

Abstract
Background
Increasing evidence supports an extensive and complex genetic contribution to Parkinson’s disease (PD). Previous genome-wide association studies (GWAS) have shed light on the genetic basis of risk for this disease. However, the genetic determinants of PD age at onset (AAO) are largely unknown.
Objectives
To identify the genetic determinants of PD AAO.
Methods
Using genetic data of 28,568 PD cases we performed a GWAS based on PD AAO.
Results
We estimated that the heritability of PD AAO due to common genetic variation was ~0.11, lower than the overall heritability of risk for PD (~0.27) likely in part because of the subjective nature of this measure. We found two genome-wide significant association signals, one at SNCA and the other a protein-coding variant in TMEM175, both of which are known PD risk loci and a Bonferroni corrected significant effect at other known PD risk loci, GBA, INPP5F/BAG3, FAM47E/SCARB2, and MCCC1. Notably, SNCA, TMEM175, SCARB2, BAG3 and GBA have all been shown to be implicated in alpha-synuclein aggregation pathways. Remarkably, other well-established PD risk loci such as GCH1 and MAPT did not show a significant effect on AAO of PD.
Conclusions
Overall, we have performed the largest AAO of PD GWAS to date and our results show that not all PD risk loci influence AAO with significant differences between risk alleles for AAO. This provides a compelling picture, both within the context of functional characterization of disease linked genetic variability and in defining differences between risk alleles for AAO, or frank risk for disease.
Keywords: Parkinson’s disease, age at onset, GBA, SNCA, TMEM175
Introduction
Parkinson disease (PD) is the most common neurodegenerative movement disorder. PD is pathologically characterized by the loss of dopaminergic neurons in the substantia nigra and α-synuclein (encoded by SNCA) protein aggregates. Current estimates are that in 2015 there were 6.9 million PD patients worldwide and this number is predicted to be 14.2 million in 20401. PD has a strong age-dependent prevalence and male-female differences, where males are ~1.5 times more likely to develop PD2, 3.
The exact cause of PD is unknown, however there is clear evidence that genetic variability plays a role in both disease development and progression. In the past two decades, mutations in several genes have been identified that cause monogenic forms of PD, accounting for about 1–5% of all PD cases4. The majority of PD cases are therefore referred to as sporadic PD. Genome-wide association studies (GWAS) have successfully identified over 40 loci robustly associated with PD. Interestingly, variability in several genes, including SNCA, LRRK2, VPS13C, GCH1 and GBA appear to play a role in both monogenic and sporadic disease5.
Substantial evidence suggests that genetic variation plays a role in age at onset (AAO) where, for example, monogenic forms of PD often present as early onset cases, but the identity of the exact genetic component in sporadic PD remains unclear6, 7. Additionally, there is some evidence that AAO may be different between males and females8, 9. Multiple studies have nominated variants and genes of interest, but replication and large cohort studies are lacking7, 10, 11. Additionally, rare coding variants in PD risk genes like GBA and LRRK2 have been associated with an earlier AAO12–17. Currently the best predictor of PD AAO is a genetic risk score (GRS) based on cumulative genetic PD risk7, 18, implying broad genetic overlap between PD susceptibility and PD AAO.
In this study, we performed the largest PD AAO GWAS to date including 28,568 PD cases. Using this large cohort, we have estimated the heritability of PD AAO and have identified several associated variants. Additionally, we have shown that the latest PD GRS remains highly correlated with of AAO.
Materials and methods
Processing of IPDGC datasets
Genotyping data (all Illumina platform based) was obtained from International Parkinson’s Disease Genomics Consortium (IPDGC) members, collaborators and public resources (Supplementary Table1). All datasets underwent quality control separately, both on individual level data and variant level data before imputation. See Supplemental Methods for a detailed description of data processing. Where possible, AAO was defined based on patient report of initial manifestation of parkinsonian motor signs (tremor, bradykinesia, rigidity or gait impairment). Where this information was not available, age of diagnosis was used as a proxy for onset age. Note that the correlation was high between age of diagnosis and AAO.
The resulting quality controlled and imputed datasets of PD cases (total n=17,415) were analyzed with the formula AGE_AT_ONSET ~ SNP + SEX + PC1-PC5. Analyses were performed per dataset with rvtests linear regression using imputed dosages19. System Genomics of Parkinson’s Disease (SGPD) data (n=581) was processed as above with the following minor changes: A linear regression model in PLINK was used to run a GWAS for AAO and relatedness cut-off of 0.05 was used. The association analysis was adjusted for sex and the first 10 eigenvectors from PC analysis.
Processing of the 23andMe dataset
As an independent second cohort we used the 23andMe PD dataset (n=10,572), which consisted of customers of the personal genetics company, 23andMe, Inc., who had consented to participate in research. PD patients were recruited through a targeted email campaign in conjunction with the Michael J. Fox Foundation or other partners, patient groups and clinics. PD cases were individuals who self-reported having been diagnosed with PD and who self-reported their age of PD diagnosis. We used age of diagnosis rather than age of symptom onset because symptom onset is often gradual and may be more difficult to self-report accurately. Note that the correlation was high between age of diagnosis and AAO (ρ = 0.917, P<1E-300) and the average difference between age of diagnosis and AAO was 2.58 years. We have previously shown that self-reported PD case status is accurate with 50 out of 50 cases confirmed via telemedicine interview20. See Supplemental Methods for a detailed description of data processing. After quality control, which included R2 > 0.5 variant removal, we analyzed 11,956,580 SNPs. Remaining PD cases (n=10,572) were analyzed using the formula AGE_AT_DIAGNOSIS ~ SNP + SEX + PC1-PC5 + genotyping platform. The genomic inflation factor was λ = 1.015.
Meta-analyzing dataset
The 17 IPDGC datasets were meta-analyzed using METAL (v.2011–03-25) fixed effects using default settings21. We excluded SNPs with an I2 statistic >50% and variants that were present in fewer than 66.7% of the datasets. One genome-wide significant variant was excluded due to this filter criteria (chr4:90711770, I2 = 58.1%. This resulted in a total of 6,850,647 variants and a genomic inflation factor of λ = 1.001. For the combined GWAS we meta-analysed all 17 IPDGC datasets with the 23andme dataset using the same quality control steps.
