

Abstract
Natural product biosynthetic pathways are composed of enzymes that use powerful chemistry to assemble complex molecules. Small molecule neurotoxins are examples of natural products with intricate scaffolds which often have high affinities for their biological targets. The focus of this review is small molecule neurotoxins targeting voltage-gated sodium channels (VGSCs) and the state of knowledge on their associated biosynthetic pathways. There are three small molecule neurotoxin receptor sites on VGSCs associated with three different classes of molecules: guanidinium toxins, alkaloid toxins, and ladder polyethers. Each of these types of toxins have unique structural features which are assembled by biosynthetic enzymes and the extent of information known about these enzymes varies among each class. The biosynthetic enzymes involved in the formation of these toxins have the potential to become useful tools in the efficient synthesis of VGSC probes.
Keywords: biosynthesis, voltage-gated sodium channels, neurotoxins, biocatalysis, enzymes
Graphical Abstract
Terrific toxins: Living organisms produce highly potent small molecule neurotoxins as forms of self defense. A subset of these toxins target voltage-gated sodium channels, a potential target for non-opioid pain management in humans. The biosynthetic pathways of these channel-disrupting ligands are discussed in the context of biocatalytic applications

1. Introduction
Natural neurotoxins are the products of Nature’s most experienced chemists and pharmacologists. These structurally complex small molecules have been isolated from bacteria, plankton, amphibians, plants, and even birds and serve as incredibly potent weapons for self-defense and prey capture. Due to the necessity of rapidly disabling potential predators and subduing prey, natural neurotoxins have evolved to target membrane-bound ion channels.[1] One such biological target is the focus of this review, voltage-gated sodium channels (VGSCs), which are located in neurons and distributed throughout cardiac, skeletal, and muscle tissues.
VGSCs are transmembrane protein complexes which facilitate cellular communication and regulate membrane potentials in response to sodium ions.[2] Fundamental structural studies on bacterial VGSCs[3] and recent cryo-EM studies on insect and human VGSCs have revealed the architecture of the protein.[4] VGSCs are composed of a large α-subunit containing the ion conducting pore and smaller regulatory β-subunits. The α-subunit is comprised of four homologous domains with each domain consisting of six transmembrane segments (Figure 1). These four domains are connected by flexible loops and together assemble the ion conducting channel, which is selective for sodium ions. The passage of sodium ions into the excitable cell is permissible when the pore is in its “open” state and not permitted when the channel is in the “closed” conformational state. Abnormal regulation of VGSCs by mutations or ligands can result in extended depolarization by excessive flow of sodium ions into the cell or loss of current.[2] Such phenotypes can have significant physiological consequences.
Figure 1.

Diagram of voltage-gated sodium channel structure and binding sites of natural small molecule neurotoxins. Receptor site I is indicated in red and is targeted by the guanidinium toxins. Receptor site II, indicated in green, interacts with alkaloid toxins and receptor site V, indicated in blue, is targeted by the ladder polyethers.
Small molecule neurotoxins are known to interact with VGSCs at three distinct locations (Figure 1).[5] Receptor site I on the flexible loops connecting segments 5 and 6 of each domain is targeted by the guanidinium toxins: tetrodotoxin (1), chiriquitoxin (2), saxitoxin (3), gonyautoxins (e.g. gonyautoxin 3, 4), and zetekitoxin AB (5). These molecules have the effect of blocking the pore and preventing ion conductance, resulting in numbness, tingling, and paralysis.[6] Alkaloid toxins such as aconitine (6), batrachotoxin (7), grayanotoxin (8), and veratridine (9) are known to interact with receptor site II, located at the top of segment 6 of domains I and IV.[7] Molecules that interact with receptor site II bind to VGSCs while in the open state, resulting in prolonged activation and cell depolarization.[8] These alkaloid toxins have several modes of disrupting channel function, including reducing ion specificity and altering the kinetics of activation and inactivation, leading to overstimulation of the central nervous system.[9] Ladder polyether compounds, including brevetoxin (10) and ciguatoxin (11), bind at receptor site V, which bridges domain I segment 6 and domain IV segment 5 (Figure 1, shown in blue). These molecules primarily alter activation and inactivation kinetics of the channel.[9]
The highly potent nature of neurotoxins for VGSC targets has inspired numerous drug design campaigns to treat central nervous system disorders, pain, and muscle spasms.[10] Small molecule neurotoxins have the benefit of target specificity observed with protein and peptide neurotoxins coupled with the added advantage of an array of drug formulation options that are unsuitable for larger, more unstable molecules.[11] However, constructing and diversifying the intricate scaffolds of natural small molecule neurotoxins remains a significant challenge and has hindered full exploration of these molecules’ therapeutic potential. Understanding the biosynthetic pathways employed by organisms to assemble these structurally complex molecules can inform and inspire synthetic and biocatalytic approaches toward these small molecule neurotoxins and related compounds. Here, we present the known elements and proposals for the biosynthetic pathway that give rise to each small molecule VGSC ligand.
2. Receptor site I
2.1. Saxitoxins & gonyautoxins
Saxitoxins and their sulfate-containing derivatives, gonyautoxins, belong to a larger class of molecules collectively referred to as paralytic shellfish toxins (PSTs). Over 50 natural products have been isolated, and these molecules share a 5-5-6 tricyclic bisguanidinium ion-containing core and vary in oxygenation pattern and incorporation of sulfate, carbamate, and acetate groups (Figure 2).[12] A subset of these compounds have been evaluated for VGSC affinity. These studies have shown that the affinity is dependent on the decoration of the scaffold, where saxitoxin (3) is often used as a benchmark with a Kd of 4.3 nM in rat brain extracts.[12a, 13] Since the elucidation of its structure in 1975,[14] saxitoxin (3) has become a fundamental tool for the study of VGSCs where it is commonly used in a tritiated form to assay the relative affinity of compounds to bind at receptor site I.[15]
Figure 2.
Examples of saxitoxin and gonyautoxin analogs isolated from natural sources.
