

Abstract
Introduction:
Charcot-Marie-Tooth (CMT) phenotypes can be distinguished by electrophysiology and genetic analysis but few can be identified by their clinical characteristics. Distinctive phenotypes are useful in identifying affected individuals and providing additional clues about the mechanism of the neuropathy. Cranial neuropathies are uncommon features of CMT, and few reports of familial hemifacial spasm (HFS) and trigeminal neuralgia (TN) have been published.
Methods:
Sixty-three members of a large CMT 1B kindred were assessed for signs of peripheral neuropathy and cranial neuropathies then tested for the G163R mutation in the myelin protein zero (MPZ) gene.
Results:
Of 27 individuals with the G163R mutation in MPZ, 10 had HFS or TN. Co-existing HFS and TN were found in 3 of these and 4 had bilateral HFS or TN.
Discussion:
This kindred exhibits a distinct CMT phenotype characterized by the development of HFS or TN decades after clinical signs of hereditary neuropathy are manifest.
Keywords: Charcot-Marie-Tooth Disease, hereditary neuropathy, trigeminal neuralgia, hemifacial spasm, phenotype, cranial neuropathy, familial
Introduction
Charcot Marie Tooth disease (CMT) or hereditary motor and sensory neuropathy is a common inherited neuromuscular disorder usually characterized by a classic phenotype of distal weakness, sensory loss, and foot deformities presenting before adulthood. CMT is caused by mutations in a growing list of genes1 but the clinical presentations overlap considerably so the inheritance pattern and clinical features are inadequate to make a precise diagnosis. Nerve conduction velocities are often useful to further clarify the phenotype but genetic heterogeneity is marked even within these groups.
Considering all cases of genetically defined CMT, it is estimated that 55% have mutations in PMP22(CMT 1A), 15% in GJB1(CMTX) and 8.5% in myelin protein zero or MPZ (CMT 1B)2, although these proportions may change with increasing use of whole exome and whole genome studies. Over 150 mutations within MPZ are reported to cause disease3 and within these, there is marked phenotypic variability from a severe, neonatal onset (Dejerine-Sottas Syndrome) to a mild form with late onset and minimally slowed conduction velocities.4 CMT 1B with MPZ mutations is dominantly inherited but frequently presents as sporadic due to de novo mutations5 and the electrophysiologic pattern ranges from very slow to nearly normal conduction velocities.6 Some MPZ mutations appear to have distinct genotype-phenotype correlations relating to the position of the variant in the protein.4
Even when limb weakness is profound, symptomatic facial weakness or sensory loss is unusual for all forms of CMT, as cranial nerves are spared with a few exceptions. Deafness has been associated with specific point mutations in the PMP22 gene7,8 ; optic atrophy with alterations in the MFN2 gene9; vocal cord paralysis with changes in several CMT genes including GDAP110; and deafness with Adie’s pupils with changes in the MPZ gene.11 The pathophysiology of these unusual associated features is unknown, however, the genotype-phenotype correlations may be relevant in distinguishing these features from acquired etiologies.
Hemifacial spasm (HFS) and trigeminal neuralgia (TN) are sporadic conditions that usually develop in mid to late adulthood and are caused by aberrant neural discharges in the facial and trigeminal cranial nerves, respectively. There are scant reports of HFS or TN associated with CMT and none have been linked to a specific mutation. This report describes a large kindred with CMT1B associated with HFS and TN.
Methods
We identified a kindred from western North Carolina descended from an 18th century founder from Sussex, England. All individuals were interviewed and examined by one of the investigators (JC, CP, or JL). Their positions in the pedigree were determined and most submitted blood samples for genetic analysis. This research was approved by the Wake Forest Health Sciences and University of Utah Health Sciences Center Institutional Review Boards.
Phenotypic analysis
A total of 63 subjects traced to this founder were interviewed regarding symptoms of neuropathy, facial pain, and facial twitching and underwent a brief neurologic examination including strength grading using medical research council (MRC) grades. Some individuals had nerve conduction testing performed. Individuals determined to be definitely affected with CMT fulfilled either clinical or electrophysiologic criteria based on guidelines set out by the European CMT Consortium.12 Subjects determined to have TN fulfilled the International Headache Society criteria for idiopathic TN.13 Individuals were determined to have HFS if they described unilateral (or bilateral but independent) painless spasms of the orbicularis oculi with spread to lower facial muscles that were evident to other persons.
