

Abstract
Cartilaginous endplate degeneration serves a key role in the process of intervertebral disc (IVD) degeneration, however, effective therapies are hindered by an incomplete understanding of the mechanisms that underlie cartilage endplate (CEP) homeostasis and degeneration. Wnt/β-catenin signalling has been reported as a major factor in regulating biological processes. Whether Wnt/β-catenin signalling engages in CEP homeostasis has not yet been investigated. The present study aimed to assess the function of CEP cells via the activation of Wnt/β-catenin signalling to examine and promote the mechanism of degeneration of CEP in vitro. Rat CEP cells were confirmed to exhibit a chondrocytic phenotype by toluidine blue staining. The increased number of senescence-associated β-galactosidase (SA-β-gal)-positive cells and reduced cellular proliferation were investigated in the presence of a β-catenin inhibitor, and the inhibitor improved the trend of senescence. An increased number of apoptotic cells was detected by lithium chloride treatment, and inhibiting Wnt/β-catenin signalling protected the cells from apoptosis. Expression of the catabolic enzymes, metalloproteinase-13 and a disintegrin and metalloproteinase with thrombospondin motifs-5, and the decreased expression of aggrecan were also observed by Wnt/β-catenin signalling activation, and a Wnt/β-catenin signalling inhibitor decreased the expression of catabolic enzymes and increased the expression of aggrecan induced by Wnt/β-catenin signalling activation. Wnt/β-catenin signalling may provide potential strategies for preventing CEP degeneration.
Keywords: Wnt/β-catenin signalling, intervertebral disc degeneration, apoptosis, senescence, cartilage endplate
Introduction
Degeneration of the intervertebral disc (IVD) is the main factor contributing to the pathogenesis of spinal disorders and constitutes an important socioeconomic burden (1,2). The cartilage endplate (CEP) is an important site for solute exchange and the import of nutrients for the IVD (3,4). Degeneration of the CEP markedly decreases the biomechanical integrity and nutrition of the disc, resulting in breakdown of the metabolic equilibrium of the extracellular matrix (ECM), which ultimately accelerates disc degeneration (5–8). The prevention of CEP degeneration is a potential therapeutic strategy for maintaining IVD health and preventing spondylopathy. However, the precise mechanisms of development, maintenance and degeneration of CEP remain to be fully clarified.
Wnt/β-catenin signalling serves a central role in regulating biological processes, including initial embryonic development, tissue organization, cell proliferation and differentiation, in addition to pathological processes (9–11). The Wnt/β-catenin signalling pathway is activated upon binding of the Wnt protein to a receptor complex, including Frizzled receptors and low-density lipoprotein receptor-related protein 5/6 on the cell surface (12,13). In the absence of Wnt ligands, glycogen synthase kinase-3β (GSK-3β)-mediated phosphorylation and proteasome-mediated degradation occur (14). However, when the Wnt ligand is present, the phosphorylation activity of GSK-3β is inhibited (15), following whichβ-catenin accumulates and translocates to the nucleus, where it combines with T-cell factor/lymphoid enhancer factor and transactivates target gene promoters (9,16). Therefore, the quantitative changes in β-catenin are an important factor in Wnt/β-catenin signalling.
Numerous studies have reported that the protein levels of β-catenin are significantly higher in IVD degenerated specimens than in normal IVD specimens (17,18), which indicates that Wnt/β-catenin signalling is involved in the process of IVD degeneration. Hiyama et al (17) reported that the Wnt/β-catenin signalling pathway enhanced IVD cellular senescence and decreased ECM synthesis. A recent study reported that activation of Wnt/β-catenin signalling promoted the apoptosis of nucleus pulposus (NP) cells induced by inflammatory cytokines, including interleukin-1β and tumour necrosis-a (19), which are abnormally secreted by NP cells and are widely recognized contributors to IVD senescence. Iwata et al (20) demonstrated that Wnt/β-catenin signalling enhanced the expression of Runt-related transcription factor 2 (Runx2) in the IVD and led to IVD calcification. However, the role of β-catenin signalling in CEP has not been fully investigated.
The present study aimed to assess the function of CEP cells with activation of Wnt/β-catenin signalling and examine the promoted degeneration mechanism of CEP.