PSTs were first identified in the Alaskan butter clam, Saxidomus giganteus, which bioaccumulates these toxins produced by cyanobacteria and dinoflagellates.[16] PST biosynthesis could present a fascinating evolutionary convergence across phyla which is not completely understood.[17] Studies investigating the biosynthetic pathway toward PSTs have been conducted in both cyanobacteria and dinoflagellates.[18] The biosynthetic pathway has been studied in significantly more detail in cyanobacteria than the dinoflagellate pathway, although there is evidence that some biosynthetic steps may be conserved. Specifically in the timing of sulfation events that convert saxitoxin into gonyautoxins[19] and the formation of the first proposed biosynthetic intermediate, 12.[20]
Several hypotheses on the biosynthetic route toward PSTs have been reported.[21] Gene clusters encoding enzymes involved in saxitoxin biosynthesis were first discovered by Neilan and coworkers in Cylindrospermopsis raciborskii.[22] Since the initial discovery, clusters have also been identified in Microseira wollei,[23] Aphanizomenon sp. NH-5,[24] Aphanizomenon gracile,[25] Dolichospermum circinale,[24] Raphidiopsis brookii[26] and Syctonema crispum.[27] These gene clusters are relatively large, ranging in size from 25.9 kb to 54 kb, and consist of genes encoding the following core enzymes present in all clusters: sxtA, sxtB, sxtD, sxtE, sxtG, sxtH, sxtI, sxtM, sxtP, sxtQ, sxtR, sxtS, sxtT, and sxtU (Table 1). The complete biosynthetic pathway has not been elucidated; however, in vitro characterization studies and intermediate analysis have begun to define crucial steps.[27–28]
Table 1.
Enzymes encoded in cyanobacterial PST biosynthetic gene clusters. Those highlighted in gray are present in all gene clusters represented.
| Enzyme | M. wollei | C. raciborskii | D. circinale | Aphanizomenon sp. | R. brookii | S. crispum | Classification |
|---|---|---|---|---|---|---|---|
| SxtA | x | x | x | x | x | x | polyketide synthase-like |
| SxtB | x | x | x | x | x | x | cytidine deaminase |
| SxtC | x | x | x | x | x | unknown | |
| SxtD | x | x | x | x | x | x | non-heme diiron |
| SxtE | x | x | x | x | x | x | unknown |
| SxtF | x | x | “toxic compound extrusion protein” | ||||
| SxtG | x | x | x | x | x | x | amidinotransferase |
| SxtH | x | x | x | x | x | x | Rieske oxygenase |
| SxtI | truncated | x | x | x | x | x | carbamoyltransferase |
| SxtJ | x | x | x | x | x | unknown | |
| SxtK | x | x | x | x | x | unknown | |
| SxtL | x | x | x | x | x | unknown | |
| SxtM | 3 proteins | x | x | x | x | x | export protein |
| SxtN | 2 proteins | x | x | x | x | x | sulfotransferase |
| SxtO | x | x | x | x | adenylylsulfate kinase | ||
| SxtP | x | x | x | x | x | x | regulator/pilli formation |
| SxtQ | x | x | x | x | x | x | unknown |
| SxtR | x | x | x | x | x | x | unknown |
| SxtS | x | x | x | x | x | x | non-heme iron, 2-oxoglutarate dependent |
| SxtT | x | x | x | x | x | x | Rieske oxygenase |
| SxtU | x | x | x | x | x | x | NADP+-dependent dehydrogenase |
| SxtV | x | x | x | x | x | FAD-dependent succinate dehydrogenase | |
| SxtW | x | x | x | x | 2[4Fe-4S] ferredoxin | ||
| SxtX | x | x | x | x | methyltransferase | ||
| SxtY | x | phosphate uptake regulator | |||||
| SxtZ | x | histidine kinase | |||||
| GxtA | x | x | x | Rieske oxygenase | |||
| SxtACT | x | acyltransferase | |||||
| SxtSUL | x | x | sulfotransferase | ||||
Identification of putative biosynthetic intermediates present in dinoflagellate and cyanobacterial culture extracts has also contributed to hypotheses on the pathway that delivers PSTs. Using mass spectrometry analysis in combination with synthetically generated standards, Yotsu-Yamashita and coworkers have identified several new potential biosynthetic intermediates, including 12 (Figure 3), and the identification of 16 in dinoflagellate extracts (Figure 3B, inset).[20, 29] Characterization of SxtA supports 12 as a biosynthetic intermediate.[28a] However, in vitro reactions with sxt oxygenases suggest that decarbamoyl 11-α-hydroxysaxitoxin (16) is not on the pathway to saxitoxin (3) and that it is more likely a shunt product of this biosynthetic pathway.[28b] The first and final steps in the pathway are well supported thus far, but the critical oxidative cyclization of the linear 12 to form the tricyclic common to the PSTs is yet to be revealed.
Figure 3.

Characterized biosynthetic steps in paralytic shellfish toxin biosynthesis. A) Substrates required for the SxtA reaction. B) Characterized late-stage steps in paralytic shellfish toxin biosynthesis. Inset: In vitro evidence for order of hydroxylation events in saxitoxin (3) biosynthesis. C) Structure of zetekitoxin AB (5).
In vitro characterization of the enzymes proposed to be involved in PST biosynthesis has been explored for 5 enzymes to date: the polyketide-like synthase SxtA[28a], Rieske oxygenases SxtT, SxtH, and GxtA[28b], and the sulfotransferase SxtN (Figure 3A–B).[27] SxtA performs the first set of reactions in the saxitoxin biosynthetic pathway, assembling intermediate 12 from arginine, S-adenosylmethionine, and malonyl-CoA (Figure 3A).[28a] Fragments of the gene encoding this enzyme have also been identified in dinoflagellates and are often used as a method of detection for PST-producing capabilities in biological samples.[30] The Rieske oxygenase SxtH was found to install the C12 β-hydroxyl group prior tricycle formation while Rieske oxygenases SxtT and GxtA perform site- and stereoselective C–H hydroxylation reactions at the C12 α-position of β-saxitoxinol (13) to form saxitoxin (3) and the C11 β-position of saxitoxin (3) to form 11-β-hydroxysaxitoxin (14), respectively (Figure 3B).[28b] The timing of installation of the C12 β-hydroxyl group present in saxitoxin (3) has not been precisely defined. However, in vitro experiments have shown that SxtH mediates this hydroxylation prior to tricycle formation.[28b] Additionally, the native redox partners necessary for catalysis by the Rieske oxygenases in cyanobacteria have not been identified; the reported study uses a non-native redox partner, VanB, from the vanillin O-demethylase Rieske oxygenase system in Pseudomonas as a surrogate.[28b] Finally, the most recently characterized enzyme, SxtN, was found to catalyze the sulfation of saxitoxin (3) to form gonyautoxin 5 (15).[27] Enzymes involved in oxidative cyclization to form the tricycle remain uncharacterized.