Mutation analysis
Fifty-eight of 63subjects submitted blood samples for genetic analysis. The exon-intron boundaries of the MPZ gene were established by sequence alignment of the H. sapiens MPZ sequence (GenBank accession NM_000530) with a chromosome 1 genomic contig, and confirmed with the Ensembl Human Geneview report (ENSG00000158887). Coding regions were confirmed by the predicted amino acid sequence alignment with the protein sequence for H. sapiens MPZ contained in GenBank (CAH70270). Intronic primers were designed to flank coding exon 4. DNA extracted from peripheral blood lymphocytes of subjects was amplified by PCR and sequenced at the University of Utah DNA Sequencing Core Facility. Sequence data were analyzed using the DNA sequencing software Sequencher (GeneCodes, Ann Arbor, Michigan). A few samples were tested at a commercial laboratory (Athena Diagnostics, Boston, Massachusetts).
Statistics
The difference in mean age between the subjects with and without HFS or TN was examined with a Student’s t-test. A Chi-Square test was used to test difference in the gender proportions. All statistical tests were two-sided and significance was determined at the 0.05 probability level.
Results
Twenty-five participants had signs and symptoms of peripheral neuropathy that fulfilled criteria for hereditary motor and sensory neuropathy (Figure 1). An additional two adolescent participants (VII: 9,10) had signs clinically suggestive of neuropathy but did not completely fulfill criteria for CMT likely due to their young age. Both children eventually tested positive for the MPZ mutation and are included in the analysis for a total of 27 affected individuals. The ratio of affected to examined (27:63) and multiple instances of male to male transmission are consistent with autosomal dominant inheritance. The age range of the participants affected with CMT was 13-73 years with an average age of 51.9 years and 16 (59%) were female. For those 27 persons affected with CMT, the self-reported age of onset of neuropathy symptoms or signs ranged from 2 years to 53 years with 15 participants stating childhood or teenage onset without providing a specific age. Five from this group believed onset was after age 30 but no outside medical records were available for review to substantiate this late onset. None reported infantile onset with delayed motor milestones. While onset of neuropathy for most was within the first 2 decades of life, there was considerable variability in the degree of distal weakness between adults in the same decade of life.
Fig.1.
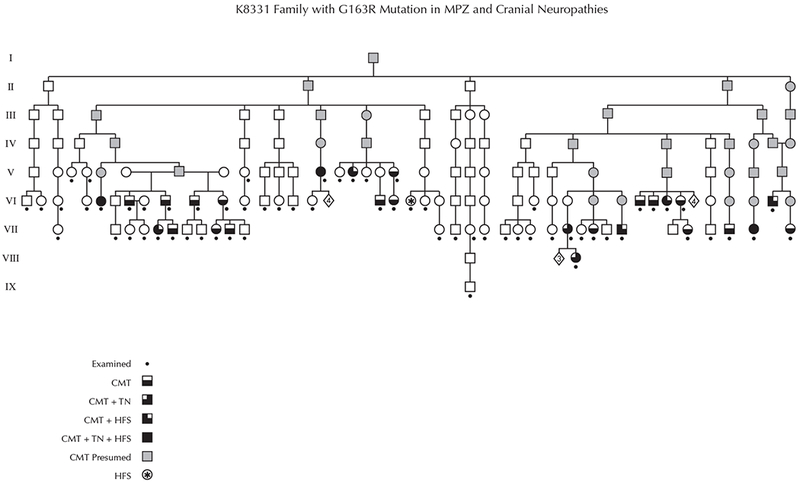
The complete pedigree ascertained through examination and genealogic records. Roman numerals indicate each generation. Gray shading indicates obligate affected individuals. The asterisk (*) indicates the individual affected with hemifacial spasm without clinical or genetic evidence for Charcot Marie Tooth disease. CMT = Charcot Marie Tooth; HFS = hemifacial spasm; TN = trigeminal neuralgia.
