

Abstract
Interferon regulatory factor 4 (IRF4), a transcription factor recognized as a key regulator of lymphoid and myeloid cell differentiation, has recently been recognized as a critical mediator of macrophage activation. Previously we have reported that IRF4 signaling is closely correlated with anti-inflammatory polarization of microglia in adult mice after stroke. However, IRF4’s role in the inflammatory response in the immature brain is unknown. Using a model of neonatal hypoxic ischemic encephalopathy (HIE) we investigated the regulatory action of IRF4 signaling in the activation of microglia and monocytes after HIE. IRF4 myeloid cell conditional knockout (CKO) postnatal day 10 (P10) male pups were subjected to a 60-minute hypoxic-ischemic insult by the Rice-Vanucci model (RVM). IRF4 gene floxed mice (IRF4fl/fl) were used as controls. Brain atrophy and behavioral deficits were measured 7 days after HIE. Flow cytometry (FC) was performed to examine central (microglial activation) and peripheral immune cell responses by both cell membrane and intracellular marker staining. Serum levels of cytokines were determined by ELISA. The results showed that IRF4 CKO pups had increased tissue loss and worse behavioral deficits than IRF4fl/fl mice seven days after HIE. FC demonstrated significantly more infiltration of monocytes and neutrophils in the ischemic brains of IRF4 CKO vs IRF4fl/fl pups. IRF4 CKO ischemic microglia were more pro-inflammatory as evidenced by higher expression of the pro-inflammatory marker CD68, and increased intracellular TNFα and IL-1β levels compared to controls. In addition, IRF4 deletion from myeloid cells resulted in increased levels of circulating pro-inflammatory cytokines and higher endothelial MMP9 expression after HIE. These data indicate that IRF4 signaling in myeloid cells plays a regulatory role in neuroinflammation and that deletion of myeloid IRF4 is detrimental to HIE injury, suggesting that IRF4 could serve as a potential therapeutic target for neonatal ischemic brain injury.
Keywords: HIE, IRF4, Microglia, Neuroinflammation, Neonate
Graphical Abstract
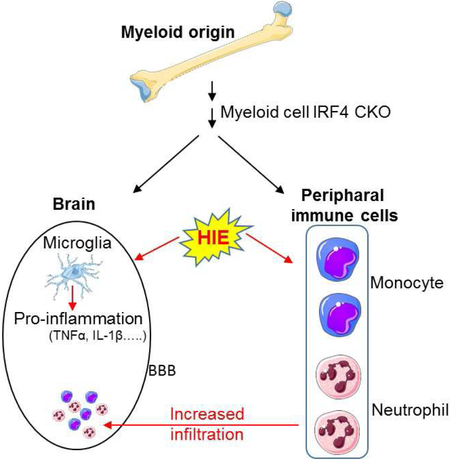
1. Introduction
Neonatal hypoxic ischemic encephalopathy (HIE) is a devastating disease that results in long-term motor and cognitive impairment in children (Lehman and Rivkin, 2014). Hypoxic ischemia is a powerful stimulus that triggers a series of events including blood brain barrier (BBB) damage, rapid activation of resident immune cells primarily the microglia (Ferrazzano et al., 2013; McRae et al., 1995), and infiltration of circulating leukocytes into the ischemic lesion, leading to secondary neuronal damage (Al Mamun et al., 2018b; Liu and McCullough, 2013; Mirza et al., 2015). Microglial activation and aggregation are pathological markers for HIE in human infants (Liu and McCullough, 2013). After ischemia, microglia and infiltrating monocytes become activated to either an M1 (pro-inflammatory) or M2 (anti-inflammatory) phenotype depending on the progression of injury and the stimuli they receive, and develop macrophage capabilities (Taylor and Sansing, 2013). M1 is a toxic cellular state associated with an increase in protein synthesis of pro-inflammatory mediators (IFNγ, IL1β, TNFα, IL6, CXCL10, etc.), ROS and NO production, and activation of proteolytic enzymes (MMP 9, MMP3) (Al Mamun et al., 2018a; del Zoppo et al., 2007; Hu et al., 2012). In contrast, M2 microglia/macrophages release anti-inflammatory mediators (IL10, TGFβ, IL4, IL13, IGF-1) (Zhou et al., 2013) and enhance expression of genes associated with inflammation resolution, scavenging, homeostasis, and basal neurogenesis (Cherry et al., 2014; Nikolakopoulou et al., 2013). Uncontrolled activation of microglia/macrophages can injure healthy neurons as well as decrease the survival of injured neurons, and a growing body of literature suggests that the regulation of microglia/macrophage activation is an important target for developing new therapeutic approaches for ischemic damage (Patel et al., 2013). Compared to adults, neonatal microglia activation happens more swiftly after ischemic insults and continues for weeks (Liu and McCullough, 2013).
Interferon regulatory factor 4 (IRF4) is a hemopoietic-specific transcription factor critical for myeloid and lymphoid lineage development and function (Tamura et al., 2008). Recently, a novel role of IRF4 in neuroinflammation has been revealed (Al Mamun et al., 2018a). We and others have found an evident link between IRF4 and anti-inflammatory responses which provide neuroprotection in young adult and aged mouse stroke models (Al Mamun et al., 2018b; Guo et al., 2014; Masuda et al., 2014; Zhao et al., 2017). IRF4 KO mice had deficits in the production of anti-inflammatory cytokines (i.e IL-4 and IL-10) and generated more pro-inflammatory cytokines like TNFα and IL-6 in a murine model of sepsis (Honma et al., 2005), indicating a beneficial effect of IRF4 signaling. However, whether the IRF4 signaling regulates the activation of microglia/macrophages in the neonatal brain and how it affects HIE induced damage are not known. In this study, we used myeloid cell IRF4 conditional knockout mice (generated by a cross between IRF4fl/fl and lysozyme Cre mice), and demonstrated that IRF4 is a potent regulator of the inflammatory response after in HIE.
