling triggered by it are crucial in a range of inflammatory disorders including sepsis, vasculitis, and trauma (1, 2). C5a exerts its effects via two seven-transmembrane receptors, namely the C5aR1 and C5aR2 (also known as C5L2) (3). Of these, C5aR1 is a prototypical G-protein–coupled receptor (GPCR)4 that is expressed in macrophages, neutrophils, and endothelial cells (3). Upon binding of C5a, C5aR1 couples to the Gαi subtype of heterotrimeric G proteins, resulting in inhibition of cAMP levels and mobilization of intracellular Ca2+ (3). Subsequently, C5a also triggers the phosphorylation of C5aR1 followed by recruitment of β-arrestins (βarrs) and receptor internalization (3).
Structurally, C5a harbors four different helices and connecting loops, and it is stabilized by the formation of three disulfide bonds (4). C5a interacts with C5aR1 through two distinct interfaces: one involves the core of C5a with the N terminus of the receptor, whereas the other involves the C terminus of C5a with the extracellular side of the transmembrane helices of C5aR1 (Fig. 1A) (5). It has been proposed that the structural determinants for high-affinity binding are provided by the first set of interaction, whereas the second set of interaction is responsible for driving functional responses through the receptor (5). Peptides derived from the C terminus of C5a can bind to C5aR1, albeit with much lower affinity compared with C5a, and they can also trigger functional responses (6, 7). Whether such peptides may induce differential coupling of G protein versus βarrs and exhibit biased functional responses remains completely unexplored.
Figure 1.

A modified C terminus C5a peptide, C5apep, is a full agonist for Gαi coupling. A, schematic representation of C5a binding to C5aR1. There are two sites of interaction, one involving the N terminus of C5aR1 and the other involving the extracellular loops. B, the primary sequence and modification of C5apep, which is derived based on the C terminus of C5a. C, C5apep behaves as a full-agonist in GloSensor-based cAMP assay. HEK-293 cells expressing C5aR1 were transfected with F22 plasmids. 24 h post-transfection, the cells were stimulated with indicated concentrations of C5a and C5apep followed by recording of bioluminescence readout. D, Gαi coupling induced by C5a and C5apep measured in CHO cells using LANCE cAMP assay. CHO cells stably expressing C5aR1 were stimulated with the indicated concentrations of respective ligands. The data presented in C and D represent the means ± S.E. of three to five independent experiments, and the EC50 values of C5a and C5apep were analyzed using unpaired t test. ***, p < 0.001.
Here, we focus on a modified hexapeptide, referred to as C5apep hereafter (Fig. 1B), which displays the highest binding affinity to C5aR1 among various C5a fragments (7), and characterize it vis-à-vis C5a with respect to Gαi and βarr coupling, functional outcomes, and cellular responses. In particular, we measure the ability of C5a and C5apep to inhibit forskolin-induced cAMP as a measure of Gαi-coupling, inducing βarr recruitment and trafficking, receptor endocytosis, ERK1/2 MAP kinase activation, IL-6 release, and neutrophil migration. We identify a significant bias in Gαi versus βarr coupling, endocytosis versus ERK1/2 activation, and IL-6 release versus neutrophil migration between the two ligands. These findings establish a framework for investigating ligand-induced functional bias at C5aR1 and pave the way for subsequent characterization of physiological outcomes arising from such ligands.
Results and discussion
C5apep is a full agonist for Gαi coupling
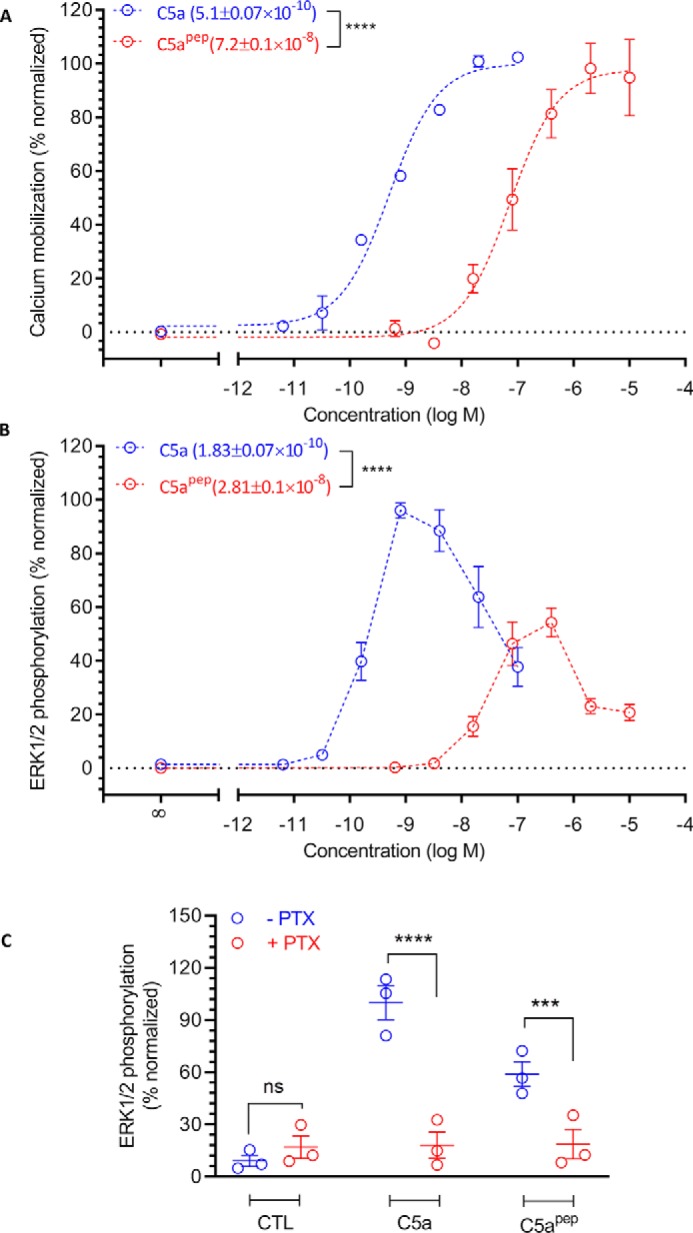
Although C5apep exhibits the highest binding affinity for C5aR1 among the peptides derived from and modified based on the C terminus of C5a, its binding affinity for C5aR1 is still significantly lower than C5a (∼70 nm for C5apep and ∼1 nm for C5a) (7). We therefore first measured the ability of C5apep to trigger Gαi coupling to C5aR1 in HEK-293 cells using the GloSensor assay (8). The cells were stimulated with forskolin to generate cAMP followed by incubation with various doses of C5a and C5apep. We observed that both C5a and C5apep inhibited cAMP level to a similar extent at saturating concentrations (Fig. 1C). As expected, based on their binding affinities for the receptor, C5apep was ∼100-fold less potent in cAMP inhibition compared with C5a (IC50 = ∼0.26 nm for C5a and IC50 = ∼16 nm for C5apep). We also measured the efficacy of C5apep in C5aR1 expressing CHO cells using the LANCE cAMP assay (9) and observed a pattern of efficacy and potency very similar to that in HEK-293 cells (Fig. 1D).