Electrophysiology was not systematically performed in all participants but our lab did collect various electrophysiologic data on 8 participants with CMT. Motor nerve conduction velocities ranged from 14-24 m/s in the lower extremities and from 22-25 m/s in the upper extremities. Two affected participants with HFS had facial nerve testing. Both had marked prolongation of distal latencies to facial muscles and of the blink responses. Electromyography of facial muscles showed motor unit enlargement with reduced recruitment and the spasms recorded were characteristic of HFS rather than myokymia.
Twenty-two participants were heterozygous for a point mutation (G to C) in exon 4 of the MPZ gene, which results in a non-synonymous exchange of a highly conserved transmembrane glycine at position 163 to arginine (G163R). An additional 5 examined patients met criteria for CMT and 2 of these underwent electrophysiologic testing which confirmed a demyelinating neuropathy. These 5 participants did not provide blood samples for mutation analysis. None of the participants without signs and symptoms of neuropathy demonstrated the G163R mutation.
The HFS/TN trait (either HFS or TN or both) was found in 10/27 (37%) of individuals with CMT. Three subjects had both HFS and TN, 5 HFS only, and 2 TN only. Half of the women (8/16) with CMT manifested the HFS/TN trait but only 2 of 11 men (18%). One patient (VI:20) without CMT also had HFS (see below). Three of the 8 participants with CMT and HFS had bilateral HFS and one of 5 participants with CMT and TN had bilateral TN (Table). The age at onset for typical neuropathy symptoms of CMT was in childhood and late teenage years for most of those affected but one person’s (VIII:3) first self-reported symptom of CMT was the development of TN at age 41. HFS presented from age 35-60 years (mean=48.8) and TN presented from age 41-56 years (mean=50.8), a non-significant difference (p=.68). Of two participants with TN who underwent vascular decompression, one had sustained improvement, and another TN participant did not respond to gamma knife irradiation. Those with HFS that had been treated with botulinum toxin injections reported consistent improvement.
Kindred members with HFS/TN
| Subject | Pedigree Position |
Sex | Age (y) | HFS,onset age (y) | TN,onset age (y) | TA |
|---|---|---|---|---|---|---|
| 1 | VII:29 | F | 68 | U, 58 | U, 56 | 4 |
| 2 | VIII:3 | F | 54 | N | U, 41 | 4 |
| 3 | VI:34 | F | 61 | U, 45 | N | 4 |
| 4 | VII:20 | F | 69 | N | U, 56 | 4 |
| 5 | V:15 | F | 67 | U, 52 | N | 4 |
| 6 | V:13 | F | 73 | B, 50 | B, 50 | 5 |
| 7 | VI:5 | F | 54 | B, 45 | U, unclear | 4 |
| 8 | VII:24 | M | 59 | B, 35 | N | 0 |
| 9 | VI:40 | M | 63 | U, 60 | N | 5 |
| 10 | VII:5 | F | 49 | U,45 | N | GW |
| 11 | VI:20 | F | 53 | U, 50 | N | 5 |
HFS=hemifacial spasm, TN=trigeminal neuralgia, U=unilateral, B=bilateral, TA=tibialis anterior power by MRC scale, N= not affected, GW=give way
At the time of examination, the CMT subjects with the HFS/TN trait were significantly older (mean=61.7 years) than subjects with CMT alone (mean = 46.7 years, p=.01). The mean age of CMT participants without the HFS/TN trait (46.7 years) was slightly younger than the mean age of onset for the HFS/TN trait (48.5 years). Tibialis anterior strength (using MRC grade) was similar (p=.96) between the two groups suggesting that typical neuropathy signs were not worse in the group with the HFS/TN trait.
None of the individuals without the G163R mutation had HFS/TN except one (VI:20). This 53 year old woman reported a history of unilateral HFS effectively treated with botulinum toxin. She did not have the G163R mutation and none of her first degree relatives (2 were examined) were affected by peripheral or cranial neuropathy. The treating neurologist confirmed the diagnosis of HFS and reported normal nerve conduction studies. An MRA of the brain revealed tortuous vertebral arteries in the area of the cranial nerve VII/VIII complex with the left vertebral artery indenting the left medulla, a possible explanation for the HFS.