2. Materials and methods
2.1. Animals
To generate myeloid cell specific IRF4 conditional knockout (CKO) mice, we crossed IRF4fl/fl with lysozyme M-Cre (LyzMCre) mice (Jackson Laboratory) in the C57BL/6 background. All the breeding pairs were housed in pathogen-free rooms and had access to food and water ad libitum. We used male P10 pups for HIE surgery. A total of 42 IRF4fl/fl and 48 IRF4 CKO pups were used in this study, including 9 pups that were excluded from further assessments because of no brain injury. All procedures were performed in accordance with NIH guidelines for the care and use of laboratory animals and were approved by the Institutional Animal care and use committee of the University of Texas Health Science Center.
2.2. Neonatal HIE model
The Rice-Vannucci Model (RVM) of neonatal hypoxia–ischemia was modified to induce HIE in P10 mice (Al Mamun et al., 2018b; Mirza et al., 2015). Briefly, P10 pups were anesthetized with isoflurane (4% for induction and 1.5–2% for maintenance). A midline cervical incision was made and the right common carotid artery (CCA) was cauterized to occlude blood flow. After surgery, the pups were returned to their dams for two hours. To induce hypoxia, the CCA-occluded mice were put in a chamber containing 10% oxygen and 90% nitrogen for 45 minutes. After that, the animals were placed on a temperature-controlled blanket for 20 min and then returned to their dams. Mice were sacrificed at 7d of surgery for histological, cellular, and molecular analysis. Sham mice underwent the same procedure except cauterization of the right CCA and induction of hypoxia in the chamber.
2.3. Behavior testing
2.3.1. Seizure activity
Seizure activity was scored according to a seizure rating scale as previously reported (Al Mamun et al., 2018b; Mirza et al., 2015). Every 5 min in 1 h at the same time of 1 day after HIE, the score corresponding to the highest level of seizure activity observed during that time period was recorded and summed to produce a total seizure score. Seizure behavior was scored as follows: 0 = normal behavior; 1 = immobility; 2 = rigid posture; 3 = repetitive scratching, circling, or head bobbing; 4 = forelimb clonus, rearing, and falling; 5 = mice that exhibited level four behaviors repeatedly; and 6 = severe tonic-clonic behavior.
2.3.2. Y-maze test
Spontaneous alternation using a Y-maze is a test for habituation and spatial working memory (Swonger and Rech, 1972). The symmetrical Y-maze consists of three white opaque plastic arms at a 120° angle from each other. After placing pups in the center, the animal is allowed to freely explore the three arms. Over the course of multiple arm entries, the subject should show a tendency to enter a less recently visited arm. The test consists of a single 5 min trial; spontaneous Alternation (%) is defined as consecutive entries in 3 different arms (arm A,B,C), divided by the number of total alternations (total arm entries minus 2) (Hsiao et al., 1995). Mice with less than 8 arm entries during the 5-min trial were excluded from the analysis. An entry occurs when all four limbs are within the arm. All the arms were cleaned after each trial.
2.4. Assessment of brain tissue atrophy by cresyl violet (CV) staining
At day 7 (P17) after HIE, the animals were anesthetized with tribromoethanol (Avertin® ip injection at a dose of 0.25mg/g body weight). Animals were perfused transcardially with ice cold 0.1M sodium phosphate buffer (pH 7.4) followed by 4% paraformaldehyde (PFA); the brain was removed from the skull and post-fixed for 18 h in 4% PFA and subsequently placed in cryoprotectant solution (30% sucrose). The brain was cut into 30-μm free-floating coronal sections on a freezing microtome and the sections were sequentially put into a 96 well plate with anti-freezing media. Eight brain sections were chosen from the middle row of the plate (row D and well #1, 3, 4, 5, 6, 7, 9, and 11) and were mounted on glass slide and stained by CV for evaluation of ischemic damage (Hsiao et al., 1995). Brain atrophy at 30d of HIE was computed by (1 – (ischemic hemisphere-ventricle-cavity) / (contralateral hemisphere-ventricle)) × 100 as previously described (Liu et al., 2012) by an investigator blinded to genotype.
2.5. Flow cytometry
Leukocytes from the brain tissue were prepared as previously described (Al Mamun et al., 2018b). Briefly, animals were anesthetized with Avertin® (2,2,2-Tribromoethanol, Sigma) and transcardially perfused with phosphate-buffered saline (PBS) for 5 min. The brain was then divided along the interhemispheric fissure into two hemispheres. Ipsilateral brains were placed in complete Roswell Park Memorial Institute (RPMI) 1640 (Lonza) medium, followed by mechanical and enzymatically digest with 150 μL collagenase/dispase (1 mg/mL) and 300 μL DNAse (10 mg/mL; both Roche Diagnostics) for 45 minutes at 37 °C with mild agitation. The cell suspension was filtered through a 70μm filter. Leukocytes were harvested from the interphase of a 70%/30% Percoll gradient. Cells were washed and blocked with mouse Fc Block (eBioscience) prior to staining with primary antibody conjugated fluorophores: CD45-eF450 (# 48-0451-82, eBioscience), CD11b-AF700 (# 101222, Biolegend), Ly6C-APC-eF780 (#47-5932-82, eBioscience), Ly6G-PE (#127608, Biolegend), CD68-APC (#107614, Biolegend) and CD206-PE-cy5.5(#141720, Biolegend). For each surface marker, 0.25 μg (1:100) of antibody was used to stain 1× 106 cells. All the antibodies were commercially purchased from Biolegend/eBioscience. For live/dead discrimination, a LIVE/DEAD Fixable Aqua Dead Cell Stain Kit was used according to manufacture instruction (Thermo Fisher Scientific, MA, USA). Cells were briefly fixed in 2% paraformaldehyde (PFA). Data were acquired on a CytoFLEX (Beckman Coulter) and analyzed using FlowJo (Treestar Inc.). No less than 100,000 events were recorded for each sample. Cell type-matched fluorescence minus one (FMO) controls were used to determine the positivity of each antibody. Gating strategy was shown in figure 4A.
