

Abstract
Collagen alpha-1(III) chain, also known as the alpha 1 chain of type III collagen, is a protein that in humans is encoded by the COL3A1 gene. Three alpha 1 chains are required to form the type III collagen molecule which has a long triple-helical domain. Type III collagen, an extracellular matrix protein, is synthesized by cells as a pre-procollagen. It is found as a major structural component in hollow organs such as large blood vessels, uterus and bowel. Other functions of type III collagen include interaction with platelets in the blood clotting cascade and it is also an important signalling molecule in would healing. Mutations in the COL3A1 gene cause the vascular type of Ehlers-Danlos syndrome (vEDS; OMIM 130050). It is the most serious form of EDS, since patients often die suddenly due to a rupture of large arteries. Inactivation of the murine Col3a1 gene leads to a shorter life span in homozygous mutant mice. The mice die prematurely from a rupture of major arteries mimicking the human vEDS phenotype. The biochemical and cellular effects of COL3A1 mutations have been studied extensively. Most of the glycine mutations lead to the synthesis of type III collagen with reduced thermal stability, which is more susceptible for proteinases. Intracellular accumulation of this normally secreted protein is also found. Ultrastructural analyses have demonstrated dilated rough endoplasmic reticulum and changes in the diameter of collagen fibers. Other clinical conditions associated with type III collagen are several types of fibroses in which increased amounts of type III collagen accumulate in the target organs.
Keywords: Extracellular matrix, Connective tissue, Vascular disease, Ehlers-Danlos syndrome, Fibrosis
1. Introduction
Type III collagen, first identified and described in 1971 (Miller et al., 1971), is an important structural protein, classified as one of the major fibrillar collagens (Prockop and Kivirikko, 1995). It constitutes about 5–20% of the entire collagen content in the human body (Miller, 1988). Its essential role in the structural integrity of arteries, uterus and bowel has been clearly demonstrated by studies on patients who harbour mutations in the COL3A1 gene (Byers, 1993 [updated 2019]; Byers et al., 2017; Malfait et al., 2017; Malfait, 2018). The cardinal clinical manifestations of these patients include spontaneous, life-threatening arterial, uterine and bowel ruptures (Byers, 1993 [updated 2019]; Byers et al., 2017; Malfait et al., 2017; Malfait, 2018). Other clinical phenotypes associated with COL3A1 missense and nonsense mutations include severe brain anomalies suggesting that COL3A1 is essential for the normal brain development.
This review summarizes the information on the human (COL3A1) and mouse (Col3a1) gene, transcripts and protein, and discusses the disease phenotypes associated with type III collagen mutations and altered protein levels, as well as mouse models. Table 1 lists the characteristics of the human (COL3A1) and mouse (Col3a1) gene, transcripts and protein, and provides links to available resources.
Table 1.
Characteristics of the COL3A1 gene, mRNA and protein in Homo sapiens and Mus musculus.
| Homo sapiens | Mus musculus | |
|---|---|---|
| Gene symbol | COL3A1 | Col3a1 |
| NCBI Gene ID | 1281 | 12825 |
| HGNC/MGI | 2201 | 88453 |
| OMIM | 120180 | |
| Chromosomal location | 2q32.2 | 1 C1.1 (23.67 cM) |
| Genome coordinates | Chr2:188974373-189012746a | Chr1:45311538-45349706 |
| GeneBank (gene) | NC_000002.12 | NC_000067.6 |
| Ensembl transcript ID | ENSG00000168542 | ENSMUST00000087883.12 |
| RefSeq (mRNA) | NM_000090.3 | NM_009930.2 |
| RefSeq (protein) | NP_000081.1 | NP_034060.2 |
| Gene length (kbp) | 38.4 | 38.2 |
| mRNA length (nt) | 5543 | 5564 |
| Number of exons | 51 | 51 |
| Number of codingexons | 51 | 51 |
| CCDS code | CCDS2297.1 | CCDS35554 |
| Uniprot name | Collagen alpha-1(III) chain | Collagen alpha-1(III) chain |
| UniProtKB/Swiss-Prot ID | P02461–1 b | P08121 |
| Length of protein in amino acids | 1466 | 1464 |
| Mass for single pro-α-chain (kDa) | 139 | 139 |
Based on GRCh38.p12.
Annotations in the Uniprot database entry P02461–1 are incomplete and do not incorporate domain expert knowledge. Also, there is no biological evidence that the Uniprot entry P02461–2 or the Ensembl entry COL3A1–202 (ENST00000317840.9) exist and they appear to be purely computational entities. Furthermore, exon coverage data based on RNA-sequencing shows no evidence for extensive alternative splicing. See Figures 1 and 2 for structural domains.
Data sources for Homo sapiens: https://www.ncbi.nlm.nih.gov/gene/1281; https://www.genecards.org/cgi-bin/carddisp.pl?gene=COL3A1; https://www.ncbi.nlm.nih.gov/CCDS/CcdsBrowse.cgi?REQUEST=GENEID&DATA=1281; https://www.uniprot.org/uniprot/P02461#P02461–1; https://www.omim.org/entry/120180?search=col3a1&highlight=col3a1;
Data sources for Mus musculus: https://www.ncbi.nlm.nih.gov/gene/12825; http://www.informatics.jax.org/marker/MGI:88453; https://www.uniprot.org/uniprot/P08121#P08121
2. Chromosomal location and intron-exon organization of COL3A1 and Col3a1
In the human genome, COL3A1 encoding the α1 chain of type III collagen is located on the long arm of chromosome 2 [2q32.2; genomic coordinates (GRCh38): Chr2:188,974,320189,012,746]. The gene is approximately 38 kb long and has 51 exons, which are numbered 152 to match the numbering of exons in the genes for other fibrillar collagens (Fig. 1) (Valkkila et al., 2001). The sizes of the exons vary from 45 nt (exons 13, 15, 18 and 30) to 1,105 (exon 52) (Valkkila et al., 2001). All exons except exon 2 begin with a complete codon, and all exons encoding the triple-helical domain start with a codon for a glycine (Valkkila et al., 2001). The first intron at approximately 11.5 kb is the largest one in the COL3A1 gene, whereas the second largest intron is 1,500 bp and the smallest intron is only 85 bp.
Fig. 1.
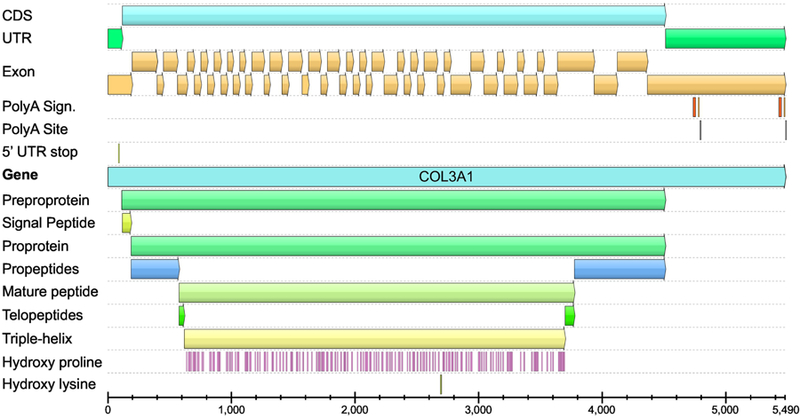
Structure of the human COL3A1 transcripts. The image shows the location of coding sequence (CDS), untranslated regions (UTR), 51 exons, polyA signals and sites, and the different functional domains. Locations of the proline residues that will be hydroxylated are also shown. The same color scheme was used in Figure 2. The annotations shown here differ slightly from those by Uniprot database (P02461–1), which are incomplete and did not incorporate domain expert knowledge. Also, there is no biological evidence that the Uniprot entry P02461–2 or the Ensembl entry COL3A1–202 exist and they appear to be purely computational entities. Furthermore, exon coverage data based on RNA-sequencing shows no evidence for extensive alternative splicing. The figure was generated using GeneBank entry NM_000090.3, and a software package UGENE (Okonechnikov et al., 2012). See Fig. 4 for schematic drawing of the different functional domains.
COL3A1 gene is in tail-to-tail orientation with a gene for another fibrillar collagen, namely COL5A2 (Valkkila et al., 2001). The sequence between these two genes is about 22 kb and it contains several repeat sequences including a LINE-1, two AT, and an Alu repeat (Valkkila et al., 2001). A phylogenetic analysis for the human COL3A1 and COL5A2 genes based on the sequences of the carboxy (C)-terminal domains indicated that these two genes likely evolved from a common ancestor (Valkkila et al., 2001).
Genetic variation present in the COL3A1 gene has been compared between four ethnic groups, namely African American, European, Mexican and Chinese (Chan et al., 2008). DNA samples from 48 unrelated individuals from each of these four groups were analysed for all the exonic and some intronic regions of four collagen genes (COL1A1, COL1A21, COL2A1 and COL3A1). A total of 114 polymorphic sites were identified in COL3A1, only four (Ala679Thr, Thr698Ala, Val1205Ile, and Gln1353His) of which led to an amino acid change. Phylogenetic trees generated using these results demonstrated that the African American group was evolutionary separated from the other three groups when COL3A1 sequences were analysed.
Two transcripts, 4.8 kb and 5.5 kb are generated from the COL3A1 gene using different polyadenylation sites (Fig. 2) (Ala-Kokko et al., 1989).
Fig. 2.
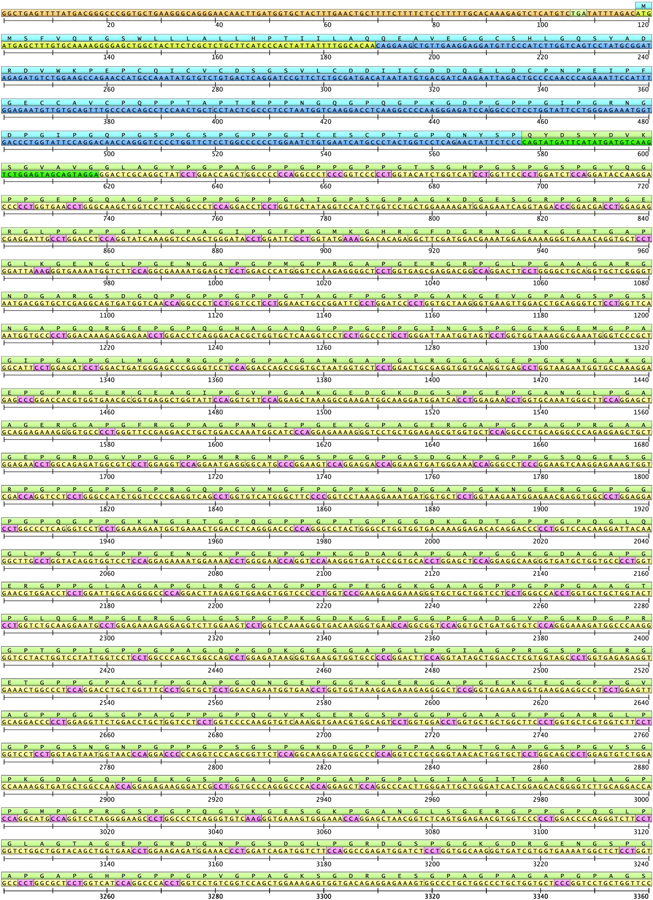
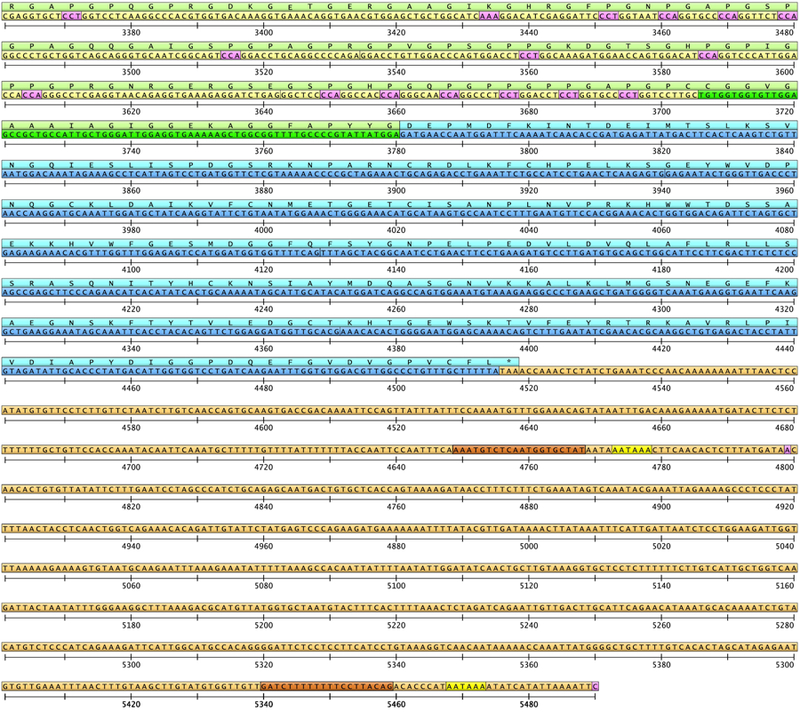
The entire COL3A1 cDNA sequence based on GeneBank entry NM_000090.3 and its translation to amino acids. Untranslated regions are shown in orange, and proline residues that will be hydroxylated in pink. For description of structural domains and the color scheme, see Fig. 1 and 4.
In the mouse genome, Col3a1 encoding the mouse α1 chain of type III collagen is located on the chromosome 1 [genomic coordinates (GRCm38): Chr1:45311538–45349706]. The gene is similar in size to the human COL3A1 gene with a length of approximately 38 kb and 51 exons. The exon-intron organization is also identical to that of the human COL3A1 gene (Toman and de Crombrugghe, 1994; Valkkila et al., 2001).
3. Protein structure
3.1. Biosynthesis of type III preprocollagen
Type III collagen is synthesized by cells as a pre-procollagen, which undergoes multiple co- and posttranslational modifications (Fig. 3). The signal peptide is cleaved off producing a procollagen molecule (Fig. 2). Three identical type III procollagen chains come together in the C-terminal ends, and the structure is stabilized by the formation of disulphide bonds. Each individual chain folds into left-handed helix and the three chains are then wrapped together into a right-handed superhelix, the triple helix (Bachinger et al., 1980; Birk and Silver, 1984; Boudko and Engel, 2004). Prior to assembling the super-helix, each monomer is subjected to a number of post-translational modifications that occur while the monomer is being translated. First, on the order of 145 prolyl residues of the 239 in the triple-helical domain are hydroxylated to 4-hydroxyproline by prolyl-4-hydroxylase. Second, some of the lysine residues are hydroxylated or glycosylated, and some lysine as well as hydroxylysine residues undergo oxidative deamination catalysed by lysyl oxidase. Other post-translational modifications occur after the triple helix is formed. The large globular domains from both ends of the molecule are removed by C- and amino(N)-terminalproteinases to generate triple-helical type III collagen monomers called tropocollagen. In addition, crosslinks form between certain lysine and hydroxylysine residues. In the extracellular space in tissues, type III collagen monomers assemble into macromolecular fibrils, which aggregate into fibers, providing a strong support structure for tissues requiring tensile strength (Fig. 3).
Fig. 3.
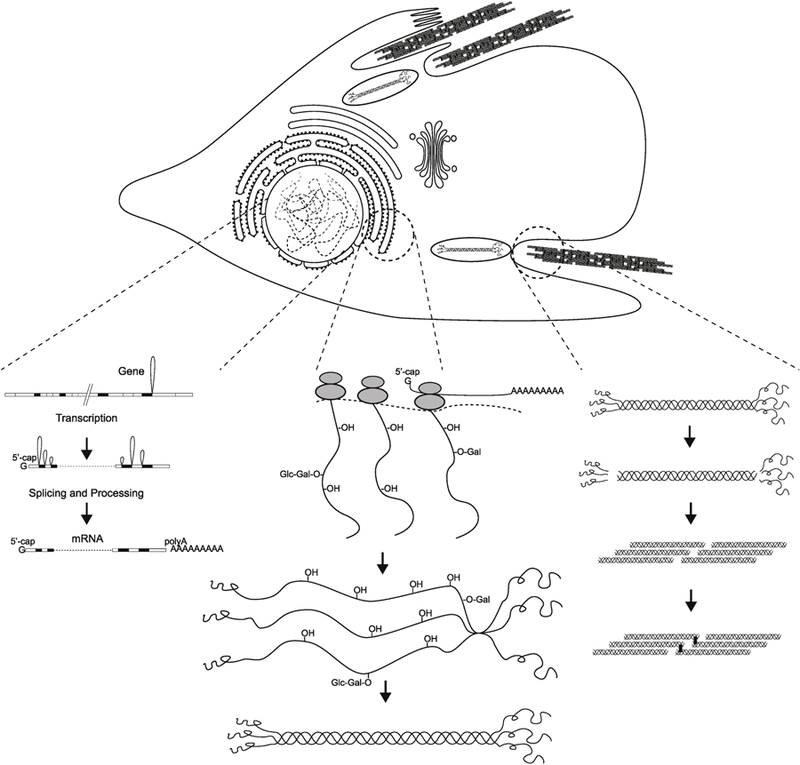
Biosynthesis of type III collagen: from COL3A1 gene to fibril assembly. The biosynthesis of type III collagen is a complex multistep process. This figure provides a schematic summary of the processes from the gene to transcription (left) to translation, posttranslational modifications (middle and right) and the assembly of the type III collagen fibrils (right). Modified with permission from (Prockop and Kivirikko, 1984). Copyright © 2019 Massachusetts Medical Society.
There are two types of hydroxylations of proline residues, namely the formation of 4hydroxyproline and 3-hydroxyproline, by two different enzymes (Weis et al., 2010). 4hydroxyprolines are known to stabilize the triple helix, whereas the exact function of 3hydroxyproline is not known, but it could be associated with inter-triple-helical interactions and supramolecular assembly. Between one and six 3-hydroxyprolines in each pro-α-chain are found in human fibrillar procollagens I, II, V, and XI, but none in the human type III procollagen although the chicken type III procollagen has one 3-hydroxyproline at position 986 (Weis et al., 2010).
The C-propeptides are important in the biosynthesis of fibrillar collagens by directing the chain selection, stabilizing the associated α chains by the formation of interchain disulphide bonds, and facilitating the formation of the triple helix (Boudko and Engel, 2004; Bourhis et al., 2012; DiChiara et al., 2018). In type III procollagen, the C-propeptide has eight cysteine residues and an N-glycosylation site. To facilitate the functional analyses, a recombinant form of the C-propeptide of type III collagen was expressed in a baculovirus system (Zafarullah et al., 1997). It was secreted into the medium and was shown to contain the expected inter- and intrachain disulphide bonds. In 2012, Bourhis et al. (2012) reported the crystal structure of the human C-propeptide of type III procollagen (Bourhis et al., 2012).This study provided the threedimensional structure of the homotrimeric propeptide and has facilitated the prediction of functional consequences of COL3A1 mutations occurring in the C-propeptide.