Materials and methods
Cell isolation and culture
All animal experiments were approved by the Ethics Committee on Animal Experiments of Fudan University (Shanghai, China). Two male Sprague-Dawley rats (age, ~12 weeks; weight, 400 g) were used in the present study and were supplied by the Shanghai Public Health Centre. The Animals were housed with free access to food and water under a 12-h light/dark cycle with constant temperature (20–23°C) and humidity (55±5%). The animals were euthanized by cervical dislocation following anaesthesia with sodium pentobarbital (50 mg/kg; New Asia Pharmaceutical Co., Ltd.). The lumbar spines of the animals were obtained within 1 h of death, and the discs were carefully dissected under a microscope to obtain only the CEP, which was minced into small pieces (<0.3 mm3) under aseptic conditions. To isolate the chondrocytes, the tissues were sequentially treated with 0.25% trypsin (Sigma-Aldrich; Merck KGaA) at 37°C for 120 min, followed by treatment with 0.02% collagenase (Sigma-Aldrich; Merck KGaA) at 37°C for 24 h. Upon enzymatic digestion, the tissues were filtered through a 70-µm mesh cell strainer (Beyotime Institute of Biotechnology) and then washed with PBS. Subsequently, the cells were released from the matrix by centrifugation at 200 × g for 5 min at room temperature following digestion and filtration. Following this, the cells were seeded into 6-well plates at 2×104 cells/well and maintained in Dulbecco's modified Eagle's medium (DMEM; Gibco; Thermo Fisher Scientific, Inc.) supplemented with 10% foetal bovine serum (FBS; Gibco; Thermo Fisher Scientific, Inc.), 100 U/ml penicillin and 100 µg/ml streptomycin (Beyotime Institute of Biotechnology) under 5% CO2 in a humidified incubator at 37°C. The primary chondrocytes were maintained in a high-density monolayer culture for 1 week at 2×105 cells/well. Toluidine blue staining (Shanghai Haoran Biotechnology Co., Ltd.) was used to confirm the chondrocytic phenotype of these cells. The cells were incubated in 6-cm plates (5×105 cells/plate) for 1 h at 37°C with the staining solution, Images of the cells were then captured under reflected light using a digital high-fidelity microscope (VH-8000; Keyence Corporation). The cells were then trypsinised and subcultured in 6-well plates, which were used in the following experiments as secondary cells.
Cell treatments
LiCl, which is a known inducer of β-catenin and an activator of Wnt signalling, was used to activate Wnt/β-catenin signalling (17,21). The CEP cells were cultured in 6-cm plates (5×105 cells/plate) at 37°C. Cells treated with 20 mM LiCl for 24 h were termed CEP-CTR (control) at 37°C. The cells treated with LiCl and the Wnt/β-catenin inhibitor ICG001 (25 µM; cat. no. SF6827; Beyotime Institute of Biotechnology) for 24 h were termed CEP-INB (inhibitor) at 37°C. An additional group treated with normal conditions was considered the normal group, and was termed NP-NOM (normal condition).
SA-β-gal staining
SA-β-gal staining was performed at 0, 24, 48 and 72 h using an SA-β-gal staining kit (Cell Signalling Technology, Inc.), according to the recommendations of the manufacturer. Briefly, the cells were washed twice with PBS and fixed with 3% formaldehyde for 15 min at 37°C. The cells were incubated overnight at 37°C with the staining solution. Images of the cells were then captured under reflected light using a digital high-fidelity microscope (VH-8000; Keyence Corporation). A total of five fields, which were distributed throughout the well, were counted (magnification, ×200), and the mean positive rate was also counted (22).
Determination of cell proliferation
The proliferation of CEP cells was assessed at 0, 24, 48 and 72 h with a Cell Counting Kit-8 (CCK-8) assay (Beyotime Institute of Biotechnology). In brief, the cells were seeded at a density of 1×104 cells/well in a 96-well plate. The cells were incubated with 10 µl CCK-8 solution at 37°C for 2 h, and then analysed with an Epoch Multi-Volume Spectrophotometer system (BioTek Instruments, Inc.) at an absorbance of 490 nm. The proliferative activities were calculated as the change in absorbance at 490 nm.