Discussion
The high penetrance of the HFS/TN trait distinguishes this CMT1B family from others with CMT. There are a few reports of HFS or TN associated with genetically uncharacterized CMT.14–16 Most of these are small pedigrees but, in 1968, Lundberg and Westerberg studied a large Swedish family with TN, HFS, optic atrophy, Argyll-Robertson pupils, and abnormal eye movements associated with a hereditary neuropathy with demyelinating electrophysiology.17 The genotype of this family is unknown but our family differs from this one as none exhibited ophthalmologic abnormalities. A small portion of our kindred was reported in 1977 and, of 13 individuals with CMT, TN was noted in six and hearing loss was present in seven.18 HFS was not reported in any of the kindred at that time. None of the affected individuals examined for this report had severe, unexplained hearing loss.
Specific genotype-phenotype correlations are well known to occur in CMT1B with some mutations resulting in infantile onset often with delayed motor milestones and other genotypes resulting in a milder, late-onset presentation with axonal electrophysiology. The kindred presented here appears to be in the smaller group of CMT1B phenotypes that have childhood onset, demyelinating electrophysiology, and the late development of ambulatory disturbance.4
The substitution of a C for a G at position 487 results in the non-synonymous substitution of a transmembrane glycine for an arginine at a position that is evolutionarily conserved.19 The G163R mutation in MPZ has been associated with CMT previously20–22 but the HFS/TN trait was not reported in these cases. The trait might have been overlooked due to a smaller number of affected persons examined (4 in the report of Street et al21 and seven in the report of Eggers et al22) or affected participants from previous reports may have been pre-symptomatic for the HFS/TN trait. There is no clinical information in Nelis et al20 but, in the Street et al21 report, the ages of the CMT-affected individuals were 30, 32, 34, and 53 years old which is below the average age of onset of HFS/TN in our kindred. Small pedigrees of familial TN with and without HFS have been reported but no candidate genes have been proposed and these families did not have polyneuropathy.24,25
The data from our kindred indicate that the HFS/TN trait shows increased penetrance with age so the HFS/TN trait frequency of 37% reported here likely underestimates the true penetrance. To underscore this point, during our study encounters, we re-examined two individuals noted to have only hereditary neuropathy in the 1977 report who had subsequently developed HFS and TN. Similarly, another person (VII-5) in the kindred had only CMT when first examined by our group in 200126 but had developed HFS when she presented to neurology clinic in 2012.
HFS and TN are clearly over-represented in this kindred since the prevalence of sporadic HFS (11/100,000) and TN (4.5/100,000) is quite low.27,28 Bilateral HFS or TN occurs in only 3% of sporadic HFS and TN cases and the co-occurrence of HFS and TN is rarely reported.29–31 In contrast, 30% of those affected with the HFS/TN trait in this kindred have bilateral involvement and 30% have both HFS and TN.
The mechanism for this extreme predilection to develop HFS/TN is unclear. As in our kindred, sporadic cases of HFS and TN demonstrate advancing incidence with age and the onset age for sporadic HFS and TN is similar to that in our kindred.32,33 Sporadic HFS/TN is believed to be caused by compression of the cranial nerves by adjacent vasculature34,35 and this risk could be increased by nerve hypertrophy. Using neuromuscular ultrasound, we have previously shown that this family has hypertrophic nerves including the vagus nerve36 and other MPZ mutations have been associated with cranial nerve thickening.37 However, the degree of nerve hypertrophy found in this CMT1B family was similar to that found by others in CMT1A38 and, as HFS/TN is rare in other forms of CMT, nerve hypertrophy alone seems to be an inadequate explanation.
The HFS/TN trait shows increasing penetrance with age which parallels the gradual axon loss that occurs with CMT so perhaps the emergence of HFS/TN is due to axon loss in the facial and trigeminal nerves. Mild facial weakness and synkinesis were noted in many family members including some without the HFS/TN trait but the degree of facial weakness was not qualitatively different than in other forms of CMT. Similarly, distal weakness was not significantly worse in the group with the trait compared with those who did not express HFS/TN and other CMT genotypes with marked axon loss do not manifest HFS/TN so axon loss is unlikely to be causative in this pedigree.