Production of a full-length recombinant COL3A1 protein has been hampered by the large number of posttranslational modifications required in the biosynthesis of collagens (Tomita et al., 1995; Shi et al., 2017). The most successful approach has been the use of a baculovirus expression system (Tomita et al., 1995; Lamberg et al., 1996), which yielded type III collagen molecules with stable triple helix, but their melting temperature was lower than that from cultured skin fibroblasts (Tomita et al., 1995). When the insect cells were co-transfected with human prolyl-4-hydroxylase, the recombinant type III procollagen was disulphide-bonded and the pepsin-digested collagen had a thermal stability of 41°C in a trypsin-chymotrypsin assay, a result comparable to that for a native type III collagen (Lamberg et al., 1996). In subsequent experiments using a yeast expression system, an intriguing finding was that co-expression of type III procollagen together with prolyl-4-hydroxylase, a key enzyme responsible for hydroxylating proline residues during collagen biosynthesis, markedly increased the amount of active prolyl-4-hydroxylase even though there was no change in the mRNA levels (Vuorela et al., 1997). The authors concluded that the increase in this activity was most likely due to an increased association of enzyme subunits to form the active enzyme tetramer and that in the absence of a collagen substrate the enzyme subunits rapidly dissociated.
The crystal structure has been determined for a synthetic triple-helical peptide with 42 amino acids (aa) that correspond to aa 991–1032 in the type III procollagen and contain the so called C-terminal cysteine knot, the region with a disulphide bond (Boudko et al., 2008). The analyses of the structure demonstrated that the multiple non-imino acids in this peptide, are part of direct intra- and interhelical contacts, and could interact with other extracellular matrix components (Boudko et al., 2008).
3.2. Structure of type III preprocollagen, procollagen and collagen
The triple-helical conformation, which is a characteristic feature of all fibrillar collagens, is possible because of the presence of a glycine as every third amino acid in the sequence of about 1,000 amino acids. This (Gly-Xaa-Yaa)n sequence is repeated 343 times in the type III collagen molecule. Proline or hydroxyproline is often found in the X- and Y-position giving the triple helix stability (Fig. 2 and Fig. 4).
Fig. 4.
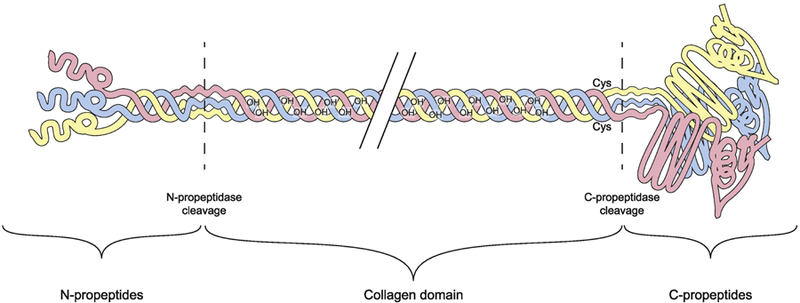
Structural domains of type III procollagen molecule. Type III collagen is synthesized by cells as a preprocollagen. The signal peptide (not shown here), which is 24 amino acids (aa) long, is cleaved off to form the procollagen chain. The total length of each of the α chains of type III procollagen is 1466 aa. Three α chains form a homotrimer as the type III procollagen molecule. The large globular domains from both ends of the molecule are removed by carboxyterminal (C) and aminoterminal (N) proteinases to form type III collagen. Several co- and post-translational modifications also take place.
The exact amino acid sequence was first determined by partial amino acid sequencing of bovine (Glanville and Fietzek, 1976; Fietzek et al., 1979; Brandt et al., 1984) and human (Gilbert et al., 1978; Seyer and Kang, 1981) type III collagen. Recombinant DNA technology then made it possible to obtain complementary DNA (cDNA) clones from chicken (Yamada et al., 1983), mouse (Wood et al., 1987), and human (Loidl et al., 1984; Chu et al., 1985; Miskulin et al., 1986; Mankoo and Dalgleish, 1988; Toman et al., 1988; Ala-Kokko et al., 1989; Janeczko and Ramirez, 1989) samples to determine the nucleotide sequence of the type III preprocollagen transcripts (Fig. 2). The cDNA and amino acid sequences between human, mouse and bovine type III collagen are very similar (Ala-Kokko et al., 1989).
Comparison of the human type III procollagen cDNA sequence to those of other two major fibrillar collagen chains, the α1 and α2 chains of type I procollagen, revealed that the triple-helical domain of the type III procollagen is 15 amino acid longer (1,029 aa) than the triplehelical domains (1,014 aa) of the two other α chains (Ala-Kokko et al., 1989). There were also differences in the lengths of the other functional domains, the largest difference being in the length of the N-propeptides, which is only 57 aa in the α2 chain of type I procollagen, but 139 and 129 aa in the α1 chains of type I and type III procollagen, respectively (Ala-Kokko et al., 1989). The codon usage for glycine, proline and alanine in the triple-helical domain was similar between the type I and type III collagen in that it favoured U as the third base (Ala-Kokko et al., 1989).
4. Tissue distribution
Type III collagen is found as a major structural component in hollow organs such as large blood vessels, uterus and bowel, tissues that must withstand stretching. It is also found in many other tissues in association with type I collagen. During the development of an embryo, type I and III collagen seem to be expressed in a coordinated manner based on a comprehensive survey of different developmental stages (E7.5 to E17.5) of the mouse embryo using in situ RNA hybridization (Niederreither et al., 1995). In another study that analysed COL3A1 mRNA levels in human fetal tissues, the expression patterns of COL1A1 and COL3A1 were quite different in the developing skeletal tissues and the authors suggested that this might mean that they are under different regulatory mechanisms (Sandberg et al., 1989).
Many early studies were not able to extract type III collagen from bone, and concluded that bone did not contain type III collagen. The first study to show that human bone does contain type III collagen was a histological analysis by Keene et al. (Keene et al., 1991) and used monoclonal antibodies. Type III collagen-containing fibers were found in all samples from donors between ages of 30 weeks to 80 years. Type III collagen was detected throughout the cortex, but was concentrated at the Haversian canal surface and the bone-periosteal interface.
A detailed mRNA expression analysis based on RNA-sequencing of 27 different human organs and tissues from 95 individuals was carried out by Fagerberg et al (Fagerberg et al., 2014). COL3A1 mRNA had a high level of expression in the gall bladder, placenta, bladder and endometrium (Fig. 5A). It was also detected in various parts of the gastrointestinal track, fat, heart, prostate, skin, spleen and testis. Unfortunately the sample collection did not contain blood vessels.
Fig. 5.
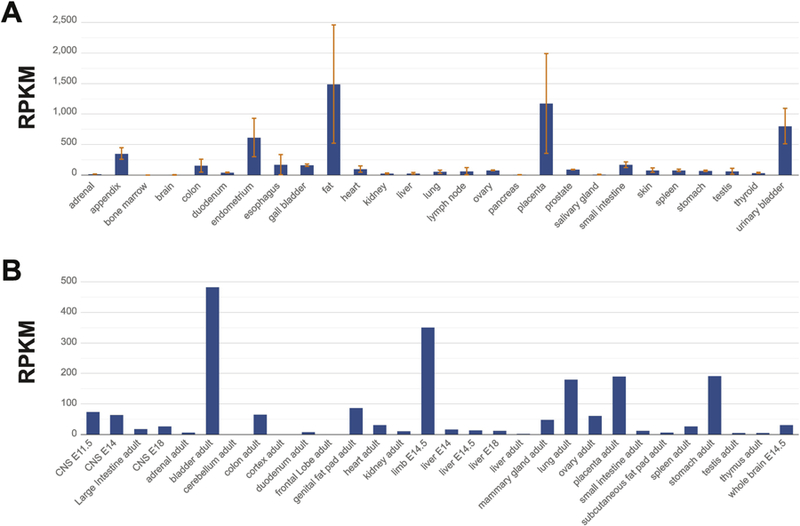
Distribution of COL3A1 mRNA in human (A) and mouse (B) tissues. RNA-sequencing data by Faderberg et al. (Fagerberg et al., 2014) for the human and Yue et al. (Yue et al., 2014) for the mouse tissues were used to generate these figures.
A similar analysis was carried out in the mouse using 69 samples from different tissues and developmental stages (Yue et al., 2014). The results showed that Col3a1 expression was high in the bladder, mammary gland, placenta, fat and in the limbs of E14.5 mouse embryos (Fig. 5B). Col3a1 mRNA was also detectable in the gastrointestinal track, heart, kidney, lung, ovary, and mouse embryonic brain and liver, but not in the adult mouse brain or liver.
Expression of type III collagen has also been studied in different benign and malignant tumors using immunostaining (d’Ardenne et al., 1984). All tumor samples were positive for type III collagen. Of the benign tumors, leiomyomas, and giant cell tumors of tendon sheaths had the highest level of type III collagen expression, whereas among the malignant tumors, leiomyosarcomas, fibrosarcomas, and sarcomas had the highest expression.
5. Function
Type III collagen provides tensile strength and integrity for many organs, but it has also been reported to have several other functions. In tissues the diameter of type III collagen fibrils is smaller than that of type I collagen (Birk and Silver, 1984). Type I and III collagens sometimes appear in the same fibrils and in that situation type III collagen regulates the fibril diameter (Fleischmajer et al., 1990; Cameron et al., 2002). Type III collagen is also found in the adult human cartilage and it has been suggested that its role is to act as a modifier of the fibril network composed of type II collagen together with other minor collagens during tissue healing (Wu et al., 2010). Type III collagen molecules appear to be cross-linked to the surface of type II collagen fibrils. Interestingly, most of the type III collagen molecules found to be present in the cartilage were not fully processed (see section 3) and contained disulphide-bonded N-terminal propeptides.
One of the earliest studies on type III collagen in 1975 showed that it influences the aggregation of human platelets (Balleisen et al., 1975). Follow-up studies demonstrated that one peptide generated by a cyanogen bromide treatment of calf skin type III collagen could facilitate platelet adhesion in human blood (Fauvel et al., 1978). The peptide was broken down further by chymotrypsin and hydroxylamine to narrow down the amino acid residues responsible for this function. Further evidence on the interaction between collagens and platelets was provided by Balleisen et al. (Balleisen et al., 1979), when they demonstrated that aggregation of platelets could be inhibited by coating the collagen fibrils with antibodies directed towards types I, II and III collagens thereby preventing the direct physical contact between platelets and collagens. Chiang et al. (Chiang et al., 1993) isolated a 47-kDa membrane protein from platelets and showed that it interacted with type III collagen. It is now known that platelets interact with type III collagen through specific glycoproteins and non-integrin receptors (Monnet and FauvelLafeve, 2000). Jarvis et al. (Jarvis et al., 2008) used 57 synthetic peptides, the sequence of which was derived from the sequence of COL3A1 and which could form a triple-helix, and tested their ability to interact with human and mouse platelets. A peptide with three hydroxyproline residues was found to bind to glycoprotein VI in platelets.
Type III collagen also functions in cell adhesion, migration, proliferation and differentiation through its interaction with integrins, which are cell surface receptors (Kim et al., 2005). The sites present in type III procollagen and showing high affinity to bind integrins are located in the C-terminal region (Kim et al., 2005).
It is likely that future research will identify additional binding sites in the type III collagen molecule for its interaction partners. This is facilitated by the Collagen Toolkit designed by the investigators at Cambridge University in the United Kingdom (https://collagentoolkit.bio.cam.ac.uk/ ). The toolkit contains 57 overlapping peptides corresponding to the triple-helical domain of type III collagen. The peptides can be used in a 96well format for mapping binding sites.
Based on measurements in the fluid collected from surgical wounds, peritoneum and serum of patients who underwent surgical procedures, the N-terminal propeptide levels of type III procollagen (PIIINP) increased 2–3 days after surgery (Haukipuro et al., 1987). At day 5 after the surgery, the serum PIIINP levels were 1000-fold higher in surgical patients than in nonsurgical patients. These findings suggest that type III collagen is required for wound healing.
6. Regulation of COL3A1 expression
Several lines of evidence suggest that COL3A1 expression is regulated also on the posttranscriptional level. The biological pathways involved in the regulation of COL3A1 expression include the transforming growth factor (TGF) β1, Wnt/β-catenin, and the p38 mitogen-activated protein kinase (MAPK) pathway. These studies have important implication for the developing new treatment strategies for fibrosis in different organs (see section 6.3).
Several studies have demonstrated that hypoxia alters type III collagen expression. Duval et al (2009) showed that hypoxia led to a decrease in both mRNA and protein levels of COL3A1 in cultured chondrocytes (Duval et al., 2009). A similar response was seen when using transient expression of the hypoxia-inducible factor 1ɑ (HIF-1ɑ), and the authors concluded that the effect is mediated by HIF-1ɑ. Zhang et al. (2018) found an opposite result when using cultured rat pulmonary microvascular endothelial cells under hypoxia (Zhang et al., 2018). In this experimental system hypoxia led to an increase in Col3a1 mRNA levels.
Samokhin et al. (2018) studied the molecular interactions in human pulmonary arterial hypertension using an in silico approach, and generated a “fibrosome” consisting of proteinprotein interactions important for lung fibrosis (Samokhin et al., 2018). Next, they incorporated into this network genes known to be regulated by aldosterone. This in silico analysis resulted in identifying NEDD9 as the key molecule in regulating gene expression that leads to pulmonary fibrosis, vascular remodeling, and pulmonary hypertension. The authors then went on to verify the findings experimentally and demonstrated that hypoxia increased NEDD9 and COL3A1 expression in human pulmonary artery endothelial cells, and that this was independent of TGFβ1 signaling. Nedd9-deficient mice exposed to hypoxia had decreased amounts of Col3a1 and decreased systolic pressure of the right ventricle. They concluded that NEDD9 targets COL3A1 to promote endothelial fibrosis and pulmonary arterial hypertension and that oxidative modification of a cysteine residue in NEDD9 and paracrine signaling of exosomes were the two mechanisms responsible for this.
In response to hypoxia, injury or metabolic stress cells release adenosine, a purine generated from ATP and ADP. Shaikh et al (2016) showed that in human skin fibroblasts adenosine acts through its receptors and stimulates COL3A1 expression via the canonical and non-canonical Wnt/β-catenin signaling pathway (Shaikh et al., 2016). Interestingly, the Wnt/β-catenin pathway was not involved in type I collagen expression in this experimental system.
When investigating the potential mechanisms leading to chronic allergic airway inflammation in a mouse model, Kilic et al (2011) discovered that a protein called the nerve growth factor induced the expression of type III collagen (Kilic et al., 2011). A treatment of the mice with an antibody against the nerve growth factor prevented this induction. The nerve growth factor was shown to signal through the MAPK pathway. Inhibition of this pathway in mouse lung fibroblasts prevented induction of COL3A1 expression by the nerve growth factor.
Yamane et al. (2018) investigated the role of branched amino acids leucine and isoleucine in the expression of Col1a1 and Col3a1 in the skin of mice deficient in branchedchain ɑ-ketoacid dehydrogenase kinase, which inactivates the enzyme involved in catabolizing branched amino acids (Yamane et al., 2018). Leucine and isoleucine levels were low in these mice. Col1a1 and Col3a1 mRNA and protein levels were also low compared to wild type mice. The authors found indirect evidence that the activity of the mammalian target of rapamycin (mTOR) was also decreased. Branched amino acids are known to signal via mTOR.
Epidermal growth factor (EGF) and basic fibroblast growth factor (bFGF; also called FGF2) enhanced the expression of COL3A1 mRNA and protein in human skin fibroblasts via MAPK signaling (Shen et al., 2018). Using human amniotic fibroblast cultures and explants of amniotic tissue, Mi et al (2018) demonstrated that cortisol decreased COL3A1 protein, but not mRNA levels (Mi et al., 2018). Further studies revealed that the reduction in protein levels was likely due to degradation mediated by the ubiquitin-proteasome pathway. Yuan et al. (2018) treated cultured human skin fibroblasts with TGFβ1 and found that the mRNA levels of COL3A1 were increased (Yuan et al., 2018).
A study by Chen et al. (2018) investigated the role of long non-coding RNAs (lncRNAs) in hypertrophic scars and found that one lncRNA had lower levels (Chen et al., 2018). In cultured skin fibroblasts overexpressing this same lncRNA reduced COL3A1 mRNA levels.
Thiele et al (2004) identified two mRNA-binding proteins called heterogeneous nuclear ribonucleoproteins (hnRNP) A1 and K that bind to sequences in the 3’UTR of COL3A1 mRNA (Thiele et al., 2004). Their experiments demonstrated that hnRNP K stabilized the COL3A1 mRNA, which resulted in increased expression. They also showed that TGFβ1 activates the synthesis of these hnRNPs as well as the synthesis of type III collagen in cardiac fibroblasts. Other studies have shown that the response to TGFβ1 varies based on the embryological origin of the cells (see Kuivaniemi et al., 2015); thus it is not possible to say if the regulatory mechanisms are the same in other vascular beds.
7. Diseases associated with COL3A1 mutations and variants, or altered levels of COL3A1 protein
Mutations in the COL3A1 gene cause the vascular type of Ehlers-Danlos syndrome (vEDS; OMIM 130050), which is a rare, life-threatening genetic disease. A few patients with arterial aneurysms without clear signs of EDS have also been found to have COL3A1 mutations (Kontusaari et al., 1990b). Other disease phenotypes associated with COL3A1 include a brain abnormality characterized by frontoparietal polymicrogyria, and many fibrotic diseases, in which increased amounts of type III collagen are found in various organs.
7.1. COL3A1 defects in patients with vEDS and other forms of EDS
vEDS (OMIM130050; ICD-10: Q79.6), previously known as the arterial-ecchymotic EDS, Sack-Barabas syndrome, EDSIV and EDS type 4, is the most serious form of EDS, since patients often die suddenly due to a rupture of large arteries or other hollow organs (Pope et al., 1977; Byers, 1993 [updated 2019]; Pope et al., 1996; Pepin et al., 2014; Byers et al., 2017; Malfait et al., 2017; Malfait, 2018). Other clinical manifestations of vEDS can include cigarette paper-like scarring of the skin, large ecchymoses over bony protuberances due to bruising, skin so thin that subcutaneous vessels are readily visible, joint laxity that is usually confined to fingers, pneumothorax, acrogeria, talipes equinovarus, keratoconus, gingival recession and fragility, and early-onset varicose veins, but the clinical picture of vEDS can be quite heterogenous even among members of the same family (Pope et al., 1977; Pope et al., 1996; Malfait et al., 2017). The median life expectancy is 50 years, and most patients develop major complications before the age of 30 years. Penetrance is 100% for mutations with autosomal dominant inheritance, but shows age-dependence. Patients with heterozygous null mutations usually have milder phenotype and these mutations have a lower penetrance of approximately 50% (Byers, 1993 [updated 2019]).
The inheritance mode of vEDS is autosomal dominant. Approximately 50% of the vEDS patients have no family history of the disease and the disease is due to de novo mutations (Byers, 1993 [updated 2019]). Prevalence estimate is 1–9/100,000, but it is likely that many vEDS cases are undiagnosed. It is estimated that there are approximately 1,500 vEDS cases in the USA (Byers, 1993 [updated 2019]).
Clinical diagnosis of vEDS is often difficult without genetic testing for COL3A1 mutations. vEDS diagnosis should be suspected in individuals with spontaneous ruptures of arteries, uterus or bowel at young age and the diagnosis confirmed by DNA sequencing. Once a mutation has been identified in the patient, testing for the at-risk relatives should also be offered (Byers, 1993 [updated 2019]).