Additional analyses and figures
PD GWAS loci were obtained from5 using Table1 and Table2 and using P.META from the provided summary stats (see Supplementary Table3). Additionally, to assess the influence of a genetic risk score (GRS) for PD case-control status on PD AAO we obtained the GRS from the most recent PD GWAS Supplementary5. The GRS was calculated and processed using PLINK for each individual as described previously7. Associations were performed using the formula AGE_AT_ONSET ~ GRS + SEX + PC1-PC5. Quantile-quantile (QQ)-plots and Manhattan plots were generated using FUMA22. Forest plots were generated in R using the package rmeta (https://cran.r-project.org/web/packages/rmeta/index.html). Locus plots23 were generated for the genome-wide significant loci and were compared to the latest published PD GWAS5. GCTA-cojo analyses were performed to identify whether there were multiple independent signals in loci of interest using all IPDGC datasets (excluding SGPD) described in Supplementary Table1 as a reference panel 24, 25. This reference panel was created using variants passing the following criteria: R2<0.8, minor allele frequency > 0.001, Hardy-Weinberg equilibrium P < 1E-6, and a maximum variant missingness of 5%. The genetic correlation between the PD AAO GWAS and the PD GWAS5 was calculated using LD Score Regression (LDSC)26 using default settings for European populations. Heritability was estimated using LDSC and GCTA based on GWAS summary statistics and individual-level genotypes, respectively.
Power calculations
We performed power calculations using the method of Brion et al.27
where is a random variable from a non-central with one degree of freedom, and is the threshold of a central distribution with one degree of freedom and a type-I error rate of 0.05. The non-centrality parameter was calculated using the method of Sham and Purcell28
is the non-centrality parameter, p is the minor allele frequency, is the effect size in years, and variance(Y) is the variance in PD AAO. We estimated variance(Y) by taking the mean variance across each cohort, weighted by their sample size. We assessed effect sizes between 0.01 to 1 years and reported the minimum effect size that yielded >80% power.
Results
Initial data overview
In total we included 18 datasets: the IPDGC data contains of 17 independent cohorts (n=17,996) and the 23andMe PD cohort (n=10,572, see Supplementary Table1 for more details). The average AAO in the IPDGC dataset was 62.14 (range 20–96, SD=12.08), while in the 23andMe dataset the average age of diagnosis was 60.71 (range 40–97, SD=9.98). We found minor differences in AAO in females and males in both the IPDGC dataset (females = 62.15, SD=11.71; males = 62.03, SD=11.95) and the 23andMe dataset (females = 59.95, SD= 9.79; males = 61.20, SD=10.07).
Heritability of Parkinson’s disease age at onset
Using LDSC, the heritability of PD AAO was h2=0.076 (SE=0.0277) in the IPDGC cohort, h2=0.0805 (SE=0.0403) in the 23andMe cohort, and 0.109 (SE=0.0255) in the complete meta-analysis. These heritability estimates for PD AAO were similar to estimates derived using GCTA in the largest IPDGC dataset (IPDGC_NeuroX, h2=0.0798, SE=0.0391, P=0.0184, N=5,428) and in the 23andMe dataset (h2=0.1235, SE=0.0341, P=1.031E−4, N=10,697). Heritability estimates in the other IPDGC datasets were considered less reliable due to the low number of included cases.
SNCA as top associated loci with Parkinson’s disease age at onset
Two loci reached genome-wide significance, SNCA and TMEM175, both of which are established PD risk loci based on PD case/control GWASes (Figure1). Common variation at the SNCA locus has clearly been established as a risk factor PD and both rare mutations and whole gene multiplications have been identified to cause monogenic PD29, 30. Several independent signals have been reported in this locus, where initially a variant in intron 4 was identified in PD31, later a second independent signal at the 5’ end was identified32, which is also associated with Dementia with Lewy bodies33 and currently at least three independent signals are present in this locus34. Here, we identified both signals to be associated with AAO (Figure2A). The 3’ end signal is the strongest with rs356203 as the most associated SNP, resulting in a reduction of ~0.6 year in AAO (P_meta=1.90E-12, beta=−0.626, SE=0.0890). Using conditional analysis, we also identified the 5’ end signal (Figure2B, rs983361, 4:90761944, P_conditional=6.82E-6, beta=−0.484, SE=0.108)
Figure1: Manhattan plot of Parkinson’s disease age at onset GWAS.

Based on meta-analyses of datasets (n=28,568) using 7,426,111 SNPs. Two genome-wide significant loci were identified: SNCA and the TMEM175/GAK. Additionally, one borderline significant locus was found, APOE.
Figure2: SNCA association with Parkinson’s disease age at onset.
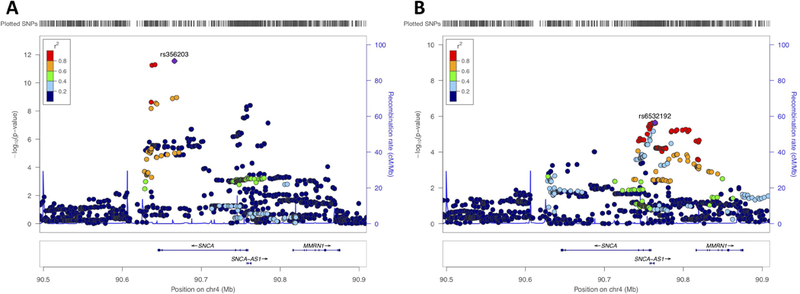
A) Locus zoom plot of the association signal of the SNCA locus. The age at onset association signal is primarily based at the 3’ end of the SNCA gene, highly similar as the Parkinson’s disease GWAS signal of Chang et al 2017 (Supplementary Figure8). B) Conditional analysis based on the most associated SNP (rs356203) shows that there is a secondary signal at the 5’ end of the SNCA gene rs6532192, as well similar as previously reported for the Parkinson’s disease GWAS Chang et al 2017.