Detection of apoptosis
The CEP cells were cultured in 6-cm plates (5×105 cells/plate) with or without 20 mM LiCl for 24 h, and apoptosis was determined by staining with Annexin V-fluorescein isothiocyanate (FITC) and propidium iodide (PI) according to the manufacturer's instructions (BD Pharmingen; BD Biosciences). The cells were stained with FITC-labelled Annexin V and PI simultaneously, and then quantified as follows: i) Annexin V-negative/PI-negative (viable cells); ii) Annexin V-positive/PI-negative (cells in the initial stages of apoptosis); iii) Annexin V-positive/PI-positive (cells in the advanced stages of apoptosis); and iv) Annexin V-negative/PI-positive (necrotic cells). To quantify apoptosis, the cells were washed with cold PBS and suspended in binding buffer. The cells were stained with 5 µl Annexin V-FITC and 5 µl PI at 4°C for 15 min, and then analysed using a FACScan flow cytometer (BDBiosciences).
Reverse transcription-quantitative polymerase chain reaction (RT-qPCR) analysis
RT-qPCR analysis was performed to detect the messenger RNA (mRNA) expression of matrix metalloproteinase (MMP)-13, a disintegrin and metalloproteinase with thrombospondin motifs (ADAMTS)-5 and aggrecan in the different treatment groups at 24 h, and GAPDH was used as the internal standard control. Briefly, total RNA was extracted using TRIzol reagent (Invitrogen; Thermo Fisher Scientific, Inc.) following the manufacturer's instructions. Single-strand complementary DNA (cDNA) templates were prepared from 1 µg total RNA using the Advantage RT-for-PCR kit (Clontech Laboratories, Inc.). Specific cDNAs were then amplified by PCR using the following primers: Aggrecan forward (Fw), 5′-CAGAAAAACAGACTGAATGGGA-3′ and reverse (Rev), 5′-GCAAGTGAAGGTGTGTATGGC-3′; MMP-13 Fw, 5′-TACGAGCATCCATCCCGAGACC-3′ and Rev, 5′-AACCGCAGCACTGAGCCTTTTC-3′; ADAMTS-5 Fw, 5′-GGCTGTGGTGTGCTGTG-3′ and Rev, 5′-CTGGTCTTTGGCTTTGAAC-3′; and GAPDH Fw, 5′-CCCCAATGTATCCGTTGTG-3′ and Rev, 5′-CTCAGTGTAGCCCAGGATGC-3′. PCR amplification from cDNA was performed using the Takara Thermal Cycler Dice Real Time System TP800 (Takara Bio, Inc.) with a final volume of 20 µl [10 µl 2X SYBR Green Mix (Invitrogen; Thermo Fisher Scientific, Inc.), 1 µl primer mix, 1 µl template DNA and 8 µl diethylpyrocarbonate-treated water]. The thermocycling conditions were as follows: Initial denaturation at 95°C for 2 min, followed by 40 cycles of denaturation at 95°C for 15 sec, annealing at 59°C for 20 sec and elongation at 72°C for 20 sec, and a final extension at 72°C for 10 min. The PCR products were subjected to amplification curve analysis and were quantified using SYBR-Green (Invitrogen; Thermo Fisher Scientific, Inc.). The data were normalized to GAPDH and quantified using the 2−ΔΔCq method (23).
Statistical analysis
All measurements were recorded using the same instrument under the same experimental conditions and were independently performed at least three times to ensure consistency. Statistical analyses were performed using SPSS 16.0 software (SPSS, Inc.). Data are expressed as the mean ± standard deviation. Significant differences were analysed with one-way analysis of variance, and the Bonferroni test was used for multiple comparisons. P<0.05 was considered to indicate a statistically significant difference.
Results
Cells cultured from the CEP maintain a chondrocytic phenotype and SA-β-gal staining
Toluidine blue staining was used to confirm the phenotype of cells cultured from the CEP. The results revealed that the cells were viable and possessed characteristic features of chondrocyte proteoglycans with light blue staining in the cytoplasm, as evidenced by typical cell morphology and ECM staining (Fig. 1A). Photomicrographs showing Staining of CEP cells for SA-β-gal indicated that the chondrocytes were large and flat with notable SA-β-gal activity (Fig. 1B).
Figure 1.