Figure 4.

Peripheral immune cell infiltration in sham and HIE brains. A representative dot plot depicts the gating strategy to differentiate the total peripharal infiltrating cells as CD45highCD11b+ (pMyeloid) and lymphocytes as CD45highCD11b− (A). In pMyeloid gate, monocytes are considered as CD45highCD11b+Ly6C+Ly6G−, and neutrophils as CD45highCD11b+Ly6G+ Ly6C−. Quantitative data of pMyeloid, lymphocytes, neutrophils and monocytes were shown in (B, C,D and E). N=5-6 for sham and 7-9/group for HIE; *P < 0.05, IRF4 fl/fl HIE vs IRF4 CKO HIE.
2.6. Intracellular cytokines staining
For intracellular cytokine staining, an ex vivo brefeldin A protocol was followed. Prior to staining, brain leukocytes (1×106 cells) were incubated with BFA (10 μg/mL, Sigma) in 1mL complete RPMI for 2h at 37 °C (5 % CO2). Afterward, cells were re-suspended in Fc Block, stained for surface antigens and washed in 100μL of fixation/permeabilization solution (BD Biosciences) for 20 minutes. Cells were then washed twice in 300μL permeabilization/wash buffer (BD Biosciences), re-suspended in an intracellular antibody cocktail (0.25μg for each antibody, 1:100 dilution) containing TNFα-PE-Cy7 (eBioscience) and IL-1β-PE (eBioscience), IL-10-APC and IL-4-PerCP-Cy5.5 (BioLegend), and subsequently fixed. Data were acquired on a CytoFLEX (Beckman Coulter) and analyzed with FlowJo (Treestar Inc.).
2.7. Immunohistochemistry
The mice were anesthetized by Avertin® and then transcardially perfused with 0.1M sodium phosphate buffer (pH 7.4) followed by 4% paraformaldehyde for post-fixation of the brains. Immunohistochemical staining of fixed-frozen sections (10 and 30μm-thickness) was performed as described previously (Liu et al., 2009). Briefly, the brains were cut and mounted onto gelatin-coated slides and allowed to air dry. The sections were then blocked in 0.1M PBS with 0.25% Triton X-100 (sigma) and 10% donkey serum for 2 hours and incubated overnight at 4 °C with the following primary antibodies: rabbit goat-MMP-9 (1:300, R&D Systems, MN, USA), rabbit anti-Iba1 (1:300, Wako, Japan), goat anti-IRF4 (sc6059; 1 : 50, Santa Cruz) and anti-von Willebrand Factor (VWF, sc-365712, Santa Cruz biotechnology Inc, TX, USA). After being washed in TBS+0.025% Triton X-100, the sections were incubated with the indicated secondary antibodies for 1 h. The following secondary antibodies was used: donkey anti-rabbit IgG Alexa Fluor 488 conjugate (A21206; 1: 500, Invitrogen, USA), donkey anti-mouse IgG Alexa Fluor 488 conjugate (A21206; 1: 500, Invitrogen, USA) donkey anti-goat IgG Alexa Fluor 594 conjugate (A11011; 1: 500, Invitrogen). The nuclei were stained with DAPI (S36939, Invitrogen). Brain slices were taken at the same distance from bregma to ensure comparison of similar structures. Eight 40× fields/animal were analyzed in the outer boundary zone of the atrophy. MMP9 fluorescence positive cells were counted by an unbiased, blinded investigator using ImageJ software (NIH).
2.8. Serum cytokine and MMP9 levels by ELISA
Tumor necrosis factor-alpha (TNF-α, Bio-legend), IL-1β (Biolegend), interleukin-10 (IL-10, e-bioscience), IL-4 (Biolegend) and mouse MMP9 (LifeSpan Bioscience, Inc) levels were measured by commercially available specific quantitative sandwich ELISA kits according to manufacturer instructions.
2.9. Statistical Analysis
Investigators were blinded to mouse strains for stroke surgery, behavioral testing, infarct, and inflammation analysis. Mice with abnormal body weights and behavior (e.g. body shaking, limp, etc.) before surgery were excluded. Data were presented as mean ± SD and analyzed with a t-test (atrophy volumes, cytokines levels, inflammatory parameters and behavior tests). P < 0.05 was considered statistically significant. All analyses were done by using GraphPad Prism software (San Diego, CA). The ordinal data of Seizure score are analyzed with Mann-Whitney U test.
3. Results
3.1. HIE outcomes in IRF4fl/fl vs IRF4 CKO pups.
We first confirmed IRF4 deletion from macrophage/microglia cells by performing Iba1 and IRF4 immunofluorescence staining in brain slices from IRF4 CKO HIE pups. We found that almost none of the Iba1 positive cells expressed IRF4 in CKO pups; however, Iba 1 and IRF4 were colocalized in IRF4fl/fl mice (Supplementary Fig. 1). Next, we examined brain tissue loss at 7 days after HIE in both IRF4fl/fl and IRF4 CKO pups, as the hypoxic-ischemic infarct becomes less visible at this time point and the ischemic brains exhibit either cavitation or atrophy. Quantitative data showed IRF4 CKO pups had significantly (p = 0.0273) more tissue loss than IRF4fl/fl pups (Fig. 1A & B). We also performed seizure scoring at acute time points and Y-maze test at 7d to examine the brain functional deficits. Corresponding with tissue loss, IRF4 CKO pups had higher seizure scores than IRF4fl/fl at either 5 min (p = 0.0006) or 24h (p = 0.0132) of HIE (Fig. 1D & E). In the Y-maze, spontaneous alteration, which measures working memory of rodents, was not different between IRF4fl/fl and IRF4 CKO groups (Fig. 1C).
Figure 1.