2.2. Zetekitoxin
Two rare saxitoxin analogs, zetekitoxin AB (5) and zetekitoxin C (structure not known), were discovered in the Panamanian golden frog, Atelopus zeteki, in 1969 (Figure 3C).[31] LD50 values were determined in mouse bioassays for the two toxins, revealing that zetekitoxin AB (5) and zetekitoxin C are significantly more potent than saxitoxin (3) with LD50s of 1.5–3 μg/kg and 80 μg/kg, respectively.[32] Direct comparison to saxitoxin (3) in Xenopus oocytes expressing human heart, rat brain IIa, and rat skeletal muscle ion channels revealed an incredible increase in potency with zetekitoxin AB (5) binding with picomolar affinity compared to the nanomolar affinity observed with saxitoxin (3).[33] Unfortunately, the Panamanian golden frog, which is classified as a toad, became critically endangered and studies on the organism ceased.[33] Zetekitoxins have not been isolated from any other biological source. The small quantity of zetekitoxin AB (5), 300 μg, from a single isolation has fueled structural studies that culminated in the elucidation of the structure of zetekitoxin AB in 2004. From extracts of toad skins that had been stored at −20 °C from 1971 to 1986, the natural products was isolated and stored and stored at −80 °C until 1999 when the material was used for detailed NMR studies that ultimately gave rise to the structure.[33] These structural studies revealed that zetekitoxin AB (5) was an analog of saxitoxin (3), with an added 1,2-oxazolidine ring-fused lactam moiety unique to zetekitoxin AB (5). This unprecedented structural feature has not been observed in any other PST. However, synthetic studies generating advanced precursors to zetekitoxin AB (5) have revealed potential stereochemical discrepancies at the carbonyl carbon relative to the original 13C NMR characterization data.[34] Further efforts to synthesize significant quantities of zetekitoxin AB (5) will likely resolve this debate.
Although Panamanian golden frogs are functionally extinct in the wild, several zoos have developed exceptional programs in efforts to repopulate the species in captivity, including the San Diego, Detroit, and Smithsonian National Zoos. It is unknown if these frogs still produce zetekitoxins in captivity. Since saxitoxin (3) has only been found to be produced by microorganisms and not by complex higher organisms,[35] it is very likely that zetekitoxins are also derived from microbial sources on the skin. Evidence of cyanobacteria, the same class of organisms known to produce saxitoxin (3), have been identified on the skin of both wild and captive populations of A. zeteki.[36] If captive toads still produce zetekitoxins, conducting metagenomic analysis on the microbial communities on the toads’ skin using saxitoxin biosynthetic gene clusters as a guide could help identify the enzymes responsible for the unique modification to the scaffold. These enzymes could be used to complement existing synthetic efforts in accessing the zetekitoxin AB (5) scaffold for further VGSC binding studies.[34, 37]
2.3. Tetrodotoxin & chiriquitoxin
The name tetrodotoxin (1) is based on the animals it is famously isolated from, the Tetraodontidae family, including pufferfish, globefish, blowfish, and porcupine fish.[38] Tetrodotoxin (1) is known for its high toxicity, with an estimated human LD50 of 10.2 μg/kg.[39] Poisoning by tetrodotoxin (1) is well recognized as a risk in consuming Japanese fugu, the pufferfish delicacy that must be harvested at certain times of the year and carefully prepared by a skilled chef to avoid poisoning the recipient.[40] The structure of the molecule was determined in 1964, which revealed a remarkable 2,4-dioxa-adamantane structure appended to a cyclic guanidine which is decorated with hydroxyl groups (Figure 4, 1).[41] Since its initial isolation from pufferfish, tetrodotoxin (1) and derivatives have also been isolated from other animals including octopuses[42] and several amphibians including populations of frogs and newts.[39] A selection of observed tetrodotoxin (1) derivatives, including chiriquitoxin (2), an analog observed in frogs,[43] are shown in Figure 4. Some of these toxins are epimers, such as 4-epi-tetrodotoxin (17) and 6-epi-tetrodotoxin (19), and are typically not isolated from the same organisms as tetrodotoxin (1).[39] This evidence of stereodivergence in isolations among different organisms implies the necessity of selective oxidation in the biosynthetic pathway, as is mirrored by enzymes the saxitoxin (3) biosynthetic pathway.[28b] The presence of these toxins across classes of organisms as well as their unique toxin profiles has drawn attention to the biosynthetic pathway, which is yet to be solved.
Figure 4.

Structures of tetrodotoxin (1) and chiriquitoxin (2) and commonly observed derivatives 4-epi-tetrodotoxin (17), 11-deoxytetrodotoxin (18), and 6-epi-tetrodotoxin (19).
A leading hypothesis on the biosynthesis of tetrodotoxin (1) and related compounds proposes that microorganisms are the true producers, synthesizing these compounds as a part of their symbiotic relationship with eukaryotic partners.[38] The most convincing evidence supporting this hypothesis is the variability of toxicity from tetrodotoxin (1)-producing organisms held in captivity, implying that some unspecified external stimulant is necessary to elicit toxin production.[44] Studies on the frog Atelopus varius[45] and the fire-bellied newt Cynops pyrrhogaster[46] indicated that toxin levels were non-detectable in captive-raised organisms, whereas studies on the rough-skinned newt, Taricha granulosa,[47] have demonstrated an increase in toxin production while held in captivity. This ambiguity and irreproducibility has motivated efforts to pursue the isolation of tetrodotoxin (1)-producing bacteria from the skin and tissues of host organisms.[39, 48] While microorganisms from producing organisms have been successfully cultured on many occasions, the common result has been inconsistent production of tetrodotoxin (1) or related metabolites in isolated culture.[49] Most notably, studies by Noguchi have unequivocally demonstrated bacterial origins for tetrodotoxin (1) production in the intestines pufferfish and xanthid crab by isolating gut microbes in pure culture and assessing them for production of tetrodotoxin (1).[50] Despite reports of many tetrodotoxin-producing bacteria, a gene cluster linked to tetrodotoxin (1) biosynthesis has not been identified. The involvement of a non-ribosomal peptide synthetase (NRPS) specific to arginine is hypothesized initiate tetrodotoxin biosynthesis, and candidate genes have been identified, but no specific activity has been confirmed.[51] Interestingly, one study in Aeromonas sp. suggested the involvement of a plasmid in the production of tetrodotoxin (1), which could account for the inconsistencies observed in tetrodotoxin (1)-containing organisms held in captivity and the distribution of the toxin among various species, as bacterial plasmids are easily transferrable.[49]
Several biosynthetic hypotheses for the assembly of tetrodotoxin (1) have been proposed. Shimizu and coworkers initially proposed two potential building blocks to combine with arginine, an apiose-type C5 sugar or isopentenyl pyrophosphate (Figure 5A).[52] In evaluating this biosynthetic hypothesis, Yotsu-Yamashita and coworkers proposed that a C10 monoterpene starter unit, geranyl guanidine (20) may be a biosynthetic precursor to the intermediate isolated from newts, 4,9-anhydro-10-hemiketal-5-deoxytetrodotoxin (21).[53] Intermediate 21 is just one oxidation state lower than 4,9-anhydrotetrodotoxin (22), which is in equilibrium with tetrodotoxin (1) in solution (Figure 5B). However, this intermediate has only been observed in samples from terrestrial animals and not the marine tetrodotoxin producers, bringing into question its validity as a biosynthetic intermediate that is universal to all producers. Conversely, Yotsu-Yamashita and coworkers also identified potential biosynthetic intermediates that were unique to marine producers.[54] Seven novel spirocyclic intermediates were extracted from pufferfish, five of which (23-27) were used to bolster a revised biosynthetic proposal requiring several additional oxidation events to achieve the complete structure of tetrodotoxin (1, Figure 5C). Intermediates 28 and 29 are predicted structures that have not been observed in the pufferfish extracts.
