

Abstract
Properdin is known as the only positive regulator of the complement system. Properdin promotes the activity of this defense system by stabilizing its key enzymatic complexes: the complement alternative pathway (AP) convertases. Besides, some studies have indicated a role for properdin as an initiator of complement activity. Though the AP is a powerful activation route of the complement system, it is also involved in a wide variety of autoimmune and inflammatory diseases, many of which affect the kidneys. The role of properdin in regulating complement in health and disease has not received as much appraisal as the many negative AP regulators, such as factor H. Historically, properdin deficiency has been strongly associated with an increased risk for meningococcal disease. Yet only recently had studies begun to link properdin to other complement-related diseases, including renal diseases. In the light of the upcoming complement-inhibiting therapies, it is interesting whether properdin can be a therapeutic target to attenuate AP-mediated injury. A full understanding of the basic concepts of properdin biology is therefore needed. Here, we first provide an overview of the function of properdin in health and disease. Then, we explore its potential as a therapeutic target for the AP-associated renal diseases C3 glomerulopathy, atypical hemolytic uremic syndrome, and proteinuria-induced tubulointerstitial injury. Considering current knowledge, properdin-inhibiting therapy seems promising in certain cases. However, knowing the complexity of properdin’s role in renal pathologies in vivo, further research is required to clarify the exact potential of properdin-targeted therapy in complement-mediated renal diseases.
Keywords: Complement system, Properdin, C3 glomerulopathy, Atypical hemolytic uremic syndrome, Proteinuria-induced tubulointerstitial injury, Complement-inhibiting therapy
Introduction
In the last decades, it has become evident that the complement system is involved in various diseases [1]. Consequently, the interest in complement-inhibiting therapy has grown immensely [2]. In principle, the complement system is a “good guy” as it is a crucial part of our innate immunity. It consists of numerous plasma and cell surface proteins that strongly interact with each other and with other regulatory (immune) systems to discriminate between foreign, altered-self, and healthy self-surfaces. In this way, complement provides the body with sophisticated immune surveillance and maintenance of homeostasis [3]. An arsenal of complement regulatory proteins keep the complement activity in control and limited to target cells. However, when the balance between the activation and regulation is disturbed, the widespread functions of complement can cause damage to healthy tissues. The kidneys are specifically vulnerable to such injury [1, 4]. In complement-mediated diseases, the blocking of complement-induced damage may outweigh the possible risks of (partly) compromising immunity. Complement inhibition can be focused at different levels of the complement activation cascades. In general, drugs can be grouped into three major functional categories: preventing complement initiation, dampening complement amplification, and blocking the complement effector molecules. Importantly, complement-inhibiting therapy must be considered with care and for each disorder, or maybe even for each patient, individually. This requires a thorough understanding of the pathological mechanisms underlying disease and of the exact functions of the molecule of target [2].
An upcoming but for a long time ignored candidate target is properdin. This complement regulator has a crucial role in promoting one of the three activation routes of the complement system, namely the alternative pathway (AP). The AP is an important contributor to overall complement activity as this pathway has a guarding function providing continuous low-level complement activity and an amplifying function that augments complement activity initiated by all three activation pathways. Properdin is well-known as the only positive regulator of the AP. However, compared to the many negative regulators that attenuate AP activity, our knowledge of the exact functions of properdin in health and disease is lagging behind. Nonetheless, properdin has regained interest in the scientific community, especially in the last 10 years, and this has initiated major advances in the complement field. In this review, a general overview of the basics of properdin biology is given first, covering the production and structure of properdin, its function in the AP, and its clinical significance. Afterwards, the possible role of properdin in renal diseases associated with complement dysregulation is examined, with focus on C3 glomerulopathy, atypical hemolytic uremic syndrome, and proteinuria-induced tubulointerstitial injury. Finally, the potential of properdin as a new therapeutic target for treatment of these diseases is discussed.
The production and structure of properdin
The complement protein properdin is a highly positively charged plasma glycoprotein. In contrast to most complement components that are being produced by the liver, properdin is mainly synthesized by leukocytes, including T cells [5], monocytes [6], and mast cells [7]. The protein is also stored in the secondary granules of neutrophils from which it is released upon stimulation [8]. The contribution of neutrophils to the total protein levels found in plasma is highlighted by the association between chemotherapy-induced neutropenia and a decline in circulating properdin levels [9]. Normal systemic levels of human properdin generally cover a large range in the healthy control group and are reported in a range from 5 to 45 μg/ml [10–24]. The large range observed among studies is likely due to differences in the methods and reagents used, e.g., detection methods, antibody combinations, and preparations used as the standard protein. Of note, systemic properdin levels are lower in healthy neonates and infants (i.e., < 1 year old) compared to adults [19, 20, 23–28].
Properdin is encoded on the X chromosome and circulates in the blood in oligomeric forms. This latter characteristic is very important for its biological function. The properdin oligomers are composed of identical rod-like monomers of ~ 53 kDa [29]. Each monomer consists of 442 amino acid residues [30] and comprises one presumed truncated and six full thrombospondin type I repeat (TSR) domains, numbered TSR0 to TSR6 from the N to C terminus [30–32]. The flexible monomeric subunits associate head-to-tail into mainly cyclic dimers, trimers, or tetramers with curly vertex structures (Fig. 1) [10, 29, 33]. In normal human plasma, the oligomers exist in a fixed ratio of approximately 1:2:1, with the trimer being the predominant form [10].
Fig. 1.

Proposed model of the structure of properdin oligomers. Properdin is composed of monomeric subunits that associate together to form dimeric, trimeric, or tetrameric oligomers with curly vertex structures. Each monomer contains six full thrombospondin type I repeat (TSR) domains named TSR1–6 and a putative truncated N-terminal TSR (indicated with the striped pattern) denoted as TSR0. This latter TSR contains all six conserved cysteine residues although overall sequence homology with the other TSRs is low
The flexible, oligomeric nature of properdin is challenging for structural and biochemical studies. For a long time, information about the functions of the individual TSRs regarding target binding and oligomerization had therefore been derived primarily from structure-function studies using mutated/truncated recombinant proteins or specific TSR-directed antibodies [31, 34, 35]. Recent advances in the elucidation of the atomic structure of properdin have now indicated that four TSR domains, originating from two monomers, are involved in vertex formation. This leaves three TSRs to form the connecting part [33, 36]. The most likely composition of properdin, based on current knowledge, is a vertex with TSR0–1/TSR5–6 connected by TSRs 2–4 (Fig. 1). A complete, high-resolution atomic structure of properdin (in complex with the convertase) still needs to be resolved.
The function of properdin in the alternative pathway of the complement system
Activation of the alternative pathway
Properdin promotes complement activity by specifically acting on the AP. In contrast to the classical (CP) and lectin (LP) complement activation pathways, the AP is constantly active at a low level under normal conditions. Spontaneous hydrolysis of C3 at a low rate enables formation of an initial, fluid-phase C3 convertase (C3(H2O)Bb) that is able to cleave C3 into its active components C3a and C3b (Fig. 2). This mechanism is known as “tick-over” and generates a persistent low level of activated C3 molecules that provide the host with constant responsiveness to potential danger. C3a is released as an anaphylatoxin to mediate inflammation, while C3b marks target cells near its activation site for phagocytosis in a process known as opsonization. Activated C3b molecules can also be generated by the activity of C3 convertases from the other two activation pathways that are formed upon recognition of immune complexes or specific carbohydrate signatures. Such pattern recognition function has also been assigned to properdin for the AP in certain contexts (see “Properdin as an initiator of alternative pathway activity” for more details). Once complement is initiated and active C3b has been formed, the AP supports an important amplification loop. By interacting with factor B (FB), factor D (FD), and properdin, target-bound C3b can form effective, stabilized AP C3 convertases (C3bBbP). These surface-bound convertases can amplify the complement response by converting many more C3 molecules into C3b, which in turn support new C3 convertase formation, C5 convertase formation, and the initiation of further downstream effects of terminal pathway activation (see Fig. 2) [3, 4, 37]. C5 convertases convert C5 into C5a, which is a strong anaphylatoxin, and C5b. This latter fragment forms the base for the assembly of the membrane attack complex (MAC; C5b-9). This pore-forming protein complex is inserted into membranes to induce direct osmotic lysis of the target cell or indirect cytotoxicity via pro-inflammatory pathways [3, 4, 37]. Thus, besides being an important immunosurveillance system, the AP is also an important amplifier of initiated immune responses as it can account for over 80% of total complement activity [38].
Fig. 2.

The alternative pathway of the complement system. Activation of the complement system can be achieved via three pathways—the classical, lectin, and alternative pathways—depending on the trigger that is recognized. The proteolytic enzyme cascades of these three pathways converge at the central event of complement activation, which is the cleavage of C3 into C3a and C3b by C3 convertases. The classical and lectin pathways are activated using pattern recognition molecules that initiate formation of the C3 convertase C4bC2a. The alternative pathway is continuously activated at a low rate by a mechanism called “tick-over.” The spontaneous hydrolysis of C3 generates the active C3(H2O) fragment which can form an initial fluid-phase C3 convertase upon association with factor B (FB) that is subsequently activated by factor D (FD) into Bb. Under normal conditions, this convertase generates small amounts of activated C3 fragments. C3a is released as an anaphylatoxin to mediate inflammation, for instance by attracting leucocytes. C3b is an opsonin; it binds to molecular or cellular target surfaces and marks them for phagocytosis. C3b can also act as a platform for formation of new C3 convertases which are effectively stabilized by properdin (P). The alternative pathway may also be initiated by properdin as this molecule recognizes target surfaces and subsequently recruits C3b and FB to form stabilized C3 convertases. Alternative pathway C3 convertases are important amplifiers of the complement reaction by converting many C3 molecules into C3b which in turn support new C3 convertase formation. Furthermore, C3b can attach to preformed C3 convertase complexes to form C5 convertases that convert C5 into C5a (an anaphylatoxin similar to C3a) and C5b to initiate terminal pathway activity. The C5b activation fragment recruits a series of other complement components, i.e., C6, C7, C8, and multiple C9 molecules, to form the membrane attack complex (MAC; C5b-9). This protein complex forms a pore that disrupts the membrane integrity and thereby can cause osmotic lysis of susceptible bacteria and cells. In sublytic amounts, MAC causes cell damage by activating still not well-understood (pro-inflammatory) signaling pathways
Regulation of alternative pathway activity
The forceful activity of the AP must be tightly regulated to avoid damage to healthy host cells and tissues. Several cell-bound and soluble complement regulatory proteins keep AP activation restricted to target surfaces and keep excessive AP activation in control [3, 4, 37]. The regulatory mechanisms are mainly aimed at inhibiting C3 convertase activity and can be divided into two ways of action. First of all, complement regulators may have cofactor activity for the circulating complement regulator factor I. This soluble protease cleaves C3b into inactive fragments that no longer support convertase formation. Secondly, regulators may have decay-accelerating activity, which means they promote the dissociation of the convertases. Regulators with this activity may also compete with FB to bind to C3b and thereby prevent convertase (re-)formation. Factor H (FH) is the most abundant and important soluble regulator. It has all of the abovementioned activities and can control AP activation both in fluid phase and on surfaces to which it is recruited, also on tissue structures that do not express membrane-bound regulators. Properdin is the only known complement regulator that promotes instead of inhibits AP activity. In addition, properdin only interacts with AP convertases, in contrast to some of the other AP complement regulators that also regulate CP/LP convertase activity [3, 4, 37].
The place of properdin in the alternative pathway
Initially, when the AP was still to be discovered, properdin was thought to be the initiator of serum-dependent complement activity in the “properdin system” defined by Louis Pillemer in 1954 [39]. His work showing a non-antibody-dependent way of complement activation was very controversial at that time and was difficult to reproduce; hence, it received much criticism. After the elucidation of the “tick-over” mechanism as an initiator of AP activity, this initiating function of properdin was replaced by its well-known function as a stabilizer of AP convertases [40]. Nonetheless, biochemical studies in the last two decades have provided new evidence for a function of properdin in directing and triggering complement activation on potentially dangerous targets.
Properdin as a stabilizer of alternative pathway convertases
Under physiological conditions, convertases are unstable enzyme complexes with a short half-life of around 90 seconds [41, 42]. In association with properdin, their stability increases up to tenfold [42, 43]. Properdin forms particularly strong interactions with the C-terminal part of C3b and binds as well to Bb [33, 36, 44, 45]. Its affinity for C3bB (the pro-convertase) and C3bBb complexes is much higher than that for C3b alone [44, 46]. Thus, properdin might stabilize these complexes by holding the two components of the convertase together. Besides, its binding induces a conformational change in C3b, which hinders both the decay-accelerating and cofactor activity of complement regulators, such as FH [33, 36, 44]. As such, properdin is an important positive regulator of the AP.