C5apep is a partial agonist for βarr recruitment
Upon C5a stimulation, C5aR1 undergoes phosphorylation and recruits βarrs, which is important for receptor desensitization and internalization, similar to other prototypical GPCRs (10, 11). Thus, we next measured the ability of C5apep to induce βarr coupling using a standard co-immunoprecipitation assay. There are two isoforms of βarrs known as βarr1 and 2 that exhibit a significant functional divergence despite a high level of sequence and structural similarity (12). We expressed either βarr1 or βarr2 with FLAG-tagged C5aR1 in HEK-293 cells and then measured their interaction upon ligand stimulation. As presented in Fig. 2 (A and B), we observed a robust recruitment of both isoforms of βarrs upon stimulation of cells with C5a. Interestingly, the levels of βarr recruitment induced by C5apep were significantly lower compared with C5a, even at saturating ligand concentrations. As a control, we stimulated cells with W54011, an antagonist of C5aR1 (13), and as expected, it did not elicit any significant levels of βarr recruitment. To probe whether there may be a temporal difference in C5aR1-βarr interaction pattern for C5a versus C5apep, we further carried out a time-course experiment for βarr recruitment. However, C5apep-induced βarr recruitment was significantly lower than C5a (Fig. 2, C and D). Taken together with the cAMP data presented above, these findings suggest that C5apep is a full-agonist for Gαi-coupling but a partial agonist for βarr recruitment.
Figure 2.

C5apep is a partial agonist for βarr recruitment. A, HEK-293 cells expressing FLAG-C5aR1 and either βarr1 or 2 were stimulated with the indicated concentrations of different ligands (W54011, 0.1 μm; C5a, 1 μm; C5apep, 10 μm) followed by cross-linking using DSP. Subsequently, FLAG-C5aR1 was immunoprecipitated using anti-FLAG antibody agarose, and co-elution of βarrs was visualized using Western blotting. B, densitometry-based quantification of data presented in A (averages ± S.E.; n = 2) normalized with respect to signal for C5a-βarr1 condition (treated as 100%) and analyzed using two-way ANOVA. ***, p < 0.001. C, a time-course co-immunoprecipitation experiment to measure the interaction of C5aR1 with βarr2. The experiment was performed following the protocol as indicated except that cells were stimulated for different time points. D, densitometry-based quantification of the data presented in C (average ± S.E.; n = 2) normalized with respect to signal for C5a at 30 min condition (treated as 100%) and analyzed using two-way ANOVA. *, p < 0.05.
C5apep triggers slower endosomal trafficking of βarrs
βarrs are normally distributed in the cytoplasm and upon agonist stimulation; they traffic to the membrane and interact with receptors (14). Subsequently, upon prolonged agonist stimulation, βarrs either dissociate from the receptor (class A pattern of βarr recruitment) or co-internalize with activated receptors in endosomal vesicles (class B pattern of βarr recruitment) (14). To probe whether C5apep might differ from C5a with respect to βarr trafficking patterns, we co-expressed C5aR1 with either βarr1-YFP or βarr2-YFP and visualized the trafficking of βarrs using confocal microscopy. We observed that C5apep was capable of promoting surface translocation of βarrs at similar levels as C5a during the early phase of agonist stimulation (Fig. 3, second column). However, we observed that C5apep was significantly slower in promoting the appearance of βarrs in endosomal punctae and vesicles compared with C5a (Fig. 3, third column), although ultimately it did induce robust endosomal localization of βarrs (Fig. 3, fourth column).
Figure 3.

C5apep induces slower endosomal trafficking of βarrs. HEK-293 cells expressing C5aR1 and βarr1/2 were stimulated with C5a (1 μm) and C5apep (10 μm), and the trafficking patterns of βarrs were visualized using confocal microscopy for indicated time points. Both C5a and C5apep induced surface localization of βarrs at early time points; however, somewhat slower endosomal trafficking of βarrs were observed for C5apep as assessed by localization of βarrs in endosomal vesicles. The figure shows representative images from three independent experiments.
C5apep exhibits a bias between ERK1/2 MAP kinase activation and receptor endocytosis
To probe whether C5apep may also exhibit differential efficacy compared with C5a in functional assays, we next measured the ability of C5apep to induce receptor endocytosis and ERK1/2 MAP kinase activation in HEK-293 cells. In agreement with our data on βarr recruitment and trafficking, we observed lower levels of receptor endocytosis induced by C5apep compared with C5a (Fig. 4A). This observation hints that βarrs may play a crucial role in endocytosis of C5aR1, and therefore, weaker βarr recruitment by C5apep translates into lower endocytosis. Interestingly however, C5apep was as efficacious as C5a in stimulating phosphorylation of ERK1/2 MAP kinase in HEK-293 cells, at least at the time points that were tested in this experiment (Fig. 4, B and C). Thus, correlation of maximal levels of endocytosis triggered by C5a and C5apep with the ERK1/2 phosphorylation reveals a bias of C5apep in these two functional responses. Full efficacy of C5apep for ERK1/2 phosphorylation, similar to that in cAMP assay, suggests that this response may be primarily driven by Gαi. To probe this possibility, we measured ERK1/2 phosphorylation after pretreatment of cells with pertussis toxin (PTX) and observed a robust inhibition of ERK1/2 phosphorylation for both C5a and C5apep (Fig. 5, A–D).
Figure 4.
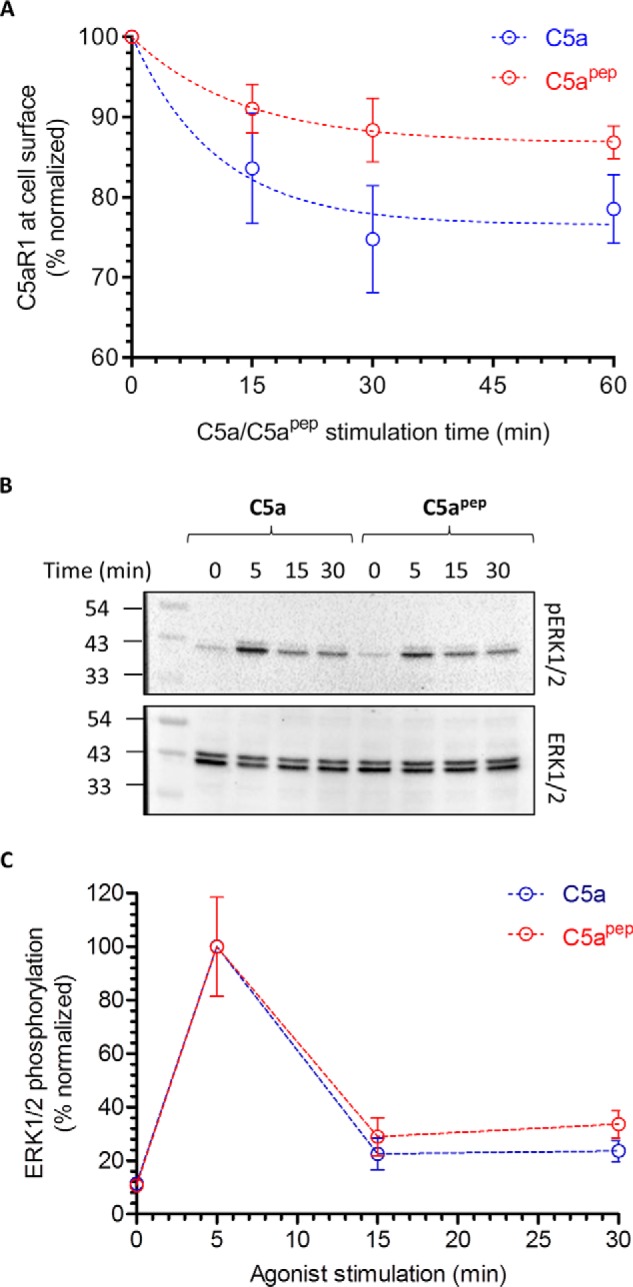
C5apep exhibits a bias between receptor endocytosis and ERK1/2 MAP kinase phosphorylation. A, HEK-293 cells expressing C5aR1 were stimulated with C5a (1 μm) and C5apep (10 μm) for indicated time points followed by the assessment of surface receptor levels using a whole-cell ELISA assay. C5apep displays a weaker efficacy in promoting C5aR1 endocytosis compared with C5a. The data represent averages ± S.E. from five independent experiments. B, C5apep induces robust phosphorylation of ERK1/2 MAP kinase at levels similar to C5a. HEK-293 cells expressing C5aR1 were stimulated with respective ligands for indicated time points followed by measurement of ERK1/2 phosphorylation using Western blotting. C, densitometry-based quantification of the ERK1/2 phosphorylation data presented in B (averages ± S.E.) of five independent experiments.