Figure 5.

Prominent biosynthetic proposals for tetrodotoxin (1) biosynthesis. A) General biosynthetic proposal by Shimizu for all producing organisms, B) Yotsu-Yamashita proposal for terrestrial tetrodotoxin (1) producers, C) Yotsu-Yamashita proposal for marine tetrodotoxin (1) producers.
Additional work is necessary to identify and link a gene cluster to tetrodotoxin biosynthesis. The elucidation of the biosynthetic pathway will provide new avenues for production of these compounds, relieving the strain on amphibian and marine sources as the sole sources for tetrodotoxin (1) and will complement synthetic techniques.
3. Receptor site II
3.1. Grayanotoxin
Grayanotoxins are produced by honey-producing plants like Rhododendron sp. and Leucothoe grayana.[55] The most abundant analogs isolated are grayanotoxin 1 (8) and grayanotoxin 3 (32).[56] Grayanotoxins have also been referred to as andromedotoxin, rhodotoxin, and asebotoxin in early studies and are often referenced as grayanane diterpenoids and grayanoids in current literature.[57] Grayanotoxins in Rhododendron honey are the primary agent of mad honey poisoning, a disease characterized by dizziness, hypotension, and bradycardia.[58] While the effects of this poisoning are serious, it is rarely fatal to humans.[59] The grayanotoxins share a characteristic 5–7-6–5 ring system bearing hydroxyl and acetate groups which modulate the compounds’ biological activity.[60]
Preliminary exploration of the grayanotoxin biosynthetic pathway has been carried out in Leucothoe grayana.[61] In 1981, Ueda and coworkers reported the incorporation of 14C-labeled mevalonic acid (31) into grayanotoxin 3 (32) when fed at the stems of the plant, consistent with a terpene-derived biosynthetic pathway (Figure 6).[61] The group also attempted 14C-labeling studies with (–)-kaurene (33), a molecule containing a 6–6-6–5 ring core, and observed low levels of incorporation (~0.0005–0.0007%) into grayanotoxin 3 (32). It is rationalized that this could be due to low uptake of (–)-kaurene (33) by the plant; however, it is still unclear whether (–)-kaurene (33) is a true biosynthetic precursor to grayanotoxin 3 (32) as no further studies have been reported. Genes responsible for the production of grayanotoxins have not been identified, likely due to the difficulty of identifying relevant genes in plant genomes. Further work is warranted, as understanding the biosynthetic machinery necessary to assemble grayanotoxins will likely highlight potential biosynthetic genes for other important alkaloids in other plant sources.
Figure 6.

Possible biosynthetic precursors obtained by labeling studies.
3.2. Aconitine
The roots and tubers of Aconitum plants are commonly used as traditional medicine in China to treat fever, inflammation, diarrhea, asthma, and certain tumors.[62] The desired pharmacological properties of Aconitum species are due to a collection of C20, C19, and C18 diterpene alkaloids.[62] The C19 diterpene aconitine (6) was the first alkaloid identified from Aconitum plants in 1833 by Geiger.[63] The aconitine core scaffold is conserved over 70 structurally unique bioactive alkaloids.[64] Despite its widespread use in China as a topical and oral analgesic, Acontium extracts containing the aconitine alkaloids have the potential to be fatally toxic.[62]
Although the specific genes for the initial processes have not been identified in Aconitum, it is understood that the biosynthesis of diterpene alkaloids typically begins via an established route to form geranylgeranyl pyrophosphate (GGPP, 34).[65] GGPP (34) is cyclized by ent-copalyl diphosphate synthase to form ent-copalyl diphosphate (35) which undergoes a rearrangement to form (–)-kaurene (33), similar to the proposed pathway for grayanotoxins.[66] Candidate genes for enzymes responsible for the diversification of the aconitine (6) scaffold have been identified through transcriptome analysis of A. carmichaelii flowers, buds, leaves, and roots, revealing the possible involvement of cytochrome P450s, methyltransferases, and acyltransferases.[66–67] Zhao and coworkers proposed a putative biosynthetic pathway based on the proposed function of each gene product assigned through BLAST analysis.[66] It is proposed that (–)-kaurene (33) is further modified by cyclases capable of a simultaneous ring expansion and contraction form the putative core intermediate (36).[66] Finally, intermediate 36 could be modified by oxygenases and acyltransferases to form aconitine (6). Knowledge of putative genes invites additional feeding studies and functional characterization of the corresponding enzymes.
3.3. Veratridine
The alkaloid veratridine (9) is isolated from the rhizomes of plants of the family Liliaceae, specifically Veratrum album, known as white hellebore.[68] Veratridine (9) is one of several alkaloids produced by V. album which confer toxicity to the medicinal plant.[69] The mode of action of veratridine (9) is the same as grayanotoxins (8, 32) and aconitine (6) where binding to receptor site II results in VGSC opening, allowing an influx of sodium into the cell.[9] The biosynthesis of veratridine has not been interrogated through feeding studies or gene analyses and no biosynthetic proposals have been reported. However, its alkaloid architecture suggests that it likely stems from similar linear terpene starting units as proposed for grayanotoxins and aconitine (6).
3.4. . Batrachotoxin
Batrachotoxin (7) is the most potent non-protein natural product neurotoxin discovered to date.[70] Its remarkable affinity for VGSCs led to its development as a tool to probe VGSCs in excitable membranes, as demonstrated by the Narahashi’s seminal study in giant squid axons.[71] The biosynthesis of this potent molecule is particularly interesting due to its variety of sources of isolation. In addition to being isolated from Columbian poison dart frogs,[70b] batrachotoxin (7) has also been isolated from two genera of passerine birds, Pitohui and Ifrita, in Papua New Guinea.[72] Interestingly, the most abundant analogs (Figure 8) are observed in both classes of organisms, suggesting a possible common biosynthetic source.[73]
Figure 8.

Structures of common batrachotoxin (7) products in poison dart frogs and birds.