Clinical management of vEDS patients often requires a multidisciplinary team of medical experts including primary care physicians, genetic counsellors, human geneticists, radiologists, vascular surgeons, general surgeons, gastroenterologists, and obstetricians. Evaluations should include blood pressure monitoring, and arterial screening by ultrasound, magnetic resonance or computed tomography imaging to detect dissections and dilatations (Byers, 1993 [updated 2019]; Eagleton, 2016; Byers et al., 2017; Malfait, 2018). Procedures such as arteriograms, colonoscopies and elective surgeries should be avoided due to tissue fragility, which could lead to complications. When surgical treatment is needed, if feasible, it should be performed by surgeons experienced in treating vEDS patients and familiar with the higher risks associated with the care of these patients (Eagleton JVS 2016).
Only one clinical trial for a potential drug therapy to prevent vascular events in vEDS patients has been published (Ong et al., 2010). In this study, carried out in France and Belgium, celiprolol, a cardioselective β-blocker with β2-agonist vasodilatation effects, was used. Altogether 53 vEDS patients, 33 (62%) of whom were later confirmed to have vEDS based on genetic testing, were randomly assigned to treatment (n=25) or no treatment (n=28) groups. The trial was ended early at 64 months due to the small number of patients available for follow-up who had not developed any of the vascular events. Although the authors concluded that celiprolol reduced the risk of arterial complication, the study has been criticized for its small size, and the fact that about third of the patients did not have COL3A1 mutations (Byers, 1993 [updated 2019]).
Female vEDS patients are at increased risk for pregnancy-related complications based on a study on 616 pregnancies of 283 women with confirmed COL3A1 defects (Murray et al., 2014) and should be followed in a high-risk obstetric program (Byers, 1993 [updated 2019]). In this study, a total of 30 (4.9%) pregnancies resulted in the death of the mother due to an arterial dissection or rupture, a uterine rupture, or complications during a cesarean section. All of the deaths occurred in women who had protein-altering COL3A1 mutations, whereas no deaths were seen in the 51 pregnancies among the 27 women with COL3A1 null mutations. The authors of this study also compared the overall death rates of these women who had been pregnant to those who had not, but had a genetically-confirmed vEDS diagnosis (n=243 nulliparous vEDS patients) and found that the overall survival rates were similar in these two groups of women (Murray et al., 2014).
7.1.1. Protein analyses with skin fibroblasts taken from patients with vEDS
Before DNA analyses became feasible, skin biopsies were taken from patients suspected of having vEDS and biochemical analyses were carried out using cultured skin fibroblasts (Pope et al., 1975; Byers et al., 1981; Stolle et al., 1985). These analyses involved culturing cells in the presence of a radioactive proline, which was incorporated into the newly synthesized collagens. It was then feasible to analyse the secreted and intracellular proteins separately and assess intracellular accumulation and secretion of collagens which have a high proline content.
Additional analyses included “stress tests” in which the proteins were subjected to a proteinase digestion at different temperatures (Stolle et al., 1985; Tromp et al., 1989a; Tromp et al., 1989b; Kontusaari et al., 1990b; Narcisi et al., 1993; Anderson et al., 1997). Properly folded type III procollagen can withstand a trypsin-chymotrypsin treatment to 41°C, but procollagen molecules containing mutations will break down at lower temperatures due to unfolding of cooperative blocks within the triple helix and expose the proteinase-sensitive sites of the molecule. Another test was to carry out digestion with collagenase, which cleaves normal type III procollagen at a very specific site at aa 781 of the triple-helical domain (Fig. 6). The procollagen can also be digested by pepsin, which will remove the globular propeptides, the end result being collagen (Kontusaari et al., 1990b; Nuytinck et al., 1992; Narcisi et al., 1993).
Fig. 6.
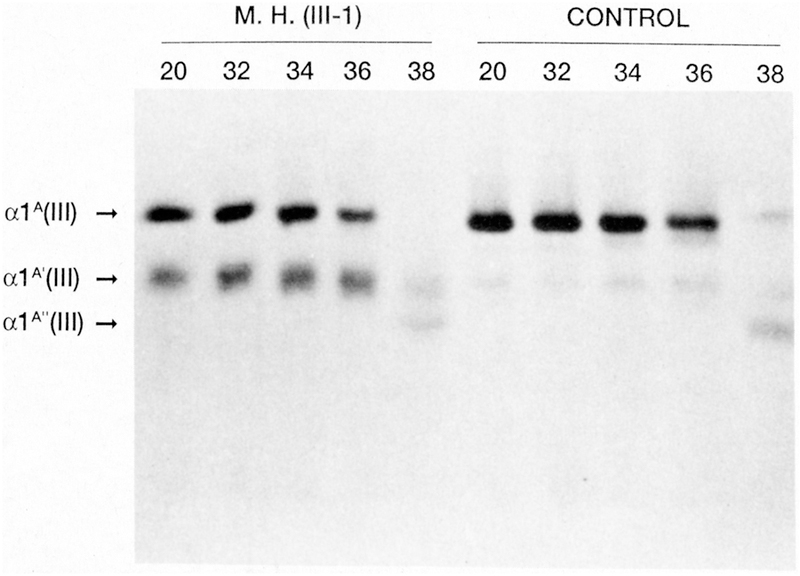
Thermal unfolding of type III collagen from a patient with a Gly619Arg mutation in COL3A1. Skin fibroblasts were obtained from the patient and a control. The effect of a single base missense mutation was tested on the type III collagen protein by using a proteinase assay. The type III collagen was first digested with collagenase which cleaves the molecule at aa 781. This was then followed by digestion with trypsin and chymotrypsin. A large fraction of the patient’s type III collagen was digested to a fragment of approximately 620 aa at temperatures 20–36°C. In comparison the type III collagen isolated from control fibroblasts stayed intact and did not break down until at 38°C. Reproduced with permission from (Kontusaari et al., 1990b).
Increased amounts and intracellular accumulation of this normally secreted protein are also often found (Anderson et al., 1997) in addition to the synthesis of an overmodified type III collagen showing retarded migration on a polyacrylamide gel (Nuytinck et al., 1992; Richards et al., 1992; Narcisi et al., 1993). In some cases, the type III collagen protein was digested with cyanogen bromide and the various peptides analysed to map the defect (Nuytinck et al., 1992; Richards et al., 1992).
7.1.2. DNA analyses and a mutation database for patients with vEDS
The first COL3A1 DNA study based on a preliminary Southern blot analysis of a vEDS patient identified an approximately 3.3-kb deletion in COL3A1 in 1988 (Superti-Furga et al., 1988). In a subsequent publication three years later, DNA sequencing of the breakpoints of the deletion demonstrated that it was much larger and included 16 exons and approximately 7.5 kb of genomic DNA (Lee et al., 1991). Based on a large number of studies that have screened for mutations in COL3A1 in patients with vEDS, large deletions and insertions are not a common type of mutations in these patients.
The first glycine mutation in the triple-helical domain was reported in 1989 in a study that was also the first detailed DNA analysis of a COL3A1 mutation using DNA sequencing both on the genomic and cDNA level (Tromp et al., 1989a). This mutation changed a glycine at aa position 790 to a serine (Gly790Ser; numbering from the beginning of the triple-helical domain) (Tromp et al., 1989a).
A total of 63 COL3A1 mutations were summarized in a review article published in 1997 (Kuivaniemi et al., 1997). About 20 years later, over 650 different mutations have been reported, which are listed in a COL3A1-specific database (see https://eds.gene.le.ac.uk/home.php?select_db=COL3A1). In 2014, 410 different mutations were reported from a single Medical Genetics Laboratory at the University of Washington, USA (Pepin et al., 2014). Based on this study and the COL3A1 mutation database, the most common type of mutation is a missense mutation substituting a bulkier amino acid for a glycine in the triple-helical domain. The second most common type of mutations in vEDS are COL3A1 RNA splicing mutations (Kuivaniemi et al., 1990; Schwarze et al., 1997).
7.1.3. Mutations in the C-propeptide of COL3A1
Missense and nonsense mutations in the C-terminal propeptide of type III procollagen possibly also cause vEDS, although the detailed functional studies to prove this are still needed (Stembridge et al., 2015).The C-propeptide is important for the assembly of the three pro-α chains (Boudko and Engel, 2004; Bourhis et al., 2012), but knowledge about the role of each aa in the sequence is limited and it is, therefore, difficult to interpret the results. One of the patients in the study by Stembridge et al. (Stembridge et al., 2015) was a female patient with a Pro1440Leu mutation in the C-terminal propeptide of type III procollagen (Stembridge et al., 2015). She had thin skin, easy bruisability, mild joint laxity, periodontal disease with gum recession, and a typical acrogeria, but no vascular or bowel ruptures by the age of 27 years. Her father, who was mosaic for the same mutation, showed mild joint laxity (see section 7.1.5.). Cultured skin fibroblasts of the patient produced less type III collagen than control fibroblasts. Protein modelling was suggestive of leucine destabilizing the structure. Another patient was a 39-year old male with easy bruisability, hyperextensible skin, ruptured ligament of the diaphragm, and bilateral inguinal hernia, but again no vascular complications (Stembridge et al., 2015). Protein analysis of cultured skin fibroblasts revealed type III collagen deficiency, and DNA sequencing of genomic DNA revealed an Arg1432Leu missense mutation. The patient’s more mildly affected sister had the same mutation. The third patient studied by Stembridge et al. (Stembridge et al., 2015) was a 26-year old female with a generalized joint laxity, easy bruisability, unusual facial appearance, and a slightly enlarged pulmonary artery. DNA sequencing found a nonsense mutation. Protein analyses demonstrated slightly reduced amounts of type III procollagen. The fourth patient had skin and joint manifestations, as well as a gastric paresis with atony, and was diagnosed with the benign hypermobility or EDS type III. DNA sequencing revealed a Lys1313Arg missense mutation, but based on protein analyses performed on cultured skin fibroblasts the amount of type III procollagen was normal. Protein modelling showed no adverse consequences. The authors concluded the variant was a benign variant.
7.1.4. RNA splicing mutations in COL3A1
The most frequently seen pattern of abnormal splicing is exon skipping, but other forms of aberrant splicing have also been detected (Fig. 7) (Kuivaniemi et al., 1990; Schwarze et al., 1997). The normal COL3A1 gene does not undergo alternative splicing. RNA splicing mutations that lead to exon skipping, and thereby remove one entire exon, produce a transcript that encodes a protein with multiple triplets of amino acids deleted, since each exon in the triplehelical region starts with a complete glycine codon. The resultant mutant protein is, therefore, shorter by the number of amino acids encoded by that exon, but the Gly-Xaa-Yaa triplets stay in frame and there are no premature termination codons.
Fig. 7.
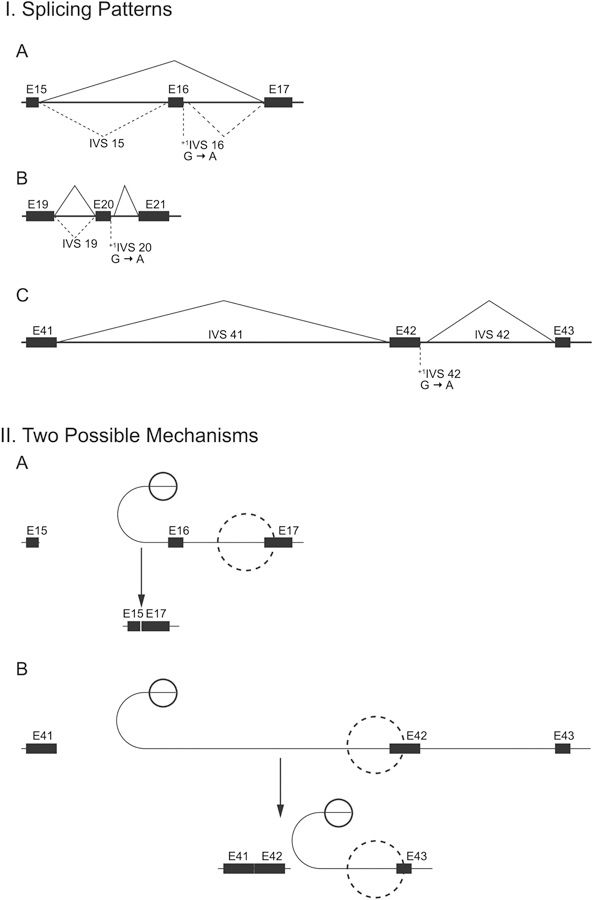
Schematic drawing of RNA splicing patterns in three different COL3A1 splicing defects (A), and two possible splicing mechanisms (B). G-to-A mutations in the first nucleotide of introns 16, 20 and 42 of COL3A1 led to different splicing patterns, which were experimentally validated by S1 nuclease probe protection experiments and cDNA sequencing. Solid lines indicate the most frequently used mode of splicing and broken lines less frequently used. The 51 exons of COL3A1 are numbered 1–52 to match the numbering of exons in the genes for other fibrillar collagens. This research was originally published in the Journal of Biological Chemistry. Kuivaniemi H, Kontusaari S, Tromp G, Zhao M, Sabol C, Prockop DJ. Identical G+1 to A mutations in three different introns of the type III procollagen gene (COL3A1) produce different patterns of RNA splicing in three variants of Ehlers-Danlos syndrome IV. An explanation for exon skipping with some mutations and not others. J. Biol. Chem. 1990; 265:12067–12074. © the American Society for Biochemistry and Molecular Biology.
In a study that described the first RNA splicing mutations in COL3A1 in 1990 (Kuivaniemi et al., 1990), three single nucleotide changes were found, each mutating the first G in a different intron (introns 16, 20 and 42) to an A (Fig. 7). The patterns of RNA splicing were studied by S1 nuclease probe protection experiments and cDNA sequencing and were found to differ(Kontusaari et al. 1990a; Kuivaniemi et al. 1990).. The intron 16 mutation led to an abnormal splicing that skipped exon 16, whereas an identical mutation in intron 20 produced two different RNA species, one of which was generated by an efficient use of a cryptic splice and the other with no splicing of intron 20 and retaining the entire intron in the resulting transcript. The third mutation located in intron 42 led to an efficient use of a near-by cryptic splice site which added only 30 nucleotides to the transcript and kept it in frame (Kuivaniemi et al., 1990). The same intron 42 mutation was later found in another unrelated patient and the splicing pattern was the same in both patients (Schwarze et al., 1997). Interestingly, two different mutations in the 5th nucleotide of intron 42 in two additional vEDS patients also produced the same abnormal splicing (Giunta and Steinmann, 2000; Okita et al., 2010) suggesting that the inclusion of 30 intronic nucleotides into the mRNA is an efficient form of splicing for intron 42. Okita et al. (Okita et al., 2010) showed that the mutation led to a decreased amount of type III collagen synthesis by the patient’s skin fibroblasts.
A single-base mutation in COL3A1 that changed the 5th nucleotide of intron 37 from G to A led to abnormal RNA splicing, which produced skipping of exon 37 (Wu et al., 1993). Quantification of the RNA splicing patterns showed that 70% of the mutant allele was spliced through exon skipping and 30% was spliced normally, when the skin fibroblasts were grown at 37°C. Interestingly, the amount of abnormal splicing increased at a lower temperature and was approximately 90% at 31°C.
Studies on RNA splicing defects in COL3A1 have led to a better understanding of the mechanism of RNA splicing in large multi-exon genes such as the collagen genes (Fig. 7). It is plausible to speculate that the order of splicing of different introns varies and that the size of the intron also influences the splicing pattern favoured when the normally occurring splice site is mutated (Kuivaniemi et al., 1990; Schwarze et al., 1997). Another factor likely to play a role is the presence of cryptic splice sites, sequences near the normally used splice sites, which are activated when the normal splice site becomes weaker through a mutation (Kuivaniemi et al., 1990). The use of cryptic splice sites in an intron leads to an incorporation of additional nucleotides into the transcript (Fig. 7). These extra sequences usually contain premature termination sites for translation and are detected by nonsense-mediated mRNA decay, the result of which is the degradation of the abnormal RNA molecules and no protein synthesis.
7.1.5. Germline and somatic cell mosaicism in the relatives of patients with COL3A1 mutations
Germline and somatic cell mosaicism refers to a situation where a mutation occurred during the embryogenesis of the individual and therefore, the mutation is not present in all the cells of that individual. Depending on the cell lineage in which the mutation occurred and the number of cells with the mutation, the disease phenotype may or may not manifest in that individual. It has been estimated that approximately 15% of the phenotypically normal parents of vEDS patients are mosaic (Byers, 1993 [updated 2019]). This is an important finding that must be taken into consideration in genetic counselling, since the risk for a mosaic parent to have another affected child is higher than that for a genetically normal parent. Prenatal testing is available for at-risk individuals (Byers, 1993 [updated 2019]).
Several of the COL3A1 mosaic cases have been studied in detail (Kontusaari et al., 1992; Richards et al., 1992; Milewicz et al., 1993; Palmeri et al., 2003; Stembridge et al., 2015). One such case was a typical female vEDS patient with arterial ruptures, pneumothorax, easy bruisability, and skin changes, who was found to have a single base mutation leading to a Gly1018Asp change in the triple-helical domain (Kontusaari et al., 1992). The parents had no signs of vEDS when last examined in their 70’s. When the inheritance of the mutation was investigated, a surprising finding was that the mother also carried the same mutation in nearly all of her blood leukocytes. Analyses of saliva and hair samples from the mother revealed that approximately 40% of the oral epithelial cells and 0 to 100% of the DNA from 28 hair roots tested individually contained the single base mutation. Based on these results, the mutation must have occurred after the first cell division of the embryo, but before the differentiation of three major germ layers, the endoderm, mesoderm and ectoderm (Kontusaari et al., 1992).
Another example of a mosaicism was in a family where the vEDS patient had a 2-kb deletion in the COL3A1, and the patient’s phenotypically normal father also had this same deletion in some of his cells (Milewicz et al., 1993). Approximately 80% of his cultured skin fibroblasts, but only 20% of his blood leukocytes, carried the deletion. The deletion was also found in his sperm cells, but due to a small sample, quantification was not possible (Milewicz et al., 1993). The authors interpreted the results to mean that the mutation occurred during his embryologic development before the lineage allocation happens.
The third example of a mosaicism was described in the unaffected 77-year old maternal grandmother of a male vEDS patient with a Gly847Glu mutation (Richards et al., 1992). The patient had a spontaneous carotid-cavernous sinus aneurysm and his mother died at the age of 50 years from a bowel rupture. The patient’s brother had thin skin and joint laxity. The amount of type III collagen synthesized by the patient, his brother and mother was reduced. Additional biochemical work on the patient’s skin fibroblasts showed that the type III collagen was overmodified. In addition, cyanogen bromide peptide mapping demonstrated differences from the normal pattern. A genetic test using allele specific hybridization on genomic DNA isolated from blood demonstrated that all three affected family members and the maternal grandmother were positive for the missense mutation. The findings on the grandmother were confirmed with a second blood sample and hair samples. Quantification of the amount of the mutant-to-normal allele ratio was approximately 0.5 suggesting that about 2/3 of the grandmother’s cells carried the mutant allele.
The fourth example of a mosaicism was reported in a study on a family in which the index patient and her mother had neurological manifestations with leg and hand contractures, epilepsy and stroke in addition to acrogeria and coiled cerebral vessels and dissections (Palmeri et al., 2003). They harboured a missense mutation at aa 883 that converted a glycine to a valine. The asymptomatic maternal grandmother with a slight ectasia of thoracic aorta was found to be a mosaic for the same mutation.
The fifth example of a mosaicism was described in the father of a female patient with a Pro1440Leu mutation in the C-terminal propeptide of type III procollagen (Stembridge et al., 2015). The father showed mild joint laxity, whereas the patient had thin skin, easy bruisability, mild joint laxity, periodontal disease with gum recession, and a typical acrogeria, but no vascular or bowel ruptures by the age of 27 years.