A coding variant in TMEM175 is associated with Parkinson’s disease age at onset
The TMEM175/GAK locus is another known PD locus where two independent signals have been identified32. While it is more parsimonious to assume that a single GWAS peak is the product of a single causal gene in the locus, there is evidence that both TMEM175 and GAK may modulate PD risk35, 36.TMEM175 and GAK share a promoter, suggesting that they may be co-regulated and may perform related cellular functions. Interestingly, the TMEM175/GAK locus is one of the few PD GWAS loci where a coding variant (TMEM175 p.M393T, exon 11, rs34311866) is amongst the highest associated variants (Figure2A). This coding variant was also the most associated variant in this locus in our PD AAO analysis (P_meta=9.62E-9, beta=−0.613, SE=0.107), resulting in an average reduction of ~0.6 years (Figure3B and Figure5). Using conditional analysis based on rs34311866 we did not identify a second independent signal in this locus (P=0.879, beta=−0.0240, SE=0.158, Supplementary Figure9).
Figure3. TMEM175/GAK locus association with Parkinson’s disease age at onset and Parkinson’s disease.
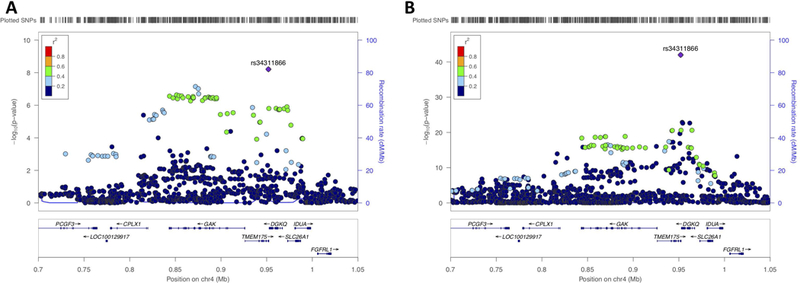
A) Locus zoom plot of the association signal of the TMEM175/GAK locus with Parkinson’s disease age at onset. The association signal is primarily based on the coding variant TMEM175 p.M393T, rs34311866. B) Locuszoom plot of the association signal of the TMEM175/GAK locus with Parkinson’s disease vs controls Chang et al 2017. Similarly, as in A the main signal is based on the coding variant TMEM175 p.M393T, rs34311866.
Figure5: Minor allele frequency differences correlated with age groups separated by case-control status.
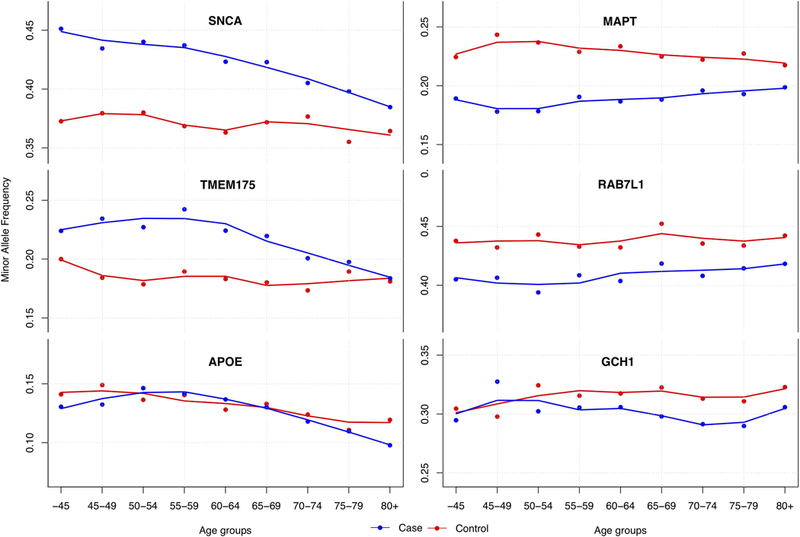
Genotype data was merged and minor allele frequency were calculated per age group and separated by case control status (IPDGC data only: N_cases=17,996 and N_controls=16,502, each age group contains >850 individuals). Lines were using LOESS regressions based on the minor allele frequency per age group. On the left the three most significant loci associated with PD age at onset are shown; SNCA=rs356203, TMEM175/GAK=rs34311866 and APOE=rs429358. On the right three significant PD case control GWAS loci are shown to have no clear effect on PD age at onset; MAPT=rs17649553, RAB7L1=rs823118, GCH1= rs11158026. Standard error-bars are not shown in figurebut are neglectable(~0.015) for each age-group.
Parkinson’s disease high risk variants in GBA and LRRK2 and age at onset
LRRK2 p.G2019S and GBA variants are the most commonly identified genetic risk factors for PD. LRRK2 p.G2019S is identified in ~1% of the general European PD population, but higher frequencies have been reported in other populations37. GBA variants are more commonly identified in Ashkenazi Jewish populations but are also present in the European population38.
LRRK2 p.G2019S (rs34637584) has already been described to have large range of AAO and reduced penetrance12, 14. Due to the low frequency and low imputation quality of this variant it did not pass the variant level QC and was only tested in 9 of the 18 datasets. Similar to previous studies37, we identified 140 carriers in the IPDGC datasets (~0.8% of all PD cases). The average AAO for p.G2019S carriers was 66.58 (range= 36–95, SD=11.66), which is ~4 years later compared to the average AAO of non-carriers in the IPDGC datasets (62.14, range 20–96, SD=12.08). However, this is based on a relatively small amount of cases and some cohorts excluded LRRK2 p.G2019S carriers pre-genotyping or excluded familial PD cases. Besides, the non-coding variant at the 5’ end of LRRK2 (rs76904798) which is also identified as a risk factor PD did not show an association with AAO (P=0.1267, beta=−0.184, SE=0.121).