Diet has been proposed as the source of batrachotoxin (7) found in amphibians and birds on different continents.[74] In 2004, Dumbacher and coworkers discovered batrachotoxin (7) in Choresine beetles in New Guinea, a likely food source for the Pitohui and Ifrita birds.[75] These same beetles are also a likely supplier of toxin for the poison dart frogs, because frogs raised in captivity do not carry batrachotoxin (7). However, the Choresine beetles are not suspected to possess the biosynthetic machinery necessary to form the batrachotoxin (7) alkaloid structure.[75] Instead, Dumbacher proposes the beetles may construct alkaloids using plant phytosterols from their own diet or using a microbial symbiont. Interestingly, the poison dart frogs which carry batrachotoxin (7) have batrachotoxin-resistant VGSCs and are therefore able to consume the toxic beetles without side effects.[76] It is unknown if Pitohui and Ifrita also have this adaptation. The mystery of batrachotoxin (7) biosynthesis is still yet to be solved as no biosynthetic intermediates have been identified. In terms of identifying new biosynthetic intermediates toward batrachotoxin (7), feeding studies could be beneficial. The possibilities of symbionts and plant sources of batrachotoxin (7) have not been ruled out and present considerable work for future studies.
4. Receptor site V
4.1. Brevetoxins & ciguatoxins
Brevetoxins (e.g. brevetoxin 1, 10) and ciguatoxins (e.g. ciguatoxin 1, 11) are referred to as ladder polyether toxins for their unique structures and are produced by the dinoflagellates Karenia brevis and Gambierdiscus species, respectively.[77] Ciguatoxins bioaccumulated by fish cause ciguatera fish poisoning, a disease characterized by a myriad of neurological and gastrointestinal symptoms.[78] Although ciguatera is rarely fatal, its effects can become chronic and persist for over a year. By contrast, brevetoxins cause similar symptoms, but the effects are rarely chronic or fatal.[79]
The biosynthesis of the brevetoxins and ciguatoxins are anticipated to be similar due to their homologous structures. Since both are produced by dinoflagellates, the genes associated with biosynthesis have not been completely identified due to the complexity of dinoflagellate genomes. However, biosynthetic routes have been proposed for ladder polyethers following the general scheme outlined in Figure 9 for ciguatoxin 1 (11).[80] Although outside of the scope of this review, the biosynthetic enzymes required for the assembly of polyether ionophore antibiotics with similar structural features to brevetoxins and ciguatoxins are known and could be used to guide biosynthetic hypotheses.[81] Murray and coworkers revealed the most comprehensive collection of polyketide synthase genes in dinoflagellates so far from the genomes of G. polynesiensis and G. excentricus, some of which could be associated with the production of ciguatoxins.[80] Concurrently, Murray and coworkers published a transcriptome from K. brevis which revealed several single and multidomain polyketide synthases potentially involved in the production of brevetoxins.[82] This cutting edge work by Murray provides new clues toward understanding the enzymes involved in assembling the complex polyether ladder scaffolds.
Figure 9.

Proposed biosynthesis of ciguatoxin 1 (11).
5. Conclusions
The natural neurotoxins discussed herein are remarkable molecules that have demonstrated significant utility to the study of VGSCs, whether they are used to probe structural features, modulate cell potential on demand, or identify new drugs. Understanding the biosynthetic pathways of these complex small molecules has the potential to provide a wealth of information for synthetic and biocatalytic efforts to avoid exploiting natural sources, such as toxin producing amphibians, insects, and plants. Further work elucidating biosynthetic pathways of small molecule neurotoxins is warranted to develop innovative new methods for the production of valuable probes and drug targets for VGSCs.
Figure 7.
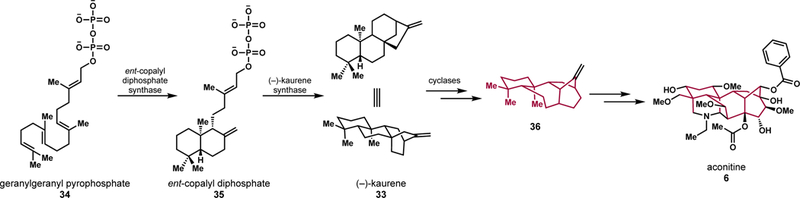
Proposed biosynthesis of aconitine (6) with geranylgeranyl pyrophosphate (34) as a precursor.
Acknowledgements
A.R.H.N. acknowledges University of Michigan Life Sciences Institute and the National Institutes of Health R35 GM124880 for funding. We thank Nicholas Denomme for helpful discussions.
Biography
 Alison Narayan earned her Ph.D. at the University of California, Berkeley (USA), where she developed novel methods for the synthesis of nitrogen heterocycles and the total synthesis of complex natural products in the group of Prof. Richmond Sarpong. As a Life Sciences Research Foundation Postdoctoral Fellow, Alison studied natural product biosynthesis and biocatalysis with Prof. David Sherman. In 32015, Alison began her independent career as an assistant professor in the Department of Chemistry and the Life Sciences Institute at the University of Michigan and focuses on elucidating the function of enzymes involved in natural product biosynthesis and the development of biocatalytic synthetic methods