From the structural studies of Alcorlo et al. [33] and Pedersen et al. [36], it became clear that the vertexes of properdin are responsible for this binding and stabilization of convertases (Fig. 3(A)). Oligomerization of at least two monomers to form these vertex structures is therefore essential for properdin’s function in vivo [33, 36]. In addition, it was shown that oligomers can use each of their vertexes for convertase binding [33, 46]. This may explain why tetrameric and trimeric properdin are reported to be more active than dimeric properdin [10, 47].
Fig. 3.

The functions of properdin in the alternative pathway. The interactions of properdin, depicted in its trimer form, with C3 convertases (C3bBb) and a surface are displayed. (A) Properdin as a stabilizer of preformed convertases on a surface. (B, C) Properdin as a platform for convertase formation after initial C3-mediated binding (B) or as a pattern recognition molecule by directly recognizing target surface structures (e.g., glycosaminoglycans and exposed DNA) and subsequently recruiting convertase components (C3bBb, C3bB, or C3b and FB)
The stabilizing action of properdin is focused predominantly on surface-bound convertase complexes, as the affinity of properdin for surface-bound C3 derivatives is much higher than that for fluid-phase fragments [44]. In line with this, it is supposed that properdin facilitates the switch from the C3 to the C5 convertase on surfaces [48]. C5 convertases are only efficiently formed on surfaces, and not in fluid phase, since their formation requires high densities of C3b [49]. There are strong indications that absence of properdin is associated with reduced C5 convertase and terminal pathway activity (also see “Unexpected findings of properdin gene knockout in C3G”) [11, 48, 50–56]. It remains unclear, however, if properdin is strictly needed for C5 convertase formation and/or if properdin stabilizes the C5 convertase complex (C3bBbC3b) itself. This latter function is generally assumed based on the high similarity between C3 and C5 convertases, and it is often stated in literature with references to studies performed in the 1970s and 1980s [42, 57, 58]. In our opinion, convincing up-to-date evidence has yet to come.
Properdin as an initiator of alternative pathway activity
In 2006, Hourcade published a study that renewed the interest in the concept of properdin as an initiator of AP activity [46]. It was demonstrated that properdin could not only stabilize C3bBb complexes but also actively accelerate the association of C3b with FB to form C3 convertases. More importantly, properdin that was bound to an artificial surface, either directly or via C3 convertase intermediates (C3b, C3bB, or C3bBb), was able to recruit components from the environment to form new convertases on its unoccupied binding sites. Thus, properdin could serve as a platform for de novo convertase assembly on a surface (Fig. 3(B, C)) [46]. Numerous studies followed which extended these findings to biological surfaces under more physiological conditions. Targets on which properdin could initiate AP activity included typical AP targets (e.g., zymosan and rabbit erythrocytes), dangerous non-self surfaces (e.g., Chlamydia pneumoniae), altered-self surfaces (e.g., apoptotic and necrotic cells), and also self surfaces such as platelets and proximal tubular epithelial cells (PTECs) [7, 17, 59–69]. Suggested binding ligands on non-microbial cells are glycosaminoglycans [60] and surface-exposed DNA [17]; both are negatively charged.
Whether endogenous properdin in serum can indeed act as a true pattern recognition molecule in the meaning of being able to recognize and directly bind surface structures without primary C3b deposition in vivo (Fig. 3(C)) is a subject of controversy. The conflicting findings on this topic depend on the following three main aspects: (1) the source of properdin that is used, e.g., purified from serum, in whole serum environment, or freshly secreted from leukocytes; (2) the experimental conditions under which the assays are performed, e.g., allowing initial C3b deposition or not; and (3) the choice of biological target surface studied. For instance, caution should be taken in interpreting results obtained through the use of purified properdin, as it can form large polymeric aggregates, especially upon freeze-thaw cycles [10, 70]. These non-physiological aggregates have different AP-activating and target-binding properties than the physiological properdin oligomers that were separated from these polymeric aggregates after purification [10, 63, 64, 67, 70]. In addition, to confirm that properdin is a true recognition molecule, conditions should be used in which no initial C3b deposition can take place [17, 71, 72]. Some studies have suggested that circulating properdin in C3-inactive serum was not able to bind freely to certain targets which purified physiological properdin oligomers [64–67, 72] or properdin freshly released from activated leukocytes [60] could bind. C3-inactive serum refers to serum in which C3 (activation) is blocked or removed, for example by adding a C3-blocking molecule, using C3-depleted serum, or using a calcium- and magnesium-chelating agent to prevent complement activation. These findings have led to the hypothesis that serum contains inhibitors that prevent or regulate the direct pattern recognition function of properdin in the circulation [73, 74]. Such a regulatory mechanism could serve to prevent unwanted properdin-mediated damage systemically and to keep the pattern recognition preserved for specific conditions of danger or disease. In a recent study of O’Flynn et al., monomeric (but not pentameric) C-reactive protein was identified as such a properdin-regulating molecule, as it was able to control properdin-mediated AP activation on the PTEC surface [75].
In conclusion, it is evident that properdin can act as a docking station for convertase assembly and further AP activation (Fig. 3(B)). However, confirming its potential as a direct pattern recognition molecule in different in vivo environments (Fig. 3(C)) demands for a critical evaluation of used properdin preparations and experimental setup. Taking these conditions into account, the most important properdin-target interactions published so far can be summarized as follows: purified physiological properdin oligomers were shown to bind directly to zymosan [63, 64], necrotic, diseased B and T cell lines [64], Chlamydia pneumoniae [65], and activated platelets [67]; freshly secreted properdin was found to bind to apoptotic T cells [60] and activated platelets [67]; and properdin in C3-inactive serum has only been shown to bind to necrotic cells so far [17]. Interestingly, recent indications of other possible AP activators have emerged, such as complement FH-related protein 4 [76] and FH-related protein 5 [77, 78]. Comparable pattern recognition and AP-activating and enhancing functions were attributed to these molecules as to properdin, but these proteins also need further investigation as to their exact role in different physiological contexts.
Properdin in the local microenvironment
Besides a systemic role in complement, properdin can also act as an important and powerful mediator in (pro-inflammatory) local microenvironments. At these sites, properdin-producing cells are abundant and the properdin they produce might escape the serum inhibition that is assumed to exist in the circulation. Thus, locally strong increases in properdin levels may be generated which may modulate diverse immune responses at these sites (reviewed in [79]). First of all, properdin may have an important role in the safe and effective clearance of dying/dead cells. Direct binding of freshly secreted properdin from neutrophils to apoptotic and necrotic cells may aid in the phagocytosis of these cells [60]. Besides, the high amounts of properdin released as a response to local stimuli may amplify the AP-mediated inflammatory events in the microenvironment. Properdin-mediated complement activation contributes to further recruitment of pro-inflammatory cells to the site of infection via generation of the potent neutrophil chemoattractant C5a. In turn, neutrophils are triggered to release properdin from their granules, which may further act in a positive feedback loop by activating complement at its own surface and by activating and recruiting more neutrophils to inflammation sites [79, 80].
Clinical significance of properdin
Properdin deficiency
Properdin deficiency is a rare X-linked disorder that mainly affects males and is strongly associated with an increased vulnerability for meningitis caused by Neisseria meningitidis strains [81, 82]. Over 100 cases have been documented in over 25 families [82, 83]. Compared to properdin-normal individuals, meningococcal infections in properdin-deficient patients are associated with higher mortality and are more frequently caused by uncommon serogroups. Besides, the infections generally occur during teenage years instead of during early childhood as in the general population [84–86]. In addition to the increased risk for meningococcal infection, a recent study found that in one family, properdin deficiency was associated with recurrent otitis media and pneumonia [87]. These findings underline the importance of properdin in the host defense against certain but apparently not all pathogens.
Three types of properdin deficiency are recognized: type I describes the complete absence of the protein and is the most common, type II encompasses the cases in which levels are very low (up to 10% of normal), and in type III, systemic properdin levels are normal, but the protein is functionally defective [82]. Female carriers present with on average half of the normal properdin levels; levels range from nearly zero to concentrations in the normal range due to an uneven inactivation of the mutated and normal X chromosome [84, 88]. The mutations underlying properdin deficiency are very heterogeneous. In type I deficient families, various nonsense and missense mutations and few small deletions have been characterized [82, 88–91]. These were located in exons 4 to 9, encoding TSR1–5 and the first part of TSR6, respectively [82, 88–91]. The genetic changes in type I deficiency typically affected highly conserved amino acids [82] and are expected to alter the conformation and/or stability of properdin in such a way that it cannot be excreted anymore and becomes catabolized intracellularly [92]. The three underlying genetic defects found in type II deficiency are two missense mutations, one in exon 4 [93] and one in exon 8 [92], and one small combined deletion/insertion in exon 5 causing a frameshift and premature stop in exon 7 [87]. As a result of these mutations, properdin is likely impaired in its oligomerization, but the effect on structure is probably less drastic than expected in type I deficiency. Rapid catabolism of abnormal properdin molecules extracellularly might explain the low systemic properdin levels [92]. Only one family with type III deficiency has been identified [94]. The affected Dutch family members contained a missense mutation in exon 9 that likely affected C3b binding by properdin [95].
Potential role for properdin in human pathologies
The involvement of properdin in other human disease settings has only recently started to become elucidated. In the last decade, multiple studies to (complement-related) diseases have emerged in which alterations in systemic properdin levels were found (see Table 1). These studies have given us an indication of diseases in which properdin might play a significant role. Importantly, as in healthy controls, properdin levels in the studied patient cohorts generally also cover a large range. Reduced systemic properdin levels as compared to controls were observed in various disease conditions, such as in the renal diseases C3 glomerulopathy (C3G) and lupus nephritis [9, 11–14, 16, 21, 96] (Table 1). This may indicate that properdin is “consumed” in the circulation due to high AP activity; i.e., properdin is repositioned from the bloodstream to complement convertases at local sites of (surface-bound) complement activation to drive the AP activity. Lowered concentrations in the blood may also indicate a problem with the properdin-producing cells as in neutropenia [9]. Few studies also found increased systemic properdin levels (Table 1), for example in IgA nephropathy and in patients on hemodialysis [15, 18, 22, 97, 98]. The observation that increased properdin levels were associated with cardiovascular events and reduced levels with chronic heart failure was somewhat contradicting and needs further examination [16, 98]. A possible explanation might be that the role of complement changes at different stages of a disease. At the initial phase, elevated properdin levels might mainly increase the risk for developing the full disease. Once the disease has advanced and is clearly manifested, properdin levels might be reduced due to consumption [16]. Furthermore, some studies have reported pathological conditions in which clear local changes in properdin were found. For example, properdin levels increased in the bronchoalveolar lavage of allergic asthma and rhinitis patients after allergen challenge, which may indicate increased production/infiltration of properdin-producing inflammatory cells [99]. Future research will define whether more AP-mediated (renal) diseases show disturbances in properdin levels systemically and/or locally or whether this is selective for certain disease mechanisms only. After all, in some AP-related disease groups, no alterations in systemic properdin levels could be discovered [100].
Table 1.