For GBA there have been several reports that variants in GBA affect AAO in PD39–41. In our large meta-analysis we identified a borderline significant hit for the GBA p.N370S variant (rs76763715, P=2.628E-6, beta=−2.600, SE=0.553) which resulted in a relatively large reduction of 2.6 year in AAO. This variant was only tested in 13 of the 18 datasets due to the low frequency or low imputation quality. Using conditional analysis, we did not identify other clear significant signals in the GBA gene (Supplementary Figure11). However, other the coding PD risk variants in GBA reached nominally significant P values (rs2230288, p.E326K: P_meta=0.00123, beta=−0.929, SE=0.287 and rs75548401, p.T369M: P_meta=0.01153, beta=−1.281, SE=0.507).
Parkinson’s disease genetic risk loci are associated with age at onset
Previously a GRS based on PD GWAS loci has been identified as the main genetic predictor for AAO7. Here we tested the association between the updated 47-SNP GRS used in the latest PD GWAS study5. For each IPDGC dataset the GRS was calculated and we identified a consistent association between the Chang et al. GRS and PD AAO. After meta-analyzing results from the individual cohorts, we found that each 1SD increase in GRS led to an earlier AAO by ~0.8 years (summary effect=−0.801, 95%CI (−0.959, −0.643), I2=11.6%) (Supplemental Figure13). Although this effect was highly significant, the genetic proportion explained by the GRS was ~0.56%, and the SNCA and TMEM175 loci combined explained ~0.3%, most likely due to the lower heritability. Furthermore, there was a significantly negative genetic correlation between the AAO GWAS summary statistics and the most recent PD GWAS 5 using LDSC (rg=−0.5511, se=0.103, p=9.027E−8).
When solely looking at the 44 SNPs that were genome-wide significant in the most recent PD GWAS5, we identified a significant effect in six loci after Bonferroni correction: SNCA, TMEM175, BST1, INPP5F/BAG3, FAM47E/SCARB2, and MCCC1 (Figure4, Supplemental Table3 and Supplementary Figure14). Interestingly, for some well-established loci no significant P-values were identified in any of the datasets, including GCH1 (P_meta= 0.9769, 80% power to detect a change in AOO >0.28 years), RAB7L1/NUCKS1 (PARK16) (P_meta=0.124, 80% power to detect a change in AOO >0.26 years), and MAPT (P_meta=0.745, 80% power to detect a change in AOO >0.32 years) (Figure5) (see Supplementary Table3 for all power calculations).
Figure4: P-value and beta value correlation plot between PD GWAS and age at onset GWAS.
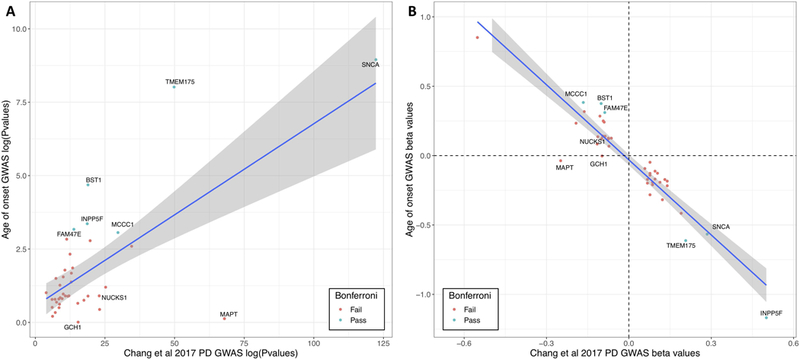
A) Log transformed P-values were plotted of the Chang et al 2017 PD GWAS (x-axis) and the current age at onset GWAS (y-axis). SNPs are annotated by their closest gene. Green dots are loci that pass Bonferroni correction and red are loci that did not pass Bonferroni correction. B) Beta-values were plotted of the Chang et al 2017 PD GWAS (x-axis) and the current age at onset GWAS (y-axis). SNPs are annotated by their closest gene. Green dots are loci that pass Bonferroni correction and red are loci that did not pass Bonferroni correction.
APOE E4 allele as positive control longevity marker
Interestingly, the variant representing the APOE E4 allele (chr19:45411941, rs429358) was among the borderline significant loci with a P_meta value of 5.696E-8 resulting in a 0.7 year earlier of AAO (beta=−0.707, SE=0.130). The APOE E4 allele is the most common genetic risk factor for Alzheimer’s disease42, but has no effect on PD (P-value in latest GWAS=0.625)32, 43. Interestingly, it has already been previously identified as an aging marker44, 45. To investigate this in our data we performed a GWAS on the reported age of controls from the IPDGC datasets and this demonstrated a result consistent with that seen in the PD cases: P value of 1.49E−5 (Effect=−0.644, SE=0.149), suggesting that associations seen at this locus are general age related effects (Figure5). Conditional tests based on rs429358 showed that the whole signal is coming from the APOE E4 allele (Supplementary Figure10). Additionally, none of the PD loci tested has a significant P value after correction for multiple testing (Supplemental Table5).
Replication of previous associated loci and potential sex effects
In the past decade, several studies have performed PD AAO analyses and nominated many genes and variants of interest (Supplemental Table2). Many of these variants and genes were identified in smaller datasets and were reported with nominal P-values or could not be replicated in other datasets. In our current GWAS we replicate the findings in SNCA, GBA and TMEM175 as described above (Supplemental Table2).
Additionally, some evidence exists there that there might be sex differences in AAO. To investigate this, we divided the IPDGC datasets (excluding the SGPD dataset) into a males-only dataset (N=11,411) and a females-only dataset (N=6,621) and performed sex-specific AAO GWASes and meta-analyses. No genome wide-significant hits were identified in either the male or the female meta-analysis and the effect sizes for the most recent PD GWAS variants Chang et al were similar (Pearson correlation=0.532, P=0.00034, Supplementary Table4). Interestingly, we did identify a similar sex-specific effect of the COMT coding variant rs4680 (p.V158M) as reported previously: P=0.0133 in males (Effect=0.370, SE=0.150) and P=0.922 in females (Effect=−0.0188, SE=0.193)46.