Alison Narayan earned her Ph.D. at the University of California, Berkeley (USA), where she developed novel methods for the synthesis of nitrogen heterocycles and the total synthesis of complex natural products in the group of Prof. Richmond Sarpong. As a Life Sciences Research Foundation Postdoctoral Fellow, Alison studied natural product biosynthesis and biocatalysis with Prof. David Sherman. In 32015, Alison began her independent career as an assistant professor in the Department of Chemistry and the Life Sciences Institute at the University of Michigan and focuses on elucidating the function of enzymes involved in natural product biosynthesis and the development of biocatalytic synthetic methods
 April Lukowski graduated from Saginaw Valley State University (USA) in 42015 with a B.S. in biochemistry with honors where she studied plant enzymology and analytical biochemistry. She then entered the Program in Chemical Biology at the University of Michigan and joined the Narayan research group as a PhD student in 2016 where she currently studies enzymes involved in paralytic shellfish toxin biosynthesis
April Lukowski graduated from Saginaw Valley State University (USA) in 42015 with a B.S. in biochemistry with honors where she studied plant enzymology and analytical biochemistry. She then entered the Program in Chemical Biology at the University of Michigan and joined the Narayan research group as a PhD student in 2016 where she currently studies enzymes involved in paralytic shellfish toxin biosynthesis
References
- [1].Adams ME, Olivera BM, Trends Neurosci. 1994, 17, 151–155. [DOI] [PubMed] [Google Scholar]
- [2].Ahern CA, Payandeh J, Bosmans F, Chanda B, J Gen Physiol 2016, 147, 1–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Payandeh J, Scheuer T, Zheng N, Catterall WA, Nature 2011, 475, 353–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].a Shen HZ, Li ZQ, Jiang Y, Pan XJ, Wu JP, Cristofori-Armstrong B, Smith JJ, Chin YKY, Lei JL, Zhou Q, King GF, Yan N, Science 2018, 362, 306-+; [DOI] [PubMed] [Google Scholar]; Pan X, Li Z, Zhou Q, Shen H, Wu K, Huang X, Chen J, Zhang J, Zhu X, Lei J, Xiong W, Gong H, Xiao B, Yan N, Science 2018, 362, eaau2486. [DOI] [PubMed] [Google Scholar]
- [5].Anger T, Madge DJ, Mulla M, Riddall D, J. Med. Chem. 2001, 44, 115–137. [DOI] [PubMed] [Google Scholar]
- [6].Suarez-Isla BA, in Marine and Freshwater Toxins (Eds.: Haddad V, Tubaro A, Kim E, Kem WR, Gopalakrishnakone P), Springer, Dordrecht, 2016, pp. 23–41. [Google Scholar]
- [7].Catterall WA, Cestele S, Yarov-Yarovoy V, Yu FH, Konoki K, Scheuer T, Toxicon 2007, 49, 124–141. [DOI] [PubMed] [Google Scholar]
- [8].Khodorov BI, Neurophysiology 1985, 17, 293–304. [PubMed] [Google Scholar]
- [9].Cestele S, Catterall WA, Biochimie 2000, 82, 883–892. [DOI] [PubMed] [Google Scholar]
- [10].a Villa FA, Gerwick L, Immunopharmacol. Immunotoxicol. 2010, 32, 228–237; [DOI] [PubMed] [Google Scholar]; b Epstein-Barash H, Shichor I, Kwon AH, Hall S, Lawlor MW, Langer R, Kohane DS, Proc. Natl. Acad. Sci. U. S. A. 2009, 106, 7125–7130; [DOI] [PMC free article] [PubMed] [Google Scholar]; c Rodriguez-Navarro AJ, Lagos M, Figueroa C, Garcia C, Recabal P, Silva P, Iglesias V, Lagos N, Neurotox. Res. 2009, 16, 408–415. [DOI] [PubMed] [Google Scholar]
- [11].a Mueller C, Altenburger U, Mohl S, J. Pharm. Pharmacol. 2018, 70, 666–674; [DOI] [PubMed] [Google Scholar]; b Frokjaer S, Otzen DE, Nat. Rev. Drug Discov. 2005, 4, 298–306. [DOI] [PubMed] [Google Scholar]
- [12].a Llewellyn LE, Nat. Prod. Rep. 2006, 23, 200–222; [DOI] [PubMed] [Google Scholar]; b Thottumkara AP, Parsons WH, Du Bois J, Angew. Chem. Int. Ed. 2014, 53, 5760–5784. [DOI] [PubMed] [Google Scholar]
- [13].a Walker JR, Novick PA, Parsons WH, McGregor M, Zablocki J, Pande VS, Du Bois J, Proc Natl Acad Sci U S A 2012, 109, 18102–18107; [DOI] [PMC free article] [PubMed] [Google Scholar]; b Koehn FE, Ghazarossian VE, Schantz EJ, Schnoes HK, Strong FM, Bioorg. Chem. 1981, 10, 412–428; [Google Scholar]; c Usup G, Leaw C, Cheah M, Amad A, Ng B, Toxicon 2004, 44, 37–43; [DOI] [PubMed] [Google Scholar]; d Moczydlowski EHS; Garber SS; Strichartz GS; Miller C, J. Gen. Physiol. 1984, 84, 687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Schantz EJ, Ghazarossian VE, Schnoes HK, Strong FM, Springer JP, Pezzanite JO, Clardy J, Journal of the American Chemical Society 1975, 97, 1238–1239. [DOI] [PubMed] [Google Scholar]
- [15].Robertson A, Negri AP, Burnell JN, Llewellyn LE, Analytical Biochemistry 2006, 356, 66–75. [DOI] [PubMed] [Google Scholar]
- [16].Schantz EJ, Magnusson HW, Journal of Protozoology 1964, 11, 239-&. [DOI] [PubMed] [Google Scholar]
- [17].Hackett JD, Wisecaver JH, Brosnahan ML, Kulis DM, Anderson DM, Bhattacharya D, Plumley FG, Erdner DL, Mol. Biol. Evol. 2013, 30, 70–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Wang DZ, Zhang SF, Zhang Y, Lin L, J. Proteomics 2016, 135, 132–140. [DOI] [PubMed] [Google Scholar]
- [19].a Sako Y, Yoshida T, Uchida A, Arakawa O, Noguchi T, Ishida Y, J. Phycol. 2001, 37, 1044–1051; [Google Scholar]; b Yoshida T, Sako Y, Uchida A, Kakutani T, Arakawa O, Noguchi T, Ishida Y, Fish. Sci. 2002, 68, 634–642. [Google Scholar]
- [20].a Cho Y, Tsuchiya S, Yoshioka R, Omura T, Konoki K, Oshima Y, Yotsu-Yamashita M, Harmful Algae 2015, 49, 58–67; [DOI] [PubMed] [Google Scholar]; b Tsuchiya S, Cho Y, Konoki K, Nagasawa K, Oshima Y, Yotsu-Yamashita M, Org. Biomol. Chem. 2014, 12, 3016–3020. [DOI] [PubMed] [Google Scholar]