Overview of human diseases and conditions associated with altered systemic properdin levels
| Disease/condition | Findings | Reference |
|---|---|---|
| a. Diseases and conditions associated with reduced systemic properdin levels | ||
| C3 glomerulopathy | Reduced P levels compared to controls. Average P levels were almost two times lower in C3GN compared with DDD, while sC5b-9 levels were elevated in C3GN compared with DDD. | Zhang et al. 2014 [12] |
| Reduced P levels compared to controls (i.e., below the mean-2sd) in 53% of the patients negative for C3NeF. C3GN was more frequent in the C3NeF-negative group, but no difference in C3GN frequency between the groups with normal versus reduced P. P consumption correlated with reduced C3 and C5 levels, with elevated sC5b-9 levels, and with a higher degree of proteinuria. | Corvillo et al. 2016 [11] | |
| Reduced P levels just below the lower limit of the reference range of controls in 4 out of 5 patients positive for C4NeF. Also decreased serum C3 and C5 levels, while C3c and sC5b-9 were increased. | Zhang et al. 2017 [13] | |
| Anti-neutrophil cytoplasmic antibody-associated vasculitis | Reduced P levels in active phase versus controls and versus remission, while plasma C3a, Bb, C5a, and sC5b-9 were elevated in active stage compared to remission. P levels inversely correlated with the proportion of crescents in the renal specimen. | Gou et al. 2013 [21] |
| Lupus nephritis | Approximately two-times reduced P levels in active lupus nephritis compared to controls, accompanied by increased plasma C3a, Bb, C5a, and C5b-9. | Gou et al. 2013 [21] |
| Human sepsis | Reduced P levels in patients on admission to the intensive care unit compared to controls. Slightly lower P levels in non-survivors compared to survivors. Low P levels correlated to increased treatment duration. | Stover et al. 2015 [14] |
| Chronic heart failure | Reduced P levels compared to controls, especially in those with a more advanced clinical disease, while FD and sC5b-9 were increased. P levels correlated with measures of cardiac function and were associated with adverse outcome. | Shahini et al. 2017 [16] |
| Viral lower respiratory tract infections | Reduced P levels in patients with severe compared to mild diseasea, although no differences found in acute versus recovery samples. | Ahout et al. 2017 [96] |
| Chemotherapy-induced neutropenia | Reduced P levels in the neutropenic state versus the preneutropenic state with normal neutrophil counts. | Tsyrkunou et al. 2017 [9] |
| b. Diseases and conditions associated with increased properdin levels | ||
| Healthy first-degree relatives of type 2 diabetes subjects | Elevated P levels in healthy first-degree relatives of type 2 diabetes subjects compared to age-matched controls. FB and sC5b-9 were also significantly higher in first-degree relatives, but no differences in C3, Bb, C3a, or FH. | Somani et al. 2012 [15] |
| Hemodialysis | Elevated P levels (by approximately factor 1.3) compared to controls, and slightly higher levels at the end of the hemodialysis session compared to the start. Also increased levels of C3d and C5b-9 after hemodialysis. | Poppelaars et al. 2016 [18] |
| Antibody-mediated rejection in heart transplant recipients | Elevated P levels in AMR patients carrying a rare AMR-associated allele in the P gene compared to control patients not carrying the rare allele and without AMR. | Marrón-Liñares et al. 2017 [97] |
| Cardiovascular events | Elevated P levels were associated with endothelial dysfunction, and with the risk of cardiovascular events. | Hertle et al. 2016 [98] |
| IgA nephropathy | Elevated P levels (by approximately factor 1.5) compared to controls. Also in the patients followed over time, P levels remained higher. | Onda et al. 2007 [22] |
aNo data on age-matched controls in this study involving very young children and no correction for age between the disease groups
P properdin, C3GN C3 glomerulonephritis, DDD dense deposit disease, C3NeF C3 nephritic factor, C4NeF C4 nephritic factor, sC5b-9 soluble C5b-9, FD factor D, FB factor B, AMR antibody-mediated rejection
Studies of properdin-deficient mouse models
Studies using properdin-deficient mouse models or mice treated with anti-properdin antibodies have provided us with indications about the beneficial and detrimental outcomes of properdin-inhibiting strategies. In general, the absence of properdin has been found to be beneficial in mouse models in which the complement activation is directed to host cells or tissues. Elimination of properdin has been shown to be promising in mouse models of renal ischemia-reperfusion injury [101, 102], arthritis [52, 53, 103], allergic airway inflammation [99], abdominal aortic aneurysm formation [104], and, very recently, atypical hemolytic uremic syndrome (aHUS) [105]. Properdin inhibition has also been shown effective in preventing complement-mediated extravascular hemolysis [62] and embryonic lethality [103] induced by the absence of an important membrane-expressed murine complement regulator. The better outcome of properdin-affected mice in these disease models is likely due to abrogated/attenuated AP activation and thus alleviation of the injuring immune responses directed to host tissues. On the contrary, in cases of infection where complement is directed to the invading pathogen, absence of properdin was often found detrimental for the host. Properdin deficiency has been associated with an exacerbated disease outcome in models of polymicrobial septic peritonitis [7], colitis [50, 51], small intestinal mucositis [106], LPS-induced non-septic shock [107], and Listeria-induced septicemia [108]. In these cases, properdin-deficient mice likely had a detrimental outcome due to compromised host defense against the microbial intruder, indicating properdin played a crucial role in this process. Nonetheless, the abovementioned concepts to predict outcome of properdin blockage do not always hold true. Whereas properdin-deficient mice indeed showed reduced survival in the model of non-septic shock induced by LPS, properdin-deficient mice were more resistant to zymosan-induced non-septic shock compared to wild-type mice [107]. In addition, in murine septicemia models, properdin deficiency worsened the outcome when the disease was induced by an infection of Listeria monocytogenes, whereas it improved the outcome when the pathology was induced by Streptococcus pneumoniae infection [108]. Also unexpected were the findings that absence of properdin in C3G, a disease characterized by glomerular injury due to excessive (fluid phase) AP activation, resulted in exacerbated renal injury [54, 109]. These studies indicate that there is a complex interplay between properdin and other (immune) effectors acting at the site of injury which determines what outcome the absence of properdin will trigger.
The role of properdin in complement-mediated renal injury
Genetic variations in complement genes and/or the presence of autoantibodies changing the function of complement components may disturb the sophisticated balance of AP activation and regulation. Especially in combination with triggering events, this may result in an overactivation of the system with subsequent damage to healthy tissues. The glomerulus is particularly vulnerable for complement attack; AP dysregulation has been associated with the disease entity C3G and with aHUS [110–115]. Besides, the renal tubular system can become prone to complement activation in diseases accompanied with proteinuria [116, 117]. The following section focuses on the role of properdin in developing these complement-associated pathologies.
C3 glomerulopathy
Clinical manifestation and pathogenesis
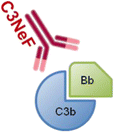
C3G describes a spectrum of severe complement-mediated renal diseases with up to 50% of patients progressing to end-stage renal disease within 10 years after first presentation [48, 115, 118–123]. C3G is characterized by the accumulation of C3 breakdown fragments in the glomeruli, without or with sparse immunoglobulin deposition [118, 124]. The causative disease process is an abnormal control of complement activation, deposition, or degradation [124]. Patients often present with low serum C3 levels as a result of the enhanced C3 turnover in the fluid phase. The main two diseases encompassed by the disease entity are dense deposit disease (DDD) and C3 glomerulonephritis (C3GN), which are distinguished from each other based on electron microscopy appearance. DDD is diagnosed on renal biopsy based on the presence of very dense ribbon-like intramembranous deposits in the glomerular basement membrane (GBM), while in C3GN these deposits appear in a less dense, more amorphous, and more diffuse pattern [124]. The most important pathogenic factors in C3G are autoantibodies stabilizing the AP C3 convertase, so-called C3 nephritic factors (C3NeF; 40–80% of cases), although pathogenic variants in AP (regulating) genes have also been reported (~ 20% of cases) [114, 115, 123]. Both C3NeF and properdin stabilize AP C3 convertases, but whereas C3NeF is strongly associated with pathogenic conditions and indicates a dysregulation of complement activity, properdin is part of healthy homeostasis and has a complement regulatory role.
Unexpected findings of properdin gene knockout in C3G
As C3G occurs as a result of overactivation of the AP leading to glomerular injury, it was hypothesized that inhibition of properdin could prevent this AP overactivity and subsequent injury. Surprisingly, however, mouse models showed a protective role for properdin in C3G (summarized in [125]). The mouse models used were FH-deficient mice (FH−/−) [54] or mice with only small amounts of truncated FH (FHm/m) [55, 109]. These two models both showed the characteristic accumulation of C3 along the GBM with morphological changes and glomerular inflammation typical for C3G. Besides, these mice had low plasma C3 and C5 levels as a result of complement consumption by the activity of the C3/C5 convertases, and thus clearly show the lack of FH-dependent complement regulation [54, 55, 109]. However, mice that were knocked out for both FH and properdin, FH−/−/P−/−, showed exacerbated injury with increased accumulation of C3 along the GBM. FHm/m/P−/− mice showed a similar detrimental effect of the absence of properdin; the renal injury was even worse since the mice died prematurely of severe glomerulonephritis. Remarkably, both these FH-affected properdin-knockout models showed a selective C3 depletion but higher intact C5 levels (less C5 consumption) compared to the mice with an intact properdin gene [54, 55, 109]. Thus, absence of properdin specifically reduced the C5 turnover and was associated with the exacerbated injury observed in these properdin-knockout mice.
Possible role for properdin in the distinction of C3G subgroups with different pathophysiology
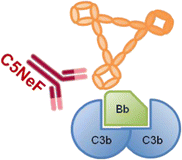
The differences in plasma C3 and C5 levels between properdin-deficient and properdin-sufficient mice are reminiscent of the complement profiles found in C3G patient groups associated with two types of C3NeF, namely properdin-dependent and properdin-independent C3NeF (Table 2) [48, 126–130]. C3NeF are a heterogeneous group of autoantibodies. All stabilize the AP convertases by preventing intrinsic and/or extrinsic decay [130], but some of them have the ability to induce both systemic C3 and C5 consumption (properdin-dependent C3NeF; resembling properdin-sufficient FH-affected mice), whereas others only seem to affect the levels of C3 (properdin-independent C3NeF; resembling properdin-deficient FH-affected mice) [48, 126–129]. A recent study suggested the new term C5NeF for C3NeF that are dependent on properdin and cause increased C5 conversion [48]. In humans, properdin-dependent types of C3NeF were more often found in C3GN [48, 126, 128]. In this C3G subtype, C5 convertase dysregulation and terminal pathway activation are more pronounced, indicated by lower C5 and higher soluble C5b-9 (sC5b-9) levels in the circulation (Table 2) [12]. Properdin-independent C3NeF are more frequently associated with DDD cases [48, 126, 128]. This subtype is characterized by a selective, more pronounced C3 convertase dysregulation, as shown by lowered C3 levels but near-normal C5 and sC5b-9 levels [12]. Thus, these biomarker profiles of C3G subtypes are in line with the functional characteristics regarding C3 and C5 conversion attributed to the properdin-dependent and properdin-independent C3NeF types.
Table 2.
Proposed underlying mechanisms in the pathophysiology of C3 glomerulopathy based on the presence of different types of convertase-stabilizing nephritic factors
|
|
|
|---|---|---|
| Type of C3NeF | Properdin-independent C3NeF | Properdin-dependent C3NeF/C5NeF |
| Associated complement profile | C3 consumption | C3 consumption |
| C5 normal or slightly consumed | C5 consumption | |
| sC5b-9 normal | sC5b-9 elevated | |
| Disease association | DDD | C3GN |
| Comparative mouse model | FH−/−/P−/− | FH−/− |
C3NeF C3 nephritic factor, C5NeF C5 nephritic factor, sC5b-9 soluble C5b-9, DDD dense deposit disease, C3GN C3 glomerulonephritis, FH factor H, P properdin
The biomarker analysis of Zhang et al. also showed direct differences in the level of properdin in C3G patients [12]. Properdin levels were found lower in C3G compared to controls, and the effect was more pronounced in the C3GN group than in the DDD group [12]. Next to the terminal pathway biomarker sC5b-9, properdin was the only AP marker that significantly differed between the two C3G subgroups. Another study also showed significantly lowered serum levels of properdin in a subset of C3G patients, namely in C3NeF-negative patients which were predominantly C3GN cases [11]. Interestingly, the properdin consumption was associated with increased C5 convertase activity leading to C5 consumption [11]. A recent cluster analysis approach performed by Iatropoulos et al. confirmed that different complement dysregulation profiles at the C3 and C5 levels exist in patient subgroups in C3G [131]. However, the identified clusters were not simply divided in C3GN and DDD but were based on shared clinical, histological, genetic, and serological complement parameters to define distinct disease entities characterized by specific pathophysiological mechanisms [131]. Properdin was not taken into account. More research is needed to investigate whether properdin, in combination with different types of nephritic factors, might be the driving force in directing the C3 or C5 convertase dysregulation and subsequent pathophysiology in C3G subgroups, and as such may be an important disease biomarker.
A role for properdin in the pathogenesis of C3G is slightly unexpected in the view of C3G being a disease caused by fluid-phase dysregulation of the AP. How properdin alters the balance of C3 and C5 consumption and how this exactly results in exacerbation of renal injury in the FH-affected mouse models requires further investigation. The current hypothesis is that properdin deficiency changes the ratio between systemic, fluid-phase (properdin-independent), and local, surface (properdin-dependent) AP activation [54–56, 109]. Depending on the presence of properdin and the availability of intact C3 and C5, deposition of systemically generated breakdown fragments and/or local AP activation may be directed to different renal structures, e.g., the endothelium, the unprotected, exposed GBM (on which properdin-independent activation may be possible), the mesangium, or the renal tubules, to cause injury to different extents. In this way, properdin may determine the intraglomerular fate of C3 breakdown fragments which are related to the different C3G subforms [54–56, 109]. Thus, further investigation into the role of properdin in C3G may aid in our understanding of the pathophysiology and distinction of C3G subforms.
Atypical hemolytic uremic syndrome
Clinical manifestation and pathogenesis
Atypical HUS is categorized as a thrombotic microangiopathy and is clinically characterized by the typical triad of hemolytic anemia, thrombocytopenia, and serious renal impairment [132, 133]. These symptoms are caused by uncontrolled AP activation focused on the glomerular endothelial cell surface. Via diverse pro-inflammatory and pro-thrombotic events, this complement activation leads to the disturbed integrity of the endothelial cell layer and subsequent renal injury [112]. In around 60% of the aHUS patients, genetic aberrations have been found in complement genes [134–136]. These aberrations include loss-of-function mutations in genes encoding complement regulatory proteins and gain-of-function mutations in genes encoding the constituents of the convertase complex. The FH gene is most frequently affected [134–136], and FH-directed autoantibodies are also a common cause of impaired complement control [137, 138]. This illustrates the importance of this regulator in protecting the glomerular structures.