HIE outcomes at 7 days after HIE in IRF4fl/fl and IRF4 CKO pups. Representative images of brain slices stained with cresyl violet (CV) (A). Quantification of brain tissue loss (B). Functional outcomes were evaluated by Y-maze (C). Seizure scores were significantly different (p< 0.05) between IRF4fl/fl and IRF4 CKO pups either at 5 min (D) or 24h after HIE (E). N=7-11/group; *P < 0.05.
3.2. CD68 was differentially expressed on microglia in IRF4fl/fl vs. IRF4 CKO ischemic brains
We next examined microglial activation after HIE with FC as microglial activation is a key element in initiating and perpetuating inflammatory responses to ischemia. CD68 is a well-established marker of M1 macrophage activation (Barros et al., 2013; Weber et al., 2015). We gated microglia as CD45intermediateCD11b+, and then quantified the median fluorescence intensity (MFI) of CD68 in microglia. Our data revealed that 7 days after HIE, the MFI of CD68 significantly increased (p = 0.0038) in IRF4 CKO microglia compared to controls (Fig. 2A & B). In contrast, there was no difference in the MFI of CD206 (M2 microglia marker) between the two strains (Fig. 2C & D). These data suggested that IRF4 CKO pup brains mounted a more robust pro-inflammatory response after HIE. Resident microglial (CD45intermediateCD11b+) cells counts were not different between two strain after HIE (data not shown).
Figure 2.

Expression of microglia cell membrane markers after HIE. Quantification of CD68 (A, B) and CD206 (C,D) expression were presented as median fluorescence intensity (MFI) in IRF4fl/fl vs IRF4 CKO pup brains. N=7-9/group; *P < 0.05.
3.3. Intracellular microglial cytokine profile in IRF4fl/fl vs. IRF4 CKO HIE pups.
To further confirm microglial phenotypes after HIE, we performed intracellular cytokine staining for both pro- and anti-inflammatory markers in microglia by flow cytometry in addition to the cell membrane markers (CD68 and CD206). There were more TNFα and IL-1β (pro-inflammatory cytokines) double positive microglia in IRF4 CKO HIE brains compared with controls (Fig. 3A, B). Of note, TNF-α (p = 0.0298) and IL-1β (p = 0.0268) MFI also significantly increased in IRF4 CKO vs. IRF4fl/fl microglia (Fig. 3C & D). However, intracellular levels of anti-inflammatory cytokines (IL-4 and IL-10) were equivalent between two groups (data not shown).
Figure 3.

Measurement of intracellular pro-inflammatory cytokines in microglia. Representative contour plots (A) showed microglial production of pro-inflammatory TNFα/IL-1β cytokine. TNFα+/IL-1β+ cells were counted in gated microglia (B). The expression levels of TNFα and IL-1β were measured as median fluorescence intensity (MFI) as shown in figure (C, D). N=7-9/group; *P < 0.05.
3.4. IRF4 CKO mice had more infiltration of peripheral leukocytes into the ischemic brains.
To evaluate the response of peripheral immune cells to HIE, we quantified infiltrating leukocytes in HIE brains with FC. Total peripheral myeloid cells were gated as CD45highCD11b+, and lymphocytes as CD45highCD11b− (Fig. 4A). Monocytes were further gated as CD45highCD11b+Ly6C+Ly6G−, and neutrophils as CD45highCD11b+Ly6G+. Quantitative data showed that there was no difference of peripheral myeloid cells infiltration between IRF4 CKO vs. fl/fl sham brains. There were significantly more peripheral myeloid cell and lymphocyte infiltration in HIE vs sham IRF4 fl/fl (p < 0.0001) and IRF4 CKO (p < 0.0001) pups. Interestingly, the number of either total peripheral myeloid cells (p < 0.001) or the breakdown monocytes (p = 0.0226)/neutrophils (p = 0.0118) was significantly higher in CKO vs. fl/fl HIE brains but not in sham brains (Fig. 4B, D, E), indicating CKO of IRF4 induced a more robust inflammatory responses in HIE brains. The total lymphocytes in peripheral blood also showed a trend towards increased levels in IRF4 CKO brains, but this did not reach statistical significance (Fig. 4C).
3.5. Serum cytokine levels after HIE in IRF4 CKO and IRF4fl/fl HIE pups
Serum cytokine levels are reflective of the level of immune responses to ischemic injury (Al Mamun et al., 2018a; Ormstad et al., 2011). To investigate how IRF4 deletion from myeloid cells affects serum cytokine levels, we measured pro-inflammatory (i.e IL-1 β, TNF-α) and anti-inflammatory (i.e IL-10, and IL-4) cytokine levels in the serum of sham and HIE mice with ELISA. We observed no difference in the serum level of pro-inflammatory (i.e IL-1β, TNF-α) and anti-inflammatory (i.e IL-10, and IL-4) cytokine between IRF4 fl/fl and IRF4 CKO sham pups. However, there was significant effect of myeloid IRF4 deletion on the aforementioned cytokine levels in HIE mice. Consistent with the microglial intracellular cytokine data, the serum pro-inflammatory cytokine TNF~ (p < 0.0001) and IL-1β (p < 0.0001) levels were significantly increased in IRF4 CKO vs. IRF4fl/fl HIE pups (Fig. 5A & B). However, there was no difference in anti-inflammatory (IL-10, and IL-4) cytokine levels between the two strains (Fig. 5C & D).
Figure 5.

Serum cytokines levels in sham and HIE pups. Pro-inflammatory (TNFα and IL-1β) (A and B) and anti-inflammatory (IL-10 and IL-4) (C and D) cytokine levels (pg/mL) were measured in the serum at 7d after sham/HIE. N=5-6 for sham and 6-10/group for HIE; ¶P < 0.05, IRF4 fl/fl sham vs IRF4 fl/fl HIE; ØP < 0.05, IRF4 CKO sham vs IRF4 CKO HIE; *P < 0.05, IRF4 fl/fl HIE vs IRF4 CKO HIE.