- [21].a Shimizu Y, Norte M, Hori A, Genenah A, Kobayashi M, J Am Chem Soc 1984, 106, 6433–6434; [Google Scholar]; b Kellmann R, Neilan BA, J. Phycol. 2007, 43, 497–508. [Google Scholar]
- [22].Kellmann R, Mihali TK, Jeon YJ, Pickford R, Pomati F, Neilan BA, Applied and Environmental Microbiology 2008, 74, 4044–4053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Mihali TK, Carmichael WW, Neilan BA, PLoS One 2011, 6, e14657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Mihali TK, Kellmann R, Neilan BA, BMC Biochem 2009, 10, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Casero MC, Ballot A, Agha R, Quesada A, Cires S, Harmful Algae 2014, 37, 28–37. [Google Scholar]
- [26].Soto-Liebe K, Murillo AA, Krock B, Stucken K, Fuentes-Valdes JJ, Trefault N, Cembella A, Vasquez M, Toxicon 2010, 56, 1350–1361. [DOI] [PubMed] [Google Scholar]
- [27].Cullen A, D’Agostino PM, Mazmouz R, Pickford R, Wood S, Neilan BA, ACS Chem. Biol. 2018, ASAPS. [DOI] [PubMed] [Google Scholar]
- [28].a Chun SW, Hinze ME, Skiba MA, Narayan ARH, Journal of the American Chemical Society 2018, 140, 2430–2433; [DOI] [PMC free article] [PubMed] [Google Scholar]; b Lukowski AL, Ellinwood DC, Hinze ME, DeLuca RJ, Du Bois J, Hall S, Narayan ARH, Journal of the American Chemical Society 2018, 140, 11863–11869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].a Tsuchiya S, Cho Y, Konoki K, Nagasawa K, Oshima Y, Yotsu-Yamashita M, Chem.-Eur. J. 2015, 21, 7835–7840; [DOI] [PubMed] [Google Scholar]; b Tsuchiya S, Cho Y, Yoshioka R, Konoki K, Nagasawa K, Oshima Y, Yotsu-Yamashita M, Angew. Chem.-Int. Edit. 2017, 56, 5327–5331. [DOI] [PubMed] [Google Scholar]
- [30].a Stuken A, Orr RJS, Kellmann R, Murray SA, Neilan BA, Jakobsen KS, Plos One 2011, 6, 12; [DOI] [PMC free article] [PubMed] [Google Scholar]; b Wiese M, Murray SA, Alvin A, Neilan BA, Toxicon 2014, 92, 102–112; [DOI] [PubMed] [Google Scholar]; c Penna A, Perini F, Dell’Aversano C, Capellacci S, Tartaglione L, Giacobbe MG, Casabianca S, Fraga S, Ciminiello P, Scardi M, Environ. Sci. Technol. 2015, 49, 14230–14238. [DOI] [PubMed] [Google Scholar]
- [31].Shindelman J, Mosher HS, Fuhrman FA, Toxicon 1969, 7, 315-+. [DOI] [PubMed] [Google Scholar]
- [32].Brown GB, Kim YH, Küntzel H, Mosher HS, Fuhrman GJ, Furhrman FA, Toxicon 1977, 15, 115–128. [DOI] [PubMed] [Google Scholar]
- [33].Yotsu-Yamashita M, Kim YH, Dudley S, Choudhary G, Pfahnl A, Oshima Y, Daly JW, Proc Natl Acad Sci 2004, 101, 4346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].a Nishikawa T, Wang C, Akimoto T, Koshino H, Nagasawa K, Asian J. Org. Chem. 2014, 3, 1308–1311; [Google Scholar]; b Nishikawa T, Urabe D, Isobe M, Heterocycles 2009, 79, 379–385. [Google Scholar]
- [35].Duran-Riveroll LM, Cembella AD, Marine Drugs 2017, 15, 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Becker MH, Richards-Zawacki CL, Gratwicke B, Belden LK, Biol. Conserv. 2014, 176, 199–206. [Google Scholar]
- [37].a Pearson AD, Williams RM, Tetrahedron 2014, 70, 7942–7949; [DOI] [PMC free article] [PubMed] [Google Scholar]; b Paladugu SR, Looper RE, Tetrahedron Letters 2015, 56, 6332–6334; [DOI] [PMC free article] [PubMed] [Google Scholar]; c Tang D, Merit J, Du Bois J, Abstr. Pap. Am. Chem. Soc. 2018, 255, 1. [Google Scholar]
- [38].Daly JW, Journal of Natural Products 2004, 67, 1211–1215. [DOI] [PubMed] [Google Scholar]
- [39].Hanifin CT, Marine Drugs 2010, 8, 577–593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Panao I, Carrascosa C, Jaber JR, Raposo A, Food Rev. Int. 2016, 32, 305–322. [Google Scholar]
- [41].a Tsuda K, Amakasu O, Ikuma S, Kawamura M, Tamura C, Tachikawa R, Sakai K, Chem. Pharm. Bull. 1964, 12, 1357-+; [DOI] [PubMed] [Google Scholar]; b Woodward RB, Gougouta Jz, Journal of the American Chemical Society 1964, 86, 5030-&. [Google Scholar]
- [42].Sheumack DD, Howden MEH, Spence I, Quinn RJ, Science 1978, 199, 188–189. [DOI] [PubMed] [Google Scholar]
- [43].Yotsui-Yamashita M, Tateki E, Toxicon 2010, 55, 153–156. [DOI] [PubMed] [Google Scholar]
- [44].Roszer T, Jozsa T, Szentmiklosi AJ, Banfalvi G, Cell Tissue Res. 2009, 336, 325–335. [DOI] [PubMed] [Google Scholar]
- [45].Daly JW, Padgett WL, Saunders RL, Cover JF, Toxicon 1997, 35, 705–709. [DOI] [PubMed] [Google Scholar]
- [46].Kudo Y, Chiba C, Konoki K, Cho Y, Yotsu-Yamashita M, Toxicon 2015, 101, 101–105. [DOI] [PubMed] [Google Scholar]
- [47].a Hanifin CT, Brodie ED, Toxicon 2002, 40, 1149–1153; [DOI] [PubMed] [Google Scholar]; b Gall BG, Stokes AN, French SS, Brodie ED III, Brodie ED Jr., Toxicon 2012, 60, 1057–1062. [DOI] [PubMed] [Google Scholar]
- [48].Chau R, Kalaitzis JA, Wood SA, Neilan BA, Marine Drugs 2013, 11, 2695–2712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Liu J, Wei F, Lu Y, Ma TL, Zhao J, Gong XL, Bao BL, Toxicon 2015, 101, 27–34. [DOI] [PubMed] [Google Scholar]
- [50].a Noguchi T, Jeon JK, Arakawa O, Sugita H, Deguchi Y, Shida Y, Hashimoto K, Journal of Biochemistry 1986, 99, 311–314; [DOI] [PubMed] [Google Scholar]; b Noguchi T, Arakawa O, Takatani T, Comp. Biochem. Physiol. Part D-Genomics Proteomics 2006, 1, 145–152. [DOI] [PubMed] [Google Scholar]