Figure 5.

C5a/C5apep-induced ERK1/2 phosphorylation is sensitive to PTX treatment. A, HEK-293 cells stably expressing C5aR1 were incubated with 100 ng/ml PTX for 12–16 h followed by serum starvation and ligand stimulation. Subsequently, ERK1/2 phosphorylation was measured by Western blotting. B, densitometry-based quantification of data presented in A from three independent experiments normalized with respect to maximal response (treated as 100%) and analyzed using one-way ANOVA. ***, p < 0.001. C, effect of PTX treatment on C5apep-induced ERK1/2 phosphorylation measured as in A above. D, densitometry-based quantification of data presented in C, normalized, and analyzed as in B.
Previous studies on several Gαi-coupled receptors have documented an interplay of βarrs and Gαi in agonist-induced ERK1/2 phosphorylation. For example, nicotinic acid induced ERK1/2 phosphorylation downstream of GPR109A can be inhibited by either PTX pretreatment or βarr knockdown (15). Similarly, carvedilol-induced ERK1/2 phosphorylation downstream of β1-adrenergic receptor requires a contribution from both Gαi and βarrs (16, 17). This interesting aspect of GPCR signaling is being actively explored and discussed in the literature (18–22). Although our data shows nearly complete inhibition of C5a/C5apep-induced ERK1/2 phosphorylation for C5aR1, evaluating the contribution of βarrs in this process would be an interesting avenue for future studies.
C5apep exhibits βarr isoform bias at a chimeric C5aR1
The interaction of βarrs with GPCRs is a biphasic process, which involves the receptor tail (i.e. phosphorylated C terminus) and the receptor core (cytoplasmic surface of the transmembrane bundle) (11, 23). Based on the stability of their interaction with βarrs, GPCRs are categorized as class A and B, which represent transient and stable interactions, respectively (14). Receptors having clusters of phosphorylatable residues in their C terminus (such as the vasopressin receptor, V2R) typically interact stably with βarrs. Because C5aR1 does not harbor such clusters, we generated a chimeric C5aR1, where we grafted the C terminus of the V2R on C5aR1 and refer to this construct as C5a-V2R (Fig. 6A). We first measured the cAMP response of C5a-V2R upon stimulation with C5a and C5apep and observed a similar efficacy and potency profile as for the WT C5aR1 (Fig. 6B). Similarly, we also observed comparable levels of ERK1/2 phosphorylation induced by both C5a and C5apep downstream of C5a-V2R (Fig. 6, C and D).
Figure 6.

C5apep exhibits full-agonist efficacy for cAMP response and ERK1/2 phosphorylation for a chimeric C5aR1, C5a-V2R. A, schematic representation of the C terminus of C5aR1 and a chimeric construct harboring the C terminus of AVPR2 (V2R), referred to as C5a-V2R. V2R tail in the chimeric construct is highlighted in red. B, C5apep behaves as a full agonist in GloSensor-based cAMP assay. HEK-293 cells expressing C5a-V2R were transfected with F22 plasmids. 24 h post-transfection, the cells were stimulated with the indicated concentrations of C5a and C5apep followed by recording of bioluminescence readout. The data represent averages ± S.E. of three independent experiments, each carried out in duplicate, and the EC50 values of C5a and C5apep were analyzed using unpaired t test. ***, p < 0.001. C, C5apep induces robust phosphorylation of ERK1/2 MAP kinase at levels similar to C5a. HEK-293 cells expressing C5a-V2R were stimulated with respective ligands (C5a, 1 μm; C5apep, 10 μm) for the indicated time points followed by measurement of ERK1/2 phosphorylation using Western blotting. D, densitometry-based quantification of ERK1/2 phosphorylation data presented in C (average ± S.E.) of five independent experiments.
We next measured the interaction of βarrs with C5a-V2R and expectedly observed an enhanced βarr recruitment for C5a-V2R compared with C5aR1 (Fig. 7, A and B). Interestingly, however, unlike C5aR1, we observed an equal recruitment of βarr1 by C5apep and C5a with C5a-V2R (Fig. 7, C and D). On the other hand, similar to C5aR1, βarr2 recruitment induced by C5apep was still significantly weaker than C5a at this chimeric receptor (Fig. 7, A and B). These findings reveal a βarr isoform recruitment bias at the chimeric receptor, a pattern that has significant implications if applicable to other GPCRs as well. For example, several high-throughput screening assays of βarr recruitment, such as the Tango assay, utilize chimeric GPCRs with V2R tail (24). This may result in miscalculation of ligand bias, and therefore, the patterns observed using chimeric receptors in primary screening should be reconfirmed using WT receptors for calculating bias profile.
Figure 7.

C5apep induces differential recruitment of βarr1 and 2 at C5aV2R. A, HEK-293 cells expressing FLAG-C5a-V2R and βarr2 were stimulated with saturating concentrations of different ligands followed by cross-linking using DSP. Subsequently, FLAG-C5a-V2R was immunoprecipitated using anti-FLAG antibody agarose, and co-elution of βarr2 was visualized using Western blotting. B, densitometry-based quantification of data presented in A (average±S.E.; n = 2) normalized with respect to signal for C5a-βarr2 30 min condition (treated as 100%) and analyzed using two-way ANOVA. **, p < 0.01; *, p < 0.05. C, interaction of C5aR1 or C5a-V22R with βarr1 measured essentially in similar fashion as described in A above. D, densitometry-based quantification of data presented in C (averages ± S.E.; n = 3) normalized with respect to signal for C5a-C5aR1 condition (treated as 100%) and analyzed using two-way ANOVA. **, p < 0.01.
C5apep is a partial agonist for βarr recruitment at C5aR2
C5a interacts with two distinct seven-transmembrane receptors, C5aR1 and C5aR2. Of these, C5aR2 does not exhibit any detectable G-protein coupling as measured in functional assays, although it robustly recruits βarrs upon agonist-stimulation (9, 25, 26). In line with previous reports in the literature, comparison of the primary sequences of C5aR1 and C5aR2 identifies mutations in highly conserved DRY and NPXXY motifs (Fig. 8A). We observed that C5apep acts as a partial agonist for βarr2 recruitment at C5aR2, similar to C5aR1 (Fig. 8B). Interestingly, however, unlike C5aR1, we did not observe agonist-induced phosphorylation of ERK1/2 downstream of C5aR2 in HEK-293 cells (Fig. 8, C and D). Although the mutations in the DRY and NPXXY motifs may be the primary determinants for the lack of functional G-protein coupling, it is also plausible C5aR2 adopts an active conformation upon agonist binding that is distinct from that of C5aR1. Such a conformation may preclude functional G-protein coupling but allow receptor phosphorylation and βarr interaction. However, future studies designed to investigate agonist-dependent conformational changes in C5aR1 and C5aR2 are required to probe such a possibility.
Figure 8.