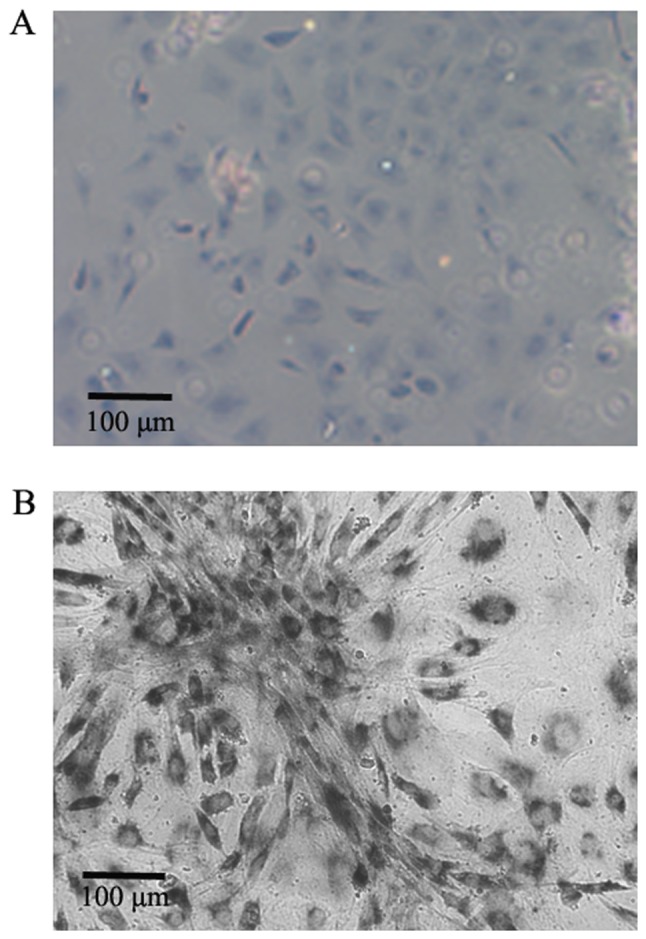
Cell phenotype. (A) A typical chondrocytic phenotype was confirmed by toluidine blue staining (magnification, ×200). Scale bar, 100 µm. (B) Representative image of senescence-associated β-galactosidase-positive cartilage endplate cells (magnification, ×200). Scale bar, 100 µm.
LiCl treatment induces CEP cellular premature senescence
To determine whether β-catenin mediates cellular senescence, the percentage of SA-β-gal-positive cells and the cellular proliferative ability were quantitatively assessed following LiCl treatment with or without a β-catenin inhibitor. The proliferative ability of CEP cells gradually decreased with increasing time, and at 72 h, the proliferative activity markedly decreased to almost half of that of the control (0.92±0.21 vs. 1.72±0.15, P<0.05). Notably, upon inhibiting the activation of Wnt/β-catenin signalling in the CEP-INB group, the protective effect was investigated, and the proliferative ability of the cells was improved compared with that of the CEP-CTR group (1.25±0.26 vs. 0.92±0.21, P<0.05; Fig. 2A). Furthermore, the premature senescence of CEP cells was investigated at 72 h. As shown in Fig. 2B, the chondrocytes become large and flat and enhanced SA-β-gal activity, the percentage of SA-β-gal-positive cells was markedly higher in the CEP-CTR group compared with that in the NP-NOM group at 72 h (28.82±2.45 vs. 7.60±1.28%, P<0.05). When the activation of Wnt/β-catenin signalling was inhibited in the CEP-INB group, the increase in the number of SA-β-gal-positive cells at 72 h was prevented; the mean positive rate was only at 19.66±2.85% in the CEP-INB group.
Figure 2.

Wnt/β-catenin signalling-mediated CEP cellular premature senescence. (A) A β-catenin inhibitor was associated with the recovery of cell proliferative ability. (B) LiCl treatment induced an increase in senescent cells, as determined by SA-β-gal staining, whereas the β-catenin inhibitor decreased the number of SA-β-gal-positive cells. *P<0.05. CEP-NOM, cells cultured under normal conditions; CEP-CTR, cells treated with LiCl; CEP-INB, cells treated with LiCl and a β-catenin inhibitor; CEP, cartilage endplate; LiCl, lithium chloride; SA-β-gal, senescence-associated β-galactosidase.