Possible role for properdin in aHUS
Although many mutations in complement regulators have been associated with aHUS development, little is known about whether properdin may be affected in patients. To our knowledge, no gain-of-function mutations in properdin have been described in patients so far. Also from our own unpublished observations, we can conclude the properdin gene is not affected in aHUS patients. Thus, to date, there is no direct evidence for a role of properdin in aHUS in humans.
Nonetheless, studies with mice have shown that properdin plays a critical role in AP activation on autologous cells. Mice were modeled for disorders in which the surface control on autologous cells was impaired by knocking out important membrane-bound complement regulatory proteins. The subsequent vulnerability of these cells to complement-mediated attack was dependent on the presence of properdin, since properdin-deficient mice showed less injury and better disease outcomes [62, 101–103]. Properdin is thus likely needed to tip the balance to AP activation on cells that are well equipped with a repertoire of complement regulatory proteins. In the absence of intact complement regulators, as is also the case in aHUS, properdin might gain in function and promote the AP on healthy self-surfaces. Since absence of properdin is beneficial in these types of modeled diseases, the interesting question arises whether properdin blockage might also prevent the surface-directed complement activation in aHUS. Despite this attractive therapeutic potential, very few studies have been performed so far using experimental (animal or cellular) models of aHUS. Very recently, an important and interesting article was published showing that properdin inhibition in a mouse model of aHUS indeed improved disease outcome as expected [105].
In addition, properdin might have a role in exacerbating the thrombotic phenotype seen in aHUS. This is another reason why properdin blockage might be beneficial in this disease. Recent studies have elucidated a central role for properdin in the complement-mediated cross-talk between platelets and neutrophils. These two cell types mutually enhance each other’s activation, directly or indirectly via AP activation, and in doing so they are important mediators of thromboinflammation (extensively reviewed in [74]). First of all, activated platelets can act as a platform for local AP amplification. Properdin released from activated neutrophils can directly bind to the surface of activated platelets and can subsequently recruit the components needed for convertase formation to further promote complement activation on this platelet surface [67]. During this AP activation on platelets at sites of vascular injury, C5a is released which is a very important chemoattractant that can recruit and activate neutrophils. As previously explained, neutrophils can enhance their own activation in a positive feedback loop by secreting properdin that in an autocrine or paracrine fashion increases properdin-mediated AP activation on its own surface [80]. In turn, the released properdin can also promote AP-mediated platelet activation and complete the vicious cycle. In line with these findings, by using whole-blood ex vivo assays, Blatt et al. showed that blockage of properdin reduced platelet-leukocyte aggregate formation by 50% [47]. Furthermore, these studies collectively showed that the properdin-mediated mechanisms contributing to thromboinflammation were enhanced when the surface-protecting function of FH was experimentally inhibited [47, 67]. In disorders of compromised complement regulation such as aHUS, it has to be investigated if properdin blockage may therefore be beneficial in interrupting the positive feedback loops responsible for the increased (complement-mediated) thromboinflammation.
Chronic proteinuric renal disease
Complement mediates proteinuria-induced tubulointerstitial injury
Glomerular dysfunction results in leakage of proteins through the glomerular filtration barrier. This proteinuria can induce tubulointerstitial injury and thereby is a strong predictor for the progression of chronic renal disease to end-stage renal disease [139, 140]. Complement activation at the surface of PTECs by filtered complement components has been proven to be a powerful mechanism underlying this proteinuria-induced tubulointerstitial injury (reviewed in [116, 117]). Under healthy conditions, complement components do not pass the glomerular filtration barrier, but in proteinuric patients complement proteins are found in the urine and deposition of complement along the tubular brush border (the apical side) is seen. Furthermore, complement inhibition attenuates the renal deterioration in proteinuric rodent models, confirming the role of complement in mediating tubulointerstitial injury [116, 117, 140].
The role of properdin in complement-mediated tubulointerstitial injury in proteinuria
It was hypothesized that properdin, as an initiator of AP activity, might play a role in triggering the tubular complement activation associated with tubulointerstitial injury in proteinuric renal disease [61]. Indeed, properdin was able to bind primary PTECs [61] and PTEC cell lines to act as a focal point for AP amplification followed by subsequent C3 and C5b-9 deposition [61, 141]. PTECs may be especially vulnerable to complement activation compared to other human cells because normally these epithelial cells do not come into contact with complement components [61] and express less complement regulatory proteins [142]. In addition, properdin was found in the urine of around 50% of proteinuric patients [143]. This urinary properdin associated with elevated sC5b-9 levels and with a worse renal function outcome but was not dependent on the degree of proteinuria [143]. Altogether, these findings advocate that properdin is an important determinant in intratubular complement activation and in this way it might play a role in the proteinuria-mediated renal damage.
The binding ligand for properdin on PTECs offers possibilities for specific therapeutic approaches
In subsequent studies, the binding ligand for properdin on renal tubular cells was identified: heparan sulfate proteoglycans. This type of proteoglycan is most abundant in renal tissues. It was found that properdin could bind specifically to certain heparan sulfate moieties of these proteoglycans in vitro and that properdin colocalized with the heparan sulfate proteoglycan syndecan-1 in vivo in proteinuric rat kidneys [68]. Interestingly, FH is also well-known for being able to bind to heparan sulfates; thus, it was speculated whether these positive and negative regulators might compete with each other in the tubular lumen under proteinuric conditions. Nonetheless, Zaferani et al. demonstrated that FH and properdin recognized different, non-overlapping epitopes of heparan sulfate chains on renal tubular epithelial cells, indicating at least no direct competition for binding sites [144].
In view of treatment strategies, this is an important and interesting finding. The authors have shown that certain low- or non-anticoagulant heparinoids could inhibit the interaction of heparan sulfates with properdin but not with FH, indicating that a specific blockage of the positive regulator properdin is feasible while leaving the inhibitory, beneficial actions of FH intact. Indeed, the in vitro experiments showed that these heparinoids inhibited C5b-9 deposition on the tubular epithelial cells and thus were able to control the AP of complement [144]. In summary, these findings illustrate that the composition of cell surfaces, and possibly their change during disease, may orchestrate the balance between AP activation and inhibition by recruiting properdin and/or FH.
Future directions
The potential of properdin as a therapeutic target in complement-mediated renal diseases
The interest in complement-targeted therapy has increased immensely in recent years, and many complement inhibitors are in the development pipeline [2]. Properdin is a relatively new kid on the block in the field of complement and immunity. Nonetheless, monoclonal antibodies against human properdin have been developed that are effective in blocking the AP in vitro [100, 145], and one anti-properdin antibody is currently at the stage of phase 2 in clinical trials [2]. Based on the role of properdin in promoting the activity of the AP, it is expected that properdin elimination can be effective in diseases of complement overactivation resulting in host tissue injury. To the best of our knowledge, no gain-of-function mutations in properdin itself have been found so far. Thus, therapy is aimed at compensating the complement dysregulation caused by other autoimmune or genetic factors (as in C3G and aHUS) or pathological conditions (as in chronic proteinuria).
In the previous sections, we have examined this potential of properdin elimination as a treatment for C3G, aHUS, and chronic proteinuric renal disease. Research so far has indicated that aHUS and chronic proteinuria might indeed benefit from properdin inhibition by limiting the properdin-mediated AP activation directed at host cell surfaces, i.e., the glomerular endothelium and proximal tubular epithelium, respectively. In addition, properdin inhibition in aHUS may decrease the thrombotic and inflammatory effects mediated by platelets and leukocytes. In contrast, properdin inhibition might not be a promising therapy in C3G, since properdin knockout in C3G mouse models clearly resulted in exacerbated renal injury. These findings might indicate that absence of properdin is advantageous in cases of uncontrolled tissue-bound activation, whereas in situations of uncontrolled fluid-phase activation, absence of properdin is unfavorable. This would be in line with the evidence for properdin being mainly a surface-directed regulator. C3G is not yet completely excluded as a potential disease that may benefit from properdin-targeted therapy. First of all, mice are not humans and the pathophysiological mechanisms may differ. The used mouse models were based on FH mutations, but such mutations are found in only a minority of C3G patients. C3NeF is a far more common abnormality found in C3G patients, and it has been shown that some C3NeF are dependent on properdin. Therefore, elimination of properdin to inhibit this C3NeF activity seems a plausible therapeutic option, but no studies on this subject have been published so far.
In summary, we are still at the beginning of understanding the effects of properdin-inhibiting therapy, and future research should help us to unravel properdin’s exact role in diverse disease settings. Its potential as a therapeutic target must be considered with care. One should always keep in mind the balance between the beneficial and detrimental functions that properdin can have in specific pathological settings. These differential effects are currently still hard to predict due to gaps in our knowledge about exact properdin biology, e.g., regarding the characteristics of the different oligomers, regarding the need of properdin for AP activation in different settings, and regarding the interaction of properdin with other immune effector molecules. Stratification of patient groups including a careful characterization of the specific AP defect and the clinical picture may aid in determining which patients may benefit from therapy. Other factors to keep in mind for safe and effective properdin inhibition are whether properdin should be inhibited locally or systemically and during which time window, i.e., acute or chronically.
Properdin inhibition in comparison to C5 inhibition and its advantages
Eculizumab, a C5-inhibiting antibody, was approved in 2007 as the first complement-specific drug, and it is a very effective therapy for aHUS patients [146, 147]. It prevents C5a release and MAC formation and in this way reduces the subsequent complement-mediated damage to the endothelium. It was expected that eculizumab would also be effective in C3G, especially in patients showing increased C5b-9 levels. However, C3G patients show a heterogeneous response to eculizumab treatment [148, 149]. Given the previously discussed associations between properdin deficiency and decreased C5 convertase activity, properdin inhibition and C5 inhibition seem functionally similar to each other. Therapeutic inhibition of properdin therefore also seems promising in aHUS. Of note, both eculizumab use and properdin deficiency increase the susceptibility for meningitis. The prophylactic vaccination that is applied in patients receiving eculizumab [150] would therefore also be needed in properdin-blocking therapy.
In general, properdin-inhibiting therapy may have several advantages over C5 inhibition or over therapies directed to other complement proteins such as C3. First, in contrast to C5 inhibition, properdin inhibition may also reduce, at least to a certain extent, adverse inflammatory effects mediated by the upstream activation products C3a and C3b. Since properdin is present in much lower concentrations than C3 (and C5), it would also be a more manageable target to block [103]. Another important advantage of properdin inhibition is the specificity of blocking the AP, which is preferred in diseases showing a specific AP defect such as aHUS. C5 inhibition blocks all terminal pathway activity, whereas properdin inhibition only affects AP activity. The functioning of the other complement activation pathways is preserved so these can still fight infection and help maintain tissue homeostasis. This is supported by the study of Heinen et al., in which it was shown, using an in vitro enzyme-linked immunosorbent assay, that properdin blockage in normal human serum specifically impaired the ability to activate the AP but not the CP and LP [151]. Nonetheless, convincing experiments should be performed to confirm whether this also holds true for in vivo situations. It is not clear yet if properdin inhibition critically affects CP- and LP-induced complement responses by impairing the AP amplification loop, since the few studies performed on this topic contradict each other [47, 62, 74, 100, 145, 151]. These findings might also indicate that the role of properdin is context-dependent and may differ between disease settings. In line with this, it has been shown that the absolute requirement of properdin for AP activation depends on the activating surface; not all microbes need the presence of properdin to activate the AP [62]. If confirmed, not all AP-mediated actions will be compromised when properdin is inhibited. This indeed seems to be the case when looking at properdin-deficient individuals which specifically show increased vulnerability to Neisseria infections but not to others. In conclusion, compared to C3 inhibition or C5 blockage by eculizumab, properdin-directed therapy may provide a more sophisticated complement inhibition that does not completely compromise host defense but keeps some complement activation on several targets intact.
Concluding remarks
Properdin is a recently discovered player in complement-related kidney diseases. We have just started to unravel its potential in therapeutics and further research is definitely needed. These studies should focus on increasing the understanding of properdin biology in general and its cross-talk with other immune pathways. Such research would benefit from validated and standardized quantitative assays for measuring properdin in serum or urine in well-defined patient groups. In combination with a diagnostic workup on other complement proteins, such data will provide us with important information on disease mechanisms in complement-mediated renal diseases and will help us to select patient groups that may benefit from properdin-directed therapy.
Questions (answers are provided following the reference list)
- Which statement about properdin is not true?
- Properdin oligomerizes in dimers, trimers, and tetramers only upon stimulation
- Properdin can recruit C3b and FB for new convertase assembly
- Properdin stabilizes AP C3/C5 convertases, but not those of the classical and lectin pathway
- Properdin oligomerization is essential for its function in vivo
- Which factors should be taken into account when the function of properdin is studied:
- Species differences
- Aggregation of properdin upon freeze-thawing
- The source of properdin: purified from serum, freshly released from cells, or in serum context
- All of the above
- Properdin is mainly produced by:
- Hepatocytes
- Leukocytes
- Hepatocytes and leukocytes
- Endothelial cells
- Which of the following statements regarding the proposed role of properdin in renal disease is not true?