Sequence alignment of C5aR2 with C5aR1, βarr2 recruitment, and ERK1/2 phosphorylation. A, the sequences of human C5aR1 and C5aR2 were retrieved from Uniprot and aligned on M-coffee server with default parameters. Alignment reliability was assessed by core/TCS and generated alignment was visualized using Espript 3. Specific mutations in the DRY and NPXXY motif are highlighted. B, HEK-293 cells expressing C5aR2-Venus and βarr2-Rluc8 constructs were first incubated with luciferase-substrate for 2 h. Subsequently, the cells were stimulated with respective ligands, and BRET signals were monitored using a dose-response curve. The data represent averages ± S.E. of three independent experiments, and the EC50 values are compared using unpaired t test. ***, p < 0.001. C, HEK-293 cells expressing C5aR1 or C5aR2 were stimulated with C5a (100 nm) for indicated time points followed by detection of ERK1/2 phosphorylation using Western blotting. D, densitometry-based quantification of data presented in C normalized with C5a response for C5aR1 (treated as 100%) and analyzed using two-way ANOVA. ***, p < 0.001.
C5apep elicits biased cellular responses
C5aR1 is endogenously expressed at high levels in macrophages and neutrophils, where it modulates multiple inflammatory responses (3). Stimulation of C5aR1 in human macrophages reduces LPS-induced release of IL-6 (2), whereas neutrophil C5aR1 activation induces rapid chemotaxis (3). To assess whether C5apep might exhibit a bias at the level of these cellular responses, we utilized primary human monocyte-derived macrophages (HMDMs) and human polymorphonuclear neutrophils (hPMNs) to measure IL-6 release and migration, respectively. Measuring Gi-mediated cAMP responses in these primary cells is technically challenging, and therefore, we measured Ca2+ mobilization in HMDMs upon stimulation by C5a and C5apep. We observed a full-agonist profile of C5apep similar to that observed in cAMP assays in HEK-293 and CHO cells, and the potency of these two agonists for Ca2+ mobilization was also similar to that observed in HEK-293 and CHO cells (Fig. 9A). Quantification and comparison of C5a and C5apep for inducing βarr recruitment in HMDMs and hPMNs at endogenous level of the receptor are technically very challenging and require development of novel sensors and assays going forward. Interestingly, however, C5apep stimulation resulted in only a submaximal phosphorylation of ERK1/2 in HMDMs compared with C5a (Fig. 9B). This is in contrast with our observation in HEK-293 cells in which C5apep stimulated ERK1/2 phosphorylation at levels similar to C5a (Fig. 4, B and C). We also found that ERK1/2 phosphorylation in HMDMs was sensitive to PTX pretreatment as in HEK-293 cells (Fig. 9C). This is particularly interesting considering that C5apep behaves as full agonist in Ca2+ mobilization experiments in HMDMs. There is growing evidence for context-specific effector coupling and functional responses downstream of several GPCRs (27, 28). These emerging findings have refined our current understanding of biased signaling by providing substantial evidence of additional levels of complexities in GPCR signaling. Our data with C5apep, especially in the context of ERK1/2 activation in HMDMs, further add to this important paradigm of GPCR signaling.
Figure 9.
C5apep exhibits bias at the level of Ca2+ mobilization and ERK1/2 phosphorylation in HMDMs. A, C5apep is a full agonist in Ca2+ mobilization assay in HMDM cells. HMDMs were first loaded with Fluo-4 calcium indicator followed by the addition of respective ligands and subsequent measurement of fluorescence intensity. The data were normalized with maximal response obtained for C5a and represent means ± S.E. of triplicate experiments performed from three independent donors, and the EC50 values of C5a and C5apep were analyzed using unpaired t test. ***, p < 0.001. B, HMDMs were stimulated with respective ligands for 10 min at room temperature before being lysed. The phospho-ERK1/2 content in the lysate was detected using AlphaLISA Surefire Ultra p-ERK1/2 kit. The data were normalized with maximal response obtained for C5a and represent the means ± S.E. of triplicate experiments performed from five independent donors, and the EC50 values of C5a and C5apep were analyzed using unpaired t test. ****, p < 0.0001. C, C5a/C5apep-induced ERK1/2 phosphorylation in HMDMs is sensitive to PTX. HMDMs seeded in tissue culture-treated 96-well plates were preincubated with 200 ng/ml PTX or vehicle in serum-free IMDM overnight. Subsequently, the cells were stimulated with C5a/C5apep, and ERK1/2 phosphorylation was measured using AlphaLISA Surefire Ultra p-ERK1/2 kit. The data are normalized with maximal response obtained for C5a and represent the means ± S.E. of triplicate experiments performed from three independent donors and analyzed using paired two-way ANOVA. ***, p < 0.001; ****, p < 0.0001. CTL, control.
We next compared the ability of C5a and C5apep to inhibit LPS-induced IL-6 release in HMDMs and to stimulate chemotaxis in hPMNs. We observed that the inhibition of LPS-induced IL-6 release in HMDMs was comparable for both C5a and C5apep (Fig. 10A) and was sensitive to PTX pretreatment (Fig. 10B). Notably, however, we observed that C5apep displayed a significantly blunted response in neutrophil chemotaxis compared with C5a even at saturating doses (Fig. 10C), although similar to IL-6 release, chemotaxis was also sensitive to PTX. These observations therefore uncover that C5apep exhibits bias at the level of cellular responses when compared with C5a in primary cells expressing endogenous levels of C5aR1. It is interesting to note here that both LPS-induced IL-6 release and hPMN chemotaxis are sensitive to PTX pretreatment, suggesting that these processes are driven primarily by Gαi coupling to C5aR1. However, because βarr knockdown or knockout studies in HMDMs and hPMNs are technically challenging and typically have suboptimal efficiency, we cannot rule out either a direct contribution of βarrs or an interplay of βarrs and Gαi in these processes, and it remains to be explored in future studies (16).
Figure 10.

C5apep exhibits bias between LPS-induced IL-6 release and neutrophil migration. A, C5apep behaves as a full-agonist for lowering of LPS-induced IL-6 release in HMDM cells. HMDMs were stimulated using 10 ng/ml LPS in the absence or presence of C5a or C5apep. Subsequently, the levels of IL-6 present in the supernatant after 24 h of stimulation were quantified using ELISA, background-corrected with the values obtained for serum-free medium/BSA, and normalized with maximal response for LPS (i.e. treated as 100%). The data represent normalized means ± S.E. of triplicate measurements conducted in cells from four independent donors, analyzed using two-way ANOVA. *, p < 0.05; ****, p < 0.0001. B, LPS-induced IL-6 response is sensitive to PTX. HMDMs seeded in 96-well plates were incubated overnight with 200 ng/ml PTX or vehicle followed by stimulation with respective ligands. Subsequently, the IL-6 content in the supernatant was quantified using ELISA kits. The data are presented, normalized, and analyzed as in A above. C, C5apep elicits only partial-agonist response in migration of human PMNs. Freshly isolated hPMNs seeded into Transwell inserts were stimulated with respective ligands added to the receiver wells and then allowed to migrate for 1 h. The number of migrated cells was recorded and normalized to the maximal C5a-induced migration. The data represent the means ± S.E. of triplicate measurements conducted in cells from three independent donors. D, C5apep-induced neutrophil migration is sensitive to PTX. Freshly isolated hPMNs were preincubated with 3 μg/ml PTX or vehicle for 4 h, and cell migration was initiated by adding respective ligands. Ligand-induced migration was measured as in C, and the data are normalized with control (i.e. no stimulation) and analyzed suing two-way ANOVA. *, p < 0.05. CTL, control.
It is important to note that chemokines, like complement C5a, also interact with their cognate GPCRs through a biphasic mechanism. Thus, it is tempting to speculate that fragments derived from the C terminus of chemokines may also exhibit biased signaling through their receptors. Our findings also suggest that a more comprehensive analysis of C5a fragments might yield additional βarr-biased ligands at C5aR1. Although crystal structures of C5aR1 bound to small molecule antagonists have been determined recently (29, 30), a C5a-bound structure is still not available. Future high-resolution data of C5a-bound C5aR1 may provide structural insights into differential engagement of C5apep compared with C5a and how these differential interactions in the ligand-binding pocket yield transducer-coupling bias.