The pathophysiology of MPZ mutations leading to neuropathy has been theorized to be due to toxic gain of function mutations that may produce the varied phenotypes of CMT1B.39 A common underlying mechanism of neuropathy appears to be protein misfolding resulting in retention of the mutant protein in the endoplasmic reticulum triggering the canonical unfolded protein response (UPR).40 Our kindred’s G163R genotype has not been examined for the UPR response but the similar G167R genotype activates the UPR more robustly than wild type MPZ.40 Like G163R, the G167R mutation is expressed in the transmembrane portion of the protein, causing MPZ to be inserted incorrectly in the membrane.41 Another MPZ mutation, Trp101X, is also predicted to result in inadequate anchoring of the protein into the myelin membrane and been associated with debilitating, chronic neuropathic pain so perhaps abnormalities in protein anchoring are common mechanisms of pain in CMT1B42. Abnormalities in myelin compaction and enhanced susceptibility to compression pathology have also been described in CMT1B and could result in a reduced threshold for aberrant discharges.37,43 It may be that both this specific transmembrane mutation and vascular compression must be present to develop HFS/TN in this family. A similar “double crush” mechanism has been proposed for multiple sclerosis associated TN where most cases demonstrate both a pontine lesion and vascular compression of the trigeminal nerve.44 Supporting this theory is the family member (VI-20) with HFS but without the G163R mutation. Her HFS was apparently due to adjacent tortuous vessels indicating that the MPZ mutation alone is not sufficient to cause HFS/TN but is acting as a strong risk factor for the trait. Another explanation for this family member’s HFS without an MPZ mutation is the possibility that the HFS/TN allele is associated with a gene loci that is independent but close to the MPZ locus. In this setting, the MPZ allele and the HFS/TN allele will co-segregate most of the time. To date, a genetic locus for familial TN or HFS has not been identified although familial clustering of these entities has been described.14,25 Therefore, a close locus, rather than a phenotypic variant of MPZ, remains in the differential in this family.
This CMT1B kindred has a distinctive phenotype of sensorimotor polyneuropathy often associated with HFS/TN. Further investigation of this family with detailed neuro-imaging would help determine whether the HFS/TN trait is due to factors intrinsic to the nerve or surrounding structures as is typical for HFS/TN. It will also be useful to determine if the HFS/TN trait is present in unrelated families with the G163R mutation or other transmembrane mutations. This description extends the spectrum of genotype-phenotype correlation for a gene commonly implicated in CMT. Understanding the mechanism of HFS and TN due to this mutation may provide insight into the pathophysiology of these cranial neuropathies in sporadic cases.
Acknowledgments
Disclosures:
This research was supported by NIH/NINDS K23 NS42713 (VL).
Dr. Caress receives research support from Orion Pharmaceuticals, Cytokinetics Inc, NIH/NINDS U01NS095388. None of these projects are relevant to the research presented in this manuscript.
Dr. Pinyan has nothing to disclose.
Dr. Lewis received limited funding from Athena Diagnostics Incorporated r travel expenses related to this research.
Dr. Lawson receives research support from the Reeves Pilot Project and Diamond Endowment Project. None of these projects are relevant to the research presented in this manuscript.
The authors would like to thank the K8331 family, Terrie Walker RN, Matt Hornsby, PT and Leah Griffin for their assistance in data collection and analysis.
Abbreviations
- CMT
Charcot Marie Tooth disease
- HFS
Hemifacial spasm
- MRC
Medical Research Council
- MPZ
Myelin Protein Zero
- TN
Trigeminal neuralgia
- UPR
Unfolded protein response
Footnotes
Statistical analysis was performed by Leah Griffin, Biostatistician II, Wake Forest Health Sciences Department of Public Health
We confirm that we have read the Journal’s position on issues involved in ethical publication and affirm that this report is consistent with those guidelines.
Contributor Information
James B. Caress, Department of Neurology, Wake Forest School of Medicine, Medical Center Boulevard, Winston-Salem, NC 27157.