3.6. MMP9 positive blood vessels increased in IRF4 CKO pups.
Increased levels of MMP9 have been detected in neurons, glia, endothelial cells and neutrophils after ischemic stroke, with each of these cell types having a unique MMP9 secretion/expression profile (Turner and Sharp, 2016). We also examined MMP9 levels in both cerebral endothelium and in circulating blood. First, we performed vWF staining to examine if there is any strain differences in the cerebral microvasculature, and found equivalent intensity of vWF staining between IRF4fl/fl and IRF4 CKO mice (Fig. 6A & B). Next we examined potential BBB damage in the cerebrovasculature by detecting MMP9 immunofluorescent signal in vWF+ blood vessels in the ipsilateral hemispheres after HIE. We found that IRF4 CKO mice had significantly higher intensity of MMP9 (p < 0.0001) in the vWF+ blood vessels at 7 days compared to their controls (Fig. 6C). Interestingly, circulating MMP9 levels were also significantly increased in the serum of IRF4 CKO vs. IRF4fl/fl mice (p = 0.0014) (Fig. 6D).
Figure 6.

MMP-9 expression at 7d after HIE. Representative images of MMP-9 and vWF double immunostaining in brains of IRF4fl/fl and IRF4 CKO pups (A).Quantification of fluorescence intensity (arbitrary unit, AU) for lectin (B) in each strain. Quantification of MMP-9+/vWF+ blood vessels in the brain (C). Serum levels of MM9 (D). N=7-11/group; *P < 0.05, IRF4 fl/fl vs IRF4 CKO.
3. Discussion
The present study demonstrated several novel findings, which not only support our hypothesis that IRF4 signaling in myeloid cells is critical for mediating neonatal stroke injury, but also suggest a unique role of IRF4 in the neuroinflammation induced by neonatal HIE. Firstly, IRF4 signaling in myeloid cells significantly contributes to HIE injury as IRF4 CKO pups had increased tissue loss and poorer functional outcomes after HIE. Secondly, IRF4 CKO only affects microglial pro-inflammatory (M1) responses to HIE, as there were surprisingly no effects on M2 microglial activation, evidenced by our microglial data using both cell membrane and intracellular markers (Fig 2 & 3). Thirdly, the deletion of IRF4 in myeloid cells promotes infiltration of peripheral leukocytes into the ischemic brains and specifically increases circulating levels of pro-inflammatory cytokines after HIE. Lastly, myeloid cell IRF4 has a crosstalk with MMP9 signaling in cerebral endothelium, and affects BBB integrity.
Cerebral ischemia induces inflammatory responses that are characterized by activation and release of cytokines, chemokines, endothelial-leukocyte adhesion molecules, and proteolytic enzymes, a series of events that lead to infiltration of leukocytes into the ischemic brain and exacerbate tissue damage (Vila et al., 2003). Previous studies have demonstrated that myeloid cells (mainly microglia and monocytes) are the major source of inflammatory cytokines in neuroinflammation (Garcia-Culebras et al., 2018; Hanisch, 2002; Mirza et al., 2015; Welser-Alves and Milner, 2013), and these cells are tightly controlled by IRFs to exhibit either pro- or anti-inflammatory phenotype in the ischemic brain depending on the molecular signals they sense (Hu et al., 2015; Hu et al., 2012). It has been reported that IRF4 is one of the major IRFs for regulation of macrophage activation in peripheral inflammation (Chistiakov et al., 2018; Satoh et al., 2010). Recently, we have found that IRF4 expression in microglia is upregulated and corresponds to anti-inflammatory responses after ischemic injury in adult animal models, suggesting a beneficial role of IRF4 signaling (Al Mamun et al., 2018a). The present study mechanistically examined IRF4 signaling in neonatal HIE, and also revealed a protective effect of IRF4 on HIE outcomes (Fig.1). Interestingly, our data showed that IRF4 signaling in neonatal cerebral ischemia exhibited age-related differences compared to our previous studies in adult ischemia. We did not see a suppressive effect of IRF4 CKO on anti-inflammatory responses; instead, our data suggested that the repercussion of IRF4 deletion in HIE is mainly mediated through augmentation of the pro-inflammatory response.
Studies of peripheral inflammation suggested that the activation of monocytes is mediated either by IRF4 augmentation of M2 (anti-inflammatory) polarization, or by IRF5 towards M1 phenotype. However, IRF4 signaling in these studies functioned through both anti- and pro-inflammatory pathways (Gunthner and Anders, 2013; Satoh et al., 2010; Schneider et al., 2018). Cytoplasmic IRF4 can not only translocate to the nucleus to mediate expression of anti-inflammatory mediators, but can also compete with IRF5 to bind to the adaptor protein MyD88 to reduce IRF5 phosphorylation leading to suppression of M1 activation (Marecki et al., 1999; Negishi et al., 2005). Our data (Fig. 2) showed that IRF4 deletion in neonatal myeloid cells only affects the M1 arm of the immune response evidenced by upregulation of CD68 in microglia, but has no effect on CD206 expression. The data suggest that, in neonatal HIE brains, IRF4 signaling has a biased effect in post-ischemia inflammatory response that aims at inhibiting IRF5. Besides IRF4, there are several other factors such as PPAR-γ, STAT-3/6, micro RNA-124, RunX that can also promote microglial M2 polarization (Crotti and Ransohoff, 2016; Essandoh et al., 2016; Hu et al., 2015). It is possible that these factors in neonates are abundant and able to compensate the effect of IRF4 CKO on production of anti-inflammatory mediators, so that the deletion of IRF4 in myeloid cells does not impact the anti-inflammatory responses (Fig. 2 and 3).