In summary, we discover C5apep as a biased C5aR1 agonist at the levels of Gαi versus βarr coupling, functional outcomes, and cellular responses. Going forward, an interesting avenue might be to evaluate the physiological responses elicited by C5apep in vivo. Given that C5a attenuates LPS-mediated cytokine production from macrophages (31) (Fig. 10), a biased ligand such as C5apep that retains this beneficial activity, although diminishing the more pro-inflammatory activities of neutrophil migration, may be a novel therapeutic approach for treating inflammatory disorders. Furthermore, as we have demonstrated the recruitment of both isoforms of βarr (i.e. βarr1 and 2) to C5aR1, it would be interesting to evaluate the shared and distinct roles of these βarr isoforms in regulation of C5aR1. It is also notable that the second receptor activated by C5a (C5aR2) does not exhibit any detectable G-protein coupling but displays robust βarr recruitment (25). Although we did not observe agonist-induced ERK1/2 phosphorylation downstream of C5aR2 upon C5a stimulation, it is plausible that it may activate other signaling pathways, and a comprehensive study is required to probe this possibility in future.
Materials and methods
General reagents, constructs, and cell culture
Most of the reagents were purchased from Sigma unless mentioned otherwise. The coding region of human C5aR1 was cloned in pcDNA3.1 vector with the N-terminal signal sequence and a FLAG tag. Coding regions of bovine βarr1 and βarr2 were cloned in pCMV vector. C5apep was synthesized from Genscript. Recombinant human C5a was either purchased from Sino Biological or purified following a previously published protocol (4). Ultrapure lipopolysaccharide from Escherichia coli K12 strain was purchased from Invivogen. BSA was purchased from Sigma. For cell culture, trypsin-EDTA, Hanks' balanced salt solution (HBSS), HEPES, Dulbecco's modified Eagle's medium (DMEM), phenol-red free DMEM, Ham's F-12 medium, Iscove's modified Dulbecco's medium (IMDM), and penicillin/streptomycin were purchased from Thermo Fisher Scientific. Dulbecco's PBS (DPBS) was purchased from Lonza.
The following cell lines were cultured as previously described (9). Chinese hamster ovary cells stably expressing the human C5aR1 (CHO-C5aR1) were maintained in Ham's F-12 medium containing 10% fetal bovine serum (FBS), 100 units/ml penicillin, 100 μg/ml streptomycin, and 400 μg/ml G418 (Invivogen). Human embryonic kidney-293 (HEK-293, ATCC) cells were maintained in DMEM containing 10% FBS, 100 IU/ml penicillin, and 100 μg/ml streptomycin. All cell lines were maintained in T175 flasks (37 °C, 5% CO2) and subcultured at 80–90% confluency using 0.05% trypsin-EDTA in DPBS. To ensure the consistency of cell function, cell morphology was continually monitored, and neither cell line was used beyond passage 20.
To generate HMDM, human buffy coat blood from anonymous healthy donors was obtained through the Australian Red Cross Blood Service. Human CD14+ monocytes were isolated from blood using Lymphoprep density centrifugation (STEMCELL) followed by CD14+ MACS magnetic bead separation (Miltenyi Biotec). The isolated monocytes were differentiated for 7 days in IMDM supplemented with 10% FBS, 100 units/ml penicillin, 100 μg/ml streptomycin, and 15 ng/ml recombinant human macrophage colony stimulating factor (Peprotech) on 10-mm square dishes (Sterilin). Nonadherent cells were removed by washing with DPBS, and the adherent differentiated HMDMs were harvested by gentle scraping.
hPMNs were obtained from venous whole blood (20 ml) collected from healthy volunteers under informed consent. The samples were collected using venepuncture into BD K2EDTA Vacutainer® blood collection tubes and processed within 5 h. For neutrophil isolation, the anticoagulated blood was firstly layered over a Lymphoprep (STEMCELL) density gradient and centrifuged (800 × g for 30 min at 22 °C), followed by residual erythrocytes removal using hypotonic lysis. Isolated PMNs were counted and resuspended in a HBSS-based migration buffer (containing calcium and magnesium, supplemented with 20 mm HEPES and 0.5% BSA).
Preparation of C5aR1, C5a-V2R, and C5aR2 expressing stable HEK-293 cell line
50–60% confluent HEK-293 cells were transfected with 7 μg of FLAG-tagged C5aR1/C5a-V2R/C5aR2 DNA complexed with 21 μg of polyethylenimine (PEI). Next day, stable selection was started with optimal dose of G418 along with untransfected cells kept as negative control. After completion of stable selection, clonal population was prepared by the limited dilution method. The highest expressing clones were propagated further and kept under G418 selection throughout the course of experiments. Surface expression of C5aR1, C5a-V2R, and C5aR2 was measured using a previously described whole-cell surface ELISA protocol (32).
ERK1/2 phosphorylation assay
Agonist-induced ERK1/2 phosphorylation was measured primarily using a previously described Western blotting–based protocol (33). C5aR1-, C5a-V2R–, or C5aR2-expressing stable cell lines were seeded into 6-well plate at a density of 1 million cells/well. The cells were serum-starved for 12 h followed by stimulation with 1 μm of C5a and 10 μm of C5apep, respectively, at selected time points. After the completion of time course, the medium was aspirated, and the cells were lysed in 100 μl of 2× SDS dye/well. The cells were heated at 95 °C for 15 min followed by centrifugation at 15,000 rpm for 10 min. 10 μl of lysate was loaded per well and separated on SDS-PAGE followed by Western blotting. The blots were blocked in 5% BSA (in TBST) for 1 h and incubated overnight with rabbit phospho-ERK (catalog no. 9101/CST) primary antibody at 1:5000 dilution. The blots were washed thrice with TBST for 10 min each and incubated with anti-rabbit HRP-coupled secondary antibody (1:10,000, catalog no. A00098/Genscript) for 1 h. The blots were washed again with TBST for three times and developed with Promega ECL solution on chemidoc (Bio-Rad). The blots were stripped with low pH stripping buffer and then reprobed for total ERK using rabbit total ERK (catalog no. 9102/CST) primary antibody at 1:5000 dilution. To measure the effect of PTX on ERK activation, HEK-293 cells stably expressing C5aR1 were seeded in 6-well plate. The cells were treated with 100 ng/ml PTX for 12–16 h followed by serum starvation. The cells were stimulated with C5a and C5apep for the indicated time points. The samples were prepared and processed as mentioned previously.
The ligand-induced phospho-ERK1/2 signaling in HMDMs was assessed using the AlphaLISA Surefire Ultra p-ERK1/2 (Thr202/Tyr204) kit (PerkinElmer) following the manufacturer's protocol. Briefly, HMDMs were seeded (50,000/well) in tissue culture-treated 96-well plates (Corning) for 24 h and serum-starved overnight. All ligand dilutions were prepared in serum-free medium containing 0.1% BSA. For stimulation, the cells were incubated with respective ligands for 10 min at room temperature and then immediately lysed using AlphaLISA lysis buffer on a microplate shaker (450 rpm, 10 min). For the detection of phospho-ERK1/2 content, cell lysate (5 μl/well) was transferred to a 384-well ProxiPlate (PerkinElmer) and added to the donor and acceptor reaction mix (2.5 μl/well, respectively) with 2-h incubation at room temperature in the dark. On a Tecan Spark 20M, following laser irradiation of donor beads at 680 nm, the chemiluminescence of acceptor beads at 615 nm was recorded. To measure the effect of PTX, culture medium was changed into serum-free IMDM containing 200 ng/ml PTX or vehicle only for overnight incubation. Afterward, ERK1/2 phosphorylation was measured as described above.