LiCl treatment is involved in cellular apoptosis
To determine whether the activation of Wnt/β-catenin signalling had any effect on the apoptosis of CEP cells, the apoptotic rate of cells stained with Annexin V-FITC and PI was qualitatively and quantitatively assessed following LiCl treatment with or without a β-catenin inhibitor. As shown in Fig. 3A and B, flow cytometric analysis revealed that the apoptotic rate of CEP cells was significantly higher following LiCl treatment compared with that of cells in the CEP-NOM group (21.80±1.35 vs. 9.42±0.42%, P<0.05). Upon incubation with the Wnt/β-catenin inhibitor, the apoptotic rate was significantly reduced compared with that in the CEP-CTR group (16.59±1.03 vs. 21.80±1.35%, P<0.05).
Figure 3.

Evaluation of the apoptosis of CEP cells following LiCl treatment. (A) Representative graphs of flow cytometric analysis. (B) Apoptotic rate of CEP cells was significantly higher following LiCl treatment, whereas inhibiting Wnt/β-catenin signalling protected against cellular apoptosis. The results are presented as the percentage of cell apoptosis. *P<0.05 CEP-CTR vs. CEP-NOM and CEP-INB. CEP-NOM, cells cultured under normal conditions; CEP-CTR, cells treated with LiCl; CEP-INB, cells treated with LiCl and a β-catenin inhibitor; CEP, cartilage endplate; LiCl, lithium chloride; FITC, fluorescein isothiocyanate.
Wnt/β-catenin signalling modulates ECM equilibrium
To determine the effects of β-catenin on the equilibrium of the critical ECM, the expression of aggrecan and catabolic enzymes (MMP-13 and ADAMTS-5) was examined by RT-qPCR analysis. As shown in Fig. 4A, LiCl treatment markedly decreased the expression of aggrecan, and this inhibitory effect was decreased by the addition of the β-catenin inhibitor. Furthermore, the levels of MMP-13 and ADAMTS-5 were increased by LiCl treatment, upon being reduced by the β-catenin inhibitor (Fig. 4B and C).
Figure 4.

Wnt/β-catenin signalling modulates the equilibrium of the extracellular matrix. Reverse transcription-quantitative polymerase chain reaction analysis of (A) aggrecan, (B) MMP-13 and (C) ADAMTS-5 mRNA expression in three groups of cells. *P<0.05 CEP-CTR vs. CEP-NOM and CEP-INB. CEP-NOM, cells cultured under normal conditions; CEP-CTR, cells treated with LiCl; CEP-INB, cells treated with LiCl and a β-catenin inhibitor; CEP, cartilage endplate; LiCl, lithium chloride; MMP-13, metalloproteinase-13; ADAMTS-5, a disintegrin and metalloproteinase with thrombospondin motifs-5; mRNA, messenger RNA.
Discussion
The ECP is a thin layer of hyaline cartilage between the vertebral body and the IVD, and contributes to an even distribution of the compressive load across the vertebral body (24). In addition, it is the gateway for nutrient transport between the vertebral marrow and the IVD (8,5). Multiple previous studies have demonstrated that cartilaginous endplate degeneration serves a key role in the process of IVD degeneration (8). The treatment and prevention of degenerative disc disease are hindered by a limited understanding of the mechanisms involved.
Various intracellular signalling pathways are involved in progression of IVD degeneration (25), and Wnt/β-catenin signalling has been reported to be one of the most important pathways (18,26), as it serves a key role in IVD development and degeneration. Numerous studies have reported that Wnt/β-catenin signalling can serve a regulatory function in cell proliferation and differentiation, and in the modulation of degeneration and regeneration processes (27,28). A previous study indicated that the number of β-catenin-positive cells increases with the progression of IVD and enhances the expression of Runx2, thus leading to IVD calcification (20). The present study aimed to examine the mechanism underlying Wnt/β-catenin signalling in CEP cells in vitro.
Several studies have reported that cellular senescence is the major cause of IVD degeneration and is positively correlated with the grade of degeneration (29). Higher levels of cell senescence have also been observed in degenerated discs compared with age-matched non-degenerated discs (30,31). Senescence decreases the cellular viability capacity for self-repair (32–34) and increases the levels of inflammatory cytokines and the secretion of catabolic enzymes, which accelerate IVD degeneration. Upon activation of Wnt/β-catenin signalling by LiCl treatment, decreased cell viability and an increased number of SA-β-gal-positive cells were observed in the present study. Additionally, it was demonstrated that a β-catenin inhibitor improved the trend of senescence, which was in accordance with the findings of Hiyama et al (17). These results suggest that Wnt/β-catenin signalling is involved in the age- and stress-induced cellular senescence of CEP cells.