7.1.6. Biochemical and cellular effects of COL3A1 mutations
The biochemical and cellular effects of COL3A1 mutations have been investigated extensively using cultured skin fibroblasts established from skin biopsies (Tromp et al., 1989b; Kontusaari et al., 1990b; Kontusaari et al., 1992; Richards et al., 1992; Narcisi et al., 1993; Anderson et al., 1997; Okita et al., 2010). This is an ideal system to study the consequences of the mutations, since skin biopsies are easy to obtain and type III procollagen is expressed in these cells (see section 7.1.1. for details).
Most patients are heterozygous for the mutation and therefore, produce both normal and abnormal α chains of the type III procollagen. Since three α chains are required to form a triplehelical structure, there can be normal and mutant homotrimers, but also trimers that have either one or two mutant α chains together with two or one normal α chains. Altogether only oneeighth of the trimers are completely normal and only one-eighth of the trimers are completely abnormal. For structural stability, the heterotrimers with both normal and mutant α chains folded together are the most problematic ones with aberrant fibril formation leading to reduced strength.
It is also possible to carry out ultrastructural analyses of skin samples taken from the patient and use light and electron microscopy for the analysis. These studies have demonstrated a dilated rough endoplasmic reticulum and changes in the diameter of collagen fibers (Nuytinck et al., 1992; Smith et al., 1997).
More recent studies have used microarray-based transcriptomics to determine which biological pathways are affected by COL3A1 mutations in cultured skin fibroblasts (Chiarelli et al., 2018). Expression profiles of skin cells from two patients who were each heterozygous for the missense mutations, Gly237Arg and Gly612Asp, and a third patient with an RNA slicing mutation in intron 14 were compared to expression profiles of nine healthy age-matched controls (Chiarelli et al., 2018). Altogether 969 genes were differentially expressed, 281 with increased and 688 with decreased expression, between the vEDS and control groups. Fibrillin-2 (FBN2) was the most differentially expressed gene with decreased expression. Several of the genes with increased expression in the patient cells were involved in transcriptional regulation and re-organization of the actin cytoskeleton. The genes with decreased expression belonged to such functional categories as cell cycle processes, cell division, histones, cell movement and intracellular trafficking, DNA damage response, and ribosomal RNA processing. A pathway enrichment analysis revealed that cell cycle was the most affected pathway. The proteasome system was also affected by having several of the differentially expressed genes.
7.1.7. Genotype-phenotype correlations among vEDS patients
In a series of 1,231 vEDS cases from a single laboratory, 630 index cases and their 601 affected relatives were analysed for clinical features and COL3A1 defects (Pepin et al., 2014). The median survival age among men and women was 46 and 54 years, respectively. The causes of deaths included vascular complications such as rupture and dissection, and bowel perforations.
In this study, a total of 410 different COL3A1 mutations deemed to be causative were found among the 572 index cases (Pepin et al., 2014). Biochemical analysis of cultured skin fibroblasts had been carried out in the remaining 58 index cases before genetic testing was available and found to produce abnormal type III procollagen. The 410 different mutations included 356 glycine substitutions, 164 RNA splicing mutations and 52 other types of mutations (insertions, duplications or deletions). In 27 vEDS patients the mutated allele led to an unstable RNA through nonsense-mediated decay, and did not produce protein (null mutations or haploinsufficiency).
Survival of vEDS patients with null mutations was 10–15 years longer than in individuals harbouring RNA splicing or glycine mutations (Pepin et al., 2014). Interestingly, survival among the patients with glycine mutations differed based on the substituting amino acid; a substitution of a valine or an aspartic acid meant worse survival than a substitution of a serine (Pepin et al., 2014).
In another smaller series from France, 215 vEDS patients from 146 different families and with 126 different variants were investigated (Frank et al., 2015). The overall findings were similar to those from the larger series described above in that patients with glycine substitutions (n=127) had poorer prognosis than patients with mutations leading to haploinsufficiency (n=14) (Frank et al., 2015). There was, however, no difference between male and female vEDS patients in the age when a major clinical complication occurred. Interestingly, in this series the vEDS patients with RNA splicing defects had earlier age at diagnosis than patients with glycine substitutions (Frank et al., 2015). The authors speculated that this could be due to differences in the clinical practices between France and the USA.
7.2. Genetic studies for other human diseases
7.2.1. COL3A1 mutations in patients with brain disorders
Several patients with severe brain anomalies in addition to connective tissue manifestations have been reported to harbour a pathogenic variant in both COL3A1 alleles (Plancke et al., 2009; Jorgensen et al., 2015; Horn et al., 2017; Vandervore et al., 2017). In human genetics this is known as a compound heterozygosity, but they are now called “biallelic variants” in the literature. Interestingly, the results from imaging and histological analyses of the brain in the affected individuals resemble those seen in patients with bilateral frontoparietal polymicrogyria (OMIM 606854), who have mutations in GRP56 (Ke et al., 2008). Since type III collagen is a ligand for GRP56, it is plausible that those mutations in COL3A1 that interfere with the receptor-ligand interaction or eliminate type III collagen altogether, manifest as this neurological phenotype. It is also noteworthy, that the Col3a1+/− mice manifest similar brain phenotype (see section 8.1). One of these patients had an Arg428Stop nonsense mutation in the paternal and a deletion of one C in Pro686 leading to premature termination in the maternal allele of COL3A1. As a consequence, the patient had no functional type III collagen, and would be expected to have a very severe phenotype. Each parent was heterozygous for one of these variants and showed no clinical phenotype (Horn et al., 2017). Four other patients, a sister and a brother, and two unrelated patients with delayed speech and motor development and frontoparietal polymicrogyria were reported to be homozygous for the same Pro49Ala variant (Horn et al., 2017; Vandervore et al., 2017). Even though the variant was classified as pathogenic or likely pathogenic based on in silico scoring algorithms, and it is intriguing that it was present in four patients, it is difficult to be certain that it is the cause of the disease, since biological functional studies did not yield any significant findings (Vandervore et al., 2017). In another study by Jorgensen et al. (Jorgensen et al., 2015), two siblings were compound heterozygotes for a mutation causing a premature termination at aa 596 and a missense mutation in the C-terminal propeptide that changed a glycine to a glutamic acid. One of these patients died suddenly at the age of 15 of arterial dissections at three different sites, and the other had a dissection in the internal carotid artery and cerebral cortical dysplasia (micropolygyria). The four family members who were heterozygous for the premature termination mutation had no clinical manifestations, whereas the one family member who was heterozygous for the missense mutation had thin, translucent skin, small joint hypermobility, pulmonary emphysema and reduced aortic elasticity, which are typical signs of vEDS (Jorgensen et al., 2015).
7.2.2. Studies for other human diseases using COL3A1 genetic variants
Since type III collagen is such an important structural component for the integrity of blood vessels, several studies have investigated the possibility that genetic variants in the COL3A1 gene increase the person’s risk for developing aneurysms. Despite of few isolated reports with positive findings (Kontusaari et al., 1990b; Anderson et al., 1996; van Keulen et al., 1999), larger studies using a comprehensive DNA sequencing analysis of the entire triple-helical domain of type III procollagen in patients with abdominal aortic aneurysms (Tromp et al., 1993) and intracranial aneurysms (Kuivaniemi et al., 1993) found no mutations that could explain the disease. Subsequently, an approach called genetic association study (Romero et al., 2002) in which the frequencies of the two alleles of COL3A1 polymorphisms were compared in cases and controls, was used. No association was found between aneurysms and the COL3A1 polymorphisms (Ogata et al., 2005; Hinterseher et al., 2011). Furthermore, family-based DNA linkage studies (Shibamura et al., 2004; Tromp et al., 2014) and genome-wide association studies (Tromp et al., 2014; Jones et al., 2017) for abdominal aortic aneurysms and intracranial aneurysms with large sample sets have found no evidence that COL3A1 variants contribute to the susceptibility of these conditions. Unlike the candidate gene approaches which rely on prior knowledge about the pathobiology of the disease and are based on educated guesses, these genome-wide approaches are unbiased and decipher the entire human genome to find statistical evidence for a linkage or an association. It should be noted that such studies require large sample sets with thousands of cases and controls to obtain reliable estimates of the disease risk (Romero et al., 2002).
COL3A1 has been tested as a potential susceptibility gene for several other clinical conditions (Muckian et al., 2002; Lv et al., 2014; Reichert et al., 2018). One recent study investigated two COL3A1 single nucleotide polymorphisms (SNPs), rs3134646 and rs1800225, among 422 patients with diverticulosis and 285 controls(Reichert et al., 2018). The rs3134646 was associated with diverticulosis, but rs1800225 not, with an odds ratio of 1.82 (95% confidence interval of 1.04–3.20) and p = 0.04. Due to the small sample size and only borderline significance, the results will need validation and replication.
In another genetic association study, three COL3A1 SNPs (rs2138533, rs11887092, and rs1800255) were analysed for their contribution to the recurrence and prognosis of stroke in the Chinese population (Lv et al., 2014). A total of 1,544 stroke patients from three different subtypes of stroke were included in the study and followed up for 4.5 years. All three variants were associated with the risk for stroke recurrence and prognosis independently from conventional risk factors. Patients who had the A allele of rs1800255, had a lower risk for reoccurrence of stroke, but an increased risk for all-cause mortality in the atherothrombotic stroke subgroup. Again, due to the small sample size, the results although statistically significant, can only be considered suggestive and need further validation.
Muckian et al. (Muckian et al., 2002) assessed the role of genetic variability in COL3A1 in coronary artery disease using a highly polymorphic intronic tandem repeat as a marker. This repeat is located in intron 25 and has between three and 13 copies of a 15-bp repeat (Mays et al., 1992). Four of the alleles are common and were used in the study. The rationale for selecting COL3A1 as the candidate gene for the study, was that it is expressed in vascular wall and is known to interact with platelets. They genotyped DNA samples obtained from individuals who were part of three different cardiovascular studies and had a total of 1,851 cases with either acute coronary syndrome and myocardial infarction, unstable coronary syndrome, or stable angina, in addition to 306 controls. The authors concluded that COL3A1 variants modulate the risk for coronary artery disease. As with all small human genetic studies, the results require confirmation in an independent data set. Furthermore, in complex phenotypes such as the coronary artery disease, the contribution of any single genetic variant is expected to be small and explain only a small fraction of the increased or decreased risk.
7.3. Altered COL3A1 mRNA and protein levels, or degradation in human diseases
Accumulation of type III collagen is a hallmark of several chronic human diseases that involve fibrotic processes including systemic sclerosis, cardiac fibrosis, lung fibrosis, liver cirrhosis and renal fibrosis (Table 2) (Jimenez et al., 1986; Krieg et al., 1986; Rosenbloom et al., 2016; Fogo et al., 2017; Karsdal et al., 2017; Ricard-Blum et al., 2018). Thus, a large number of studies have investigated the potential of using the determination of the PIIINP levels (Risteli et al., 1988) in the urine and serum as a biomarker for the presence of fibrosis. The N-terminal propeptide is cleaved off enzymatically during the biosynthesis of type III collagen (Fig. 3) and it has been proposed that its levels are a surrogate marker for the amount of type III collagen in a given tissue. Another serum test (C3M), developed more recently, measures the levels of a 10aa neoepitope of type III collagen generated by digestion with matrix metalloproteinase 9 (Barascuk et al., 2010). This test does not recognize intact type III collagen, but is specific for the short peptide, and can thus be used to measure type III collagen degradation. A third test (C3C) was developed to measure fragments of type III collagen generated by cathepsins B, L, S and K by ELISA and was tested in patients with chronic obstructive pulmonary disease (COPD) (Rasmussen et al., 2017). This test recognizes a neo-epitope located in aa 642–651 of the αchain of type III collagen. COPD patients (n=68) had significantly elevated levels of C3C in their serum compared to healthy controls (n=20). Serum tests of these three assays and any new tests to be developed would provide an important clinical advancement for diagnostics and monitoring treatment responses since they are minimally invasive tests.
Table 2.
Human diseases associated with altered levels of COL3A1 mRNA, protein or its degradation products.
| Condition | Tissue | Method | Results | Studya (PMID) |
|---|---|---|---|---|
| Dilated cardiomyopathy | Heart | Gel electrophoresis, IHC | COL3A1 protein increased | Marijianowski et al (JACC 1995); 7722119 |
| Idiopathic restrictive cardiomyopathy | Heart | IHC | COL3A1 protein increased | Hayashi et al Int J Cardiology 1998; 9688428 |
| Hypertrophic cardiomyopathy | Heart | IHC | COL3A1 protein increased | Kitamura et al Clin Cardiol 2001;11303702 |
| Ischemic cardiomyopathy | Heart | IHC | COL3A1 protein increased | Herpel et al Histopathology 2006; 16681691 |
| Valvular cardiomyopathy | Heart | IHC | COL3A1 protein increased | Herpel et al Histopathology 2006; 16681691 |
| Myocardial infarction | Serum | RIA | PIIINP increased 2 days after infarction, but level is within normal limits | Manhenke et al (Eur Heart J 2014); 24255130 |
| Renal fibrosis | Urine and serum | RIA | PIIINP increased | (Soylemezoglu et al., 1997); 9306339 |
| Progression of kidney disease | Urine | RIA | PIIINP increased with progression of disease | (Ix et al., 2015); 25655067 |
| Diabetic nephropathy | Kidney tissue | IHC, in situ RNA hybridization | COL3A1 mRNA and protein increased | (Razzaque et al., 1995); 8822111 |
| Type III collagen glomerulopathy | Kidney tissue | IHC, EM | COL3A1 protein increased | (Fogo et al., 2017); 28532638 |
| Liver fibrosis | Serum | RIA | ELF increased | (Tanwar et al., 2017); 27906753 |
| Chronic obstructive pulmonary disease | Serum | ELISA | C3C increased | (Rasmussen et al., 2017); 28076408 |
| Cryptogenic fibrosing alveolitis | Lung tissue and serum | Gel electrophoresis, RIA | COL3A1 protein and PIIINP increased. Higher PIIINP correlated with response to treatment | (Kirk et al., 1984); 6495240 |
| Progressive systemic sclerosis | Fibroblasts isolated from skin | Gel electrophoresis, Northern, dot-blot | Total collagen increased; COL3A1 mRNA increased | (Jimenez et al., 1986); 3800922 |
| Systemic sclerosis | Serum | RIA | ELF and PIIIINP increased | (Abignano et al., 2019); 30239834 |
| Dupuytren’s disease | Aponeurosis | COL3A1 protein increased | 391658 | |
| Peyronie’s disease | Penis | Gel electrophoresis | COL3A1 protein increased | (Somers et al., 1989); 2918606 |
| Crohn’s disease | Fibroblasts isolated from intestine | COL3A1 protein increased | (Stallmach et al., 1992); 1587410 | |
| Crohn’s disease subtypes | Serum | ELISA | C3M levels higher in penetrating disease | (van Haaften et al., 2017); 28481042 |
| Osteoarthritis | Cartilage | IHC, in situ RNA hybridization | Present in osteoarthritic, but not in normal cartilage | (Aigner et al., 1993); 7680669 |
| Hyperthyroidism | Serum | RIA | PIIINP increased | (Inui et al., 1992); 1576745 |
| Glioma | Tumor tissue | qRT-PCR, IHC | COL3A1 mRNA increased | (Gao et al., 2018); 30008884 |
| Bladder cancer progression | Tumor tissue | Microarray; qRT-PCR; IHC | COL3A1 mRNA and protein increased | (Yuan et al., 2017); 29050298 |
| Breast cancer | Tumor tissue | IHC | PIIINP | (Kauppila et al., 1998); 10211114 |
| Breast cancer: progression and survival | Serum | ELISA | PIIINP increased with progression of disease and shorter survival | (Lipton et al., 2018); 29923614 |
C3C, test measuring fragments of type III collagen generated by cathepsins B, L, S and K; C3M, test for measuring levels of a 10-aa neoepitope of type III collagen generated by digestion with matrix metalloproteinase 9; ELF, enhanced liver fibrosis, a panel of three markers including PIIINP, hyaluronic acid and fibronectin; ELISA, enzyme-linked immunosorbent assay; EM, electron microscopy; IHC, immunohistochemistry; PIIINP, N-terminal propeptide of type III procollagen; qRT-PCR, quantitative reverse transcriptase PCR; RIA, radioimmunoassay
Only one representative study for each disease, since a large number of studies have been reported.
7.3.1. Cardiac diseases
The role of extracellular matrix proteins in the pathophysiology of some of the major cardiovascular diseases has been studied extensively (Prabhu and Frangogiannis, 2016; Frangogiannis, 2017). Extracellular matrix accumulation in the heart tissue leads to cardiac fibrosis, scarring that results from an injury, such as a myocardial infarction, and leads to impaired function of the heart muscle (Krenning et al., 2010). Many studies, however, do not distinguish what collagen type was studied and just refer to collagen. Studies that have investigated type III collagen specifically include those on the hypertrophic (Hayashi et al., 1998; Kitamura et al., 2001) and dilated cardiomyopathy (Marijianowski et al., 1995; Hayashi et al., 1998; Bonapace et al., 2006; Herpel et al., 2006; Sivakumar et al., 2008), as well as pulmonary vascular disease (Frangogiannis, 2017), and replacement fibrosis following myocardial infarction (Herpel et al., 2006; Prabhu and Frangogiannis, 2016). Type III collagen changes have also been studied in a rare cardiomyopathy called idiopathic restrictive cardiomyopathy (Hayashi et al., 1998).
Marijianowski et al (1995) studied the amount and distribution of type I and III collagen in heart tissue samples obtained from patients with dilated cardiomyopathy (n=19) and agematched controls (n=17) who died of non-cardiovascular diseases (Marijianowski et al., 1995). Using biochemical and histological methods, they found that both collagen types were increased in the diseased heart tissues, but the increase was more pronounced in type I collagen and therefore the type I-to-III –ratio increased. The authors speculated that this finding could be clinically relevant and explain part of the disease characteristics, since type III collagen gives more elastic properties to the tissue and type I collagen is more rigid. The findings of increased type III collagen levels being associated with dilated cardiomyopathy have subsequently been validated in other studies (Hayashi et al., 1998; Sivakumar et al., 2008).
Herpel et al (2006) investigated several extracellular matrix proteins in heart tissue samples obtained from patients diagnosed with dilated (n=15), ischemic (n=17) and valvular cardiomyopathy (n=7) using immunostaining (Herpel et al., 2006). They showed that the different types of cardiomyopathies all had myocardial interstial fibrosis, but that they differed in the distribution of various extracellular matrix proteins. The amount of type III collagen was the greatest in samples from patients with dilated cardiomyopathy, and lowest in those from patients with valvular disease. Bonapace et al (2006) studied aortic stiffness in 89 patients diagnosed with dilated cardiomyopathy and found that serum PIIINP levels were correlated with clinical parameters measuring aortic stiffness (Bonapace et al., 2006).