Receptor internalization assay
50–60% confluent HEK-293 cells were transfected with 3.5 μg of FLAG-tagged C5aR1 DNA by polyethylenimine method of transfection at DNA:PEI ratio of 1:3. 24 h post-transfection, 0.15 million cells/well were seeded in 24-well plate (precoated with 0.01% poly-d-lysine). After 24 h, the cells were serum-starved for 6 h followed by stimulation with 1 μm of C5a and 10 μm C5apep, respectively, for selected time points. After stimulation, the cells were washed once with ice-cold 1× TBS. The cells were then fixed with 4% (w/v) paraformaldehyde on ice for 20 min and washed thrice with TBS to remove paraformaldehyde. Blocking was done with 1% BSA prepared in 1× TBS for 1.5 h. This was followed by incubation of cells with HRP-conjugated anti-FLAG M2 antibody (Sigma) at a dilution of 1:2000 prepared in 1% BSA + 1× TBS for 1.5 h. Afterward, the cells were washed thrice with 1% BSA + 1× TBS. Surface expression was measured by incubating cells with 200 μl of 3,3′,5,5′-tetramethylbenzidine (Genscript) per well, and reaction was stopped by transferring 100 μl of developed colored solution to a 96-well plate already containing 100 μl of 1 m H2SO4. Absorbance was read at 450 nm in a multiplate reader (Victor X4). For normalization, cell density was measured using janus green. Briefly, 3,3′,5,5′-tetramethylbenzidine was removed, and the cells were washed twice with 1× TBS followed by incubation with 0.2% (w/v) janus green for 15 min. Destaining was done with three washes of 1 ml of distilled water. Stain was eluted by adding 800 μl of 0.5 n HCl per well. 200 μl of this solution was transferred to a 96-well plate, and absorbance was read at 595 nm. Data normalization was done by dividing A450 by A595 values.
GloSensor assay for cAMP measurement
50–60% confluent HEK-293 cells were co-transfected with 3.5 μg each of C5aR1 or C5a-V2R and 22F (Promega) plasmids. 24 h post-transfection, the cells were trypsinized and harvested by centrifugation at 1500 rpm for 10 min. The medium was aspirated, and cells were resuspended in luciferin sodium solution (0.5 mg/ml) (Gold Biotech) prepared in 1× HBSS (Gibco) and 20 mm HEPES, pH 7.4. The cells were then seeded in a 96-well plate at a density of 0.4 million cells/well and kept at 37 °C for 1.5 h in the CO2 incubator followed by incubation at room temperature for 30 min. Basal reading was read on luminescence mode of multiplate reader (Victor X4), and cycles were adjusted until basal values were stabilized. The cells were then incubated with 1 μm forskolin, and readings were recorded until maximum luminescence values were obtained. This was followed by stimulation of cells with specified concentrations of C5a and C5apep, and values were recorded for 1 h. The data were normalized with respect to minimal stimulation dose of ligand after basal correction.
Cross-linking and co-immunoprecipitation
50–60% confluent HEK-293 cells were co-transfected with C5aR1 or C5a-V2R and βarr1/βarr2 plasmids by PEI (as mentioned earlier). 48 h post-transfection, the cells were serum-starved for 6 h and stimulated with respective doses of C5a/C5apep, harvested, and proceeded for cross-linking experiment. The cells were lysed by Dounce homogenization in 20 mm HEPES, pH 7.4, 100 mm NaCl, 1× phosphatase inhibitor mixture (Roche), 2 mm benzamidine hydrochloride, and 1 mm phenylmethylsulfonyl fluoride. This was followed by the addition of 1 mm dithiobis(succinimidyl-propionate) from a freshly prepared 100 mm stock in DMSO. Lysate was tumbled at room temperature for 40 min, and the reaction was quenched by adding 1 m Tris, pH 8.5. Lysates were solublized in 1%(v/v) MNG for 1 h at room temperature followed by centrifugation at 15,000 rpm for 15 min. Cleared supernatant was transferred to a separate tube already containing pre-equilibrated M1-FLAG beads supplemented with 2 mm CaCl2. The solution was tumbled for 2 h at 4 °C and washed alternately with low salt buffer (20 mm HEPES, pH 7.4, 150 mm NaCl, 0.01% MNG, 2 mm CaCl2) and high salt buffer (20 mm HEPES, pH 7.4, 350 mm NaCl, 0.01% MNG, 2 mm CaCl2), respectively. The bound proteins were eluted in FLAG-elution buffer containing 20 mm HEPES, pH 7.4, 150 mm NaCl, 2 mm EDTA, 0.01% MNG, and 250 μg/ml FLAG peptide. Co-immunoprecipitated βarr was detected by Western blotting using rabbit anti-βarr mAb (1:5000, CST catalog no. D24H9). The blots were stripped and reprobed for receptor with HRP-conjugated anti-FLAG M2 antibody (1:5000). The blots were developed on Chemidoc (Bio-Rad) and quantified using ImageLab software (Bio-Rad).
Sequence alignment of C5aR1 and C5aR2 sequences
Reviewed sequences of human C5aR1 and C5aR2 (also referred to as C5L2) were retrieved from Uniprot. Sequences were aligned on M-coffee server with default parameters. Alignment reliability was assessed by core/TCS, and generated alignment was visualized using Espript 3.
BRET assay for measuring the interaction of βarr2 with C5aR2
C5a-mediated βarr2 recruitment to C5aR2 was measured using bioluminescent resonance energy transfer (BRET) as previously described (9). Briefly, HEK-293 cells were transiently transfected with C5aR2-Venus and βarr2-Rluc8 constructs using XTG9 (Roche). At 24 h post-transfection, the cells were gently detached using 0.05% trypsin-EDTA and seeded (100,000/well) onto white 96-well TC plates (Corning) in phenol-red free DMEM containing 5% FBS. On the following day, the cells were firstly incubated with the substrate EnduRen (30 μm; Promega) for 2 h (37 °C, 5% CO2). On a Tecan Spark 20M microplate reader (37 °C), the BRET light emissions (460–485 and 520–545 nm) were continuously monitored for 25 reads with respective ligands added after the first 5 reads. The ligand-induced BRET ratio was calculated by subtracting the Venus (520–545 nm) over Rluc8 (460–485 nm) emission ratio of the vehicle-treated wells from that of the ligand-treated wells.
Confocal microscopy
For visualization of ligand-induced βarr recruitment, HEK-293 cells were co-transfected with C5aR1 and βarr1-YFP or βarr2-YFP plasmids in 1:1 ratio (total 7 μg) by PEI. 24 h post-transfection, 1 million cells were seeded in 35-mm glass bottom dish precoated with 0.01% poly-d-lysine. After 24 h, the cells were serum-starved for 6 h and stimulated with respective doses of C5a (1 μm) and C5apep (10 μm). For live-cell imaging, images were acquired using Carl Zeiss LSM780NLO confocal microscope for specified time intervals, and image processing was done in ZEN lite (Zen-blue/ZEN-black) software from Zeiss. Confocal microscopy experiments were performed on two independent set of cells (i.e. independent transfections), and multiple cells were imaged and analyzed.
Intracellular calcium mobilization assays
Ligand-induced intracellular calcium mobilization was assessed using Fluo-4 NW Calcium Assay kit (Thermo Fisher Scientific) following the manufacturer's instructions. Briefly, HMDMs were seeded (50,000/well) in black clear-bottom 96-well TC plates (Corning) for 24 h before the assay. The cells were first stained with the Fluo-4 dye in assay buffer (1× HBSS, 20 mm HEPES) for 45 min (37 °C, 5% CO2). Respective ligands were prepared in assay buffer containing 0.5% BSA. On a Flexstation 3 platform, the fluorescence (excitation/emission, 494/516 nm) was continually monitored for a total of 100 s with ligand addition performed at 16 s.