James A. Lewis, The Neil Group, Winston-Salem, NC, Treehaus38@aol.com.
Clark W. Pinyan, Wake Forest School of Medicine, Winston-Salem, NC, cpinyan@wakehealth.edu.
Victoria H. Lawson, Dartmouth Hitchcock Medical Center, Geisel School of Medicine at Dartmouth, One Medical Center Drive, Lebanon, NH 03756-0001, Victoria.H.Lawson@hitchcock.edu.
References
- 1. http://www.molgen.ua.ac.be/CMTMutations/Mutations/MutByGene.cfm.
- 2.Saporta AS, Sottile SL, Miller LJ, Feely SM, Siskind CE, Shy ME. Charcot-Marie-Tooth disease subtypes and genetic testing strategies. Ann Neurol 2011;69:22–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. http://www.hgmd.cf.ac.uk/ac/index.php.
- 4.Sanmaneechai O, Feely SM, Scherer SS, Herrmann DN, Burns J, et al. Phenotype-genotype characteristics and baseline natural history of heritable neuropathies caused by mutations in the myelin protein zero gene. Brain 2015;138:3180–3192. - [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shy ME, Jani A, Krajewski KM, Lewis RA, Li J, Shy RR, et al. Phenotypic clustering in MPZ mutations. Brain 2004;127:371–84. [DOI] [PubMed] [Google Scholar]
- 6.DeJonghe P, Timmerman V, Ceuterick C, Nelis E, De Vriendt E, et al. The Thr124Met mutation in the peripheral myelin protein zero (MPZ) gene is associated with a clinically distinct Charcot-Marie-Tooth phenotype. Brain 1999;122:281–90. [DOI] [PubMed] [Google Scholar]
- 7.Kovach MJ, Lin JP, Boyadjiev S, Campbell K, et al. A Unique Point Mutation in the PMP22 Gene Is Associated with Charcot-Marie-Tooth Disease and Deafness Am J Hum Genet 1999;64:1580–1593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sambuughin N, de Bantel A, McWilliams S, Sivakumar K Deafness and CMT disease associated with a novel four amino acid deletion in the PMP22 gene. Neurology 2003;60:506–508. [DOI] [PubMed] [Google Scholar]
- 9.Züchner S, De Jonghe P, Jordanova A, Claeys KG, Guergueltcheva V, Cherninkova S, Hamilton SR, Van Stavern G, Krajewski KM, Stajich J, Tournev I, Verhoeven K, Langerhorst CT, de Visser M, Baas F, Bird T, Timmerman V, Shy M, Vance JM. Axonal neuropathy with optic atrophy is caused by mutations in mitofusin 2. Ann Neurol 2006;59:276–81. [DOI] [PubMed] [Google Scholar]
- 10.Sevilla T, Cuesta A, Chumillas MJ, Mayordomo F, Pedrola L, Palau F,et al. Clinical, electrophysiological and morphological findings of Charcot-Marie-Tooth neuropathy with vocal cord palsy and mutations in the GDAP1 gene. Brain 2003; 126: 2023–33. [DOI] [PubMed] [Google Scholar]
- 11.Misu K, Yoshihara T, Yamamoto M, Hattori N, Nagamatu M, Mukai E, et al. Two families of Charcot-Marie-Tooth disease with Adie’s pupil, axonal neuropahy and the Thr124Met mutation in the peripheral myelin protein zero gene. Rinsho Shinkeigaku 2000;40(2):149–54. [PubMed] [Google Scholar]
- 12.De Jonghe P, Timmerman V, Van Broeckhoven C. 2nd Workshop of the European CMT Consortium: 53rd ENMC International Workshop on Classification and Diagnostic Guidelines for Charcot-Marie-Tooth Type 2 (CMT2–HMSN II) and Distal Hereditary Motor Neuropathy (Distal HMN–Spinal CMT): 26–28 September 1997, Naarden, The Netherlands. Neuromuscul Disord 1998;8:426–431. [PubMed] [Google Scholar]