Discussion
Here we performed a genome-wide AAO analysis using 28,568 PD cases and discovered several PD loci associated with AAO. We identified two genome-wide significantly associated loci—SNCA (P_meta = 1.90E-12) and TMEM175 (P_meta = 9.62E-9)—as well as several sub-significant loci including GBA, SCARB2, and BAG3. Notably, this is the first study reporting genome-wide significance for PD GWAS loci for PD AAO. The heritability estimate of PD AAO was ~0.11, which is much less than the heritability of PD case-control status ~0.2747. Our heritability estimate was lower than an estimate from a previous study in familial PD, although this was based on a relatively small dataset (n=504 families)6. This is likely, in large part, due to the subjective nature of this phenotype.
It has been shown that PD case-control GWAS loci are associated with PD AAO7. We found that a GRS based on the latest PD GWAS5 was also highly associated with an earlier age of PD onset (Figure6). Furthermore, there was a significant negative genetic correlation between the PD case-control GWAS and the PD AAO GWAS. Six individual loci remain significant after Bonferroni correction, including two well-established PD loci—SNCA and TMEM175—and four less studied PD loci—BST1, INPP5F/BAG3, FAM47E/SCARB2, and MCCC1. Interestingly, no effect was identified for several well-established loci including GCH1, RAB7L1 (PARK16) and MAPT (Supplemental Table3). For these three loci, our study had 80% power (at P=0.05) to detect changes greater than 0.28, 0.26, and 0.32 years, respectively. For comparison, the two genome-wide significant hits in SNCA and TMEM175 modified AAO by 0.57 and 0.61 years. We believe this provides compelling evidence that only a subset of PD case-control risk SNPs also modulates PD AAO, and notably that variability at MAPT, a major GWA identified risk factor for PD does not influence AAO. We believe these data may suggest that different PD loci modulate risk via different pathways and that this likely has therapeutic consequences.
Interestingly, three of our top hits (SNCA, TMEM175, and GBA) are strongly implicated in α-synuclein mechanisms of PD pathogenesis pathology. SNCA encodes for the α-synuclein protein which is the major constituent of Lewy bodies, the defining pathology of PD. Both common and rare variants at SNCA are associated with increased PD risk, including duplications or triplications of the genomic locus29. One of the distinguishing features of autosomal dominant PD caused by SNCA gene multiplications is an early AAO48, 49. The common variants at SNCA associated with PD risk and AAO have also been demonstrated to enhance SNCA gene expression50. Thus, based on the genetics of both Mendelian and sporadic PD SNCA dosage appears to be a major driver of disease onset age. TMEM175 has been shown to impair lysosomal and mitochondrial function and increases α-synuclein aggregation36. GBA has been shown to be involved in several α-synuclein aggregation mechanisms, including autophagy and enhancing α-synuclein cell to cell transmission51–53. Other loci identified by our PD AAO analyses have also been linked to alpha-synuclein clearance, for example for SCARB2, encoding the ER-to-lysosome transporter of GBA, and BAG354, 55. Taken together, it is tempting to speculate about the direct link between PD AAO loci influencing the α-synuclein accumulation and clearance pathways and the other non-significant PD loci acting in another pathway by a different mechanism, but more experiments are needed to confirm this.
Several previous studies have performed PD AAO analyses in smaller datasets. We were unable to replicate the majority of these previous reported variants, likely because of the lack of power to detect a true signal in the smaller studies (<2,000 samples) (see Supplementary Table2). We may have been unable to replicate previous work by Hill-Burns et al. since much of their analysis was restricted to familial PD cases11. Their analysis of sporadic PD included almost 4,000 cases, but unlike our study and several others7, 40, 56 they did not find significant associations with SNCA (rs356203, P=0.355) or TMEM175 (rs34311866, P=0.525).
Although we included a very large amount of data there were still several limitations to our study. First, we had limited power to detect rare variants that modulate PD AAO due to the use of genotyping arrays and large number of cohorts with a relatively small size. Several reports have shown that rare variants may influence AAO of PD. For example, a recent report showed that rare variants in LRRK2 lowered AAO16. This likely also explains the relatively moderate P-values for rare coding variants in GBA and LRRK2 on age onset.
Second, due to the lack of reliable genotype data we excluded chromosomes X and Y from our analysis. Both of these limitations could be addressed by using genome sequencing data and large genome sequencing projects are currently underway. Third, the heritability of PD AAO (~0.11) was much lower than the heritability of PD case-control status47, which in part explains the lack of genome-wide significantly associated loci in our study. Fourth, there was a reasonable amount of heterogeneity in the mean AAO our different cohorts. Age of PD diagnosis was self-reported in some of our cohorts and assessed by physicians in other cohorts. Some cohorts were specifically designed to include younger onset cases. Other cohorts provided age of diagnosis while others used AAO, although these are likely to be highly correlated (r>0.9 in our data). By analyzing each cohort separately, we were able to mitigate many of these inter-study sources of variation. Indeed, there was relatively little heterogeneity of SNP effect size estimates between studies (Supplementary Table3). However, future data collection and studies would probably benefit from more specific and more predefined structured and symptom specific AAO diagnostic criteria. Implementation of such criteria in large studies and cohorts or even in healthcare systems could significantly improve the understanding of the genetics of PD AAO.
Overall, we have performed the largest AAO of PD GWAS to date and our results reveal an interesting and significant genetic component. Our results show that not all PD risk loci influence AAO with significant differences between risk alleles for AAO, which implies different mechanisms for risk for developing PD and PD AAO. This provides a compelling picture, both within the context of functional characterization of disease linked genetic variability and in defining differences between risk alleles for AAO, or frank risk for disease. The significantly associated variability is centered on the gene encoding alpha-synuclein and on variability in several other lysosomal proteins that have been shown to directly influence alpha-synuclein aggregation or clearance. Thus, these data support the notion that alpha-synuclein is an important target for future disease modifying or preventive therapies and that drugs targeting alpha-synuclein production and clearance may be most valuable.