The presence of IRF4 has also been reported in neurons, and can rescue neurons from ischemia/reperfusion-induced death through serum response factor (SRF) signaling (Guo et al., 2014). Whether the neuronal IRF4 also contributes to inflammatory responses is not clear. Interestingly, IRF4 over expression in Th2 cells resulted in increased anti-inflammatory cytokines (IL-4 and IL-10) expression (Ahyi et al., 2009). In addition, whole body IRF4 deficient mice were unable to produce anti-inflammatory cytokines (i.e IL-4 and IL-10) and had high levels of pro-inflammatory cytokines (i.e TNFα and IL-6) in models of chronic inflammation (Honma et al., 2005). These previous studies suggest IRF4 in non-myeloid cells may play a non-negligible role in producing anti-inflammatory cytokines. Nevertheless, our present study showed IRF4 deletion from myeloid cells is sufficient to exacerbate immune responses and negatively affect HIE outcomes, highlighting the significance of myeloid cells.
Our data showed conditional deletion of IRF4 in myeloid cells facilitates infiltration of peripheral leukocytes including monocytes, neutrophils, and lymphocytes into the brain after HIE, probably due to the increased production of pro-inflammatory cytokines/chemokines. Neutrophils and monocytes are the most abundant leukocytes present at the site of injury with a peak influx between 1 to 3 days after ischemia and can be seen in the ipsilateral hemisphere for up to 30 days after HIE (Al Mamun et al., 2018b; Gronberg et al., 2013). Exacerbated infiltration of neutrophils and monocytes resulted in enhanced inflammatory responses and tissue injury (Jin et al., 2010; Jones et al., 2018). Previous studies have demonstrated that neutrophils are one of the first peripheral cell type to respond after ischemic stroke, contributing to BBB disruption (Jickling et al., 2015; Price et al., 2004). It has been reported that neutrophils serve as potential secretory cells for MMP9 after ischemic stroke, and neutrophil infiltration increased MMP-9 levels in the ischemic brain (Justicia et al., 2003; Rosell et al., 2008). In the present study, we also found that the expression of MMP9 (Fig. 6), a marker of BBB compromise, was up-regulated along with the increased infiltration of neutrophils in IRF4 CKO mice (Fig. 4D). MMP9 alone can degrade almost all components of the extracellular matrix and basal lamina such as laminins, fibronectin, or type IV collagen, predisposing BBB to rupture and increase the infiltration of peripheral myeloid cells and lymphocytes into the brain (Nagase et al., 2006; Turner and Sharp, 2016). The production of MMP9 is negatively regulated by anti-inflammatory cytokines IL-4 and IL-10 (Konnecke and Bechmann, 2013); however, our IRF4 CKO model generated a pro-inflammatory microenvironment that does not favor MMP9 down-regulation.
In conclusion, our data provide evidence that IRF4 deletion from myeloid cells is detrimental, contributing to a brain environment that is more pro-inflammatory in response to neonatal HIE. The role of IRF4 signaling is unique in neonates in that, although IRF4 directly mediates expression of anti-inflammatory cytokines, it plays a more important role in suppressing the production of pro-inflammatory mediators. The involvement of IRF4 signaling in HIE is not limited to recruited leukocytes; it also has important crosstalk with BBB MMP expression. Research into IRF4’s role in HIE is still in its infancy. Further studies are warranted to elucidate the pathophysiological function of this transcription factor and to explore its potential for development into a therapeutic strategy to treat the lifelong damage induced by HIE.
Supplementary Material
Supplementary figure 1. IRF4 expression in activated microglia/macrophages at 7d after HIE. Representative images of co-expression of IRF4 (red) with Iba1 (green) in activated microglia/macrophages were shown. RF4+Iba1+ double positive cells (inset in the merged image) were only seen in IRF4fl/fl brain but not in IRF4 CKO brains. Scale bar = 100μm.
{kind=link}
Supplementary figure 2. Brain leukocytes were gated on a forward scatter (FSC)/side scatter (SSC) plot. In live cell population, microglia and infiltrating myeloid cells were gated based on relative expression of CD45 and CD11b. The central immune cells (microglia) were considered as CD45intermediate CD11b+; infiltrating peripheral myeloid cells (pMyeloid) and infiltrating lymphocytes (Lymphocytes) were identified as CD45high CD11b+ and CD45high CD11b− respectively.
{kind=link}
Highlights.
Interferon regulatory factor 4 (IRF4) CKO boosts pro-inflammatory responses to Hypoxic Ischemic Encephalopathy (HIE)
IRF4 has a cross-talk with Blood Brain Barrier function
Age-related differences exist in IRF4 signaling in cerebral ischemia
4. Acknowledgement
This work was supported by the National Institutes of Health (grants NS093042/NS091794 to Fudong Liu)
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest
None of the authors have a conflict of interest relevant to this work.