Idiopathic restrictive cardiomyopathy is a rare disease manifesting as a diastolic dysfunction. Histologically the disease is characterized by fibrosis in the heart tissue and hypertrophy of myocytes making the heart tissue stiff. Hayashi et al (Hayashi et al., 1998) carried out a histological analysis of endomyocardial biopsy samples (n=7) from these patients and compared the findings to those from patients diagnosed with coronary artery disease (n=5), hypertrophic cardiopathy (n=11) and dilated cardiomyopathy (n=14). The results revealed a marked increase in the amount of type III collagen deposited in the myocardial interstitium. Kitamura et al (2001) analyzed histologically 35 heart tissue specimens from 35 patients with hypertrophic cardiomyopathy (Kitamura et al., 2001). The amount of type III collagen protein detected in immunostaining correlated with several cardiac metrics measuring diastolic function and demonstrated that increased amount of fibrosis was associated with poorer cardiac function. Interestingly, the amount of type I collagen did not correlate with these metrics.
Thiele et al (Thiele et al., 2004) found that the mRNA levels of hnRNP E1, K and A1 were elevated in heart tissue samples from patients diagnosed with aortic stenosis (n=18), and E1 and K were elevated in patients with ischemic cardiomyopathy (n=6), compared to control heart tissue samples (n=20) taken from hearts harvested for transplantation. As described in section 6, A1 and K bind to sequences in the 3’UTR of COL3A1 mRNA to stabilize the transcript.
PIIINP has been tested as a potential marker for heart disease, but the results have been contradictory. In a large community-based study of 967 well-characterized participants, no correlation was found between serum PIIINP levels and various cardiovascular risk factors such as hypertension or diabetes (Wang et al., 2007). There was also no correlation between echocardiographically-determined left ventricular or atrial mass and PIIINP levels (Wang et al., 2007). Manhenke et al (2011) measured several biomarkers, including PIIINP, in serum samples from 233 patients who had suffered a myocardial infarction within the past three days (Manhenke et al., 2014). The patients were then followed up for two years. Several of the other biomarkers including type I collagen markers, but not PIIINP, changed during the follow-up that included treatment with either losartan or captopril. In this study PIIINP showed no prognostic value. The same authors (Manhenke et al., 2014) analyzed 42 patients with first-time mmyocardial infarction that produced ST-elevation in an electrocardiogram. The patients underwent percutaneous coronary intervention with stent implantation. In agreement with their earlier study, PIIINP levels stayed within normal limits during the entire time. There was, however, an increase in PIIINP levels during the first two days after the infarction, but the levels did not exceed the normal limits.
In 2012, Fertin et al (2012) carried out a systematic review on 59 published studies (Fertin et al., 2012) investigating the usefulness of biomarkers predicting which myocardial infarction patients develop left ventricular remodeling that leads to heart failure. PIIINP was one of the biomarkers studied, but it was measured in only three studies for a total of 210 patients. In all three studies the serum PIIINP levels were correlated with left ventricular remodeling.
7.3.2. Other diseases
The histological hallmarks of chronic renal failure are glomerulosclerosis and tubulointerstial fibrosis due to the accumulation of connective tissue proteins (Soylemezoglu et al., 1997). Based on histological analyses tubular epithelial cells in patients with diabetic nephropathy synthesize increased amounts of type III collagen (Razzaque et al., 1995). Individual studies on renal fibrosis have concluded that PIIINP levels in the urine and serum correlate with the amount of fibrosis present in the kidneys (Soylemezoglu et al., 1997). A recent systematic review (Mansour et al., 2017) that included only studies in which renal biopsy was used to verify the kidney disease, however, concluded that PIIINP levels were not independently associated with a chronic kidney disease after adjusting for proteinuria. In another communitybased study of 958 individuals ≥65 years, urine PIIINP levels were found to increase with the progression of the kidney disease and higher PIIINP levels were also associated with death (Ix et al., 2015).
For liver diseases, PIIINP assay is part of the “Enhanced Liver Fibrosis” (ELF) panel, which includes assays for three molecules: hyaluronic acid, PIIINP, and tissue inhibitor of matrix metalloproteinase-1 (Tanwar et al., 2017). The ELF panel has been used as a surrogate marker of liver fibrosis allowing a minimally invasive blood sampling method to be used instead of obtaining a liver biopsy (Tanwar et al., 2017). ELF score and PIIINP levels were also shown to be correlated with skin and lung fibrosis in patients with systemic sclerosis (Abignano et al., 2019).
Collagen accumulation and metabolism has also been studied extensively in fibrotic lung diseases. For example, Kirk et al. (Kirk et al., 1984) carried out a study on patients diagnosed with cryptogenic fibrosing alveolitis. They showed that the serum PIIINP concentrations were increased in these patients compared to normal controls or patients with non-fibrotic lung diseases. With biochemical analyses of lung tissue samples they were able to demonstrate that the serum PIIINP levels correlated with the type III to type I collagen ratios in the lung. Interestingly, those patients with the highest serum PIIINP levels before the treatment had the best response to treatment measured by improvements in lung function tests forced vital capacity and transfer coefficient values.
Another disease with type III collagen accumulation is Dupuytren’s disease, in which type III collagen is present in increased amounts in the aponeurosis (Menzel et al., 1979; Somers et al., 1989). Collagen III accumulation has been seen in yet another fibrotic condition called Peyronie’s disease, in which scar tissue forms in the tunica albuginea of the penis as a result of currently unknown mechanism and leads to a severely impaired function (Somers et al., 1989).
Elevated levels of type III collagen have also been seen in patients with Crohn’s disease. One study showed increased amounts of type III collagen in fibroblast cultures originating from patients’ intestine (Stallmach et al., 1992). In another study C3M levels were higher in serum samples from patients with the penetrating form of Crohn’s disease indicating increased breakdown of type III collagen by matrix metalloproteinase 9 (van Haaften et al., 2017). The best marker to differentiate between the different subtypes of Crohn’s disease was the ratio of type III collagen to C3M, reflecting an imbalance between the synthesis and the breakdown of type III collagen. Another study was able to differentiate between Crohn’s disease and ulcerative colitis using serum biomarkers including the C3M assay (Mortensen et al., 2015).
Type III collagen expression has also been studied in non-fibrotic diseases. In a study that investigated 33 patients with hyperthyroidism, the serum levels of PIIINP were found to be elevated compared to 26 controls (Inui et al., 1992). The authors pointed out that the liver function of these patients was normal.
Aigner et al (Aigner et al., 1993) had an unexpected finding in that type III collagen was present in dedifferentiated chondrocytes in cartilage samples taken from patients with late stage osteoarthritis, when the well-established expression profile of chondrocytes includes type II, IX and XI collagens, but no type III collagen. In this study immunostaining was used to investigate protein expression and in situ RNA hybridization to determine the location and extent of mRNA expression. Interestingly, these cells also expressed type II collagen, but no type I collagen. Normal chondrocytes did not express any type III collagen (Aigner et al., 1993). Another group, however, detected type III collagen even in normal human articular cartilage specimens taken from individuals between ages 17 and 81 years (Wotton and Duance, 1994).
Type III collagen expression has also been evaluated in cancer. In one study where 33 glioma tissues samples were analyzed, a significant increase in COL3A1 mRNA levels was found and the increased expression correlated with the tumor grade with high grade gliomas showing the highest levels of expression (Gao et al., 2018). Immunohistological staining of glioma tissues demonstrated the same results on the protein level. In follow-up studies in which COL3A1 expression in a cell culture system was silenced by siRNA, the proliferation and migration of glioma cells was suppressed.
In another cancer study, overexpression of COL3A1 was found to be associated with a poor prognosis of bladder cancer (Yuan et al., 2017). Similarly, increased levels of PIIINP were associated with a poor prognosis and a shorter overall survival of breast cancer patients (Lipton et al., 2018).
8. Col3a1 mutant mice
Currently there are four different Col3a1 mouse models (Liu et al., 1997; Smith et al., 2011; Long et al., 2015; D’Hondt et al., 2018). As can be seen in Table 3, which summarizes the characteristics of these models, none of them are ideal models of the human vEDS. They have, however, become useful models for studying the function of type III collagen in various tissues and organs.
Table 3.
Comparison of Col3a1 models
| Feature | Col3a1 KO | Col3a1 deletion | Transgenic with Gly182Ser mutation | Tsk/+ (Cys-to-Ser in PIIINP) |
|---|---|---|---|---|
| Original publication | Liu et al. 1997 (Liu et al., 1997) | Smith et al. 2011 (Smith et al., 2011) | D’hondt et al. 2018 (D’Hondt et al., 2018) | Long et al. 2015 (Long et al., 2015) |
| Method used to create model | Homologous recombination with a targeting vector that contains neo resulting in a deletion of the promoter and exon 1 sequences | Deletion occurred spontaneously; 185 kb; removes Col3a1 promoter and exons 1–39 | Transgenic mouse lines with and without Gly182Ser mutation to overexpress Col3a1 | Mutagenesis with ethylnitrosurea |
| Phenotype in homozygous mice | Early death in 95%; remaining 5% had arterial aneurysm and rupture at age 6 months | Early death | Mice overexpressing Col3a1 with Gly182Ser mutation die at 13–14 weeks due to severe skin wounds | Embryonic death |
| Phenotype in heterozygous mice | Appeared grossly normal; but stress test showed decreased strength in aorta and bowel | 28% have thoracic aortic dissection | Tight skin resembling systemic sclerosis in humans | |
| Vascular phenotype | Arterial aneurysm and rupture at age 6 months | Thoracic aortic dissection (no aneurysm) in 28% of heterozygote mice | Grossly normal, but in stress test aorta showed reduced tensile strength | |
| Bowel phenotype | Enlarged with spontaneous rupture | Normal | Normal | Normal |
| Skin phenotype | Severe wounds | Normal | Thin and fragile with severe wounds in male mice | Tight skin resembling systemic sclerosis in humans |
| Light microscopy findings | Heterozygote mice have reduced amount of collagen and fragmentation of internal elastic lamina in aorta | Heterozygote mice have reduced amount of collagen | Thinner aortic adventitia | Excessive deposition of ECM proteins in skin |
| Electron microscopy findings | Abnormal type I collagen fibrils | Abnormal collagen fibrils | Abnormal collagen fibrils | Thick collagen fibrils |
| Biochemical analyses | Col3a1 mRNA levels decreased | Col5a1 and Tgfβ mRNA levels normal | Less Col3a1 protein in transgenic Gly182Ser mice; thermal stability normal | Col3a1 mRNA levels increased |
| Differences between male and female mice | Males more often affected | Male mice died of dissection twice as frequently as female mice | Male transgenic mice express more Col3a1 |
EMC, extracellular matrix; KO, knockout
8.1. Inactivation by knockout of the murine Col3a1 gene
Inactivation of the murine Col3a1 gene was carried out by homologous recombination in embryonic stem cells using a targeting vector in which the Col3a1 promoter and the first exon were deleted and replaced by the neomycin gene (Liu et al., 1997). Approximately 95% of the homozygous mutant mice died early on, with most deaths occurring in the first two days after birth, but heterozygous mice appeared phenotypically normal (Liu et al., 1997). The homozygous mice that survived were smaller than their wild-type littermates, and they died at the age of about 6 months from a rupture of major arteries or the bowel, mimicking the human vEDS phenotype. These mice also had skin lesions, such as open wounds. Histological analysis using light microscopy did not reveal any obvious abnormalities in the analyzed tissues. Electron microscopy, however, demonstrated abnormal collagen fibers in the skin, aorta, liver, lung, heart and bowel (Liu et al., 1997). These studies provided important information to our basic understanding of the fibrillogenesis of collagen by demonstrating that lack of type III collagen led to disorganized type I collagen fibrils with variable diameters (Liu et al., 1997). Type III collagen thus appears to regulate the type I collagen fibril formation.
The homozygous Col3a1−/− mice were also studied for potential brain defects, since COL3A1 is a ligand for GPR56, which harbors mutations in patients with bilateral frontoparietal polymicrogyria, a severe brain malformation (Jeong et al., 2012). In histological analyses all Col3a1−/− mice showed a severe malformation of the brain cortex at E18.5. The defects were described as cobblestone like cortical malformations and included pial breakdown in the basement membrane, neuronal overmigration, radial glial detachment, and formation of marginal zone heterotopias. None of the heterozygous Col3a1+/− or wildtype mice had these malformations. Based on these findings, type III collagen appears to be important for brain development.
The heterozygous mutant mice of the same Col3a1 knock-out mouse model were studied further by Cooper et al. (Cooper et al., 2010). Less collagen and reduced amounts of Col3a1 mRNA were found in the abdominal aorta and the wall strength was reduced. Similarly, the bowel walls from these mice showed increased compliance and decreased strength, which was tested by applying pressure and determining when the bowel wall burst. Histological lesions found in the internal elastic lamina of the aortae were more severe in older mice and more common in male mice (Cooper et al., 2010). In conclusion, the heterozygous mice lack the clinical features of vEDS, but do display several histological signs of Col3a1 deficiency. A 9month doxycycline treatment of heterozygous (Col3a1+/−) mice reduced the number of histological lesions in the aorta (Tae et al., 2012). This was most likely due to a decrease in Mmp9 expression, since doxycycline is a known inhibitor of MMPs.
The Col3a1 knock-out mouse model has been studied for many different organ systems and biological processes including the bladder function and healing of bone fractures. The amount of type III collagen was reduced and the collagen fiber diameter more varied in the bladder tissues of Col3a1+/−mice compared to wild-type mice (Stevenson et al., 2006). In functional tests using bladder muscle strips removed from the mice, the muscle removed from heterozygous mice generated less tension and were more compliant suggesting that there is a critical level of type III collagen required for normal bladder function.
A detailed analysis of the skeletons of the wild-type, Col3a1+/− and Col3a1−/− mice at age 6–8 weeks was carried out using microcomputer tomography (Volk et al., 2014). The results demonstrated a reduction in several bone phenotypes including the bone volume, bone volume fraction, connectivity density, structure model index and trabecular thickness in the Col3a1+/− compared to the wild-type mice. In cell culture experiments, mesenchymal progenitor cells isolated from the Col3a1−/− mice had decreased activity of alkaline phosphatase and impaired mineralization. The authors concluded that these findings implied that type III collagen is involved in the formation of the trabecular bone. Furthermore, using a bilateral tibial fracture model in the heterozygous (Col3a1+/−) mice reduced levels of Col3a1 were shown to impair the bone formation and alter remodelling during the healing of the fractures (Miedel et al., 2015). Based on immunofluorescent staining, fracture callus contained considerable amounts of type III collagen, whereas very little or no type III collagen was detected in the uninjured cortical bone.
8.2. A large deletion of the murine Col3a1 gene
Another mouse model with a large 185-kb deletion encompassing the promoter region and the first 39 exons of the Col3a1 gene was first discovered by its phenotype of sudden deaths due to thoracic aortic dissections (Smith et al., 2011). The underlying genetic defect in these mice was detected by genetic mapping and DNA sequencing. In follow-up studies investigating 225 mice heterozygous for the deletion, aortic dissection was seen in 28% and it was twice as common in male as female mice. No skin or bowel problems were detected in any of the mice. Mice homozygous for the deletion appeared to die before birth, since no homozygous mice were found. Histological analyses of the aortic wall showed reduced amounts of collagen in the media, but not in the adventitia layer. Transmission electron microscopy images demonstrated abnormal collagen fibers (Smith et al., 2011).
8.3. Transgenic mice overexpressing a mutated form of Col3a1
The third currently available mouse model includes transgenic mice overexpressing Col3a1 with a Gly182Ser mutation (D’Hondt et al., 2018). A control transgenic mouse line overexpressing the normal Col3a1 was also produced. The mice with the Gly182Ser transgene developed severe skin wounds, demonstrated vascular fragility in the form of reduced tensile strength and died prematurely at the age of 13–14 weeks (D’Hondt et al., 2018). Both skin and aorta contained abnormal collagen fibrils, and there was less type III collagen in the aortic wall (D’Hondt et al., 2018). The collagen type III:I ratio in the skin changed from 0.5 in the nontransgenic mice to 1.0 in the transgenic mice overexpressing normal Col3a1, to 0.3 in the transgenic mice with the Gly182Ser mutation. There were no differences in the extent of posttranslational modifications or the thermal stability of the collagens produced between the two transgenic mouse lines (D’Hondt et al., 2018). The diameter of collagen fibers was more variable and the fibers were generally thicker in the Gly182Ser transgenic mice than in the other mouse lines.
8.4. Tsk2/+ mice with a missense mutation
The tight skin (Tsk2/+) mice resemble the human disease systemic sclerosis. They were identified in a screening after mutagenic agent ethylnitrosurea was used to generate mutations in the mouse genome (Long et al., 2015). The defect was genetically mapped to mouse chromosome 1, which harbors Col3a1. Sequencing of RNA and genomic DNA identified a total of 265 sequence variants in both wild-type and Tsk2/+ mice and they were thus excluded from further analyses. Thirteen sequence variants were found in all four Tsk2/+ mice, but ten of them were also found in another strain of mice and were excluded. Of the remaining three sequence variants present only in the Tsk2/+ mice, two were in introns, and one was a missense variant in Col3a1 that converted a cysteine to a serine in the N-terminal propeptide. This Cys-to-Ser change mutates one of the cysteine residues that are normally part of intrachain disulphide bonds. Based on the RNA-sequencing data, Col3a1 had the highest number of reads from the genes located in the candidate interval, indicating that its expression was increased, which is in agreement with previous biochemical studies. Complementation experiments were carried out in which the Tsk2/+ mice were crossed with Col3a1−/+ mice to prove that the sequence variant found in Col3a1 is Tsk2. The result showed that the Tsk2 gene was not able to rescue the Col3a1-knockout phenotype.
9. Conclusions
Nearly 50 years of research into the structure and function of type III collagen has demonstrated that it is an essential structural component of blood vessels, uterus and bowel. Without type III collagen mice die in utero and with mutated forms of it humans develop serious clinical manifestations leading to a premature death due to a spontaneous rupture of an artery, bowel or uterus. Furthermore, some COL3A1 mutations lead to a severe brain abnormality and developmental delay. Increased amounts of type III collagen are found in many acquired human fibrotic diseases such as kidney and liver fibrosis, and systemic sclerosis. Type III collagen is known to interact with type I and II collagens in the fibril formation and is an important regulator of fibril diameter; increase in type III collagen content will lead to the formation of thinner fibrils. Type III collagen is also a critical component in platelet aggregation and thus initiating the blood clotting cascade.
There are still several unanswered questions about the function of type III collagen, e.g., what are the molecules it interacts with, is it possible to create a computational model that predicts the effects of a COL3A1 mutation accurately, and how do we prevent overexpression of type III collagen and fibrotic processes. It is also not clear why some COL3A1 mutations lead to vEDS, whereas others manifest as a severe brain abnormality. The classic vEDS manifests as an autosomal dominant disease, but in the case of the brain abnormality, all reported cases have had two mutant COL3A1 alleles suggesting an autosomal recessive inheritance pattern. To facilitate answering these important research questions with implications to human health, additional animal models are required.
Supplementary Material
Highlights.
Type III collagen is a structural component of blood vessels, uterus and bowel.
Type III collagen regulates type I collagen fibril formation.
Mutations in COL3A1 cause vascular Ehlers-Danlos syndrome.
Mutations in COL3A1 can also cause frontoparietal polymicrogyria.
Accumulation of type III collagen is found in fibrotic diseases.
Acknowledgments
This review and the corresponding Gene Wiki article are written as part of the Gene Wiki Review series--a series resulting from a collaboration between the journal GENE and the Gene Wiki Initiative. The Gene Wiki Initiative is supported by National Institutes of Health (GM089820). Additional support for Gene Wiki Reviews is provided by Elsevier, the publisher of GENE.