- In proteinuria, properdin initiates and amplifies local complement activation on proximal tubular epithelial cells and thereby contributes to tubulointerstitial injury
- Properdin prevents C5 conversion and thereby it has protecting roles in C3G
- In aHUS properdin may both be involved in triggering the onset of disease as well as in exacerbating the thromboinflammatory course of disease
- Properdin can be important for clearance of dangerous pathogens and altered self-cells, but its requirement for AP activation is not equal for all targets.
- Which of the following statements regarding properdin-dependent and -independent C3NeF is true?
- Properdin-dependent C3NeF are associated with a selective C3 consumption, which resembles the complement profile of FH-/-/P-/- knockout mice
- Properdin-independent C3NeF are associated with a selective C3 consumption and are most often found in C3GN
- Properdin-dependent C3NeF are associated with elevated terminal pathway activation markers and are mainly found in C3GN
- Properdin-independent C3NeF are associated with elevated terminal pathway activation markers and are most often found in C3GN
Abbreviations
- aHUS
Atypical hemolytic uremic syndrome
- AP
Alternative pathway
- C3G
C3 glomerulopathy
- C3GN
C3 glomerulonephritis
- C3NeF
C3 nephritic factor
- CP
Classical pathway
- DDD
Dense deposit disease
- FB
Factor B
- FD
Factor D
- FH
Factor H
- FI
Factor I
- GBM
Glomerular basement membrane
- LP
Lectin pathway
- MAC
Membrane attack complex
- PTEC
Proximal tubular epithelial cells
- sC5b-9
Soluble C5b-9
- TSR
Thrombospondin type 1 repeat
Compliance with ethical standards
Conflict of interest
The authors declare no conflict of interest. Dr. N. van de Kar is a member of the International aHUS Advisory Board of Alexion. This affiliation had no influence on the content of this manuscript.
Footnotes
Answers
1.a; 2.d; 3.b; 4.b; 5.c
References
- 1.Ricklin D, Reis ES, Lambris JD. Complement in disease: a defence system turning offensive. Nat Rev Nephrol. 2016;12:383–401. doi: 10.1038/nrneph.2016.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ricklin D, Mastellos DC, Reis ES, Lambris JD. The renaissance of complement therapeutics. Nat Rev Nephrol. 2018;14:26–47. doi: 10.1038/nrneph.2017.156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ricklin D, Hajishengallis G, Yang K, Lambris JD. Complement: a key system for immune surveillance and homeostasis. Nat Immunol. 2010;11:785–797. doi: 10.1038/ni.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Noris M, Remuzzi G. Overview of complement activation and regulation. Semin Nephrol. 2013;33:479–492. doi: 10.1016/j.semnephrol.2013.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schwaeble W, Dippold WG, Schafer MK, Pohla H, Jonas D, Luttig B, Weihe E, Huemer HP, Dierich MP, Reid KB. Properdin, a positive regulator of complement activation, is expressed in human T cell lines and peripheral blood T cells. J Immunol. 1993;151:2521–2528. [PubMed] [Google Scholar]
- 6.Whaley K. Biosynthesis of the complement components and the regulatory proteins of the alternative complement pathway by human peripheral blood monocytes. J Exp Med. 1980;151:501–516. doi: 10.1084/jem.151.3.501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Stover CM, Luckett JC, Echtenacher B, Dupont A, Figgitt SE, Brown J, Mannel DN, Schwaeble WJ. Properdin plays a protective role in polymicrobial septic peritonitis. J Immunol. 2008;180:3313–3318. doi: 10.4049/jimmunol.180.5.3313. [DOI] [PubMed] [Google Scholar]
- 8.Wirthmueller U, Dewald B, Thelen M, Schafer MK, Stover C, Whaley K, North J, Eggleton P, Reid KB, Schwaeble WJ. Properdin, a positive regulator of complement activation, is released from secondary granules of stimulated peripheral blood neutrophils. J Immunol. 1997;158:4444–4451. [PubMed] [Google Scholar]
- 9.Tsyrkunou A, Agarwal S, Koirala B, Finberg RW, Nath R, Barton B, Levitz SM, Wang JP, Ram S (2017) Properdin levels in individuals with chemotherapy-induced neutropenia. Open Forum Infect Dis. 10.1093/ofid/ofw250 [DOI] [PMC free article] [PubMed]
- 10.Pangburn MK. Analysis of the natural polymeric forms of human properdin and their functions in complement activation. J Immunol. 1989;142:202–207. [PubMed] [Google Scholar]
- 11.Corvillo F, Bravo Garcia-Morato M, Nozal P, Garrido S, Tortajada A, Rodriguez de Cordoba S, Lopez-Trascasa M. Serum properdin consumption as a biomarker of C5 convertase dysregulation in C3 glomerulopathy. Clin Exp Immunol. 2016;184:118–125. doi: 10.1111/cei.12754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang Y, Nester CM, Martin B, Skjoedt MO, Meyer NC, Shao D, Borsa N, Palarasah Y, Smith RJ. Defining the complement biomarker profile of C3 glomerulopathy. Clin J Am Soc Nephrol. 2014;9:1876–1882. doi: 10.2215/CJN.01820214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang Y, Meyer NC, Fervenza FC, Lau W, Keenan A, Cara-Fuentes G, Shao D, Akber A, Fremeaux-Bacchi V, Sethi S, Nester CM, Smith RJH. C4 nephritic factors in C3 glomerulopathy: a case series. Am J Kidney Dis. 2017;70:834–843. doi: 10.1053/j.ajkd.2017.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Stover CM, McDonald J, Byrne S, Lambert DG, Thompson JP. Properdin levels in human sepsis. Front Immunol. 2015;6:24. doi: 10.3389/fimmu.2015.00024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Somani R, Richardson VR, Standeven KF, Grant PJ, Carter AM. Elevated properdin and enhanced complement activation in first-degree relatives of South Asian subjects with type 2 diabetes. Diabetes Care. 2012;35:894–899. doi: 10.2337/dc11-1483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shahini N, Michelsen AE, Nilsson PH, Ekholt K, Gullestad L, Broch K, Dahl CP, Aukrust P, Ueland T, Mollnes TE, Yndestad A, Louwe MC. The alternative complement pathway is dysregulated in patients with chronic heart failure. Sci Rep. 2017;7:42532. doi: 10.1038/srep42532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Xu W, Berger SP, Trouw LA, de Boer HC, Schlagwein N, Mutsaers C, Daha MR, van Kooten C. Properdin binds to late apoptotic and necrotic cells independently of C3b and regulates alternative pathway complement activation. J Immunol. 2008;180:7613–7621. doi: 10.4049/jimmunol.180.11.7613. [DOI] [PubMed] [Google Scholar]
- 18.Poppelaars F, Gaya da Costa M, Berger SP, Assa S, Meter-Arkema AH, Daha MR, van Son WJ, Franssen CF, Seelen MA. Strong predictive value of mannose-binding lectin levels for cardiovascular risk of hemodialysis patients. J Transl Med. 2016;14:236. doi: 10.1186/s12967-016-0995-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.de Paula PF, Barbosa JE, Junior PR, Ferriani VP, Latorre MR, Nudelman V, Isaac L. Ontogeny of complement regulatory proteins - concentrations of factor h, factor I, c4b-binding protein, properdin and vitronectin in healthy children of different ages and in adults. Scand J Immunol. 2003;58:572–577. doi: 10.1046/j.1365-3083.2003.01326.x. [DOI] [PubMed] [Google Scholar]
- 20.Grumach AS, Ceccon ME, Rutz R, Fertig A, Kirschfink M. Complement profile in neonates of different gestational ages. Scand J Immunol. 2014;79:276–281. doi: 10.1111/sji.12154. [DOI] [PubMed] [Google Scholar]
- 21.Gou SJ, Yuan J, Chen M, Yu F, Zhao MH. Circulating complement activation in patients with anti-neutrophil cytoplasmic antibody-associated vasculitis. Kidney Int. 2013;83:129–137. doi: 10.1038/ki.2012.313. [DOI] [PubMed] [Google Scholar]
- 22.Onda K, Ohi H, Tamano M, Ohsawa I, Wakabayashi M, Horikoshi S, Fujita T, Tomino Y. Hypercomplementemia in adult patients with IgA nephropathy. J Clin Lab Anal. 2007;21:77–84. doi: 10.1002/jcla.20154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Minta JO, Jezyk PD, Lepow IH. Distribution and levels of properdin in human body fluids. Clin Immunol Immunopathol. 1976;5:84–90. doi: 10.1016/0090-1229(76)90152-5. [DOI] [PubMed] [Google Scholar]
- 24.Sonntag J, Brandenburg U, Polzehl D, Strauss E, Vogel M, Dudenhausen JW, Obladen M. Complement system in healthy term newborns: reference values in umbilical cord blood. Pediatr Dev Pathol. 1998;1:131–135. doi: 10.1007/s100249900016. [DOI] [PubMed] [Google Scholar]
- 25.Davis CA, Vallota EH, Forristal J. Serum complement levels in infancy: age related changes. Pediatr Res. 1979;13:1043–1046. doi: 10.1203/00006450-197909000-00019. [DOI] [PubMed] [Google Scholar]
- 26.Wolach B, Dolfin T, Regev R, Gilboa S, Schlesinger M. The development of the complement system after 28 weeks’ gestation. Acta Paediatr Scand. 1997;86:523–527. doi: 10.1111/j.1651-2227.1997.tb08924.x. [DOI] [PubMed] [Google Scholar]
- 27.Prellner K, Sjoholm AG, Truedsson L. Concentrations of C1q, factor B, factor D and properdin in healthy children, and the age-related presence of circulating C1r-C1s complexes. Acta Paediatr Scand. 1987;76:939–943. doi: 10.1111/j.1651-2227.1987.tb17268.x. [DOI] [PubMed] [Google Scholar]
- 28.Drew JH, Arroyave CM. The complement system of the newborn infant. Biol Neonate. 1980;37:209–217. doi: 10.1159/000241276. [DOI] [PubMed] [Google Scholar]
- 29.Smith CA, Pangburn MK, Vogel CW, Muller-Eberhard HJ. Molecular architecture of human properdin, a positive regulator of the alternative pathway of complement. J Biol Chem. 1984;259:4582–4588. [PubMed] [Google Scholar]
- 30.Nolan KF, Schwaeble W, Kaluz S, Dierich MP, Reid KB. Molecular cloning of the cDNA coding for properdin, a positive regulator of the alternative pathway of human complement. Eur J Immunol. 1991;21:771–776. doi: 10.1002/eji.1830210333. [DOI] [PubMed] [Google Scholar]
- 31.Higgins JM, Wiedemann H, Timpl R, Reid KB. Characterization of mutant forms of recombinant human properdin lacking single thrombospondin type I repeats. Identification of modules important for function. J Immunol. 1995;155:5777–5785. [PubMed] [Google Scholar]
- 32.Sun Z, Reid KB, Perkins SJ. The dimeric and trimeric solution structures of the multidomain complement protein properdin by X-ray scattering, analytical ultracentrifugation and constrained modelling. J Mol Biol. 2004;343:1327–1343. doi: 10.1016/j.jmb.2004.09.001. [DOI] [PubMed] [Google Scholar]
- 33.Alcorlo M, Tortajada A, Rodriguez de Cordoba S, Llorca O. Structural basis for the stabilization of the complement alternative pathway C3 convertase by properdin. Proc Natl Acad Sci U S A. 2013;110:13504–13509. doi: 10.1073/pnas.1309618110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bertram P, Akk AM, Zhou HF, Mitchell LM, Pham CT, Hourcade DE. Anti-mouse properdin TSR 5/6 monoclonal antibodies block complement alternative pathway-dependent pathogenesis. Monoclon Antib Immunodiagn Immunother. 2015;34:1–6. doi: 10.1089/mab.2014.0066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Perdikoulis MV, Kishore U, Reid KB. Expression and characterisation of the thrombospondin type I repeats of human properdin. Biochim Biophys Acta. 2001;1548:265–277. doi: 10.1016/s0167-4838(01)00238-2. [DOI] [PubMed] [Google Scholar]