- 13.Anonymous. Classification and diagnostic criteria for headache disorders, cranial neuralgias and facial pain. Headache Classification Committee of the International Headache Society. Cephalalgia 1988;8(7):1–96. [PubMed] [Google Scholar]
- 14.Coffey RJ and Fromm GH. Familial trigeminal neuralgia and Charcot-Marie-Tooth neuropathy. Surg Neurol 1991;25:49–53. [DOI] [PubMed] [Google Scholar]
- 15.Testa D, Milanese C, La Mantia L, Mastrangelo M, Crenna P, Negri S. Familial trigeminal neuralgia in Charcot-Marie-Tooth disease. J Neurol 1981;225:283–287. [DOI] [PubMed] [Google Scholar]
- 16.Kalyanaraman K, Smith BH, Schlagenhauff RE. Hereditary hypertrophic neuropathy with facial and trigeminal involvement. Arch Neurol 1974;31:15–17. [DOI] [PubMed] [Google Scholar]
- 17.Lundberg PO and Westerberg CE. A hereditary neurological disease with facial spasm. J Neurol Sci 1968;8:85–100. [DOI] [PubMed] [Google Scholar]
- 18.Cruse RP, Conomy JP, Wilbourn AJ, Hanson MR. Hereditary hypertrophic neuropathy combining features of tic douloureus, Charcot-Marie-Tooth disease, and deafness. Cleve Clin Q 1977;44:107–111. [DOI] [PubMed] [Google Scholar]
- 19.Spiryda LB. Myelin protein zero and membrane adhesion. J Neurosci Res 1998;54:137–146. [DOI] [PubMed] [Google Scholar]
- 20.Nelis E, Haites N, Van Broekhoven C. Mutations in the peripheral myelin genes and associated genes in inherited peripheral neuropathies. Hum Mutat 1999;13:11–28. [DOI] [PubMed] [Google Scholar]
- 21.Street VA, Meekins G, Lipe HP, Seltzer WK, Carter GT, Kraft GH, Bird TD. Charcot-Marie-Tooth neuropathy: clinical phenotypes of four novel mutations in the MPZ and Cx 32 genes. Neuromuscul Disord 2002;12:643–650. [DOI] [PubMed] [Google Scholar]
- 22.Eggers SC, Keswani SC, Melli G, Cornblath DR. Clinical and genetic description of a family with Charcot-Marie-Tooth disease type 1B from a transmembrane MPZ mutation. Muscle Nerve 2004;29:867–869. [DOI] [PubMed] [Google Scholar]
- 23.Numakura C, Lin C, Ikegami T, Guldberg P, Hayasaka K. Molecular analysis in Japanese patients with Charcot-Marie-Tooth disease: DGGE analysis for PMP22, MPZ, and Cx32/GJB1 mutations. Hum Mutat 2002;20:392–8. [DOI] [PubMed] [Google Scholar]
- 24.Duff JM, Spinner RJ, Lindor NM, Dodick DW, Atkinson JLD. Familial trigeminal neuralgia and contralateral hemifacial spasm. Neurology 1999;53:216. [DOI] [PubMed] [Google Scholar]
- 25.Fleetwood IG, Innes AM, Hansen SR, Steinberg GK. Familial trigeminal neuralgia. J Neurosurg 2001;95:513–517. [DOI] [PubMed] [Google Scholar]
- 26.Lewis JA, Allen WA, Barbee CL, Seltzer W. Nantahala neuropathy: CMT, trigeminal neuralgia and hemifacial spasm caused by an MPZ mutation. J Neurol Sci 2001;187 (suppl 1):S479 Abstract [Google Scholar]
- 27.Love S and Coakham HB. Trigeminal neuralgia pathology and pathogenesis. Brain 2001;124:2347–2360 [DOI] [PubMed] [Google Scholar]
- 28.Auger RG and Whisnant JP. Hemifacial spasm in Rochester and Olmsted County, Minnesota 1960-1984. Arch Neurol 1990;47:1233–1234. [DOI] [PubMed] [Google Scholar]
- 29.Tan EK and Jankovic J. Bilateral hemifacial spasm: a report of five cases and a literature review. Mov Disord 1999;14(2):345–349. [DOI] [PubMed] [Google Scholar]
- 30.Zakrzewska JM. Diagnosis and differential diagnosis of trigeminal neuralgia. Clin J Pain 2002;18:14–21. [DOI] [PubMed] [Google Scholar]