HK is supported by the Faculty of Medicine and Health Sciences, Stellenbosch University, South Africa. GT is supported by the South African Tuberculosis Bioinformatics Initiative (SATBBI), a Strategic Health Innovation Partnership grant from the South African Medical Research Council and South African Department of Science and Technology.
The authors are grateful to the two anonymous expert reviewers for helpful suggestions.
The corresponding Gene Wiki entry for this review can be found here: https://en.wikipedia.org/wiki/Collagen,_type_III,_alpha_1
List of abbreviations
- A
adenosine
- aa
amino acid(s)
- Alu
a type of repeat sequence in the genome
- Arg428Stop
an arginine at amino acid position 428 has changed to a stop codon
- Arg1432Leu
an arginine at amino acid position 1432 has changed to a leucine
- AT
a type of repeat sequence in the genome
- bFGF
basic fibroblast growth factor
- bp
base pair(s)
- C
cytidine
- C3C
fragments of type III collagen generated by digestion with cathepsins B, L, S, and K
- C3M
a 10-aa neopeptide of type III collagen generated by digestion with matrixmetalloproteinase 9
- CCDS
Consensus Coding Sequence
- CDS
coding seqeunce
- COPD
chronic obstructive pulmonary disease
- C-terminal
carboxyterminal
- cDNA
DNA complementary to RNA
- E18.5
developmental stage of a mouse embryo at 18.5 days
- ECM
extracellular matrix
- EGF
epidermal growth factor
- ELF
enhanced liver fibrosis, a panel of three biomarkers for liver fibrosis
- ELISA
enzyme-linked immunosorbent assay
- EM
electron microscope
- G
guanosine
- Gly182Ser
a glycine at amino acid position 182 has changed to a serine
- Gly237Arg
a glycine at amino acid position 237 has changed to an arginine
- Gly612Asp
a glycine at amino acid position 612 has changed to an aspartic acid
- Gly-Xaa-Yaa
a triplet in the triple-helical domain of type III collagen showing glycine as every third amino acid
- GRCh38
Genome Reference Consortium Human Build 38
- GRCm38
Genome Reference Consortium Mouse Build 38
- GRP56
G protein-coupled receptor 56
- HGNC
Human Genome Nomenclature Committee
- HIF-1α
hypoxia-inducible factor alpha 1
- hnRNP
heterogeneous nuclear ribonucleoprotein
- ICD-10
International Classification of Diseases 10th revision
- IHC
immunohistochemistry
- kb
kilobase(s) or 1000 bp
- kDa
kilodalton(s)
- KO
knock-out
- LINE
a type of repeat sequence in the genome
- Lys1313Arg
a lysine at amino acid position 1313 has changed to an arginine
- MAPK
mitogen-activated protein kinase
- MGI
Mouse Genome Informatics
- mTOR
mammalian target of rapamycin
- NCBI
National Center for Biotechnology Information
- NEDD9
neural precursor cell expressed, developmentally down-regulated 9
- nt
nucleotide(s)
- N-terminal
aminoterminal
- OMIM
online inheritance in man database
- PIIINP
aminoterminal propeptide of type III procollagen
- polyA
polyadenylation site
- Pro49Ala
a proline at amino acid position 49 has changed to an alanine
- Pro686
a proline at amino acid position 686
- Pro1440Leu
a proline at amino acid position 1440 has changed to a leucine
- qRT-PCR
quantitative reverse transcriptase polymerase chain reaction
- RIA
radioimmunoassay
- T
thymidine
- TGFβ
transforming growth factor beta
- Tsk2/+
tight skin mouse with a mutation in Col3a1
- UTR
untranslated region(s)
- vEDS
vascular Ehlers-Danlos syndrome
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest statement
The authors declare that they have no commercial or financial relationships that could be construed as a potential conflict of interest.
References
- Abignano G, Blagojevic J, Bissell LA, Dumitru RB, Eng S, Allanore Y, Avouac J, Bosello S, Denton CP, Distler O, Ferraccioli G, Jordan S, Matucci-Cerinic M, Ong V, Messenger M, Hutchinson M, Buch MH, Emery P and Del Galdo F, 2019. European multicentre study validates enhanced liver fibrosis test as biomarker of fibrosis in systemic sclerosis. Rheumatology (Oxford) 58, 254–259. [DOI] [PubMed] [Google Scholar]
- Aigner T, Bertling W, Stoss H, Weseloh G and von der Mark K, 1993. Independent expression of fibril-forming collagens I, II, and III in chondrocytes of human osteoarthritic cartilage. J Clin Invest 91, 829–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ala-Kokko L, Kontusaari S, Baldwin CT, Kuivaniemi H and Prockop DJ, 1989. Structure of cDNA clones coding for the entire prepro alpha 1 (III) chain of human type III procollagen. Differences in protein structure from type I procollagen and conservation of codon preferences. Biochem J 260, 509–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson DW, Edwards TK, Ricketts MH, Kuivaniemi H, Tromp G, Stolle CA, Deak SB and Boyd CD, 1996. Multiple defects in type III collagen synthesis are associated with the pathogenesis of abdominal aortic aneurysms. Ann N Y Acad Sci 800, 216–28. [DOI] [PubMed] [Google Scholar]
- Anderson DW, Thakker-Varia S, Tromp G, Kuivaniemi H and Stolle CA, 1997. A glycine (415)-to-serine substitution results in impaired secretion and decreased thermal stability of type III procollagen in a patient with Ehlers-Danlos syndrome type IV. Hum Mutat 9, 62–3. [DOI] [PubMed] [Google Scholar]
- Bachinger HP, Bruckner P, Timpl R, Prockop DJ and Engel J, 1980. Folding mechanism of the triple helix in type-III collagen and type-III pN-collagen. Role of disulfide bridges and peptide bond isomerization. Eur J Biochem 106, 619–32. [DOI] [PubMed] [Google Scholar]
- Balleisen L, Gay S, Marx R and Kuhn K, 1975. Comparative investigation on the influence of human and bovine collagen types I, II and III on the aggregation of human platelets. Klin Wochenschr 53, 903–5. [DOI] [PubMed] [Google Scholar]
- Balleisen L, Nowack H, Gay S and Timpl R, 1979. Inhibition of collagen-induced platelet aggregation by antibodies to distinct types of collagens. Biochem J 184, 683–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barascuk N, Veidal SS, Larsen L, Larsen DV, Larsen MR, Wang J, Zheng Q, Xing R, Cao Y, Rasmussen LM and Karsdal MA, 2010. A novel assay for extracellular matrix remodeling associated with liver fibrosis: An enzyme-linked immunosorbent assay (ELISA) for a MMP-9 proteolytically revealed neo-epitope of type III collagen. Clin Biochem 43, 899–904. [DOI] [PubMed] [Google Scholar]
- Birk DE and Silver FH, 1984. Collagen fibrillogenesis in vitro: comparison of types I, II, and III. Arch Biochem Biophys 235, 178–85. [DOI] [PubMed] [Google Scholar]
- Bonapace S, Rossi A, Cicoira M, Golia G, Zanolla L, Franceschini L, Conte L, Marino P, Zardini P and Vassanelli C, 2006. Aortic stiffness correlates with an increased extracellular matrix turnover in patients with dilated cardiomyopathy. Am Heart J 152, 93.e1–6. [DOI] [PubMed] [Google Scholar]
- Boudko SP and Engel J, 2004. Structure formation in the C terminus of type III collagen guides disulfide cross-linking. J Mol Biol 335, 1289–97. [DOI] [PubMed] [Google Scholar]
- Boudko SP, Engel J, Okuyama K, Mizuno K, Bachinger HP and Schumacher MA, 2008. Crystal structure of human type III collagen Gly991-Gly1032 cystine knot-containing peptide shows both 7/2 and 10/3 triple helical symmetries. J Biol Chem 283, 32580–9. [DOI] [PubMed] [Google Scholar]
- Bourhis JM, Mariano N, Zhao Y, Harlos K, Exposito JY, Jones EY, Moali C, Aghajari N and Hulmes DJ, 2012. Structural basis of fibrillar collagen trimerization and related genetic disorders. Nat Struct Mol Biol 19, 1031–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandt A, Glanville RW, Horlein D, Bruckner P, Timpl R, Fietzek PP and Kuhn K, 1984. Complete amino acid sequence of the N-terminal extension of calf skin type III procollagen. Biochem J 219, 625–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byers PH, 1993. [updated 2019]. Vascular Ehlers-Danlos Syndrome, in: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Stephens K and Amemiya A (Eds.), GeneReviews® University of Washington, Seattle, Seattle (WA). [PubMed] [Google Scholar]
- Byers PH, Belmont J, Black J, De Backer J, Frank M, Jeunemaitre X, Johnson D, Pepin M, Robert L, Sanders L and Wheeldon N, 2017. Diagnosis, natural history, and management in vascular Ehlers-Danlos syndrome. Am J Med Genet C Semin Med Genet 175, 40–47. [DOI] [PubMed] [Google Scholar]
- Byers PH, Holbrook KA, Barsh GS, Smith LT and Bornstein P, 1981. Altered secretion of type III procollagen in a form of type IV Ehlers-Danlos syndrome. Biochemical studies in cultured fibroblasts. Lab Invest 44, 336–41. [PubMed] [Google Scholar]
- Cameron GJ, Alberts IL, Laing JH and Wess TJ, 2002. Structure of type I and type III heterotypic collagen fibrils: an X-ray diffraction study. J Struct Biol 137, 15–22. [DOI] [PubMed] [Google Scholar]
- Chan TF, Poon A, Basu A, Addleman NR, Chen J, Phong A, Byers PH, Klein TE and Kwok PY, 2008. Natural variation in four human collagen genes across an ethnically diverse population. Genomics 91, 307–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Li J, Li Q, Li X, Gao Y, Hua X, Zhou B and Li J, 2018. Overexpression of LncRNA AC067945.2 Down-Regulates Collagen Expression in Skin Fibroblasts and Possibly Correlates with the VEGF and Wnt Signalling Pathways. Cell Physiol Biochem 45, 761–771. [DOI] [PubMed] [Google Scholar]
- Chiang TM, Seyer JM and Kang AH, 1993. Collagen-platelet interaction: separate receptor sites for types I and III collagen. Thromb Res 71, 443–56. [DOI] [PubMed] [Google Scholar]
- Chiarelli N, Carini G, Zoppi N, Ritelli M and Colombi M, 2018. Transcriptome analysis of skin fibroblasts with dominant negative COL3A1 mutations provides molecular insights into the etiopathology of vascular Ehlers-Danlos syndrome. PLoS One 13, e0191220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu ML, Weil D, de Wet W, Bernard M, Sippola M and Ramirez F, 1985. Isolation of cDNA and genomic clones encoding human pro-alpha 1 (III) collagen. Partial characterization of the 3’ end region of the gene. J Biol Chem 260, 4357–63. [PubMed] [Google Scholar]
- Cooper TK, Zhong Q, Krawczyk M, Tae HJ, Muller GA, Schubert R, Myers LA, Dietz HC, Talan MI and Briest W, 2010. The haploinsufficient Col3a1 mouse as a model for vascular EhlersDanlos syndrome. Vet Pathol 47, 1028–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- d’Ardenne AJ, Kirkpatrick P and Sykes BC, 1984. Distribution of laminin, fibronectin, and interstitial collagen type III in soft tissue tumours. J Clin Pathol 37, 895–904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Hondt S, Guillemyn B, Syx D, Symoens S, De Rycke R, Vanhoutte L, Toussaint W, Lambrecht BN, De Paepe A, Keene DR, Ishikawa Y, Bachinger HP, Janssens S, Bertrand MJM and Malfait F, 2018. Type III collagen affects dermal and vascular collagen fibrillogenesis and tissue integrity in a mutant Col3a1 transgenic mouse model. Matrix Biol 70, 72–83. [DOI] [PubMed] [Google Scholar]
- DiChiara AS, Li RC, Suen PH, Hosseini AS, Taylor RJ, Weickhardt AF, Malhotra D, McCaslin DR and Shoulders MD, 2018. A cysteine-based molecular code informs collagen C-propeptide assembly. Nat Commun 9, 4206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duval E, Leclercq S, Elissalde JM, Demoor M, Galera P and Boumediene K, 2009. Hypoxia-inducible factor 1alpha inhibits the fibroblast-like markers type I and type III collagen during hypoxia-induced chondrocyte redifferentiation: hypoxia not only induces type II collagen and aggrecan, but it also inhibits type I and type III collagen in the hypoxia-inducible factor 1alphadependent redifferentiation of chondrocytes. Arthritis Rheum 60, 3038–48. [DOI] [PubMed] [Google Scholar]
- Eagleton MJ, 2016. Arterial complications of vascular Ehlers-Danlos syndrome. J Vasc Surg 64, 1869–1880. [DOI] [PubMed] [Google Scholar]
- Fagerberg L, Hallstrom BM, Oksvold P, Kampf C, Djureinovic D, Odeberg J, Habuka M, Tahmasebpoor S, Danielsson A, Edlund K, Asplund A, Sjostedt E, Lundberg E, Szigyarto CA, Skogs M, Takanen JO, Berling H, Tegel H, Mulder J, Nilsson P, Schwenk JM, Lindskog C, Danielsson F, Mardinoglu A, Sivertsson A, von Feilitzen K, Forsberg M, Zwahlen M, Olsson I, Navani S, Huss M, Nielsen J, Ponten F and Uhlen M, 2014. Analysis of the human tissue-specific expression by genome-wide integration of transcriptomics and antibody-based proteomics. Mol Cell Proteomics 13, 397–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fauvel F, Legrand YJ, Bentz H, Fietzek PP, Kuhn K and Caen JP, 1978. Platelet-collagen interaction: adhesion of human blood platelets to purified (CB4) peptide from type III collagen. Thromb Res 12, 841–50. [DOI] [PubMed] [Google Scholar]
- Fertin M, Dubois E, Belliard A, Amouyel P, Pinet F and Bauters C, 2012. Usefulness of circulating biomarkers for the prediction of left ventricular remodeling after myocardial infarction. Am J Cardiol 110, 277–83. [DOI] [PubMed] [Google Scholar]
- Fietzek PP, Allmann H, Rauterberg J, Henkel W, Wachter E and Kuhn K, 1979. The covalent structure of calf skin type III collagen. I. The amino acid sequence of the amino terminal region of the alpha 1(III) chain (position 1−−222). Hoppe Seylers Z Physiol Chem 360, 809–20. [DOI] [PubMed] [Google Scholar]
- Fleischmajer R, MacDonald ED, Perlish JS, Burgeson RE and Fisher LW, 1990. Dermal collagen fibrils are hybrids of type I and type III collagen molecules. J Struct Biol 105, 162–9. [DOI] [PubMed] [Google Scholar]
- Fogo AB, Lusco MA, Najafian B and Alpers CE, 2017. AJKD Atlas of Renal Pathology: Type III Collagen Glomerulopathy. Am J Kidney Dis 69, e25–e26. [DOI] [PubMed] [Google Scholar]
- Frangogiannis NG, 2017. Fibroblasts and the extracellular matrix in right ventricular disease. Cardiovasc Res 113, 1453–1464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank M, Albuisson J, Ranque B, Golmard L, Mazzella JM, Bal-Theoleyre L, Fauret AL, Mirault T, Denarie N, Mousseaux E, Boutouyrie P, Fiessinger JN, Emmerich J, Messas E and Jeunemaitre X, 2015. The type of variants at the COL3A1 gene associates with the phenotype and severity of vascular Ehlers-Danlos syndrome. Eur J Hum Genet 23, 1657–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao YF, Zhu T, Chen J, Liu L and Ouyang R, 2018. Knockdown of collagen alpha-1(III) inhibits glioma cell proliferation and migration and is regulated by miR128–3p. Oncol Lett 16, 1917–1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert EH, Glatstein E, Goris ML and Earle JD, 1978. Value of 111Indium chloride bone marrow scanning in the differential diagnosis of blood count depression in lymphoma. Cancer 41, 143–52. [DOI] [PubMed] [Google Scholar]
- Giunta C and Steinmann B, 2000. Characterization of 11 new mutations in COL3A1 of individuals with Ehlers-Danlos syndrome type IV: preliminary comparison of RNase cleavage, EMC and DHPLC assays. Hum Mutat 16, 176–7. [DOI] [PubMed] [Google Scholar]
- Glanville RW and Fietzek PP, 1976. Amino acid sequence to the N-terminal non-triple helical cross link region of type III collagen. FEBS Lett 72, 99–102. [DOI] [PubMed] [Google Scholar]
- Haukipuro K, Risteli L, Kairaluoma MI and Risteli J, 1987. Aminoterminal propeptide of type III procollagen in healing wound in humans. Ann Surg 206, 752–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi T, Shimomura H, Terasaki F, Toko H, Okabe M, Deguchi H, Hirota Y, Kitaura Y and Kawamura K, 1998. Collagen subtypes and matrix metalloproteinase in idiopathic restrictive cardiomyopathy. Int J Cardiol 64, 109–16. [DOI] [PubMed] [Google Scholar]
- Herpel E, Pritsch M, Koch A, Dengler TJ, Schirmacher P and Schnabel PA, 2006. Interstitial fibrosis in the heart: differences in extracellular matrix proteins and matrix metalloproteinases in end-stage dilated, ischaemic and valvular cardiomyopathy. Histopathology 48, 736–47. [DOI] [PubMed] [Google Scholar]
- Hinterseher I, Tromp G and Kuivaniemi H, 2011. Genes and abdominal aortic aneurysm. Ann Vasc Surg 25, 388–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horn D, Siebert E, Seidel U, Rost I, Mayer K, Abou Jamra R, Mitter D and Kornak U, 2017. Biallelic COL3A1 mutations result in a clinical spectrum of specific structural brain anomalies and connective tissue abnormalities. Am J Med Genet A 173, 2534–2538. [DOI] [PubMed] [Google Scholar]
- Inui T, Ochi Y, Chen W, Nakajima Y and Kajita Y, 1992. Increased serum concentration of type IV collagen peptide and type III collagen peptide in hyperthyroidism. Clin Chim Acta 205, 181–6. [DOI] [PubMed] [Google Scholar]