- 36.Pedersen DV, Roumenina L, Jensen RK, Gadeberg TA, Marinozzi C, Picard C, Rybkine T, Thiel S, Sorensen UB, Stover C, Fremeaux-Bacchi V, Andersen GR. Functional and structural insight into properdin control of complement alternative pathway amplification. EMBO J. 2017;36:1084–1099. doi: 10.15252/embj.201696173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Merle NS, Church SE, Fremeaux-Bacchi V, Roumenina LT. Complement system part I - molecular mechanisms of activation and regulation. Front Immunol. 2015;6:262. doi: 10.3389/fimmu.2015.00262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Harboe M, Ulvund G, Vien L, Fung M, Mollnes TE. The quantitative role of alternative pathway amplification in classical pathway induced terminal complement activation. Clin Exp Immunol. 2004;38:439–446. doi: 10.1111/j.1365-2249.2004.02627.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pillemer L, Blum L, Lepow IH, Ross OA, Todd EW, Wardlaw AC. The properdin system and immunity. I. Demonstration and isolation of a new serum protein, properdin, and its role in immune phenomena. Science. 1954;120:279–285. doi: 10.1126/science.120.3112.279. [DOI] [PubMed] [Google Scholar]
- 40.Maves KK, Weiler JM. Properdin: approaching four decades of research. Immunol Res. 1993;12:233–243. doi: 10.1007/BF02918255. [DOI] [PubMed] [Google Scholar]
- 41.Pangburn MK, Muller-Eberhard HJ. The C3 convertase of the alternative pathway of human complement. Enzymic properties of the bimolecular proteinase. Biochem J. 1986;235:723–730. doi: 10.1042/bj2350723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Medicus RG, Gotze O, Muller-Eberhard HJ. Alternative pathway of complement: recruitment of precursor properdin by the labile C3/C5 convertase and the potentiation of the pathway. J Exp Med. 1976;144:1076–1093. doi: 10.1084/jem.144.4.1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fearon DT, Austen KF. Properdin: binding to C3b and stabilization of the C3b-dependent C3 convertase. J Exp Med. 1975;142:856–863. doi: 10.1084/jem.142.4.856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Farries TC, Lachmann PJ, Harrison RA. Analysis of the interactions between properdin, the third component of complement (C3), and its physiological activation products. Biochem J. 1988;252:47–54. doi: 10.1042/bj2520047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Farries TC, Lachmann PJ, Harrison RA. Analysis of the interaction between properdin and factor B, components of the alternative-pathway C3 convertase of complement. Biochem J. 1988;253:667–675. doi: 10.1042/bj2530667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hourcade DE. The role of properdin in the assembly of the alternative pathway C3 convertases of complement. J Biol Chem. 2006;281:2128–2132. doi: 10.1074/jbc.M508928200. [DOI] [PubMed] [Google Scholar]
- 47.Blatt AZ, Saggu G, Kulkarni KV, Cortes C, Thurman JM, Ricklin D, Lambris JD, Valenzuela JG, Ferreira VP. Properdin-mediated C5a production enhances stable binding of platelets to granulocytes in human whole blood. J Immunol. 2016;196:4671–4680. doi: 10.4049/jimmunol.1600040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Marinozzi MC, Chauvet S, Le Quintrec M, Mignotet M, Petitprez F, Legendre C, Cailliez M, Deschenes G, Fischbach M, Karras A, Nobili F, Pietrement C, Dragon-Durey MA, Fakhouri F, Roumenina LT, Fremeaux-Bacchi V. C5 nephritic factors drive the biological phenotype of C3 glomerulopathies. Kidney Int. 2017;92:1232–1241. doi: 10.1016/j.kint.2017.04.017. [DOI] [PubMed] [Google Scholar]
- 49.Berends ET, Gorham RD, Jr, Ruyken M, Soppe JA, Orhan H, Aerts PC, de Haas CJ, Gros P, Rooijakkers SH. Molecular insights into the surface-specific arrangement of complement C5 convertase enzymes. BMC Biol. 2015;13:93. doi: 10.1186/s12915-015-0203-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jain U, Cao Q, Thomas NA, Woodruff TM, Schwaeble WJ, Stover CM, Stadnyk AW. Properdin provides protection from Citrobacter rodentium-induced intestinal inflammation in a C5a/IL-6-dependent manner. J Immunol. 2015;194:3414–3421. doi: 10.4049/jimmunol.1401814. [DOI] [PubMed] [Google Scholar]
- 51.Jain U, Midgen CA, Schwaeble WJ, Stover CM, Stadnyk AW. Properdin regulation of complement activation affects colitis in interleukin 10 gene-deficient mice. Inflamm Bowel Dis. 2015;21:1519–1528. doi: 10.1097/MIB.0000000000000398. [DOI] [PubMed] [Google Scholar]
- 52.Dimitrova P, Ivanovska N, Belenska L, Milanova V, Schwaeble W, Stover C. Abrogated RANKL expression in properdin-deficient mice is associated with better outcome from collagen-antibody-induced arthritis. Arthritis Res Ther. 2012;14:R173. doi: 10.1186/ar3926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Dimitrova P, Ivanovska N, Schwaeble W, Gyurkovska V, Stover C. The role of properdin in murine zymosan-induced arthritis. Mol Immunol. 2010;47:1458–1466. doi: 10.1016/j.molimm.2010.02.007. [DOI] [PubMed] [Google Scholar]
- 54.Ruseva MM, Vernon KA, Lesher AM, Schwaeble WJ, Ali YM, Botto M, Cook T, Song W, Stover CM, Pickering MC. Loss of properdin exacerbates C3 glomerulopathy resulting from factor H deficiency. J Am Soc Nephrol. 2013;24:43–52. doi: 10.1681/ASN.2012060571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Williams AL, Gullipalli D, Ueda Y, Sato S, Zhou L, Miwa T, Tung KS, Song WC. C5 inhibition prevents renal failure in a mouse model of lethal C3 glomerulopathy. Kidney Int. 2017;91:1386–1397. doi: 10.1016/j.kint.2016.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lesher AM, Nilsson B, Song WC. Properdin in complement activation and tissue injury. Mol Immunol. 2013;56:191–198. doi: 10.1016/j.molimm.2013.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Fischer E, Kazatchkine MD. Surface-dependent modulation by H of C5 cleavage by the cell-bound alternative pathway C5 convertase of human complement. J Immunol. 1983;130:2821–2824. [PubMed] [Google Scholar]
- 58.Medicus RG, Schreiber RD, Gotze O, Muller-Eberhard HJ. A molecular concept of the properdin pathway. Proc Natl Acad Sci US A. 1976;73:612–616. doi: 10.1073/pnas.73.2.612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Spitzer D, Mitchell LM, Atkinson JP, Hourcade DE. Properdin can initiate complement activation by binding specific target surfaces and providing a platform for de novo convertase assembly. J Immunol. 2007;179:2600–2608. doi: 10.4049/jimmunol.179.4.2600. [DOI] [PubMed] [Google Scholar]
- 60.Kemper C, Mitchell LM, Zhang L, Hourcade DE. The complement protein properdin binds apoptotic T cells and promotes complement activation and phagocytosis. Proc Natl Acad Sci US A. 2008;105:9023–9028. doi: 10.1073/pnas.0801015105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Gaarkeuken H, Siezenga MA, Zuidwijk K, van Kooten C, Rabelink TJ, Daha MR, Berger SP. Complement activation by tubular cells is mediated by properdin binding. Am J Physiol Renal Physiol. 2008;295:F1397–F1403. doi: 10.1152/ajprenal.90313.2008. [DOI] [PubMed] [Google Scholar]
- 62.Kimura Y, Miwa T, Zhou L, Song WC. Activator-specific requirement of properdin in the initiation and amplification of the alternative pathway complement. Blood. 2008;111:732–740. doi: 10.1182/blood-2007-05-089821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Agarwal S, Ferreira VP, Cortes C, Pangburn MK, Rice PA, Ram S. An evaluation of the role of properdin in alternative pathway activation on Neisseria meningitidis and Neisseria gonorrhoeae. J Immunol. 2010;185:507–516. doi: 10.4049/jimmunol.0903598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ferreira VP, Cortes C, Pangburn MK. Native polymeric forms of properdin selectively bind to targets and promote activation of the alternative pathway of complement. Immunobiology. 2010;215:932–940. doi: 10.1016/j.imbio.2010.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Cortes C, Ferreira VP, Pangburn MK. Native properdin binds to Chlamydia pneumoniae and promotes complement activation. Infect Immun. 2011;79:724–731. doi: 10.1128/IAI.00980-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Agarwal S, Specht CA, Haibin H, Ostroff GR, Ram S, Rice PA, Levitz SM (2011) Linkage specificity and role of properdin in activation of the alternative complement pathway by fungal glycans. mBio 2: pii: e00178-11 [DOI] [PMC free article] [PubMed]
- 67.Saggu G, Cortes C, Emch HN, Ramirez G, Worth RG, Ferreira VP. Identification of a novel mode of complement activation on stimulated platelets mediated by properdin and C3(H2O) J Immunol. 2013;190:6457–6467. doi: 10.4049/jimmunol.1300610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zaferani A, Vives RR, van der Pol P, Hakvoort JJ, Navis GJ, van Goor H, Daha MR, Lortat-Jacob H, Seelen MA, van den Born J. Identification of tubular heparan sulfate as a docking platform for the alternative complement component properdin in proteinuric renal disease. J Biol Chem. 2011;286:5359–5367. doi: 10.1074/jbc.M110.167825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.O'Flynn J, Dixon KO, Faber Krol MC, Daha MR, van Kooten C. Myeloperoxidase directs properdin-mediated complement activation. J Innate Immun. 2014;6:417–425. doi: 10.1159/000356980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Farries TC, Finch JT, Lachmann PJ, Harrison RA. Resolution and analysis of ‘native’ and ‘activated’ properdin. Biochem J. 1987;243:507–517. doi: 10.1042/bj2430507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Harboe M, Johnson C, Nymo S, Ekholt K, Schjalm C, Lindstad JK, Pharo A, Hellerud BC, Nilsson Ekdahl K, Mollnes TE, Nilsson PH. Properdin binding to complement activating surfaces depends on initial C3b deposition. Proc Natl Acadl Sci US A. 2017;114:E534–e539. doi: 10.1073/pnas.1612385114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Harboe M, Garred P, Lindstad JK, Pharo A, Muller F, Stahl GL, Lambris JD, Mollnes TE. The role of properdin in zymosan- and Escherichia coli-induced complement activation. J Immunol. 2012;189:2606–2613. doi: 10.4049/jimmunol.1200269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kemper C, Atkinson JP, Hourcade DE. Properdin: emerging roles of a pattern-recognition molecule. Ann Rev Immunol. 2010;28:131–155. doi: 10.1146/annurev-immunol-030409-101250. [DOI] [PubMed] [Google Scholar]
- 74.Blatt AZ, Pathan S, Ferreira VP. Properdin: a tightly regulated critical inflammatory modulator. Immunol Rev. 2016;274:172–190. doi: 10.1111/imr.12466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.O'Flynn J, van der Pol P, Dixon KO, Prohaszka Z, Daha MR, van Kooten C. Monomeric C-reactive protein inhibits renal cell-directed complement activation mediated by properdin. Am J Physiol Renal Physiol. 2016;310:F1308–F1316. doi: 10.1152/ajprenal.00645.2014. [DOI] [PubMed] [Google Scholar]