Apoptotic cells have been identified in degenerated endplates, and their quantity has a positive correlation with the grade of disease degeneration (6). Shirazi-Adl et al (6) reported that apoptosis was particularly noticeable in the CEP of advanced-age subjects, and was more evident in a surgically treated group than in a naturally aged group. In the present study, a higher number of apoptotic cells was detected following LiCl treatment, and inhibiting Wnt/β-catenin signalling protected against cellular apoptosis. This finding was similar to previous studies on NP and chondrocyte apoptosis (17,21). These results suggest that Wnt/β-catenin signalling is involved in the regulation of CEP cell numbers, which is essential for maintaining the basic homeostasis of IVD.
Equilibrium of the ECM is regulated by anabolic and catabolic enzymes, and a break in the balance will accelerate IVD degeneration. Aggrecans are the most important components of endplate ECM, and a decrease in aggrecans will result in the compression, or even calcification, of the ECM, followed by accelerated disc degeneration (35). MMP-13 and ADAMTS-5 are known to be important catabolic enzymes that decrease collagen and aggrecan levels, thus deteriorating the microenvironment of the IVD. In the present study, LiCl treatment enhanced the expression of MMP-13 and ADAMTS-5 and decreased the expression of aggrecan. Notably, a Wnt/β-catenin signalling inhibitor decreased the expression of catabolic enzymes (MMP-13 and ADAMTS-5) and increased the expression of aggrecan induced by Wnt/β-catenin signalling activation, which provide novel insights into the role of Wnt/β-catenin signalling in mediating the regulation of ECM metabolism in the CEP. The findings if the present study may provide potential strategies for preventing CEP degeneration.
However, the present study had several limitations, including the lack of a control group treated with NaCl and a group treated only with ICG001, which would allow more precise results to be obtained; however the reliability of the results obtained were not affected. Another limitation is the lack of data in animal models in vivo, and future in vivo experiments are required to determine whether the activation of Wnt/β-catenin signalling can accelerate degeneration of the IVD.
Acknowledgements
Not applicable.
Funding
This study was supported by the Shanghai Municipality Health Bureau (grant no. 201640101) and the Jinshan Science and Technology Committee (grant nos. 2015-3-6 and 2016-3-06).
Availability of data and materials
The datasets used and/or analysed in the present study are available from the corresponding author on reasonable request.
Authors' contributions
LD, ZJ and JW designed the study. DL, HW and WL collected the data. QZ and GX analysed the data. LD and ZJ prepared the manuscript. LD, ZJ and JW revised the manuscript. All authors read and approved the final manuscript.
Ethics approval and consent to participate
The present study was approved by the Ethics Committee on Animal Experiments of Fudan University.
Patients consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
References
- 1.Hoy D, Brooks P, Woolf A, Blyth F, March L, Bain C, Baker P, Smith E, Buchbinder R. Assessing risk of bias in prevalence studies: Modification of an existing tool and evidence of interrater agreement. J Clin Epidemiol. 2012;65:934–939. doi: 10.1016/j.jclinepi.2011.11.014. [DOI] [PubMed] [Google Scholar]
- 2.de Schepper EI, Damen J, van Meurs JB, Ginai AZ, Popham M, Hofman A, Koes BW, Bierma-Zeinstra SM. The association between lumbar disc degeneration and low back pain: The influence of age, gender, and individual radiographic features. Spine (Phila Pa 1976) 2010;35:531–536. doi: 10.1097/BRS.0b013e3181aa5b33. [DOI] [PubMed] [Google Scholar]
- 3.Magnier C, Boiron O, Wendling-Mansuy S, Chabrand P, Deplano V. Nutrient distribution and metabolism in the intervertebral disc in the unloaded state: A parametric study. J Biomech. 2009;42:100–108. doi: 10.1016/j.jbiomech.2008.10.034. [DOI] [PubMed] [Google Scholar]