- Ix JH, Biggs ML, Mukamal K, Djousse L, Siscovick D, Tracy R, Katz R, Delaney JA, Chaves P, Rifkin DE, Hughes-Austin JM, Garimella PS, Sarnak MJ, Shlipak MG and Kizer JR, 2015. Urine Collagen Fragments and CKD Progression-The Cardiovascular Health Study. J Am Soc Nephrol 26, 2494–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janeczko RA and Ramirez F, 1989. Nucleotide and amino acid sequences of the entire human alpha 1 (III) collagen. Nucleic Acids Res 17, 6742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarvis GE, Raynal N, Langford JP, Onley DJ, Andrews A, Smethurst PA and Farndale RW, 2008. Identification of a major GpVI-binding locus in human type III collagen. Blood 111, 4986–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeong SJ, Li S, Luo R, Strokes N and Piao X, 2012. Loss of Col3a1, the gene for Ehlers-Danlos syndrome type IV, results in neocortical dyslamination. PLoS One 7, e29767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jimenez SA, Feldman G, Bashey RI, Bienkowski R and Rosenbloom J, 1986. Co-ordinate increase in the expression of type I and type III collagen genes in progressive systemic sclerosis fibroblasts. Biochem J 237, 837–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones GT, Tromp G, Kuivaniemi H, Gretarsdottir S, Baas AF, Giusti B, Strauss E, Van’t Hof FN, Webb TR, Erdman R, Ritchie MD, Elmore JR, Verma A, Pendergrass S, Kullo IJ, Ye Z, Peissig PL, Gottesman O, Verma SS, Malinowski J, Rasmussen-Torvik LJ, Borthwick KM, Smelser DT, Crosslin DR, de Andrade M, Ryer EJ, McCarty CA, Bottinger EP, Pacheco JA, Crawford DC, Carrell DS, Gerhard GS, Franklin DP, Carey DJ, Phillips VL, Williams MJ, Wei W, Blair R, Hill AA, Vasudevan TM, Lewis DR, Thomson IA, Krysa J, Hill GB, Roake J, Merriman TR, Oszkinis G, Galora S, Saracini C, Abbate R, Pulli R, Pratesi C, Saratzis A, Verissimo AR, Bumpstead S, Badger SA, Clough RE, Cockerill G, Hafez H, Scott DJ, Futers TS, Romaine SP, Bridge K, Griffin KJ, Bailey MA, Smith A, Thompson MM, van Bockxmeer FM, Matthiasson SE, Thorleifsson G, Thorsteinsdottir U, Blankensteijn JD, Teijink JA, Wijmenga C, de Graaf J, Kiemeney LA, Lindholt JS, Hughes A, Bradley DT, Stirrups K, Golledge J, Norman PE, Powell JT, Humphries SE, Hamby SE, Goodall AH, Nelson CP, Sakalihasan N, Courtois A, Ferrell RE, Eriksson P, Folkersen L, Franco-Cereceda A, Eicher JD, Johnson AD, Betsholtz C, Ruusalepp A, Franzen O, Schadt EE, Bjorkegren JL, et al. , 2017. Meta-Analysis of Genome-Wide Association Studies for Abdominal Aortic Aneurysm Identifies Four New Disease-Specific Risk Loci. Circ Res 120, 341353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorgensen A, Fagerheim T, Rand-Hendriksen S, Lunde PI, Vorren TO, Pepin MG, Leistritz DF and Byers PH, 2015. Vascular Ehlers-Danlos Syndrome in siblings with biallelic COL3A1 sequence variants and marked clinical variability in the extended family. Eur J Hum Genet 23, 796–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karsdal MA, Nielsen SH, Leeming DJ, Langholm LL, Nielsen MJ, Manon-Jensen T, Siebuhr A, Gudmann NS, Ronnow S, Sand JM, Daniels SJ, Mortensen JH and Schuppan D, 2017. The good and the bad collagens of fibrosis - Their role in signaling and organ function. Adv Drug Deliv Rev 121, 43–56. [DOI] [PubMed] [Google Scholar]
- Kauppila S, Stenback F, Risteli J, Jukkola A and Risteli L, 1998. Aberrant type I and type III collagen gene expression in human breast cancer in vivo. J Pathol 186, 262–8. [DOI] [PubMed] [Google Scholar]
- Ke N, Ma H, Diedrich G, Chionis J, Liu G, Yu DH, Wong-Staal F and Li QX, 2008. Biochemical characterization of genetic mutations of GPR56 in patients with bilateral frontoparietal polymicrogyria (BFPP). Biochem Biophys Res Commun 366, 314–20. [DOI] [PubMed] [Google Scholar]
- Keene DR, Sakai LY and Burgeson RE, 1991. Human bone contains type III collagen, type VI collagen, and fibrillin: type III collagen is present on specific fibers that may mediate attachment of tendons, ligaments, and periosteum to calcified bone cortex. J Histochem Cytochem 39, 59–69. [DOI] [PubMed] [Google Scholar]
- Kilic A, Sonar SS, Yildirim AO, Fehrenbach H, Nockher WA and Renz H, 2011. Nerve growth factor induces type III collagen production in chronic allergic airway inflammation. J Allergy Clin Immunol 128, 1058–66.e1–4. [DOI] [PubMed] [Google Scholar]
- Kim JK, Xu Y, Xu X, Keene DR, Gurusiddappa S, Liang X, Wary KK and Hook M, 2005. A novel binding site in collagen type III for integrins alpha1beta1 and alpha2beta1. J Biol Chem 280, 32512–20. [DOI] [PubMed] [Google Scholar]
- Kirk JM, Bateman ED, Haslam PL, Laurent GJ and Turner-Warwick M, 1984. Serum type III procollagen peptide concentration in cryptogenic fibrosing alveolitis and its clinical relevance. Thorax 39, 726–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitamura M, Shimizu M, Ino H, Okeie K, Yamaguchi M, Funjno N, Mabuchi H and Nakanishi I, 2001. Collagen remodeling and cardiac dysfunction in patients with hypertrophic cardiomyopathy: the significance of type III and VI collagens. Clin Cardiol 24, 325–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kontusaari S, Tromp G, Kuivaniemi H, Ladda RL and Prockop DJ, 1990a. Inheritance of an RNA splicing mutation (G+ 1 IVS20) in the type III procollagen gene (COL3A1) in a family having aortic aneurysms and easy bruisability: phenotypic overlap between familial arterial aneurysms and Ehlers-Danlos syndrome type IV. Am J Hum Genet 47, 112–20. [PMC free article] [PubMed] [Google Scholar]
- Kontusaari S, Tromp G, Kuivaniemi H, Romanic AM and Prockop DJ, 1990b. A mutation in the gene for type III procollagen (COL3A1) in a family with aortic aneurysms. J Clin Invest 86, 1465–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kontusaari S, Tromp G, Kuivaniemi H, Stolle C, Pope FM and Prockop DJ, 1992. Substitution of aspartate for glycine 1018 in the type III procollagen (COL3A1) gene causes type IV Ehlers-Danlos syndrome: the mutated allele is present in most blood leukocytes of the asymptomatic and mosaic mother. Am J Hum Genet 51, 497–507. [PMC free article] [PubMed] [Google Scholar]
- Krenning G, Zeisberg EM and Kalluri R, 2010. The origin of fibroblasts and mechanism of cardiac fibrosis. J Cell Physiol 225, 631–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krieg T, Langer I, Gerstmeier H, Keller J, Mensing H, Goerz G and Timpl R, 1986. Type III collagen aminopropeptide levels in serum of patients with progressive systemic scleroderma. J Invest Dermatol 87, 788–91. [DOI] [PubMed] [Google Scholar]
- Kuivaniemi H, Kontusaari S, Tromp G, Zhao MJ, Sabol C and Prockop DJ, 1990. Identical G+1 to A mutations in three different introns of the type III procollagen gene (COL3A1) produce different patterns of RNA splicing in three variants of Ehlers-Danlos syndrome. IV. An explanation for exon skipping some mutations and not others. J Biol Chem 265, 12067–74. [PubMed] [Google Scholar]
- Kuivaniemi H, Prockop DJ, Wu Y, Madhatheri SL, Kleinert C, Earley JJ, Jokinen A, Stolle C, Majamaa K, Myllyla VV and et al. , 1993. Exclusion of mutations in the gene for type III collagen (COL3A1) as a common cause of intracranial aneurysms or cervical artery dissections: results from sequence analysis of the coding sequences of type III collagen from 55 unrelated patients. Neurology 43, 2652–8. [DOI] [PubMed] [Google Scholar]
- Kuivaniemi H, Ryer EJ, Elmore JR and Tromp G, 2015. Understanding the pathogenesis of abdominal aortic aneurysms. Expert Rev Cardiovasc Ther 13, 975–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuivaniemi H, Tromp G and Prockop DJ, 1997. Mutations in fibrillar collagens (types I, II, III, and XI), fibril-associated collagen (type IX), and network-forming collagen (type X) cause a spectrum of diseases of bone, cartilage, and blood vessels. Hum Mutat 9, 300–15. [DOI] [PubMed] [Google Scholar]
- Lamberg A, Helaakoski T, Myllyharju J, Peltonen S, Notbohm H, Pihlajaniemi T and Kivirikko KI, 1996. Characterization of human type III collagen expressed in a baculovirus system. Production of a protein with a stable triple helix requires coexpression with the two types of recombinant prolyl 4-hydroxylase subunit. J Biol Chem 271, 11988–95. [DOI] [PubMed] [Google Scholar]
- Lee B, D’Alessio M, Vissing H, Ramirez F, Steinmann B and Superti-Furga A, 1991. Characterization of a large deletion associated with a polymorphic block of repeated dinucleotides in the type III procollagen gene (COL3A1) of a patient with Ehlers-Danlos syndrome type IV. Am J Hum Genet 48, 511–7. [PMC free article] [PubMed] [Google Scholar]
- Lipton A, Leitzel K, Ali SM, Polimera HV, Nagabhairu V, Marks E, Richardson AE, Krecko L, Ali A, Koestler W, Esteva FJ, Leeming DJ, Karsdal MA and Willumsen N, 2018. High turnover of extracellular matrix reflected by specific protein fragments measured in serum is associated with poor outcomes in two metastatic breast cancer cohorts. Int J Cancer 143, 3027–3034. [DOI] [PubMed] [Google Scholar]
- Liu X, Wu H, Byrne M, Krane S and Jaenisch R, 1997. Type III collagen is crucial for collagen I fibrillogenesis and for normal cardiovascular development. Proc Natl Acad Sci U S A 94, 1852–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loidl HR, Brinker JM, May M, Pihlajaniemi T, Morrow S, Rosenbloom J and Myers JC, 1984. Molecular cloning and carboxyl-propeptide analysis of human type III procollagen. Nucleic Acids Res 12, 9383–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long KB, Li Z, Burgwin CM, Choe SG, Martyanov V, Sassi-Gaha S, Earl JP, Eutsey RA, Ahmed A, Ehrlich GD, Artlett CM, Whitfield ML and Blankenhorn EP, 2015. The Tsk2/+ mouse fibrotic phenotype is due to a gain-of-function mutation in the PIIINP segment of the Col3a1 gene. J Invest Dermatol 135, 718–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lv W, Lin Y, Song W, Sun K, Yu H, Zhang Y, Zhang C, Li L, Suo M, Hui R and Chen J, 2014. Variants of COL3A1 are associated with the risk of stroke recurrence and prognosis in the Chinese population: a prospective study. J Mol Neurosci 53, 196–203. [DOI] [PubMed] [Google Scholar]
- Malfait F, 2018. Vascular aspects of the Ehlers-Danlos Syndromes. Matrix Biol 71–72, 380–395. [DOI] [PubMed] [Google Scholar]
- Malfait F, Francomano C, Byers P, Belmont J, Berglund B, Black J, Bloom L, Bowen JM, Brady AF, Burrows NP, Castori M, Cohen H, Colombi M, Demirdas S, De Backer J, De Paepe A, Fournel-Gigleux S, Frank M, Ghali N, Giunta C, Grahame R, Hakim A, Jeunemaitre X, Johnson D, Juul-Kristensen B, Kapferer-Seebacher I, Kazkaz H, Kosho T, Lavallee ME, Levy H, Mendoza-Londono R, Pepin M, Pope FM, Reinstein E, Robert L, Rohrbach M, Sanders L, Sobey GJ, Van Damme T, Vandersteen A, van Mourik C, Voermans N, Wheeldon N, Zschocke J and Tinkle B, 2017. The 2017 international classification of the Ehlers-Danlos syndromes. Am J Med Genet C Semin Med Genet 175, 8–26. [DOI] [PubMed] [Google Scholar]
- Manhenke C, Ueland T, Jugdutt BI, Godang K, Aukrust P, Dickstein K and Orn S, 2014. The relationship between markers of extracellular cardiac matrix turnover: infarct healing and left ventricular remodelling following primary PCI in patients with first-time STEMI. Eur Heart J 35, 395–402. [DOI] [PubMed] [Google Scholar]
- Mankoo BS and Dalgleish R, 1988. Human pro alpha 1(III) collagen: cDNA sequence for the 3’ end. Nucleic Acids Res 16, 2337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mansour SG, Puthumana J, Coca SG, Gentry M and Parikh CR, 2017. Biomarkers for the detection of renal fibrosis and prediction of renal outcomes: a systematic review. BMC Nephrol 18, 72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marijianowski MM, Teeling P, Mann J and Becker AE, 1995. Dilated cardiomyopathy is associated with an increase in the type I/type III collagen ratio: a quantitative assessment. J Am Coll Cardiol 25, 1263–72. [DOI] [PubMed] [Google Scholar]
- Mays PK, Tromp G, Kuivaniemi H, Ryynanen M and Prockop DJ, 1992. A 15 base-pair AT-rich variable number tandem repeat in the type III procollagen gene (COL3A1) as an informative marker for 2q31–2q32.3. Matrix 12, 44–9. [DOI] [PubMed] [Google Scholar]
- Menzel EJ, Piza H, Zielinski C, Endler AT, Steffen C and Millesi H, 1979. Collagen types and anticollagen-antibodies in Dupuytren’s disease. Hand 11, 243–8. [DOI] [PubMed] [Google Scholar]
- Mi Y, Wang W, Lu J, Zhang C, Wang Y, Ying H and Sun K, 2018. Proteasome-mediated degradation of collagen III by cortisol in amnion fibroblasts. J Mol Endocrinol 60, 45–54. [DOI] [PubMed] [Google Scholar]
- Miedel EL, Brisson BK, Hamilton T, Gleason H, Swain GP, Lopas L, Dopkin D, Perosky JE, Kozloff KM, Hankenson KD and Volk SW, 2015. Type III collagen modulates fracture callus bone formation and early remodeling. J Orthop Res 33, 675–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milewicz DM, Witz AM, Smith AC, Manchester DK, Waldstein G and Byers PH, 1993. Parental somatic and germ-line mosaicism for a multiexon deletion with unusual endpoints in a type III collagen (COL3A1) allele produces Ehlers-Danlos syndrome type IV in the heterozygous offspring. Am J Hum Genet 53, 62–70. [PMC free article] [PubMed] [Google Scholar]
- Miller EJ, 1988. Collagen types: structure, distribution and functions., in: Nimni ME (Ed.), Collagen Vol. 1 CRC Press, Boca Raton, FL, pp. 139–156. [Google Scholar]
- Miller EJ, Epstein EH Jr. and Piez KA, 1971. Identification of three genetically distinct collagens by cyanogen bromide cleavage of insoluble human skin and cartilage collagen. Biochem Biophys Res Commun 42, 1024–9. [DOI] [PubMed] [Google Scholar]
- Miskulin M, Dalgleish R, Kluve-Beckerman B, Rennard SI, Tolstoshev P, Brantly M and Crystal RG, 1986. Human type III collagen gene expression is coordinately modulated with the type I collagen genes during fibroblast growth. Biochemistry 25, 1408–13. [DOI] [PubMed] [Google Scholar]
- Monnet E and Fauvel-Lafeve F, 2000. A new platelet receptor specific to type III collagen. Type III collagen-binding protein. J Biol Chem 275, 10912–7. [DOI] [PubMed] [Google Scholar]
- Mortensen JH, Godskesen LE, Jensen MD, Van Haaften WT, Klinge LG, Olinga P, Dijkstra G, Kjeldsen J, Karsdal MA, Bay-Jensen AC and Krag A, 2015. Fragments of Citrullinated and MMP-degraded Vimentin and MMP-degraded Type III Collagen Are Novel Serological Biomarkers to Differentiate Crohn’s Disease from Ulcerative Colitis. J Crohns Colitis 9, 863–72. [DOI] [PubMed] [Google Scholar]
- Muckian C, Fitzgerald A, O’Neill A, O’Byrne A, Fitzgerald DJ and Shields DC, 2002. Genetic variability in the extracellular matrix as a determinant of cardiovascular risk: association of type III collagen COL3A1 polymorphisms with coronary artery disease. Blood 100, 1220–3. [DOI] [PubMed] [Google Scholar]
- Murray ML, Pepin M, Peterson S and Byers PH, 2014. Pregnancy-related deaths and complications in women with vascular Ehlers-Danlos syndrome. Genet Med 16, 874–80. [DOI] [PubMed] [Google Scholar]
- Narcisi P, Wu Y, Tromp G, Earley JJ, Richards AJ, Pope FM and Kuivaniemi H, 1993. Single base mutation that substitutes glutamic acid for glycine 1021 in the COL3A1 gene and causes Ehlers-Danlos syndrome type IV. Am J Med Genet 46, 278–83. [DOI] [PubMed] [Google Scholar]
- Niederreither K, D’Souza R, Metsaranta M, Eberspaecher H, Toman PD, Vuorio E and De Crombrugghe B, 1995. Coordinate patterns of expression of type I and III collagens during mouse development. Matrix Biol 14, 705–13. [DOI] [PubMed] [Google Scholar]
- Nuytinck L, Narcisi P, Nicholls A, Renard JP, Pope FM and De Paepe A, 1992. Detection and characterisation of an overmodified type III collagen by analysis of non-cutaneous connective tissues in a patient with Ehlers-Danlos syndrome IV. J Med Genet 29, 375–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogata T, Shibamura H, Tromp G, Sinha M, Goddard KA, Sakalihasan N, Limet R, MacKean GL, Arthur C, Sueda T, Land S and Kuivaniemi H, 2005. Genetic analysis of polymorphisms in biologically relevant candidate genes in patients with abdominal aortic aneurysms. J Vasc Surg 41, 1036–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okita H, Ikeda Y, Mitsuhashi Y, Namikawa H, Kitamura Y, Hamasaki Y, Yamazaki S and Hatamochi A, 2010. A novel point mutation at donor splice-site in intron 42 of type III collagen gene resulting in the inclusion of 30 nucleotides into the mature mRNA in a case of vascular type of Ehlers-Danlos syndrome. Arch Dermatol Res 302, 395–9. [DOI] [PubMed] [Google Scholar]
- Okonechnikov K, Golosova O, Fursov M and team U, 2012. Unipro UGENE: a unified bioinformatics toolkit. Bioinformatics 28, 1166–7. [DOI] [PubMed] [Google Scholar]
- Ong KT, Perdu J, De Backer J, Bozec E, Collignon P, Emmerich J, Fauret AL, Fiessinger JN, Germain DP, Georgesco G, Hulot JS, De Paepe A, Plauchu H, Jeunemaitre X, Laurent S and Boutouyrie P, 2010. Effect of celiprolol on prevention of cardiovascular events in vascular Ehlers-Danlos syndrome: a prospective randomised, open, blinded-endpoints trial. Lancet 376, 1476–84. [DOI] [PubMed] [Google Scholar]
- Palmeri S, Mari F, Meloni I, Malandrini A, Ariani F, Villanova M, Pompilio A, Schwarze U, Byers PH and Renieri A, 2003. Neurological presentation of Ehlers-Danlos syndrome type IV in a family with parental mosaicism. Clin Genet 63, 510–5. [DOI] [PubMed] [Google Scholar]
- Pepin MG, Schwarze U, Rice KM, Liu M, Leistritz D and Byers PH, 2014. Survival is affected by mutation type and molecular mechanism in vascular Ehlers-Danlos syndrome (EDS type IV). Genet Med 16, 881–8. [DOI] [PubMed] [Google Scholar]