References
- Ahyi AN, Chang HC, Dent AL, Nutt SL, Kaplan MH, 2009. IFN regulatory factor 4 regulates the expression of a subset of Th2 cytokines. J Immunol 183,1598–1606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al Mamun A, Chauhan A, Yu H, Xu Y, Sharmeen R, Liu F, 2018a. Interferon regulatory factor 4/5 signaling impacts on microglial activation after ischemic stroke in mice. Eur J Neurosci 47, 140–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al Mamun A, Yu H, Romana S, Liu F, 2018b. Inflammatory Responses are Sex Specific in Chronic Hypoxic-Ischemic Encephalopathy. Cell Transplant, 963689718766362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barros MH, Hauck F, Dreyer JH, Kempkes B, Niedobitek G, 2013. Macrophage polarisation: an immunohistochemical approach for identifying M1 and M2 macrophages. PLoS One 8, e80908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherry JD, Olschowka JA, O'Banion MK, 2014. Neuroinflammation and M2 microglia: the good, the bad, and the inflamed. J Neuroinflammation 11, 98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chistiakov DA, Myasoedova VA, Revin VV, Orekhov AN, Bobryshev YV, 2018. The impact of interferon-regulatory factors to macrophage differentiation and polarization into M1 and M2. Immunobiology 223,101–111. [DOI] [PubMed] [Google Scholar]
- Crotti A, Ransohoff RM, 2016. Microglial Physiology and Pathophysiology: Insights from Genome-wide Transcriptional Profiling. Immunity 44, 505–515. [DOI] [PubMed] [Google Scholar]
- del Zoppo GJ, Milner R, Mabuchi T, Hung S, Wang X, Berg GI, Koziol JA, 2007. Microglial activation and matrix protease generation during focal cerebral ischemia. Stroke 38, 646–651. [DOI] [PubMed] [Google Scholar]
- Essandoh K, Li Y, Huo J, Fan GC, 2016. MiRNA-Mediated Macrophage Polarization and its Potential Role in the Regulation of Inflammatory Response. Shock 46,122–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrazzano P, Chanana V, Uluc K, Fidan E, Akture E, Kintner DB, Cengiz P, Sun D, 2013. Age-dependent microglial activation in immature brains after hypoxia- ischemia. CNS Neurol Disord Drug Targets 12, 338–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Culebras A, Duran-Laforet V, Pena-Martinez C, Ballesteros I, Pradillo JM, Diaz-Guzman J, Lizasoain I, Moro MA, 2018. Myeloid cells as therapeutic targets in neuroinflammation after stroke: Specific roles of neutrophils and neutrophil-platelet interactions. J Cereb Blood Flow Metab, 271678X18795789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gronberg NV, Johansen FF, Kristiansen U, Hasseldam H, 2013. Leukocyte infiltration in experimental stroke. J Neuroinflammation 10, 115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunthner R, Anders HJ, 2013. Interferon-regulatory factors determine macrophage phenotype polarization. Mediators Inflamm 2013, 731023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo S, Li ZZ, Jiang DS, Lu YY, Liu Y, Gao L, Zhang SM, Lei H, Zhu LH, Zhang XD, Liu DP, Li H, 2014. IRF4 is a novel mediator for neuronal survival in ischaemic stroke. Cell Death Differ 21, 888–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanisch UK, 2002. Microglia as a source and target of cytokines. Glia 40, 140–155. [DOI] [PubMed] [Google Scholar]
- Honma K, Udono H, Kohno T, Yamamoto K, Ogawa A, Takemori T, Kumatori A, Suzuki S, Matsuyama T, Yui K, 2005. Interferon regulatory factor 4 negatively regulates the production of proinflammatory cytokines by macrophages in response to LPS. Proc Natl Acad Sci U S A 102, 16001–16006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsiao KK, Borchelt DR, Olson K, Johannsdottir R, Kitt C, Yunis W, Xu S, Eckman C, Younkin S, Price D, et al. , 1995. Age-related CNS disorder and early death in transgenic FVB/N mice overexpressing Alzheimer amyloid precursor proteins. Neuron 15, 1203–1218. [DOI] [PubMed] [Google Scholar]
- Hu X, Leak RK, Shi Y, Suenaga J, Gao Y, Zheng P, Chen J, 2015. Microglial and macrophage polarization-new prospects for brain repair. Nat Rev Neurol 11, 56–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu X, Li P, Guo Y, Wang H, Leak RK, Chen S, Gao Y, Chen J, 2012. Microglia/macrophage polarization dynamics reveal novel mechanism of injury expansion after focal cerebral ischemia. Stroke 43, 3063–3070. [DOI] [PubMed] [Google Scholar]
- Jickling GC, Liu D, Ander BP, Stamova B, Zhan X, Sharp FR, 2015. Targeting neutrophils in ischemic stroke: translational insights from experimental studies. J Cereb Blood Flow Metab 35, 888–901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin R, Yang G, Li G, 2010. Inflammatory mechanisms in ischemic stroke: role of inflammatory cells. J Leukoc Biol 87, 779–789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones KA, Maltby S, Plank MW, Kluge M, Nilsson M, Foster PS, Walker FR, 2018. Peripheral immune cells infiltrate into sites of secondary neurodegeneration after ischemic stroke. Brain Behav Immun 67, 299–307. [DOI] [PubMed] [Google Scholar]
- Justicia C, Panes J, Sole S, Cervera A, Deulofeu R, Chamorro A, Planas AM, 2003. Neutrophil infiltration increases matrix metalloproteinase-9 in the ischemic brain after occlusion/reperfusion of the middle cerebral artery in rats. J Cereb Blood Flow Metab 23, 1430–1440. [DOI] [PubMed] [Google Scholar]
- Konnecke H, Bechmann I, 2013. The role of microglia and matrix metalloproteinases involvement in neuroinflammation and gliomas. Clin Dev Immunol 2013, 914104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehman LL, Rivkin MJ, 2014. Perinatal arterial ischemic stroke: presentation, risk factors, evaluation, and outcome. Pediatr Neurol 51, 760–768. [DOI] [PubMed] [Google Scholar]
- Liu F, Benashski SE, Persky R, Xu Y, Li J, McCullough LD, 2012. Age-related changes in AMP-activated protein kinase after stroke. Age (Dordr) 34, 157–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu F, McCullough LD, 2013. Inflammatory responses in hypoxic ischemic encephalopathy. Acta Pharmacol Sin 34, 1121–1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marecki S, Atchison ML, Fenton MJ, 1999. Differential expression and distinct functions of IFN regulatory factor 4 and IFN consensus sequence binding protein in macrophages. J Immunol 163, 2713–2722. [PubMed] [Google Scholar]