- 76.Hebecker M, Jozsi M. Factor H-related protein 4 activates complement by serving as a platform for the assembly of alternative pathway C3 convertase via its interaction with C3b protein. J Biol Chem. 2012;287:19528–19536. doi: 10.1074/jbc.M112.364471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Csincsi AI, Kopp A, Zoldi M, Banlaki Z, Uzonyi B, Hebecker M, Caesar JJ, Pickering MC, Daigo K, Hamakubo T, Lea SM, Goicoechea de Jorge E, Jozsi M. Factor H-related protein 5 interacts with pentraxin 3 and the extracellular matrix and modulates complement activation. J Immunol. 2015;194:4963–4973. doi: 10.4049/jimmunol.1403121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Chen Q, Manzke M, Hartmann A, Buttner M, Amann K, Pauly D, Wiesener M, Skerka C, Zipfel PF. Complement factor H-related 5-hybrid proteins anchor properdin and activate complement at self-surfaces. J Am Soc Nephrol. 2016;27:1413–1425. doi: 10.1681/ASN.2015020212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Cortes C, Ohtola JA, Saggu G, Ferreira VP. Local release of properdin in the cellular microenvironment: role in pattern recognition and amplification of the alternative pathway of complement. Front Immunol. 2012;3:412. doi: 10.3389/fimmu.2012.00412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Camous L, Roumenina L, Bigot S, Brachemi S, Fremeaux-Bacchi V, Lesavre P, Halbwachs-Mecarelli L. Complement alternative pathway acts as a positive feedback amplification of neutrophil activation. Blood. 2011;117:1340–1349. doi: 10.1182/blood-2010-05-283564. [DOI] [PubMed] [Google Scholar]
- 81.Sjoholm AG, Braconier JH, Soderstrom C. Properdin deficiency in a family with fulminant meningococcal infections. Clin Exp Immunol. 1982;50:291–297. [PMC free article] [PubMed] [Google Scholar]
- 82.Fijen CA, van den Bogaard R, Schipper M, Mannens M, Schlesinger M, Nordin FG, Dankert J, Daha MR, Sjoholm AG, Truedsson L, Kuijper EJ. Properdin deficiency: molecular basis and disease association. Mol Immunol. 1999;36:863–867. doi: 10.1016/s0161-5890(99)00107-8. [DOI] [PubMed] [Google Scholar]
- 83.Grumach AS, Kirschfink M. Are complement deficiencies really rare? Overview on prevalence, clinical importance and modern diagnostic approach. Mol Immunol. 2014;61:110–117. doi: 10.1016/j.molimm.2014.06.030. [DOI] [PubMed] [Google Scholar]
- 84.Fijen CA, van den Bogaard R, Daha MR, Dankert J, Mannens M, Kuijper EJ. Carrier detection by microsatellite haplotyping in 10 properdin type 1-deficient families. Eur J Clin Investig. 1996;26:902–906. doi: 10.1111/j.1365-2362.1996.tb02136.x. [DOI] [PubMed] [Google Scholar]
- 85.Fijen CA, Kuijper EJ, te Bulte MT, Daha MR, Dankert J. Assessment of complement deficiency in patients with meningococcal disease in The Netherlands. Clin Infect Dis. 1999;28:98–105. doi: 10.1086/515075. [DOI] [PubMed] [Google Scholar]
- 86.Figueroa JE, Densen P. Infectious diseases associated with complement deficiencies. Clini Microbiol Rev. 1991;4:359–395. doi: 10.1128/cmr.4.3.359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Schejbel L, Rosenfeldt V, Marquart H, Valerius NH, Garred P. Properdin deficiency associated with recurrent otitis media and pneumonia, and identification of male carrier with Klinefelter syndrome. Clin Immunol. 2009;131:456–462. doi: 10.1016/j.clim.2009.02.008. [DOI] [PubMed] [Google Scholar]
- 88.van den Bogaard R, Fijen CA, Schipper MG, de Galan L, Kuijper EJ, Mannens MM. Molecular characterisation of 10 Dutch properdin type I deficient families: mutation analysis and X-inactivation studies. Eur J Hum Genet. 2000;8:513–518. doi: 10.1038/sj.ejhg.5200496. [DOI] [PubMed] [Google Scholar]
- 89.Truedsson L, Westberg J, Fredrikson GN, Sjoholm AG, Kuijper EJ, Fijen CA, Spath PJ, Uhlen M. Human properdin deficiency has a heterogeneous genetic background. Immunopharmacology. 1997;38:203–206. doi: 10.1016/s0162-3109(97)00087-8. [DOI] [PubMed] [Google Scholar]
- 90.Helminen M, Seitsonen S, Jarva H, Meri S, Jarvela IE. A novel mutation W388X underlying properdin deficiency in a Finnish family. Scand J Immunol. 2012;75:445–448. doi: 10.1111/j.1365-3083.2012.02674.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Spath PJ, Sjoholm AG, Fredrikson GN, Misiano G, Scherz R, Schaad UB, Uhring-Lambert B, Hauptmann G, Westberg J, Uhlen M, Wadelius C, Truedsson L. Properdin deficiency in a large Swiss family: identification of a stop codon in the properdin gene, and association of meningococcal disease with lack of the IgG2 allotype marker G2m(n) Clin Exp Immunol. 1999;118:278–284. doi: 10.1046/j.1365-2249.1999.01056.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Fredrikson GN, Gullstrand B, Westberg J, Sjoholm AG, Uhlen M, Truedsson L. Expression of properdin in complete and incomplete deficiency: normal in vitro synthesis by monocytes in two cases with properdin deficiency type II due to distinct mutations. J Clin Immunol. 1998;18:272–282. doi: 10.1023/a:1027385806871. [DOI] [PubMed] [Google Scholar]
- 93.Westberg J, Fredrikson GN, Truedsson L, Sjoholm AG, Uhlen M. Sequence-based analysis of properdin deficiency: identification of point mutations in two phenotypic forms of an X-linked immunodeficiency. Genomics. 1995;29:1–8. doi: 10.1006/geno.1995.1208. [DOI] [PubMed] [Google Scholar]
- 94.Sjoholm AG, Kuijper EJ, Tijssen CC, Jansz A, Bol P, Spanjaard L, Zanen HC. Dysfunctional properdin in a Dutch family with meningococcal disease. N Engl J Med. 1988;319:33–37. doi: 10.1056/NEJM198807073190106. [DOI] [PubMed] [Google Scholar]
- 95.Fredrikson GN, Westberg J, Kuijper EJ, Tijssen CC, Sjoholm AG, Uhlen M, Truedsson L. Molecular characterization of properdin deficiency type III: dysfunction produced by a single point mutation in exon 9 of the structural gene causing a tyrosine to aspartic acid interchange. J Immunol. 1996;157:3666–3671. [PubMed] [Google Scholar]
- 96.Ahout IML, Brand KH, Zomer A, van den Hurk WH, Schilders G, Brouwer ML, Neeleman C, Groot R, Ferwerda G. Prospective observational study in two Dutch hospitals to assess the performance of inflammatory plasma markers to determine disease severity of viral respiratory tract infections in children. BMJ Open. 2017;7:e014596. doi: 10.1136/bmjopen-2016-014596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Marron-Linares GM, Nunez L, Crespo-Leiro MG, Barge-Caballero E, Pombo J, Paniagua-Martin MJ, Suarez-Fuentetaja N, Cid J, Grille-Cancela Z, Muniz-Garcia J, Tan CD, Rodriguez ER, Vazquez-Rodriguez JM, Hermida-Prieto M. Polymorphisms in genes related to the complement system and antibody-mediated cardiac allograft rejection. J Heart Lung Transplant. 2017;37:477–485. doi: 10.1016/j.healun.2017.07.006. [DOI] [PubMed] [Google Scholar]
- 98.Hertle E, Arts IC, van der Kallen CJ, Feskens EJ, Schalkwijk CG, Stehouwer CD, van Greevenbroek MM. The alternative complement pathway is longitudinally associated with adverse cardiovascular outcomes. The CODAM study. Thromb Haemost. 2016;115:446–457. doi: 10.1160/TH15-05-0439. [DOI] [PubMed] [Google Scholar]
- 99.Wang Y, Miwa T, Ducka-Kokalari B, Redai IG, Sato S, Gullipalli D, Zangrilli JG, Haczku A, Song WC. Properdin contributes to allergic airway inflammation through local C3a generation. J Immunol. 2015;195:1171–1181. doi: 10.4049/jimmunol.1401819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Pauly D, Nagel BM, Reinders J, Killian T, Wulf M, Ackermann S, Ehrenstein B, Zipfel PF, Skerka C, Weber BH. A novel antibody against human properdin inhibits the alternative complement system and specifically detects properdin from blood samples. PLoS One. 2014;9:e96371. doi: 10.1371/journal.pone.0096371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Miwa T, Sato S, Gullipalli D, Nangaku M, Song WC. Blocking properdin, the alternative pathway, and anaphylatoxin receptors ameliorates renal ischemia-reperfusion injury in decay-accelerating factor and CD59 double-knockout mice. J Immunol. 2013;190:3552–3559. doi: 10.4049/jimmunol.1202275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Miao J, Lesher AM, Miwa T, Sato S, Gullipalli D, Song WC. Tissue-specific deletion of Crry from mouse proximal tubular epithelial cells increases susceptibility to renal ischemia-reperfusion injury. Kidney Int. 2014;86:726–737. doi: 10.1038/ki.2014.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Kimura Y, Zhou L, Miwa T, Song WC. Genetic and therapeutic targeting of properdin in mice prevents complement-mediated tissue injury. J Clin Invest. 2010;120:3545–3554. doi: 10.1172/JCI41782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Zhou HF, Yan H, Stover CM, Fernandez TM, Rodriguez de Cordoba S, Song WC, Wu X, Thompson RW, Schwaeble WJ, Atkinson JP, Hourcade DE, Pham CT. Antibody directs properdin-dependent activation of the complement alternative pathway in a mouse model of abdominal aortic aneurysm. Proc Natl Acad Sci U S A. 2012;109:E415–E422. doi: 10.1073/pnas.1119000109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Ueda Y, Miwa T, Gullipalli D, Sato S, Ito D, Kim H, Palmer M, Song WC. Blocking properdin prevents complement-mediated hemolytic uremic syndrome and systemic thrombophilia. J Am Soc Nephrol. 2018;29:1928–1937. doi: 10.1681/ASN.2017121244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Jain U, Midgen CA, Woodruff TM, Schwaeble WJ, Stover CM, Stadnyk AW. Properdin deficiency protects from 5-fluorouracil-induced small intestinal mucositis in a complement activation-independent, interleukin-10-dependent mechanism. Clin Exp Immunol. 2017;188:36–44. doi: 10.1111/cei.12922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Ivanovska ND, Dimitrova PA, Luckett JC, El-Rachkidy Lonnen R, Schwaeble WJ, Stover CM. Properdin deficiency in murine models of nonseptic shock. J Immunol. 2008;180:6962–6969. doi: 10.4049/jimmunol.180.10.6962. [DOI] [PubMed] [Google Scholar]
- 108.Dupont A, Mohamed F, Salehen N, Glenn S, Francescut L, Adib R, Byrne S, Brewin H, Elliott I, Richards L, Dimitrova P, Schwaeble W, Ivanovska N, Kadioglu A, Machado LR, Andrew PW, Stover C. Septicaemia models using Streptococcus pneumoniae and Listeria monocytogenes: understanding the role of complement properdin. Med Microbiol Immunol. 2014;203:257–271. doi: 10.1007/s00430-013-0324-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Lesher AM, Zhou L, Kimura Y, Sato S, Gullipalli D, Herbert AP, Barlow PN, Eberhardt HU, Skerka C, Zipfel PF, Hamano T, Miwa T, Tung KS, Song WC. Combination of factor H mutation and properdin deficiency causes severe C3 glomerulonephritis. J Am Soc Nephrol. 2013;24:53–65. doi: 10.1681/ASN.2012060570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Zipfel PF, Heinen S, Jozsi M, Skerka C. Complement and diseases: defective alternative pathway control results in kidney and eye diseases. Mol Immunol. 2006;43:97–106. doi: 10.1016/j.molimm.2005.06.015. [DOI] [PubMed] [Google Scholar]
- 111.Holers VM. The spectrum of complement alternative pathway-mediated diseases. Immunol Rev. 2008;223:300–316. doi: 10.1111/j.1600-065X.2008.00641.x. [DOI] [PubMed] [Google Scholar]
- 112.Noris M, Remuzzi G. Glomerular diseases dependent on complement activation, including atypical hemolytic uremic syndrome, membranoproliferative glomerulonephritis, and C3 glomerulopathy: core curriculum 2015. Am J Kidney Dis. 2015;66:359–375. doi: 10.1053/j.ajkd.2015.03.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.de Cordoba SR, de Jorge EG. Translational mini-review series on complement factor H: genetics and disease associations of human complement factor H. Clin Exp Immunol. 2008;151:1–13. doi: 10.1111/j.1365-2249.2007.03552.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Zipfel PF, Skerka C, Chen Q, Wiech T, Goodship T, Johnson S, Fremeaux-Bacchi V, Nester C, de Cordoba SR, Noris M, Pickering M, Smith R. The role of complement in C3 glomerulopathy. Mol Immunol. 2015;67:21–30. doi: 10.1016/j.molimm.2015.03.012. [DOI] [PubMed] [Google Scholar]