Supplementary Material
Acknowledgements
We would like to thank all of the subjects who donated their time and biological samples to be a part of this study. We also would like to thank all members of the International Parkinson Disease Genomics Consortium (IPDGC). See for a complete overview of members, acknowledgements and funding http://pdgenetics.org/partners. We also thank the research participants and employees of 23andMe for making this work possible. This work was supported in part by the Intramural Research Programs of the National Institute of Neurological Disorders and Stroke (NINDS), the National Institute on Aging (NIA), and the National Institute of Environmental Health Sciences both part of the National Institutes of Health, Department of Health and Human Services; project numbers 1ZIA-NS003154, Z01-AG000949–02 and Z01-ES101986. In addition, this work was supported by the Department of Defense (award W81XWH-09–2-0128), and The Michael J Fox Foundation for Parkinson’s Research. This work was supported by National Institutes of Health grants R01NS037167, R01CA141668, P50NS071674, American Parkinson Disease Association (APDA); Barnes Jewish Hospital Foundation; Greater St Louis Chapter of the APDA. The KORA (Cooperative Research in the Region of Augsburg) research platform was started and financed by the Forschungszentrum für Umwelt und Gesundheit, which is funded by the German Federal Ministry of Education, Science, Research, and Technology and by the State of Bavaria. This study was also funded by the German Federal Ministry of Education and Research (BMBF) under the funding code 031A430A, the EU Joint Programme-Neurodegenerative Diseases Research (JPND) project under the aegis of JPND -www.jpnd.eu-through Germany, BMBF, funding code 01ED1406 and iMed-the Helmholtz Initiative on Personalized Medicine. This study is funded by the German National Foundation grant (DFG SH599/6–1) (grant to M.S), Michael J Fox Foundation, and MSA Coalition, USA (to M.S). The McGill study was funded by the Michael J. Fox Foundation and the Canadian Consortium on Neurodegeneration in Aging (CCNA). This study utilized the high-performance computational capabilities of the Biowulf Linux cluster at the National Institutes of Health, Bethesda, MD, USA. (http://biowulf.nih.gov), and DNA panels, samples, and clinical data from the National Institute of Neurological Disorders and Stroke Human Genetics Resource Center DNA and Cell Line Repository. People who contributed samples are acknowledged in descriptions of every panel on the repository website. We thank P Tienari (Molecular Neurology Programme, Biomedicum, University of Helsinki), T Peuralinna (Department of Neurology, Helsinki University Central Hospital), L Myllykangas (Folkhalsan Institute of Genetics and Department of Pathology, University of Helsinki), and R Sulkava (Department of Public Health and General Practice Division of Geriatrics, University of Eastern Finland) for the Finnish controls (Vantaa85+ GWAS data). We used genome-wide association data generated by the Wellcome Trust Case-Control Consortium 2 (WTCCC2) from UK patients with Parkinson’s disease and UK control individuals from the 1958 Birth Cohort and National Blood Service. Genotyping of UK replication cases on ImmunoChip was part of the WTCCC2 project, which was funded by the Wellcome Trust (083948/Z/07/Z). UK population control data was made available through WTCCC1. This study was supported by the Medical Research Council and Wellcome Trust disease centre (grant WT089698/Z/09/Z to NW, JHa, and ASc). As with previous IPDGC efforts, this study makes use of data generated by the Wellcome Trust Case-Control Consortium. A full list of the investigators who contributed to the generation of the data is available from www.wtccc.org.uk. Funding for the project was provided by the Wellcome Trust under award 076113, 085475 and 090355. This study was also supported by Parkinson’s UK (grants 8047 and J-0804) and the Medical Research Council (G0700943 and G1100643). Sequencing and genotyping done in McGill University was supported by grants from the Michael J. Fox Foundation, the Canadian Consortium on Neurodegeneration in Aging (CCNA) and in part thanks to funding from the Canada First Research Excellence Fund (CFREF), awarded to McGill University for the Healthy Brains for Healthy Lives (HBHL) program. PRoBaND data was funded by Parkinson’s UK (Grant ref J-1101) and supported by NHS Greater Glasgow & Clyde and University of Glasgow. DNA extraction work that was done in the UK was undertaken at University College London Hospitals, University College London, who received a proportion of funding from the Department of Health’s National Institute for Health Research Biomedical Research Centres funding. This study was supported in part by the Wellcome Trust/Medical Research Council Joint Call in Neurodegeneration award (WT089698) to the Parkinson’s Disease Consortium (UKPDC), whose members are from the UCL Institute of Neurology, University of Sheffield, and the Medical Research Council Protein Phosphorylation Unit at the University of Dundee. We thank the Quebec Parkinson’s Network (http://rpq-qpn.org) and its members. This work was supported by the Medical Research Council grant MR/N026004/1. Data used in the preparation of this article were obtained from the Parkinson’s Progression Markers Initiative (PPMI) database (www.ppmi-info.org/data). For up-to-date information on the study, visit www.ppmi-info.org.
PPMI, a public-private partnership, is funded by the Michael J. Fox Foundation for Parkinson’s Research and funding partners, including AbbVie, Avid, Biogen, Bristol-Myers Squibb, Covance, GE Healthcare, Genentech, GlaxoSmithKline, Lilly, Lundbeck, Merck, Meso Scale Discovery, Pfizer, Piramal, Roche, Servier, Teva, UCB, and Golub Capital. Data and biospecimens used in preparation of this manuscript were obtained from the Parkinson’s Disease Biomarkers Program (PDBP) Consortium, part of the National Institute of Neurological Disorders and Stroke at the National Institutes of Health. Investigators include: Roger Albin, Roy Alcalay, Alberto Ascherio, DuBois Bowman, Alice Chen-Plotkin, Ted Dawson, Richard Dewey, Dwight German, Xuemei Huang, Rachel Saunders-Pullman, Liana Rosenthal, Clemens Scherzer, David Vaillancourt, Vladislav Petyuk, Andy West and Jing Zhang. The PDBP Investigators have not participated in reviewing the data analysis or content of the manuscript. A full acknowledgement is available in the Supplementary data.