- 4.Raj PP. Intervertebral disc: Anatomy-physiology-pathophysiology-treatment. Pain Pract. 2008;8:18–44. doi: 10.1111/j.1533-2500.2007.00171.x. [DOI] [PubMed] [Google Scholar]
- 5.Grunhagen T, Shirazi-Adl A, Fairbank JC, Urban JP. Intervertebral disk nutrition: A review of factors influencing concentrations of nutrients and metabolites. Orthop Clin North Am. 2011;42(vii):465–477. doi: 10.1016/j.ocl.2011.07.010. [DOI] [PubMed] [Google Scholar]
- 6.Shirazi-Adl A, Taheri M, Urban JP. Analysis of cell viability in intervertebral disc: Effect of endplate permeability on cell population. J Biomech. 2010;43:1330–1336. doi: 10.1016/j.jbiomech.2010.01.023. [DOI] [PubMed] [Google Scholar]
- 7.Ding F, Shao ZW, Xiong LM. Cell death in intervertebral disc degeneration. Apoptosis. 2013;18:777–785. doi: 10.1007/s10495-013-0839-1. [DOI] [PubMed] [Google Scholar]
- 8.Kang R, Li H, Ringgaard S, Rickers K, Sun H, Chen M, Xie L, Bünger C. Interference in the endplate nutritional pathway causes intervertebral disc degeneration in an immature porcine model. Int Orthop. 2014;38:1011–1017. doi: 10.1007/s00264-014-2319-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nusse R, Clevers H. Wnt/β-catenin signaling, disease, and emerging therapeutic modalities. Cell. 2017;169:985–999. doi: 10.1016/j.cell.2017.05.016. [DOI] [PubMed] [Google Scholar]
- 10.Ring A, Kim YM, Kahn M. Wnt/catenin signaling in adult stem cell physiology and disease. Stem Cell Rev. 2014;10:512–525. doi: 10.1007/s12015-014-9515-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang Y, Li YP, Paulson C, Shao JZ, Zhang X, Wu M, Chen W. Wnt and the Wnt signaling pathway in bone development and disease. Front Biosci (Landmark Ed) 2014;19:379–407. doi: 10.2741/4214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lara-Castillo N, Johnson ML. LRP receptor family member associated bone disease. Rev Endocr Metab Disord. 2015;16:141–148. doi: 10.1007/s11154-015-9315-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Joiner DM, Ke J, Zhong Z, Xu HE, Williams BO. LRP5 and LRP6 in development and disease. Trends Endocrinol Metab. 2013;24:31–39. doi: 10.1016/j.tem.2012.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Aberle H, Bauer A, Stappert J, Kispert A, Kemler R. Beta-catenin is a target for the ubiquitin-proteasome pathway. EMBO J. 1997;16:3797–3804. doi: 10.1093/emboj/16.13.3797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Clevers H, Nusse R. Wnt/β-catenin signaling and disease. Cell. 2012;149:1192–1205. doi: 10.1016/j.cell.2012.05.012. [DOI] [PubMed] [Google Scholar]
- 16.MacDonald BT, Tamai K, He X. Wnt/beta-catenin signaling: Components, mechanisms, and diseases. Dev Cell. 2009;17:9–26. doi: 10.1016/j.devcel.2009.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hiyama A, Sakai D, Risbud MV, Tanaka M, Arai F, Abe K, Mochida J. Enhancement of intervertebral disc cell senescence by WNT/β-catenin signaling-induced matrix metalloproteinase expression. Arthritis Rheum. 2010;62:3036–3047. doi: 10.1002/art.27599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chen J, Jia YS, Liu GZ, Sun Q, Zhang F, Ma S, Wang YJ. Role of LncRNA TUG1 in intervertebral disc degeneration and nucleus pulposus cells via regulating Wnt/β-catenin signaling pathway. Biochem Biophys Res Commun. 2017;491:668–674. doi: 10.1016/j.bbrc.2017.07.146. [DOI] [PubMed] [Google Scholar]
- 19.Wang J, Chen H, Cao P, Wu X, Zang F, Shi L, Liang L, Yuan W. Inflammatory cytokines induce caveolin-1/β-catenin signalling in rat nucleus pulposus cell apoptosis through the p38 MAPK pathway. Cell Prolif. 2016;49:362–372. doi: 10.1111/cpr.12254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Iwata M, Aikawa T, Hakozaki T, Arai K, Ochi H, Haro H, Tagawa M, Asou Y, Hara Y. Enhancement of Runx2 expression is potentially linked to β-catenin accumulation in canine intervertebral disc degeneration. J Cell Physiol. 2015;230:180–190. doi: 10.1002/jcp.24697. [DOI] [PubMed] [Google Scholar]