Chemotaxis assays
Ligand-induced hPMN migration was assessed using 6.5-mm Transwell polycarbonate membrane inserts with 3.0-μm pore (Corning) to create a modified Boyden chamber (31). Freshly isolated hPMNs were seeded onto inserts (500,000/well) for 20 min (37 °C, 5% CO2) in a HBSS-based migration buffer as described in previous section. To initiate cell migration, respective ligands prepared in migration buffer were added to the receiver wells in duplicates. After 60-min migration (37 °C, 5% CO2), the inserts were gently washed once with DPBS, and the residual cells on the upper side of the membrane were removed using a cotton swab. Migrated cells were detached by adding 500 μl/well Accumax solution (Invitrogen) to the receiver wells (10 min, room temperature) and then counted using a Bio-Rad TC20TM automated cell counter. For PTX treatment experiments, freshly isolated hPMNs were incubated with 3 μg/ml pertussis toxin (Tocris) or vehicle only and seeded onto inserts (500,000/well) (37 °C, 5% CO2) in migration buffer. Upon 4-h incubation with PTX, cell migration was measured as described above.
Measurement of cytokines release using ELISA
The immunomodulatory effect of respective C5aR1 ligands on LPS-induced cytokine release was assessed in primary human macrophages as previously described (34). HMDMs were seeded in 96-well TC plates (100,000/well) for 24 h before treatment. All ligands were prepared in serum-free IMDM containing 0.1% BSA. For stimulation, the cells were co-treated with LPS and respective C5aR1 ligands for 24 h (37 °C, 5% C02). The supernatant was collected and stored at −20 °C till use. IL-6 levels in the supernatant were quantified using respective human ELISA kits (BD OptEIA) as per the manufacturer's protocol.
Data collection, processing, and analysis
All experiments were conducted in triplicate and repeated on at least three separate days (for cell lines) or using cells from at least three donors (for HMDMs) unless otherwise specified. The data were analyzed using GraphPad software (Prism 8.0) and expressed as means ± S.E. The data from each repeat were normalized accordingly before being combined. For all dose-response assays, logarithmic concentration-response curves were plotted using combined data and analyzed to determine the respective potency values.
Author contributions
S. P. and A. K. S. investigation; S. P., X. X. L., T. M. W., and A. K. S. writing-review and editing; X. X. L., A. S., M. B., P. K., H. D., M. C., E. G., T. M. W., and A. K. S. methodology; T. M. W. and A. K. S. conceptualization; T. M. W. and A. K. S. supervision; A. K. S. resources; A. K. S. funding acquisition; A. K. S. writing-original draft; A. K. S. project administration.
Acknowledgments
We thank the members of our laboratories for critical reading of the manuscript. We thank Ravi Ranjan for helping with C5aR1 and C5a-V2R cloning and optimization of C5a purification.
This work was supported by DBT Wellcome Trust India Alliance Intermediate Fellowship IA/I/14/1/501285 (to A. K. S.), Department of Biotechnology of the Government of India Innovative Young Biotechnologist Award BT/08/IYBA/2014-3 (to A. K. S.), a LADY TATA Memorial Trust Young Researcher Award (to A. K. S.), Science and Engineering Research Board Grant SB/SO/BB-121/2013, Council of Scientific and Industrial Research Grant 37[1637]14/EMR-II, and National Health and Medical Research Council of Australia Project Grant 1082271. The authors declare that they have no conflicts of interest with the contents of this article.
- GPCR
- G-protein–coupled receptor
- βarr
- β-arrestins
- ERK
- extracellular signal-regulated kinase
- MAP
- mitogen-activated protein
- LPS
- lipopolysaccharide
- PTX
- pertussis toxin
- IL
- interleukin
- HMDM
- human monocyte-derived macrophages
- hPMN
- human polymorphonuclear neutrophil
- HBSS
- Hanks' balanced salt solution
- DMEM
- Dulbecco's modified Eagle's medium
- IMDM
- Iscove's modified Dulbecco's medium
- DPBS
- Dulbecco's PBS
- FBS
- fetal bovine serum
- PEI
- polyethylenimine
- BRET
- bioluminescent resonance energy transfer
- MNG
- maltose neopentyl glycol.
References
- 1. Manthey H. D., Woodruff T. M., Taylor S. M., and Monk P. N. (2009) Complement component 5a (C5a). Int. J. Biochem. Cell Biol. 41, 2114–2117 10.1016/j.biocel.2009.04.005 [DOI] [PubMed] [Google Scholar]
- 2. Ward P. A. (2004) The dark side of C5a in sepsis. Nat. Rev. Immunol. 4, 133–142 10.1038/nri1269 [DOI] [PubMed] [Google Scholar]
- 3. Klos A., Wende E., Wareham K. J., and Monk P. N. (2013) International Union of Basic and Clinical Pharmacology [corrected]: LXXXVII. Complement peptide C5a, C4a, and C3a receptors. Pharmacol. Rev. 65, 500–543 10.1124/pr.111.005223 [DOI] [PubMed] [Google Scholar]
- 4. Schatz-Jakobsen J. A., Yatime L., Larsen C., Petersen S. V., Klos A., and Andersen G. R. (2014) Structural and functional characterization of human and murine C5a anaphylatoxins. Acta Crystallogr. D Biol. Crystallogr. 70, 1704–1717 10.1107/S139900471400844X [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Siciliano S. J., Rollins T. E., DeMartino J., Konteatis Z., Malkowitz L., Van Riper G., Bondy S., Rosen H., and Springer M. S. (1994) Two-site binding of C5a by its receptor: an alternative binding paradigm for G protein-coupled receptors. Proc. Natl. Acad. Sci. U.S.A. 91, 1214–1218 10.1073/pnas.91.4.1214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kawai M., Quincy D. A., Lane B., Mollison K. W., Or Y. S., Luly J. R., and Carter G. W. (1992) Structure-function studies in a series of carboxyl-terminal octapeptide analogues of anaphylatoxin C5a. J. Med. Chem. 35, 220–223 10.1021/jm00080a004 [DOI] [PubMed] [Google Scholar]
- 7. Konteatis Z. D., Siciliano S. J., Van Riper G., Molineaux C. J., Pandya S., Fischer P., Rosen H., Mumford R. A., and Springer M. S. (1994) Development of C5a receptor antagonists: differential loss of functional responses. J. Immunol. 153, 4200–4205 [PubMed] [Google Scholar]
- 8. Kumar B. A., Kumari P., Sona C., and Yadav P. N. (2017) GloSensor assay for discovery of GPCR-selective ligands. Methods Cell Biol. 142, 27–50 10.1016/bs.mcb.2017.07.012 [DOI] [PubMed] [Google Scholar]
- 9. Croker D. E., Halai R., Fairlie D. P., and Cooper M. A. (2013) C5a, but not C5a-des Arg, induces upregulation of heteromer formation between complement C5a receptors C5aR and C5L2. Immunol. Cell Biol. 91, 625–633 10.1038/icb.2013.48 [DOI] [PubMed] [Google Scholar]
- 10. Braun L., Christophe T., and Boulay F. (2003) Phosphorylation of key serine residues is required for internalization of the complement 5a (C5a) anaphylatoxin receptor via a β-arrestin, dynamin, and clathrin-dependent pathway. J. Biol. Chem. 278, 4277–4285 10.1074/jbc.M210120200 [DOI] [PubMed] [Google Scholar]