Footnotes
Declaration of Interests
Dr Nalls’ participation is supported by a consulting contract between Data Tecnica International LLC and the National Institute on Aging, NIH, Bethesda, MD, USA. Dr Nalls also consults for Genoom Health, Illumina Inc, The Michael J. Fox Foundation for Parkinson’s Research and University of California Healthcare. The other authors declare no competing interests. Ziv Gan-Or is consulting for Lysosomal Therapeutics Inc., Denali, Idorsia, Prevail Therapeutics and Inception Sciences. JC Corvol has received grants from the French Ministry of Health, Agence National pour la Recherche, INSERM, Actelion, Michael J Fox Foundation, Ipsen; consulting fees from BrainEver, Theranexus, BMS, Zambon, Abbvie, Clevexel unrelated to this research.
Data availability
IPDGC GWAS summary statistics are available (after publication) on the IPDGC website (http://pdgenetics.org/resources) and 23andMe GWAS summary statistics will be made available to qualified researchers under an agreement with 23andMe that protects the privacy of the 23andMe participants. Please visit http://research.23andme.com/collaborate/#publication for more information and to apply to access the data.”
References
- 1.Dorsey ER, Bloem BR. The Parkinson Pandemic-A Call to Action. JAMA Neurol 2018;75(1):9–10. [DOI] [PubMed] [Google Scholar]
- 2.Reeve A, Simcox E, Turnbull D. Ageing and Parkinson’s disease: why is advancing age the biggest risk factor? Ageing Res Rev 2014;14:19–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Moisan F, Kab S, Mohamed F, et al. Parkinson disease male-to-female ratios increase with age: French nationwide study and meta-analysis. J Neurol Neurosurg Psychiatry 2016;87(9):952–957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Singleton A, Hardy J. The Evolution of Genetics: Alzheimer’s and Parkinson’s Diseases. Neuron 2016;90(6):1154–1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chang D, Nalls MA, Hallgrímsdóttir IB, et al. A meta-analysis of genome-wide association studies identifies 17 new Parkinson’s disease risk loci. Nat Genet 2017;49(10):1511–1516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hamza TH, Payami H. The heritability of risk and age at onset of Parkinson’s disease after accounting for known genetic risk factors. J Hum Genet 2010;55(4):241–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nalls MA, Escott-Price V, Williams NM, et al. Genetic risk and age in Parkinson’s disease: Continuum not stratum. Mov Disord 2015;30(6):850–854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Alves G, Müller B, Herlofson K, et al. Incidence of Parkinson’s disease in Norway: the Norwegian ParkWest study. J Neurol Neurosurg Psychiatry 2009;80(8):851–857. [DOI] [PubMed] [Google Scholar]
- 9.Haaxma CA, Bloem BR, Borm GF, et al. Gender differences in Parkinson’s disease. J Neurol Neurosurg Psychiatry 2007;78(8):819–824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Latourelle JC, Pankratz N, Dumitriu A, et al. Genomewide association study for onset age in Parkinson disease. BMC Med Genet 2009;10:98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hill-Burns EM, Ross OA, Wissemann WT, et al. Identification of genetic modifiers of age-at-onset for familial Parkinson’s disease. Hum Mol Genet 2016;25(17):3849–3862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lee AJ, Wang Y, Alcalay RN, et al. Penetrance estimate of LRRK2 p.G2019S mutation in individuals of non-Ashkenazi Jewish ancestry. Mov Disord 2017;32(10):1432–1438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Malek N, Weil RS, Bresner C, et al. Features of -associated Parkinson’s disease at presentation in the UK study. J Neurol Neurosurg Psychiatry 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Marder K, Wang Y, Alcalay RN, et al. Age-specific penetrance ofLRRK2G2019S in the Michael J. Fox Ashkenazi Jewish LRRK2 Consortium. Neurology 2015;85(1):89–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nichols WC, Pankratz N, Marek DK, et al. Mutations in GBA are associated with familial Parkinson disease susceptibility and age at onset. Neurology 2009;72(4):310–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Xiao B, Deng X, Ng EY-L, et al. Association of LRRK2 Haplotype With Age at Onset in Parkinson Disease. JAMA Neurol 2018;75(1):127–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gan-Or Z, Amshalom I, Kilarski LL, et al. Differential effects of severe vs mild GBA mutations on Parkinson disease. Neurology 2015;84(9):880–887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Escott-Price V, International Parkinson’s Disease Genomics C, Nalls MA, et al. Polygenic risk of Parkinson disease is correlated with disease age at onset. Ann Neurol 2015;77(4):582–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhan X, Hu Y, Li B, Abecasis GR, Liu DJ. RVTESTS: an efficient and comprehensive tool for rare variant association analysis using sequence data. Bioinformatics 2016;32(9):1423–1426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dorsey ER, Ray Dorsey E, Darwin KC, et al. Virtual research visits and direct-to-consumer genetic testing in Parkinson’s disease. DIGITAL HEALTH 2015;1:205520761559299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Willer CJ, Li Y, Abecasis GR. METAL: fast and efficient meta-analysis of genomewide association scans. Bioinformatics 2010;26(17):2190–2191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Watanabe K, Taskesen E, van Bochoven A, Posthuma D. Functional mapping and annotation of genetic associations with FUMA. Nat Commun 2017;8(1):1826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pruim RJ, Welch RP, Sanna S, et al. LocusZoom: regional visualization of genome-wide association scan results. Bioinformatics 2010;26(18):2336–2337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yang J, Ferreira T, Morris AP, et al. Conditional and joint multiple-SNP analysis of GWAS summary statistics identifies additional variants influencing complex traits. Nat Genet 2012;44(4):369–375, S361–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yang J, Lee SH, Goddard ME, Visscher PM. GCTA: a tool for genome-wide complex trait analysis. Am J Hum Genet 2011;88(1):76–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bulik-Sullivan BK, Loh P-R, Finucane HK, et al. LD Score regression distinguishes confounding from polygenicity in genome-wide association studies. Nat Genet 2015;47(3):291–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Brion MJ, Shakhbazov K, Visscher PM. Calculating statistical power in Mendelian randomization studies. Int J Epidemiol 2013;42(5):1497–1501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sham PC, Purcell SM. Statistical power and significance testing in large-scale genetic studies. Nat Rev Genet 2014;15(5):335–346. [DOI] [PubMed] [Google Scholar]