- Plancke A, Holder-Espinasse M, Rigau V, Manouvrier S, Claustres M and Khau Van Kien P, 2009. Homozygosity for a null allele of COL3A1 results in recessive Ehlers-Danlos syndrome. Eur J Hum Genet 17, 1411–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pope FM, Martin GR, Lichtenstein JR, Penttinen R, Gerson B, Rowe DW and McKusick VA, 1975. Patients with Ehlers-Danlos syndrome type IV lack type III collagen. Proc Natl Acad Sci U S A 72, 1314–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pope FM, Martin GR and McKusick VA, 1977. Inheritance of Ehlers-Danlos type IV syndrome. J Med Genet 14, 200–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pope FM, Narcisi P, Nicholls AC, Germaine D, Pals G and Richards AJ, 1996. COL3A1 mutations cause variable clinical phenotypes including acrogeria and vascular rupture. Br J Dermatol 135, 163–81. [PubMed] [Google Scholar]
- Prabhu SD and Frangogiannis NG, 2016. The Biological Basis for Cardiac Repair After Myocardial Infarction: From Inflammation to Fibrosis. Circ Res 119, 91–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prockop DJ and Kivirikko KI, 1984. Heritable diseases of collagen. N Engl J Med 311, 376–86. [DOI] [PubMed] [Google Scholar]
- Prockop DJ and Kivirikko KI, 1995. Collagens: molecular biology, diseases, and potentials for therapy. Annu Rev Biochem 64, 403–34. [DOI] [PubMed] [Google Scholar]
- Rasmussen DG, Sand JM, Karsdal MA and Genovese F, 2017. Development of a Novel Enzyme-Linked Immunosorbent Assay Targeting a Neo-Epitope Generated by Cathepsin-Mediated Turnover of Type III Collagen and Its Application in Chronic Obstructive Pulmonary Disease. PLoS One 12, e0170023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Razzaque MS, Koji T, Horita Y, Nishihara M, Harada T, Nakane PK and Taguchi T, 1995. Synthesis of type III collagen and type IV collagen by tubular epithelial cells in diabetic nephropathy. Pathol Res Pract 191, 1099–104. [DOI] [PubMed] [Google Scholar]
- Reichert MC, Kupcinskas J, Krawczyk M, Jungst C, Casper M, Grunhage F, Appenrodt B, Zimmer V, Weber SN, Tamelis A, Lukosiene JI, Pauziene N, Kiudelis G, Jonaitis L, Schramm C, Goeser T, Schulz A, Malinowski M, Glanemann M, Kupcinskas L and Lammert F, 2018. A Variant of COL3A1 (rs3134646) Is Associated With Risk of Developing Diverticulosis in White Men. Dis Colon Rectum 61, 604–611. [DOI] [PubMed] [Google Scholar]
- Ricard-Blum S, Baffet G and Theret N, 2018. Molecular and tissue alterations of collagens in fibrosis. Matrix Biol 68–69, 122–149. [DOI] [PubMed] [Google Scholar]
- Richards AJ, Ward PN, Narcisi P, Nicholls AC, Lloyd JC and Pope FM, 1992. A single base mutation in the gene for type III collagen (COL3A1) converts glycine 847 to glutamic acid in a family with Ehlers-Danlos syndrome type IV. An unaffected family member is mosaic for the mutation. Hum Genet 89, 414–8. [DOI] [PubMed] [Google Scholar]
- Risteli J, Niemi S, Trivedi P, Maentausta O, Mowat AP and Risteli L, 1988. Rapid equilibrium radioimmunoassay for the amino-terminal propeptide of human type III procollagen. Clin Chem 34, 715–8. [PubMed] [Google Scholar]
- Romero R, Kuivaniemi H, Tromp G and Olson J, 2002. The design, execution, and interpretation of genetic association studies to decipher complex diseases. Am J Obstet Gynecol 187, 1299–312. [DOI] [PubMed] [Google Scholar]
- Rosenbloom J, Ren S and Macarak E, 2016. New frontiers in fibrotic disease therapies: The focus of the Joan and Joel Rosenbloom Center for Fibrotic Diseases at Thomas Jefferson University. Matrix Biol 51, 14–25. [DOI] [PubMed] [Google Scholar]
- Samokhin AO, Stephens T, Wertheim BM, Wang RS, Vargas SO, Yung LM, Cao M, Brown M, Arons E, Dieffenbach PB, Fewell JG, Matar M, Bowman FP, Haley KJ, Alba GA, Marino SM, Kumar R, Rosas IO, Waxman AB, Oldham WM, Khanna D, Graham BB, Seo S, Gladyshev VN, Yu PB, Fredenburgh LE, Loscalzo J, Leopold JA and Maron BA, 2018. NEDD9 targets COL3A1 to promote endothelial fibrosis and pulmonary arterial hypertension. Sci Transl Med 10, eaap7294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandberg M, Makela JK, Multimaki P, Vuorio T and Vuorio E, 1989. Construction of a human pro alpha 1(III) collagen cDNA clone and localization of type III collagen expression in human fetal tissues. Matrix 9, 82–91. [DOI] [PubMed] [Google Scholar]
- Schwarze U, Goldstein JA and Byers PH, 1997. Splicing defects in the COL3A1 gene: marked preference for 5’ (donor) spice-site mutations in patients with exon-skipping mutations and Ehlers-Danlos syndrome type IV. Am J Hum Genet 61, 1276–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seyer JM and Kang AH, 1981. Covalent structure of collagen: amino acid sequence of alpha 1(III)-CB9 from type III collagen of human liver. Biochemistry 20, 2621–7. [DOI] [PubMed] [Google Scholar]
- Shaikh G, Zhang J, Perez-Aso M, Mediero A and Cronstein B, 2016. Adenosine A2A receptor promotes collagen type III synthesis via beta-catenin activation in human dermal fibroblasts. Br J Pharmacol 173, 3279–3291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen T, Gao K, Miao Y and Hu Z, 2018. Exogenous growth factors enhance the expression of cola1, cola3, and Elastin in fibroblasts via activating MAPK signaling pathway. Mol Cell Biochem 442, 203–210. [DOI] [PubMed] [Google Scholar]
- Shi J, Ma X, Gao Y, Fan D, Zhu C, Mi Y and Xue W, 2017. Hydroxylation of Human Type III Collagen Alpha Chain by Recombinant Coexpression with a Viral Prolyl 4-Hydroxylase in Escherichia coli. Protein J 36, 322–331. [DOI] [PubMed] [Google Scholar]
- Shibamura H, Olson JM, van Vlijmen-Van Keulen C, Buxbaum SG, Dudek DM, Tromp G, Ogata T, Skunca M, Sakalihasan N, Pals G, Limet R, MacKean GL, Defawe O, Verloes A, Arthur C, Lossing AG, Burnett M, Sueda T and Kuivaniemi H, 2004. Genome scan for familial abdominal aortic aneurysm using sex and family history as covariates suggests genetic heterogeneity and identifies linkage to chromosome 19q13. Circulation 109, 2103–8. [DOI] [PubMed] [Google Scholar]
- Sivakumar P, Gupta S, Sarkar S and Sen S, 2008. Upregulation of lysyl oxidase and MMPs during cardiac remodeling in human dilated cardiomyopathy. Mol Cell Biochem 307, 159–67. [DOI] [PubMed] [Google Scholar]
- Smith LB, Hadoke PW, Dyer E, Denvir MA, Brownstein D, Miller E, Nelson N, Wells S, Cheeseman M and Greenfield A, 2011. Haploinsufficiency of the murine Col3a1 locus causes aortic dissection: a novel model of the vascular type of Ehlers-Danlos syndrome. Cardiovasc Res 90, 182–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith LT, Schwarze U, Goldstein J and Byers PH, 1997. Mutations in the COL3A1 gene result in the Ehlers-Danlos syndrome type IV and alterations in the size and distribution of the major collagen fibrils of the dermis. J Invest Dermatol 108, 241–7. [DOI] [PubMed] [Google Scholar]
- Somers KD, Sismour EN, Wright GL Jr., Devine CJ Jr., Gilbert DA and Horton CE, 1989. Isolation and characterization of collagen in Peyronie’s disease. J Urol 141, 629–31. [DOI] [PubMed] [Google Scholar]
- Soylemezoglu O, Wild G, Dalley AJ, MacNeil S, Milford-Ward A, Brown CB and el Nahas AM, 1997. Urinary and serum type III collagen: markers of renal fibrosis. Nephrol Dial Transplant 12, 1883–9. [DOI] [PubMed] [Google Scholar]
- Stallmach A, Schuppan D, Riese HH, Matthes H and Riecken EO, 1992. Increased collagen type III synthesis by fibroblasts isolated from strictures of patients with Crohn’s disease. Gastroenterology 102, 1920–9. [DOI] [PubMed] [Google Scholar]
- Stembridge NS, Vandersteen AM, Ghali N, Sawle P, Nesbitt M, Pollitt RC, Ferguson DJ, Holden S, Elmslie F, Henderson A, Hulmes DJ and Pope FM, 2015. Clinical, structural, biochemical and X-ray crystallographic correlates of pathogenicity for variants in the Cpropeptide region of the COL3A1 gene. Am J Med Genet A 167a, 1763–72. [DOI] [PubMed] [Google Scholar]
- Stevenson K, Kucich U, Whitbeck C, Levin RM and Howard PS, 2006. Functional changes in bladder tissue from type III collagen-deficient mice. Mol Cell Biochem 283, 107–14. [DOI] [PubMed] [Google Scholar]
- Stolle CA, Pyeritz RE, Myers JC and Prockop DJ, 1985. Synthesis of an altered type III procollagen in a patient with type IV Ehlers-Danlos syndrome. A structural change in the alpha 1(III) chain which makes the protein more susceptible to proteinases. J Biol Chem 260, 1937–44. [PubMed] [Google Scholar]
- Superti-Furga A, Gugler E, Gitzelmann R and Steinmann B, 1988. Ehlers-Danlos syndrome type IV: a multi-exon deletion in one of the two COL3A1 alleles affecting structure, stability, and processing of type III procollagen. J Biol Chem 263, 6226–32. [PubMed] [Google Scholar]
- Tae HJ, Marshall S, Zhang J, Wang M, Briest W and Talan MI, 2012. Chronic treatment with a broad-spectrum metalloproteinase inhibitor, doxycycline, prevents the development of spontaneous aortic lesions in a mouse model of vascular Ehlers-Danlos syndrome. J Pharmacol Exp Ther 343, 246–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanwar S, Trembling PM, Hogan BJ, Srivastava A, Parkes J, Harris S, Grant P, Nastouli E, Ocker M, Wehr K, Herold C, Neureiter D, Schuppan D and Rosenberg WM, 2017. Noninvasive markers of liver fibrosis: on-treatment changes of serum markers predict the outcome of antifibrotic therapy. Eur J Gastroenterol Hepatol 29, 289–296. [DOI] [PubMed] [Google Scholar]
- Thiele BJ, Doller A, Kahne T, Pregla R, Hetzer R and Regitz-Zagrosek V, 2004. RNA-binding proteins heterogeneous nuclear ribonucleoprotein A1, E1, and K are involved in posttranscriptional control of collagen I and III synthesis. Circ Res 95, 1058–66. [DOI] [PubMed] [Google Scholar]
- Toman PD and de Crombrugghe B, 1994. The mouse type-III procollagen-encoding gene: genomic cloning and complete DNA sequence. Gene 147, 161–8. [DOI] [PubMed] [Google Scholar]
- Toman PD, Ricca GA and de Crombrugghe B, 1988. Nucleotide sequence of a cDNA coding for the amino-terminal region of human prepro alpha 1(III) collagen. Nucleic Acids Res 16, 7201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomita M, Ohkura N, Ito M, Kato T, Royce PM and Kitajima T, 1995. Biosynthesis of recombinant human pro-alpha 1(III) chains in a baculovirus expression system: production of disulphidebonded and non-disulphide-bonded species containing full-length triple helices. Biochem J 312 ( Pt 3), 847–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tromp G, Kuivaniemi H, Shikata H and Prockop DJ, 1989a. A single base mutation that substitutes serine for glycine 790 of the alpha 1 (III) chain of type III procollagen exposes an arginine and causes Ehlers-Danlos syndrome IV. J Biol Chem 264, 1349–52. [PubMed] [Google Scholar]
- Tromp G, Kuivaniemi H, Stolle C, Pope FM and Prockop DJ, 1989b. Single base mutation in the type III procollagen gene that converts the codon for glycine 883 to aspartate in a mild variant of Ehlers-Danlos syndrome IV. J Biol Chem 264, 19313–7. [PubMed] [Google Scholar]
- Tromp G, Weinsheimer S, Ronkainen A and Kuivaniemi H, 2014. Molecular basis and genetic predisposition to intracranial aneurysm. Ann Med 46, 597–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tromp G, Wu Y, Prockop DJ, Madhatheri SL, Kleinert C, Earley JJ, Zhuang J, Norrgard O, Darling RC, Abbott WM and et al. , 1993. Sequencing of cDNA from 50 unrelated patients reveals that mutations in the triple-helical domain of type III procollagen are an infrequent cause of aortic aneurysms. J Clin Invest 91, 2539–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valkkila M, Melkoniemi M, Kvist L, Kuivaniemi H, Tromp G and Ala-Kokko L, 2001. Genomic organization of the human COL3A1 and COL5A2 genes: COL5A2 has evolved differently than the other minor fibrillar collagen genes. Matrix Biol 20, 357–66. [DOI] [PubMed] [Google Scholar]
- van Haaften WT, Mortensen JH, Karsdal MA, Bay-Jensen AC, Dijkstra G and Olinga P, 2017. Misbalance in type III collagen formation/degradation as a novel serological biomarker for penetrating (Montreal B3) Crohn’s disease. Aliment Pharmacol Ther 46, 26–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Keulen CJ, van de Akker E, Pals G and Rauwerda JA, 1999. The role of type III collagen in the development of familial abdominal aortic aneurysms. Eur J Vasc Endovasc Surg 18, 65–70. [DOI] [PubMed] [Google Scholar]
- Vandervore L, Stouffs K, Tanyalcin I, Vanderhasselt T, Roelens F, Holder-Espinasse M, Jorgensen A, Pepin MG, Petit F, Khau Van Kien P, Bahi-Buisson N, Lissens W, Gheldof A, Byers PH and Jansen AC, 2017. Bi-allelic variants in COL3A1 encoding the ligand to GPR56 are associated with cobblestone-like cortical malformation, white matter changes and cerebellar cysts. J Med Genet 54, 432–440. [DOI] [PubMed] [Google Scholar]
- Volk SW, Shah SR, Cohen AJ, Wang Y, Brisson BK, Vogel LK, Hankenson KD and Adams SL, 2014. Type III collagen regulates osteoblastogenesis and the quantity of trabecular bone. Calcif Tissue Int 94, 621–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vuorela A, Myllyharju J, Nissi R, Pihlajaniemi T and Kivirikko KI, 1997. Assembly of human prolyl 4hydroxylase and type III collagen in the yeast pichia pastoris: formation of a stable enzyme tetramer requires coexpression with collagen and assembly of a stable collagen requires coexpression with prolyl 4-hydroxylase. Embo j 16, 6702–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang TJ, Larson MG, Benjamin EJ, Siwik DA, Safa R, Guo CY, Corey D, Sundstrom J, Sawyer DB, Colucci WS and Vasan RS, 2007. Clinical and echocardiographic correlates of plasma procollagen type III amino-terminal peptide levels in the community. Am Heart J 154, 291–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weis MA, Hudson DM, Kim L, Scott M, Wu JJ and Eyre DR, 2010. Location of 3-hydroxyproline residues in collagen types I, II, III, and V/XI implies a role in fibril supramolecular assembly. J Biol Chem 285, 2580–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood L, Theriault N and Vogeli G, 1987. Complete nucleotide sequence of the N-terminal domains of the murine alpha-1 type-III collagen chain. Gene 61, 225–30. [DOI] [PubMed] [Google Scholar]
- Wotton SF and Duance VC, 1994. Type III collagen in normal human articular cartilage. Histochem J 26, 412–6. [DOI] [PubMed] [Google Scholar]
- Wu JJ, Weis MA, Kim LS and Eyre DR, 2010. Type III collagen, a fibril network modifier in articular cartilage. J Biol Chem 285, 18537–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y, Kuivaniemi H, Tromp G, Strobel D, Romanic AM and Prockop DJ, 1993. Temperature sensitivity of aberrant RNA splicing with a mutation in the G+5 position of intron 37 of the gene for type III procollagen from a patient with Ehlers-Danlos syndrome type IV. Hum Mutat 2, 28–36. [DOI] [PubMed] [Google Scholar]
- Yamada Y, Mudryj M and de Crombrugghe B, 1983. A uniquely conserved regulatory signal is found around the translation initiation site in three different collagen genes. J Biol Chem 258, 14914–9. [PubMed] [Google Scholar]
- Yamane T, Morioka Y, Kitaura Y, Iwatsuki K, Shimomura Y and Oishi Y, 2018. Branched-chain amino acids regulate type I tropocollagen and type III tropocollagen syntheses via modulation of mTOR in the skin. Biosci Biotechnol Biochem 82, 611–615. [DOI] [PubMed] [Google Scholar]
- Yuan B, Broadbent JA, Huan J and Yang H, 2018. The Effects of Adipose Stem Cell-Conditioned Media on Fibrogenesis of Dermal Fibroblasts Stimulated by Transforming Growth Factor-beta1. J Burn Care Res 39, 129–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan L, Shu B, Chen L, Qian K, Wang Y, Qian G, Zhu Y, Cao X, Xie C, Xiao Y and Wang X, 2017. Overexpression of COL3A1 confers a poor prognosis in human bladder cancer identified by co-expression analysis. Oncotarget 8, 70508–70520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yue F, Cheng Y, Breschi A, Vierstra J, Wu W, Ryba T, Sandstrom R, Ma Z, Davis C, Pope BD, Shen Y, Pervouchine DD, Djebali S, Thurman RE, Kaul R, Rynes E, Kirilusha A, Marinov GK, Williams BA, Trout D, Amrhein H, Fisher-Aylor K, Antoshechkin I, DeSalvo G, See LH, Fastuca M, Drenkow J, Zaleski C, Dobin A, Prieto P, Lagarde J, Bussotti G, Tanzer A, Denas O, Li K, Bender MA, Zhang M, Byron R, Groudine MT, McCleary D, Pham L, Ye Z, Kuan S, Edsall L, Wu YC, Rasmussen MD, Bansal MS, Kellis M, Keller CA, Morrissey CS, Mishra T, Jain D, Dogan N, Harris RS, Cayting P, Kawli T, Boyle AP, Euskirchen G, Kundaje A, Lin S, Lin Y, Jansen C, Malladi VS, Cline MS, Erickson DT, Kirkup VM, Learned K, Sloan CA, Rosenbloom KR, Lacerda de Sousa B, Beal K, Pignatelli M, Flicek P, Lian J, Kahveci T, Lee D, Kent WJ, Ramalho Santos M, Herrero J, Notredame C, Johnson A, Vong S, Lee K, Bates D, Neri F, Diegel M, Canfield T, Sabo PJ, Wilken MS, Reh TA, Giste E, Shafer A, Kutyavin T, Haugen E, Dunn D, Reynolds AP, Neph S, Humbert R, Hansen RS, De Bruijn M, et al. , 2014. A comparative encyclopedia of DNA elements in the mouse genome. Nature 515, 355–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zafarullah K, Brown EM, Kuivaniemi H, Tromp G, Sieron AL, Fertala A and Prockop DJ, 1997. Synthesis and conformational properties of a recombinant C-propeptide of human type III procollagen. Matrix Biol 16, 201–9. [DOI] [PubMed] [Google Scholar]
- Zhang B, Niu W, Dong HY, Liu ML, Luo Y and Li ZC, 2018. Hypoxia induces endothelialmesenchymal transition in pulmonary vascular remodeling. Int J Mol Med 42, 270278. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.