- 11. Ranjan R., Dwivedi H., Baidya M., Kumar M., and Shukla A. K. (2017) Novel structural insights into GPCR–β-arrestin interaction and signaling. Trends Cell Biol. 27, 851–862 10.1016/j.tcb.2017.05.008 [DOI] [PubMed] [Google Scholar]
- 12. Srivastava A., Gupta B., Gupta C., and Shukla A. K. (2015) Emerging functional divergence of β-arrestin isoforms in GPCR function. Trends Endocrinol. Metab. 26, 628–642 10.1016/j.tem.2015.09.001 [DOI] [PubMed] [Google Scholar]
- 13. Sumichika H., Sakata K., Sato N., Takeshita S., Ishibuchi S., Nakamura M., Kamahori T., Ehara S., Itoh K., Ohtsuka T., Ohbora T., Mishina T., Komatsu H., and Naka Y. (2002) Identification of a potent and orally active non-peptide C5a receptor antagonist. J. Biol. Chem. 277, 49403–49407 10.1074/jbc.M209672200 [DOI] [PubMed] [Google Scholar]
- 14. Oakley R. H., Laporte S. A., Holt J. A., Caron M. G., and Barak L. S. (2000) Differential affinities of visual arrestin, β-arrestin1, and β-arrestin2 for G protein-coupled receptors delineate two major classes of receptors. J. Biol. Chem. 275, 17201–17210 10.1074/jbc.M910348199 [DOI] [PubMed] [Google Scholar]
- 15. Walters R. W., Shukla A. K., Kovacs J. J., Violin J. D., DeWire S. M., Lam C. M., Chen J. R., Muehlbauer M. J., Whalen E. J., and Lefkowitz R. J. (2009) β-Arrestin1 mediates nicotinic acid-induced flushing, but not its antilipolytic effect, in mice. J. Clin. Invest. 119, 1312–1321 10.1172/JCI36806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Dwivedi H., Baidya M., and Shukla A. K. (2018) GPCR signaling: the interplay of Gαi and β-arrestin. Curr. Biol. 28, R324–R327 10.1016/j.cub.2018.02.027 [DOI] [PubMed] [Google Scholar]
- 17. Wang J., Hanada K., Staus D. P., Makara M. A., Dahal G. R., Chen Q., Ahles A., Engelhardt S., and Rockman H. A. (2017) Gαi is required for carvedilol-induced β1 adrenergic receptor β-arrestin biased signaling. Nat. Commun. 8, 1706 10.1038/s41467-017-01855-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Grundmann M., Merten N., Malfacini D., Inoue A., Preis P., Simon K., Rüttiger N., Ziegler N., Benkel T., Schmitt N. K., Ishida S., Müller I., Reher R., Kawakami K., Inoue A., et al. (2018) Lack of β-arrestin signaling in the absence of active G proteins. Nat. Commun. 9, 341 10.1038/s41467-017-02661-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Gutkind J. S., and Kostenis E. (2018) Arrestins as rheostats of GPCR signalling. Nat. Rev. Mol. Cell Biol. 19, 615–616 10.1038/s41580-018-0041-y [DOI] [PubMed] [Google Scholar]
- 20. Milligan G., and Inoue A. (2018) Genome editing provides new insights into receptor-controlled signalling pathways. Trends Pharmacol. Sci. 39, 481–493 10.1016/j.tips.2018.02.005 [DOI] [PubMed] [Google Scholar]
- 21. Alvarez-Curto E., Inoue A., Jenkins L., Raihan S. Z., Prihandoko R., Tobin A. B., and Milligan G. (2016) Targeted elimination of G proteins and arrestins defines their specific contributions to both intensity and duration of G protein-coupled receptor signaling. J. Biol. Chem. 291, 27147–27159 10.1074/jbc.M116.754887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Gurevich V. V., and Gurevich E. V. (2018) Arrestin-mediated signaling: is there a controversy? World J. Biol. Chem. 9, 25–35 10.4331/wjbc.v9.i3.25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Shukla A. K., Westfield G. H., Xiao K., Reis R. I., Huang L. Y., Tripathi-Shukla P., Qian J., Li S., Blanc A., Oleskie A. N., Dosey A. M., Su M., Liang C. R., Gu L. L., Shan J. M., et al. (2014) Visualization of arrestin recruitment by a G-protein-coupled receptor. Nature 512, 218–222 10.1038/nature13430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Barnea G., Strapps W., Herrada G., Berman Y., Ong J., Kloss B., Axel R., and Lee K. J. (2008) The genetic design of signaling cascades to record receptor activation. Proc. Natl. Acad. Sci. U.S.A. 105, 64–69 10.1073/pnas.0710487105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Li R., Coulthard L. G., Wu M. C., Taylor S. M., and Woodruff T. M. (2013) C5L2: a controversial receptor of complement anaphylatoxin, C5a. FASEB J. 27, 855–864 10.1096/fj.12-220509 [DOI] [PubMed] [Google Scholar]
- 26. Van Lith L. H., Oosterom J., Van Elsas A., and Zaman G. J. (2009) C5a-stimulated recruitment of β-arrestin2 to the nonsignaling 7-transmembrane decoy receptor C5L2. J. Biomol. Screen 14, 1067–1075 10.1177/1087057109341407 [DOI] [PubMed] [Google Scholar]
- 27. Gundry J., Glenn R., Alagesan P., and Rajagopal S. (2017) A practical guide to approaching biased agonism at G protein coupled receptors. Front Neurosci. 11, 17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ho J. H., Stahl E. L., Schmid C. L., Scarry S. M., Aubé J., and Bohn L. M. (2018) G protein signaling-biased agonism at the κ-opioid receptor is maintained in striatal neurons. Sci. Signal. 11, eaar4309 10.1126/scisignal.aar4309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Liu H., Kim H. R., Deepak R., Wang L., Chung K. Y., Fan H., Wei Z., and Zhang C. (2018) Orthosteric and allosteric action of the C5a receptor antagonists. Nat. Struct. Mol. Biol. 25, 472–481 10.1038/s41594-018-0067-z [DOI] [PubMed] [Google Scholar]
- 30. Robertson N., Rappas M., Doré A. S., Brown J., Bottegoni G., Koglin M., Cansfield J., Jazayeri A., Cooke R. M., and Marshall F. H. (2018) Structure of the complement C5a receptor bound to the extra-helical antagonist NDT9513727. Nature 553, 111–114 10.1038/nature25025 [DOI] [PubMed] [Google Scholar]
- 31. Seow V., Lim J., Cotterell A. J., Yau M. K., Xu W., Lohman R. J., Kok W. M., Stoermer M. J., Sweet M. J., Reid R. C., Suen J. Y., and Fairlie D. P. (2016) Receptor residence time trumps drug-likeness and oral bioavailability in determining efficacy of complement C5a antagonists. Sci. Rep. 6, 24575 10.1038/srep24575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Pandey S., Roy D., and Shukla A. K. (2019) Measuring surface expression and endocytosis of GPCRs using whole-cell ELISA. Methods Cell Biol. 149, 131–140 10.1016/bs.mcb.2018.09.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kumari P., Dwivedi H., Baidya M., and Shukla A. K. (2019) Measuring agonist-induced ERK MAP kinase phosphorylation for G-protein-coupled receptors. Methods Cell Biol. 149, 141–153 10.1016/bs.mcb.2018.09.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Croker D. E., Monk P. N., Halai R., Kaeslin G., Schofield Z., Wu M. C., Clark R. J., Blaskovich M. A., Morikis D., Floudas C. A., Cooper M. A., and Woodruff T. M. (2016) Discovery of functionally selective C5aR2 ligands: novel modulators of C5a signalling. Immunol. Cell Biol. 94, 787–795 10.1038/icb.2016.43 [DOI] [PubMed] [Google Scholar]