- 29.Singleton AB, Farrer M, Johnson J, et al. alpha-Synuclein locus triplication causes Parkinson’s disease. Science 2003;302(5646):841. [DOI] [PubMed] [Google Scholar]
- 30.Polymeropoulos MH, Lavedan C, Leroy E, et al. Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science 1997;276(5321):2045–2047. [DOI] [PubMed] [Google Scholar]
- 31.Simón-Sánchez J, Schulte C, Bras JM, et al. Genome-wide association study reveals genetic risk underlying Parkinson’s disease. Nat Genet 2009;41(12):1308–1312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nalls MA, Pankratz N, Lill CM, et al. Large-scale meta-analysis of genome-wide association data identifies six new risk loci for Parkinson’s disease. Nat Genet 2014;46(9):989–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Guerreiro R, Ross OA, Kun-Rodrigues C, et al. Investigating the genetic architecture of dementia with Lewy bodies: a two-stage genome-wide association study. Lancet Neurol 2018;17(1):64–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pihlstrom L, Blauwendraat C, Cappelletti C, et al. A comprehensive analysis of SNCA-related genetic risk in sporadic parkinson disease. Ann Neurol 2018;84(1):117–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dumitriu A, Pacheco CD, Wilk JB, et al. Cyclin-G-associated kinase modifies α-synuclein expression levels and toxicity in Parkinson’s disease: results from the GenePD Study. Hum Mol Genet 2011;20(8):1478–1487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jinn S, Drolet RE, Cramer PE, et al. TMEM175 deficiency impairs lysosomal and mitochondrial function and increases α-synuclein aggregation. Proc Natl Acad Sci U S A 2017;114(9):2389–2394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Correia Guedes L, Ferreira JJ, Rosa MM, Coelho M, Bonifati V, Sampaio C. Worldwide frequency of G2019S LRRK2 mutation in Parkinson’s disease: a systematic review. Parkinsonism Relat Disord 2010;16(4):237–242. [DOI] [PubMed] [Google Scholar]
- 38.Sidransky E, Nalls MA, Aasly JO, et al. Multicenter analysis of glucocerebrosidase mutations in Parkinson’s disease. N Engl J Med 2009;361(17):1651–1661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gan-Or Z, Giladi N, Rozovski U, et al. Genotype-phenotype correlations between GBA mutations and Parkinson disease risk and onset. Neurology 2008;70(24):2277–2283. [DOI] [PubMed] [Google Scholar]
- 40.Lill CM, Hansen J, Olsen JH, Binder H, Ritz B, Bertram L. Impact of Parkinson’s disease risk loci on age at onset. Mov Disord 2015;30(6):847–850. [DOI] [PubMed] [Google Scholar]
- 41.Davis AA, Andruska KM, Benitez BA, Racette BA, Perlmutter JS, Cruchaga C. Variants in GBA, SNCA, and MAPT influence Parkinson disease risk, age at onset, and progression. Neurobiol Aging 2016;37:209.e201–209.e207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Liu C-C, Liu C-C, Kanekiyo T, Xu H, Bu G. Apolipoprotein E and Alzheimer disease: risk, mechanisms and therapy. Nat Rev Neurol 2013;9(2):106–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Federoff M, Jimenez-Rolando B, Nalls MA, Singleton AB. A large study reveals no association between APOE and Parkinson’s disease. Neurobiol Dis 2012;46(2):389–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Garatachea N, Emanuele E, Calero M, et al. ApoE gene and exceptional longevity: Insights from three independent cohorts. Exp Gerontol 2014;53:16–23. [DOI] [PubMed] [Google Scholar]
- 45.Broer L, Buchman AS, Deelen J, et al. GWAS of longevity in CHARGE consortium confirms APOE and FOXO3 candidacy. J Gerontol A Biol Sci Med Sci 2015;70(1):110–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Klebe S, Golmard J-L, Nalls MA, et al. The Val158Met COMT polymorphism is a modifier of the age at onset in Parkinson’s disease with a sexual dimorphism. J Neurol Neurosurg Psychiatry 2013;84(6):666–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Keller MF, Saad M, Bras J, et al. Using genome-wide complex trait analysis to quantify ‘missing heritability’ in Parkinson’s disease. Hum Mol Genet 2012;21(22):4996–5009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Konno T, Ross OA, Puschmann A, Dickson DW, Wszolek ZK. Autosomal dominant Parkinson’s disease caused by SNCA duplications. Parkinsonism Relat Disord 2016;22 Suppl 1:S1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ross OA, Braithwaite AT, Skipper LM, et al. Genomic investigation of α-synuclein multiplication and parkinsonism. Ann Neurol 2008;63(6):743–750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Soldner F, Stelzer Y, Shivalila CS, et al. Parkinson-associated risk variant in distal enhancer of α-synuclein modulates target gene expression. Nature 2016;533(7601):95–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Du T-T, Wang L, Duan C-L, et al. GBA deficiency promotes SNCA/α-synuclein accumulation through autophagic inhibition by inactivated PPP2A. Autophagy 2015;11(10):1803–1820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mazzulli JR, Xu Y-H, Sun Y, et al. Gaucher disease glucocerebrosidase and α-synuclein form a bidirectional pathogenic loop in synucleinopathies. Cell 2011;146(1):37–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bae E-J, Yang N-Y, Song M, et al. Glucocerebrosidase depletion enhances cell-to-cell transmission of α-synuclein. Nat Commun 2014;5:4755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cao Y-L, Yang Y-P, Mao C-J, et al. A role of BAG3 in regulating SNCA/α-synuclein clearance via selective macroautophagy. Neurobiol Aging 2017;60:104–115. [DOI] [PubMed] [Google Scholar]
- 55.Rothaug M, Zunke F, Mazzulli JR, et al. LIMP-2 expression is critical for β-glucocerebrosidase activity and α-synuclein clearance. Proc Natl Acad Sci U S A 2014;111(43):15573–15578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Brockmann K, Schulte C, Hauser A-K, et al. SNCA: major genetic modifier of age at onset of Parkinson’s disease. Mov Disord 2013;28(9):1217–1221. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.