- Masuda T, Iwamoto S, Yoshinaga R, Tozaki-Saitoh H, Nishiyama A, Mak TW, Tamura T, Tsuda M, Inoue K, 2014. Transcription factor IRF5 drives P2X4R+−reactive microglia gating neuropathic pain. Nat Commun 5, 3771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McRae A, Gilland E, Bona E, Hagberg H, 1995. Microglia activation after neonatal hypoxic-ischemia. Brain Res Dev Brain Res 84, 245–252. [DOI] [PubMed] [Google Scholar]
- Mirza MA, Ritzel R, Xu Y, McCullough LD, Liu F, 2015. Sexually dimorphic outcomes and inflammatory responses in hypoxic-ischemic encephalopathy. J Neuroinflammation 12, 32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagase H, Visse R, Murphy G, 2006. Structure and function of matrix metalloproteinases and TIMPs. Cardiovasc Res 69, 562–573. [DOI] [PubMed] [Google Scholar]
- Negishi H, Ohba Y, Yanai H, Takaoka A, Honma K, Yui K, Matsuyama T, Taniguchi T, Honda K, 2005. Negative regulation of Toll-like-receptor signaling by IRF-4. Proc Natl Acad Sci U S A 102, 15989–15994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikolakopoulou AM, Dutta R, Chen Z, Miller RH, Trapp BD, 2013. Activated microglia enhance neurogenesis via trypsinogen secretion. Proc Natl Acad Sci U S A 110, 8714–8719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ormstad H, Aass HC, Lund-Sorensen N, Amthor KF, Sandvik L, 2011. Serum levels of cytokines and C-reactive protein in acute ischemic stroke patients, and their relationship to stroke lateralization, type, and infarct volume. J Neurol 258, 677–685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel AR, Ritzel R, McCullough LD, Liu F, 2013. Microglia and ischemic stroke: a double-edged sword. Int J Physiol Pathophysiol Pharmacol 5, 73–90. [PMC free article] [PubMed] [Google Scholar]
- Price CJ, Menon DK, Peters AM, Ballinger JR, Barber RW, Balan KK, Lynch A, Xuereb JH, Fryer T, Guadagno JV, Warburton EA, 2004. Cerebral neutrophil recruitment, histology, and outcome in acute ischemic stroke: an imaging-based study. Stroke 35, 1659–1664. [DOI] [PubMed] [Google Scholar]
- Rosell A, Cuadrado E, Ortega-Aznar A, Hernandez-Guillamon M, Lo EH, Montaner J, 2008. MMP-9-positive neutrophil infiltration is associated to blood-brain barrier breakdown and basal lamina type IV collagen degradation during hemorrhagic transformation after human ischemic stroke. Stroke 39, 1121–1126. [DOI] [PubMed] [Google Scholar]
- Satoh T, Takeuchi O, Vandenbon A, Yasuda K, Tanaka Y, Kumagai Y, Miyake T, Matsushita K, Okazaki T, Saitoh T, Honma K, Matsuyama T, Yui K, Tsujimura T, Standley DM, Nakanishi K, Nakai K, Akira S, 2010. The Jmjd3-Irf4 axis regulates M2 macrophage polarization and host responses against helminth infection. Nat Immunol 11, 936–944. [DOI] [PubMed] [Google Scholar]
- Schneider A, Weier M, Herderschee J, Perreau M, Calandra T, Roger T, Giannoni E, 2018. IRF5 Is a Key Regulator of Macrophage Response to Lipopolysaccharide in Newborns. Front Immunol 9, 1597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swonger AK, Rech RH, 1972. Serotonergic and cholinergic involvement in habituation of activity and spontaneous alternation of rats in a Y maze. J Comp Physiol Psychol 81, 509–522. [DOI] [PubMed] [Google Scholar]
- Tamura T, Yanai H, Savitsky D, Taniguchi T, 2008. The IRF family transcription factors in immunity and oncogenesis. Annu Rev Immunol 26, 535–584. [DOI] [PubMed] [Google Scholar]
- Taylor RA, Sansing LH, 2013. Microglial responses after ischemic stroke and intracerebral hemorrhage. Clin Dev Immunol 2013, 746068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner RJ, Sharp FR, 2016. Implications of MMP9 for Blood Brain Barrier Disruption and Hemorrhagic Transformation Following Ischemic Stroke. Front Cell Neurosci 10, 56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vila N, Castillo J, Davalos A, Esteve A, Planas AM, Chamorro A, 2003. Levels of anti-inflammatory cytokines and neurological worsening in acute ischemic stroke. Stroke 34, 671–675. [DOI] [PubMed] [Google Scholar]
- Weber M, Moebius P, Buttner-Herold M, Amann K, Preidl R, Neukam FW, Wehrhan F, 2015. Macrophage polarisation changes within the time between diagnostic biopsy and tumour resection in oral squamous cell carcinomas--an immunohistochemical study. Br J Cancer 113, 510–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welser-Alves JV, Milner R, 2013. Microglia are the major source of TNF-alpha and TGF-beta1 in postnatal glial cultures; regulation by cytokines, lipopolysaccharide, and vitronectin. Neurochem Int 63, 47–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao SC, Wang C, Xu H, Wu WQ, Chu ZH, Ma LS, Zhang YD, Liu F, 2017. Age-related differences in interferon regulatory factor-4 and -5 signaling in ischemic brains of mice. Acta Pharmacol Sin 38, 1425–1434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou R, Yang Z, Tang X, Tan Y, Wu X, Liu F, 2013. Propofol protects against focal cerebral ischemia via inhibition of microglia-mediated proinflammatory cytokines in a rat model of experimental stroke. PLoS One 8, e82729. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary figure 1. IRF4 expression in activated microglia/macrophages at 7d after HIE. Representative images of co-expression of IRF4 (red) with Iba1 (green) in activated microglia/macrophages were shown. RF4+Iba1+ double positive cells (inset in the merged image) were only seen in IRF4fl/fl brain but not in IRF4 CKO brains. Scale bar = 100μm.
Supplementary figure 2. Brain leukocytes were gated on a forward scatter (FSC)/side scatter (SSC) plot. In live cell population, microglia and infiltrating myeloid cells were gated based on relative expression of CD45 and CD11b. The central immune cells (microglia) were considered as CD45intermediate CD11b+; infiltrating peripheral myeloid cells (pMyeloid) and infiltrating lymphocytes (Lymphocytes) were identified as CD45high CD11b+ and CD45high CD11b− respectively.