- 115.Servais A, Noel LH, Roumenina LT, Le Quintrec M, Ngo S, Dragon-Durey MA, Macher MA, Zuber J, Karras A, Provot F, Moulin B, Grunfeld JP, Niaudet P, Lesavre P, Fremeaux-Bacchi V. Acquired and genetic complement abnormalities play a critical role in dense deposit disease and other C3 glomerulopathies. Kidney Int. 2012;82:454–464. doi: 10.1038/ki.2012.63. [DOI] [PubMed] [Google Scholar]
- 116.Tang S, Lai KN, Sacks SH. Role of complement in tubulointerstitial injury from proteinuria. Kidney Blood Press Res. 2002;25:120–126. doi: 10.1159/000063520. [DOI] [PubMed] [Google Scholar]
- 117.Hsu SI, Couser WG. Chronic progression of tubulointerstitial damage in proteinuric renal disease is mediated by complement activation: a therapeutic role for complement inhibitors? J Am Soc Nephrol. 2003;14:S186–S191. doi: 10.1097/01.asn.0000070032.58017.20. [DOI] [PubMed] [Google Scholar]
- 118.Riedl M, Thorner P, Licht C. C3 glomerulopathy. Pediatr Nephrol. 2017;32:43–57. doi: 10.1007/s00467-015-3310-4. [DOI] [PubMed] [Google Scholar]
- 119.Lu DF, Moon M, Lanning LD, McCarthy AM, Smith RJ. Clinical features and outcomes of 98 children and adults with dense deposit disease. Pediatr Nephrol. 2012;27:773–781. doi: 10.1007/s00467-011-2059-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Bomback AS, Santoriello D, Avasare RS, Regunathan-Shenk R, Canetta PA, Ahn W, Radhakrishnan J, Marasa M, Rosenstiel PE, Herlitz LC, Markowitz GS, D'Agati VD, Appel GB. C3 glomerulonephritis and dense deposit disease share a similar disease course in a large United States cohort of patients with C3 glomerulopathy. Kidney Int. 2018;93:977–985. doi: 10.1016/j.kint.2017.10.022. [DOI] [PubMed] [Google Scholar]
- 121.Rabasco C, Cavero T, Roman E, Rojas-Rivera J, Olea T, Espinosa M, Cabello V, Fernandez-Juarez G, Gonzalez F, Avila A, Baltar JM, Diaz M, Alegre R, Elias S, Anton M, Frutos MA, Pobes A, Blasco M, Martin F, Bernis C, Macias M, Barroso S, de Lorenzo A, Ariceta G, Lopez-Mendoza M, Rivas B, Lopez-Revuelta K, Campistol JM, Mendizabal S, de Cordoba SR, Praga M. Effectiveness of mycophenolate mofetil in C3 glomerulonephritis. Kidney Int. 2015;88:1153–1160. doi: 10.1038/ki.2015.227. [DOI] [PubMed] [Google Scholar]
- 122.Medjeral-Thomas NR, O'Shaughnessy MM, O'Regan JA, Traynor C, Flanagan M, Wong L, Teoh CW, Awan A, Waldron M, Cairns T, O'Kelly P, Dorman AM, Pickering MC, Conlon PJ, Cook HT. C3 glomerulopathy: clinicopathologic features and predictors of outcome. Clini J Am Soc Nephrol. 2014;9:46–53. doi: 10.2215/CJN.04700513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Iatropoulos P, Noris M, Mele C, Piras R, Valoti E, Bresin E, Curreri M, Mondo E, Zito A, Gamba S, Bettoni S, Murer L, Fremeaux-Bacchi V, Vivarelli M, Emma F, Daina E, Remuzzi G. Complement gene variants determine the risk of immunoglobulin-associated MPGN and C3 glomerulopathy and predict long-term renal outcome. Mol Immunol. 2016;71:131–142. doi: 10.1016/j.molimm.2016.01.010. [DOI] [PubMed] [Google Scholar]
- 124.Pickering MC, D'Agati VD, Nester CM, Smith RJ, Haas M, Appel GB, Alpers CE, Bajema IM, Bedrosian C, Braun M, Doyle M, Fakhouri F, Fervenza FC, Fogo AB, Fremeaux-Bacchi V, Gale DP, Goicoechea de Jorge E, Griffin G, Harris CL, Holers VM, Johnson S, Lavin PJ, Medjeral-Thomas N, Paul Morgan B, Nast CC, Noel LH, Peters DK, Rodriguez de Cordoba S, Servais A, Sethi S, Song WC, Tamburini P, Thurman JM, Zavros M, Cook HT. C3 glomerulopathy: consensus report. Kidney Int. 2013;84:1079–1089. doi: 10.1038/ki.2013.377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Daha MR. Unexpected role for properdin in complement C3 glomerulopathies. J Am Soc Nephrol. 2013;24:5–7. doi: 10.1681/ASN.2012111110. [DOI] [PubMed] [Google Scholar]
- 126.Varade WS, Forristal J, West CD. Patterns of complement activation in idiopathic membranoproliferative glomerulonephritis, types I, II, and III. Am J Kidney Dis. 1990;16:196–206. doi: 10.1016/s0272-6386(12)81018-3. [DOI] [PubMed] [Google Scholar]
- 127.Tanuma Y, Ohi H, Hatano M. Two types of C3 nephritic factor: properdin-dependent C3NeF and properdin-independent C3NeF. Clin Immunol Immunopathol. 1990;56:226–238. doi: 10.1016/0090-1229(90)90144-f. [DOI] [PubMed] [Google Scholar]
- 128.Clardy CW, Forristal J, Strife CF, West CD. A properdin dependent nephritic factor slowly activating C3, C5, and C9 in membranoproliferative glomerulonephritis, types I and III. Clin Immunol Immunopathol. 1989;50:333–347. doi: 10.1016/0090-1229(89)90141-4. [DOI] [PubMed] [Google Scholar]
- 129.Mollnes TE, Ng YC, Peters DK, Lea T, Tschopp J, Harboe M. Effect of nephritic factor on C3 and on the terminal pathway of complement in vivo and in vitro. Clin Exp Immunol. 1986;65:73–79. [PMC free article] [PubMed] [Google Scholar]
- 130.Paixao-Cavalcante D, Lopez-Trascasa M, Skattum L, Giclas PC, Goodship TH, de Cordoba SR, Truedsson L, Morgan BP, Harris CL. Sensitive and specific assays for C3 nephritic factors clarify mechanisms underlying complement dysregulation. Kidney Int. 2012;82:1084–1092. doi: 10.1038/ki.2012.250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Iatropoulos P, Daina E, Curreri M, Piras R, Valoti E, Mele C, Bresin E, Gamba S, Alberti M, Breno M, Perna A, Bettoni S, Sabadini E, Murer L, Vivarelli M, Noris M, Remuzzi G. Cluster analysis identifies distinct pathogenetic patterns in C3 glomerulopathies/immune complex-mediated membranoproliferative GN. J Am Soc Nephrol. 2018;29:283–294. doi: 10.1681/ASN.2017030258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.George JN, Nester CM. Syndromes of thrombotic microangiopathy. N Engl J Med. 2014;371:654–666. doi: 10.1056/NEJMra1312353. [DOI] [PubMed] [Google Scholar]
- 133.Noris M, Remuzzi G. Atypical hemolytic-uremic syndrome. N Engl J Med. 2009;361:1676–1687. doi: 10.1056/NEJMra0902814. [DOI] [PubMed] [Google Scholar]
- 134.Fremeaux-Bacchi V, Fakhouri F, Garnier A, Bienaime F, Dragon-Durey MA, Ngo S, Moulin B, Servais A, Provot F, Rostaing L, Burtey S, Niaudet P, Deschenes G, Lebranchu Y, Zuber J, Loirat C. Genetics and outcome of atypical hemolytic uremic syndrome: a nationwide French series comparing children and adults. Clin J Am Soc Nephrol. 2013;8:554–562. doi: 10.2215/CJN.04760512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Maga TK, Nishimura CJ, Weaver AE, Frees KL, Smith RJ. Mutations in alternative pathway complement proteins in American patients with atypical hemolytic uremic syndrome. Hum Mutat. 2010;31:E1445–E1460. doi: 10.1002/humu.21256. [DOI] [PubMed] [Google Scholar]
- 136.Noris M, Caprioli J, Bresin E, Mossali C, Pianetti G, Gamba S, Daina E, Fenili C, Castelletti F, Sorosina A, Piras R, Donadelli R, Maranta R, van der Meer I, Conway EM, Zipfel PF, Goodship TH, Remuzzi G. Relative role of genetic complement abnormalities in sporadic and familial aHUS and their impact on clinical phenotype. Clin J Am Soc Nephrol. 2010;5:1844–1859. doi: 10.2215/CJN.02210310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Dragon-Durey MA, Loirat C, Cloarec S, Macher MA, Blouin J, Nivet H, Weiss L, Fridman WH, Fremeaux-Bacchi V. Anti-factor H autoantibodies associated with atypical hemolytic uremic syndrome. J Am Soc Nephrol. 2005;16:555–563. doi: 10.1681/ASN.2004050380. [DOI] [PubMed] [Google Scholar]
- 138.Hofer J, Janecke AR, Zimmerhackl LB, Riedl M, Rosales A, Giner T, Cortina G, Haindl CJ, Petzelberger B, Pawlik M, Jeller V, Vester U, Gadner B, van Husen M, Moritz ML, Wurzner R, Jungraithmayr T. Complement factor H-related protein 1 deficiency and factor H antibodies in pediatric patients with atypical hemolytic uremic syndrome. Clin J Am Soc Nephrol. 2013;8:407–415. doi: 10.2215/CJN.01260212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Remuzzi G, Bertani T. Pathophysiology of progressive nephropathies. N Engl J Med. 1998;339:1448–1456. doi: 10.1056/NEJM199811123392007. [DOI] [PubMed] [Google Scholar]
- 140.Abbate M, Zoja C, Remuzzi G. How does proteinuria cause progressive renal damage? J Am Soc Nephrol. 2006;17:2974–2984. doi: 10.1681/ASN.2006040377. [DOI] [PubMed] [Google Scholar]
- 141.Nagamachi S, Ohsawa I, Suzuki H, Sato N, Inoshita H, Hisada A, Honda D, Shimamoto M, Shimizu Y, Horikoshi S, Tomino Y. Properdin has an ascendancy over factor H regulation in complement-mediated renal tubular damage. BMC Nephrol. 2014;15:82. doi: 10.1186/1471-2369-15-82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Ichida S, Yuzawa Y, Okada H, Yoshioka K, Matsuo S. Localization of the complement regulatory proteins in the normal human kidney. Kidney Int. 1994;46:89–96. doi: 10.1038/ki.1994.247. [DOI] [PubMed] [Google Scholar]
- 143.Siezenga MA, van der Geest RN, Mallat MJ, Rabelink TJ, Daha MR, Berger SP. Urinary properdin excretion is associated with intrarenal complement activation and poor renal function. Nephrol Dial Transplant. 2010;25:1157–1161. doi: 10.1093/ndt/gfp630. [DOI] [PubMed] [Google Scholar]
- 144.Zaferani A, Vives RR, van der Pol P, Navis GJ, Daha MR, van Kooten C, Lortat-Jacob H, Seelen MA, van den Born J. Factor H and properdin recognize different epitopes on renal tubular epithelial heparan sulfate. J Biol Chem. 2012;287:31471–31481. doi: 10.1074/jbc.M112.380386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Gupta-Bansal R, Parent JB, Brunden KR. Inhibition of complement alternative pathway function with anti-properdin monoclonal antibodies. Mol Immunol. 2000;37:191–201. doi: 10.1016/s0161-5890(00)00047-x. [DOI] [PubMed] [Google Scholar]
- 146.Zuber J, Fakhouri F, Roumenina LT, Loirat C, Fremeaux-Bacchi V. Use of eculizumab for atypical haemolytic uraemic syndrome and C3 glomerulopathies. Nat Rev Nephrol. 2012;8:643–657. doi: 10.1038/nrneph.2012.214. [DOI] [PubMed] [Google Scholar]
- 147.Legendre CM, Licht C, Muus P, Greenbaum LA, Babu S, Bedrosian C, Bingham C, Cohen DJ, Delmas Y, Douglas K, Eitner F, Feldkamp T, Fouque D, Furman RR, Gaber O, Herthelius M, Hourmant M, Karpman D, Lebranchu Y, Mariat C, Menne J, Moulin B, Nurnberger J, Ogawa M, Remuzzi G, Richard T, Sberro-Soussan R, Severino B, Sheerin NS, Trivelli A, Zimmerhackl LB, Goodship T, Loirat C. Terminal complement inhibitor eculizumab in atypical hemolytic-uremic syndrome. N Engl J Med. 2013;368:2169–2181. doi: 10.1056/NEJMoa1208981. [DOI] [PubMed] [Google Scholar]
- 148.Le Quintrec M, Lapeyraque AL, Lionet A, Sellier-Leclerc AL, Delmas Y, Baudouin V, Daugas E, Decramer S, Tricot L, Cailliez M, Dubot P, Servais A, Mourey-Epron C, Pourcine F, Loirat C, Fremeaux-Bacchi V, Fakhouri F. Patterns of clinical response to eculizumab in patients with C3 glomerulopathy. Am J Kidney Dis. 2018;72:84–92. doi: 10.1053/j.ajkd.2017.11.019. [DOI] [PubMed] [Google Scholar]
- 149.Vivarelli M, Emma F. Treatment of C3 glomerulopathy with complement blockers. Semin Thromb Hemost. 2014;40:472–477. doi: 10.1055/s-0034-1375299. [DOI] [PubMed] [Google Scholar]
- 150.Dmytrijuk A, Robie-Suh K, Cohen MH, Rieves D, Weiss K, Pazdur R. FDA report: eculizumab (Soliris) for the treatment of patients with paroxysmal nocturnal hemoglobinuria. Oncologist. 2008;13:993–1000. doi: 10.1634/theoncologist.2008-0086. [DOI] [PubMed] [Google Scholar]
- 151.Heinen S, Pluthero FG, van Eimeren VF, Quaggin SE, Licht C. Monitoring and modeling treatment of atypical hemolytic uremic syndrome. Mol Immunol. 2013;54:84–88. doi: 10.1016/j.molimm.2012.10.044. [DOI] [PubMed] [Google Scholar]