- [51].a Pratheepa V, Alex A, Silva M, Vasconcelos V, Toxicon 2016, 119, 186–193; [DOI] [PubMed] [Google Scholar]; b Leao JM, Lozano-Leon A, Giraldez J, Vilarino O, Gago-Martinez A, Marine Drugs 2018, 16, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Kotaki YSY, J Am Chem Soc 1993, 115, 827. [Google Scholar]
- [53].a Kudo Y, Yamashita Y, Mebs D, Cho Y, Konoki K, Yasumoto T, Yotsu-Yamashita M, Angew. Chem.-Int. Edit. 2014, 53, 14546–14549; [DOI] [PubMed] [Google Scholar]; b Kudo Y, Yasumoto T, Mebs D, Cho Y, Konoki K, Yotsu-Yamashita M, Angew. Chem.-Int. Edit. 2016, 55, 8728–8731. [DOI] [PubMed] [Google Scholar]
- [54].Ueyama N, Sugimoto K, Kudo Y, Onodera K, Cho Y, Konoki K, Nishikawa T, Yotsu-Yamashita M, Chem.-Eur. J. 2018, 24, 7250–7258. [DOI] [PubMed] [Google Scholar]
- [55].a Kubo O, Archiv Fur Experimentelle Pathologie Und Pharmakologie 1912, 67, 111–117; [Google Scholar]; b Li Y, Liu YB, Yu SS, Phytochem. Rev. 2013, 12, 305–325. [Google Scholar]
- [56].a Kakisawa H, Kurono M, Takahashi S, Hirata Y, Tetrahedron Letters 1961, 59–67; [Google Scholar]; b Kumazawa Z, Iriye R, Tetrahedron Letters 1970, 927-&; [DOI] [PubMed] [Google Scholar]; c Zhang MK, Xie YY, Zhan GQ, Lei L, Shu PH, Chen YL, Xue YB, Luo ZW, Wan Q, Yao GM, Zhang YH, Phytochemistry 2015, 117, 107–115. [DOI] [PubMed] [Google Scholar]
- [57].a Kohanawa M, Jpn. J. Pharmacol. 1956, 6, 46–56; [DOI] [PubMed] [Google Scholar]; b Li CH, Yan XT, Zhang AL, Gao JM, J. Agric. Food Chem. 2017, 65, 9934–9949. [DOI] [PubMed] [Google Scholar]
- [58].Gunduz A, Turedi S, Uzun H, Topbas M, Am. J. Emerg. Med. 2006, 24, 595–598. [DOI] [PubMed] [Google Scholar]
- [59].Koca I, Koca AF, Food Chem. Toxicol. 2007, 45, 1315–1318. [DOI] [PubMed] [Google Scholar]
- [60].Tsuji K, Kawanishi T, Handa S, Kamano H, Iwasa J, Seyama I, J. Pharmacol. Exp. Ther. 1991, 257, 788–794. [PubMed] [Google Scholar]
- [61].Masutani T, Hamada M, Kawano E, Iwasa J, Kumazawa Z, Ueda H, Agricultural and Biological Chemistry 1981, 45, 1281–1282. [Google Scholar]
- [62].Nyirimigabo E, Xu Y, Li Y, Wang Y, Agyemang K, Zhang Y, J Pharm Pharmacol 2015, 67, 1–19. [DOI] [PubMed] [Google Scholar]
- [63].Glasby J, Encyclopedia of the Alkaloids, Plenum Press, New York, London, 1975. [Google Scholar]
- [64].Zhou G, Tang L, Zhou X, Wang T, Kou Z, Wang Z, J. Ethnopharmacol. 2015, 160, 173–193. [DOI] [PubMed] [Google Scholar]
- [65].Vranová E, Coman D, Gruissem W, Molecular Plant 2012, 5, 318–333. [DOI] [PubMed] [Google Scholar]
- [66].Zhao DK, Shen Y, Shi YN, Shi XQ, Qiao Q, Zi SH, Zhao EQ, Yu DQ, Kennelly EJ, Phytochemistry 2018, 152, 113–124. [DOI] [PubMed] [Google Scholar]
- [67].Rai M, Rai A, Kawano N, Yoshimatsu K, Takahashi H, Suzuki H, Kawahara N, Saito K, Yamazaki M, Molecules 2017, 22, 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Attaurrahman, Ali RA, Gilani A, Choudhary MI, Aftab K, Sener B, Turkoz S, Planta Med. 1993, 59, 569–571. [DOI] [PubMed] [Google Scholar]
- [69].Alamgir ANM, in Therapeutic Use of Medicinal Plants and Their Extracts: Vol 1: Pharmacognosy, Vol. 73, Birkhauser Verlag Ag, Basel, 2017, pp. 61–104. [Google Scholar]
- [70].a Daly JW, Witkop B, Bommer P, Biemann K, Journal of the American Chemical Society 1965, 87, 124-&; [DOI] [PubMed] [Google Scholar]; b Tokuyama T, Daly J, Witkop B, Journal of the American Chemical Society 1969, 91, 3931-+. [DOI] [PubMed] [Google Scholar]
- [71].a Khodorov BI, Hoppe-Seylers Zeitschrift Fur Physiologische Chemie 1983, 364, 614–615; [Google Scholar]; b Narahashi T, Albuquerque EX, Deguchi T, Journal of General Physiology 1971, 58, 54-+; [DOI] [PMC free article] [PubMed] [Google Scholar]; c Narahashi T, Deguchi T, Albuquerque EX, Nature-New Biology 1971, 229, 221-+. [DOI] [PubMed] [Google Scholar]
- [72].Dumbacher JP, Spande TF, Daly JW, Proc. Natl. Acad. Sci. U. S. A. 2000, 97, 12970–12975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Daly JW, Spande TF, Garraffo HM, Journal of Natural Products 2005, 68, 1556–1575. [DOI] [PubMed] [Google Scholar]
- [74].Saporito RA, Spande TF, Garraffo HM, Donnelly MA, Heterocycles 2009, 79, 277–297. [Google Scholar]
- [75].Dumbacher JP, Wako A, Derrickson SR, Samuelson A, Spande TF, Daly JW, Proc. Natl. Acad. Sci. U. S. A. 2004, 101, 15857–15860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Daly JW, Myers CW, Warnick JE, Albuquerque EX, Science 1980, 208, 1383–1385. [DOI] [PubMed] [Google Scholar]
- [77].a Snyder RV, Guerrero MA, Sinigalliano CD, Winshell J, Perez R, Lopez JV, Rein KS, Phytochemistry 2005, 66, 1767–1780; [DOI] [PMC free article] [PubMed] [Google Scholar]; b Bhakuni DS, J. Sci. Ind. Res. 1995, 54, 702–716. [Google Scholar]
- [78].Morabito S, Silvestro S, Faggio C, Nat. Prod. Res. 2018, 32, 621–631. [DOI] [PubMed] [Google Scholar]
- [79].Zhang F, Xu X, Li T, Liu Z, Mar. Drugs 2013, 11, 4698–4723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Kohli GS, Campbell K, John U, Smith KF, Fraga S, Rhodes LL, Murray SA, J. Eukaryot. Microbiol. 2017, 64, 691–706. [DOI] [PubMed] [Google Scholar]
- [81].Liu TG, Cane DE, Deng ZX, in Complex Enzymes in Microbial Natural Product Biosynthesis, Part B: Polyketides, Aminocoumarins and Carbohydrates, Vol. 459 (Ed.: Hopwood DA), Elsevier Academic Press Inc, San Diego, 2009, pp. 187–214. [DOI] [PubMed] [Google Scholar]
- [82].Van Dolah FM, Kohli GS, Morey JS, Murray SA, J. Phycol. 2017, 53, 1325–1339. [DOI] [PMC free article] [PubMed] [Google Scholar]