- 21.Ning B, Wang P, Pei X, Kang Y, Song J, Wang D, Zhang W, Ma R. Dual function of β-catenin in articular cartilage growth and degeneration at different stages of postnatal cartilage development. Int Orthop. 2012;36:655–664. doi: 10.1007/s00264-011-1315-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ding L, Zeng Q, Wu J, Li D, Wang H, Lu W, Jiang Z, Xu G. Caveolin1 regulates oxidative stress-induced senescence in nucleus pulposus cells primarily via the p53/p21 signaling pathway in vitro. Mol Med Rep. 2017;16:9521–9527. doi: 10.3892/mmr.2017.7789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 24.Xu HG, Zheng Q, Song JX, Li J, Wang H, Liu P, Wang J, Wang CD, Zhang XL. Intermittent cyclic mechanical tension promotes endplate cartilage degeneration via canonical Wnt signaling pathway and E-cadherin/β-catenin complex cross-talk. Osteoarthritis Cartilage. 2016;24:158–168. doi: 10.1016/j.joca.2015.07.019. [DOI] [PubMed] [Google Scholar]
- 25.Yang H, Yuan C, Wu C, Qian J, Shi Q, Li X, Zhu X, Zou J. The role of TGF-β1/Smad2/3 pathway in platelet-rich plasma in retarding intervertebral disc degeneration. J Cell Mol Med. 2016;20:1542–1549. doi: 10.1111/jcmm.12847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Xie H, Jing Y, Xia J, Wang X, You C, Yan J. Aquaporin 3 protects against lumbar intervertebral disc degeneration via the Wnt/β-catenin pathway. Int J Mol Med. 2016;37:859–864. doi: 10.3892/ijmm.2016.2470. [DOI] [PubMed] [Google Scholar]
- 27.Smolders LA, Meij BP, Onis D, Riemers FM, Bergknut N, Wubbolts R, Grinwis GC, Houweling M, Groot Koerkamp MJ, van Leenen D, et al. Gene expression profiling of early intervertebral disc degeneration reveals a down-regulation of canonical Wnt signaling and caveolin-1 expression: Implications for development of regenerative strategies. Arthritis Res Ther. 2013;15:R23. doi: 10.1186/ar4157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sanges D, Romo N, Simonte G, Di Vicino U, Tahoces AD, Fernandez E, Cosma MP. Wnt/β-catenin signaling triggers neuron reprogramming and regeneration in the mouse retina. Cell Rep. 2013;4:271–286. doi: 10.1016/j.celrep.2013.06.015. [DOI] [PubMed] [Google Scholar]
- 29.Gruber HE, Ingram JA, Davis DE, Hanley EJ., Jr Increased cell senescence is associated with decreased cell proliferation in vivo in the degenerating human annulus. Spine J. 2009;9:210–215. doi: 10.1016/j.spinee.2008.01.012. [DOI] [PubMed] [Google Scholar]
- 30.Jeong SW, Lee JS, Kim KW. In vitro lifespan and senescence mechanisms of human nucleus pulposus chondrocytes. Spine J. 2014;14:499–504. doi: 10.1016/j.spinee.2013.06.099. [DOI] [PubMed] [Google Scholar]
- 31.Feng C, Liu H, Yang M, Zhang Y, Huang B, Zhou Y. Disc cell senescence in intervertebral disc degeneration: Causes and molecular pathways. Cell Cycle. 2016;15:1674–1684. doi: 10.1080/15384101.2016.1152433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.van Deursen JM. The role of senescent cells in ageing. Nature. 2014;509:439–446. doi: 10.1038/nature13193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Munoz-Espin D, Serrano M. Cellular senescence: From physiology to pathology. Nat Rev Mol Cell Biol. 2014;15:482–496. doi: 10.1038/nrm3823. [DOI] [PubMed] [Google Scholar]
- 34.Sharpless NE, Sherr CJ. Forging a signature of in vivo senescence. Nat Rev Cancer. 2015;15:397–408. doi: 10.1038/nrc3988. [DOI] [PubMed] [Google Scholar]
- 35.Hee HT, Chuah YJ, Tan BH, Setiobudi T, Wong HK. Vascularization and morphological changes of the endplate after axial compression and distraction of the intervertebral disc. Spine (Phila Pa 1976) 2011;36:505–511. doi: 10.1097/BRS.0b013e3181d32410. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets used and/or analysed in the present study are available from the corresponding author on reasonable request.