- 31.Colosimo C, Bologna M, Lamberti S et al. A comparative study of primary and secondary hemifacial spasm. Arch Neurol 2006;63:441–444. [DOI] [PubMed] [Google Scholar]
- 32.Nilsen B, Le KD, Dietrichs E. Prevalence of hemifacial spasm in Oslo, Norway. Neurology 2004;63:1532–1533. [DOI] [PubMed] [Google Scholar]
- 33.Yadav YR, Nishtha Y, Sonjjay P, Vijay P, Shailendra R, Yatin K. Trigeminal neuralgia. Asian J Neurosurg 2017;12:585–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jannetta PJ, Abassy M, Maroon JC, Ramos FM, Albin MS. Etiology and definitive microsurgical treatment of hemifacial spasm. Operative techniques and results in 47 patients. JNeurosurg 1977;47(3);321–328. [DOI] [PubMed] [Google Scholar]
- 35.Maarbjerg S, Wolfram F, Gozalov A. Significance of neurovascular contact in classical trigeminal neuralgia. Brain 2014;138:311–319 [DOI] [PubMed] [Google Scholar]
- 36.Cartwright MS, Brown ME, Eulitt P, Walker FO, Lawson VH, Caress JB. Diagnostic nerve ultrasound in Charcot-Marie-Tooth disease type 1B. Muscle Nerve 2009;40:98–102. [DOI] [PubMed] [Google Scholar]
- 37.Shizuka M, Ikeda Y, Watanabe M, Okamoto K, Shoji M, Ikegami T, Hayasaka K. A novel mutation of the myelin P0 gene segregating Charcot-Marie-Tooth disease type 1B manifesting as trigeminal nerve thickening. J Neurol Neurosurg Psychiatry 1999;67(2):250–1. Letter [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Martinoli C, Schenone A, Bianchi S, Mandich P, Caponetto C, Abbruzzese M, et al. Sonography of the median nerve in Charcot-Marie-Tooth disease. Am J Roentgenol 2002; 178: 1553–1556. [DOI] [PubMed] [Google Scholar]
- 39.Wrabetz L, D’Antonio M, Pennuto M, Dati G, Tinelli E, Fratta P et al. Different intracellular pathmechanisms produce diverse Myelin Protein Zero neuropathies in transgenic mice. J Neurosci 2006;26:2358–2368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bai Y, Wu X, Brennan KM, Wang DS, D’Antonio M, Moran J et al. Myelin protein zero mutations and the unfolded protein response in Charcot Marie Tooth disease type 1B. Ann Clin Transl Neurol 2018;5:445–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shames I, Fraser A, Colby J, Orfali W, Jackson Snipes G. Phenotypic differences between peripheral myelin protein-22 (PMP22) and myelin protein zero (P0) mutations associated with Charcot-Marie-Tooth-related diseases. J Neuropathol Exp Neurol 2003;62(7):751–64. [DOI] [PubMed] [Google Scholar]
- 42.Ramirez JD, Barnes PRJ, Mills KR, Bennett DLH. Intermediate Charcot-Marie-Tooth disease due to a novel Trp101stop myelin protein zero mutation associated with debilitating neuropathic pain. Pain 2012;153:1763–1768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Magot A, Latour P, Mussini, JM, Mourtada R, Guihenuec P, Pereon Y. A new MPZ mutation associated with a mild CMT1 phenotype presenting with recurrent nerve compression. Muscle Nerve 2008;38: 1055–1059. [DOI] [PubMed] [Google Scholar]
- 44.Truini A, Prosperini L, Calistri V, Fiorelli M, Pozzilli C, Millefiorini E, Frontoni M, Cortese A, Caramia F, Cruccu G. A dual concurrent mechanism explains trigeminal neuralgia in patients with multiple sclerosis. Neurology 2016;86(22): 2094–2099. [DOI] [PubMed] [Google Scholar]