

Abstract
Mitochondrial fragmentation frequently occurs in chronic pathological conditions as seen in various human diseases. In fact, abnormal mitochondrial morphology and mitochondrial dysfunction are hallmarks of heart failure (HF) in both human patients and HF animal models. A link between mitochondrial fragmentation and cardiac pathologies has been widely proposed, but the physiological relevance of mitochondrial fission and fusion in the heart is still unclear. Recent studies have increasingly shown that posttranslational modifications (PTMs) of fission and fusion proteins are capable of directly modulating the stability, localization, and/or activity of these proteins. These PTMs include phosphorylation, acetylation, ubiquitination, conjugation of small ubiquitin-like modifier proteins, O-linked-N-acetyl-glucosamine glycosylation, and proteolysis. Thus, understanding the PTMs of fission and fusion proteins may allow us to understand the complexities that determine the balance of mitochondrial fission and fusion as well as mitochondrial function in various cell types and organs including cardiomyocytes and the heart. In this review, we summarize present knowledge regarding the function and regulation of mitochondrial fission and fusion in cardiomyocytes, specifically focusing on the PTMs of each mitochondrial fission/fusion protein. We also discuss the molecular mechanisms underlying abnormal mitochondrial morphology in HF and their contributions to the development of cardiac diseases, highlighting the crucial roles of PTMs of mitochondrial fission and fusion proteins. Finally, we discuss the future potential of manipulating PTMs of fission and fusion proteins as a therapeutic strategy for preventing and/or treating HF.
Keywords: DLP1, Drp1, Mfn, mitophagy, OPA1
INTRODUCTION
Mitochondria are highly dynamic organelles with the ability to divide (fission), combine (fusion), and move along the cytoskeleton to interact with other organelles, which determines cell-specific mitochondrial morphology, intracellular distribution, and function in the mammalian cells (54, 109, 197, 202). These mitochondrial dynamics are mainly controlled by the balance between fission and fusion, both processes regulated by the relative activities of mitochondrial fission and fusion machineries. The core components of the mammalian machineries are members of the dynamin family of large GTPases that can bind and hydrolyze guanosine triphosphate (GTP). A GTPase, dynamin-related protein 1 (Drp1, also called DLP1), and its receptors [mitochondrial fission factor (Mff; 65, 138) and mitochondrial dynamics proteins of 49 and 51 kDa (MiD49 and MiD51, respectively; 141, 210)] dictate mitochondrial fission, whereas the GTPases mitofusin 1 and 2 (Mfn1 and Mfn2, respectively) and optic atrophy protein-1 (OPA1) mediate mitochondrial fusion (18, 150, 192).
The relative activities of fission and fusion proteins are likely determined by several factors: 1) the total expression levels of these proteins, controlled by transcription, translation, protein stability, and degradation; 2) the expression levels of these proteins at the mitochondria [e.g., Drp1 can be recruited from the cytosol to the outer mitochondria membrane (OMM) in response to cellular stimuli]; and 3) the overall activities of the GTPases, which likely depend on GTP availability, inter/intra-protein-protein interactions, and/or protein expression levels present at the mitochondria. These three factors interdependently modulate the balance of fission and fusion (Fig. 1). For instance, under physiological conditions, the expression levels of these proteins may influence their localization and GTPase activity at the mitochondria, whereas the localizations of the proteins affect their activities at the mitochondria. Thus, this interdependence points to overall protein activities as the integrative regulators of mitochondrial morphology. Recent studies have increasingly shown that posttranslational modifications (PTMs) of fission and fusion proteins are capable of directly modulating their stability, localization, and/or activity (19, 55, 126, 136). Therefore, understanding the PTMs of fission and fusion proteins may allow us to understand the complexities that determine the balance of fission and fusion that ultimately dictate mitochondrial morphology in various cell types and organs including cardiomyocytes (CMs) and the heart (Fig. 2, A–D).
Fig. 1.
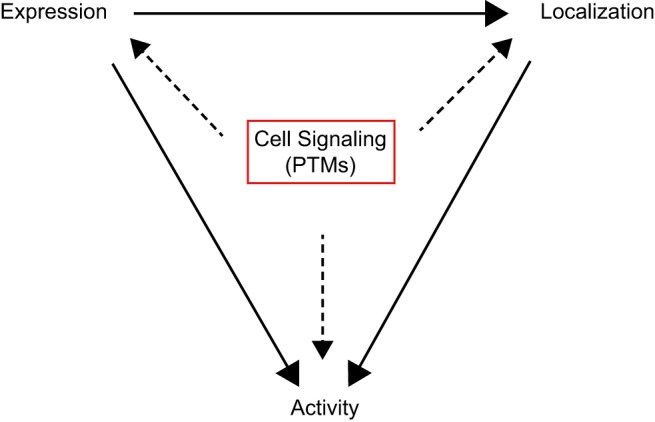
Regulation of mitochondrial morphology by posttranslational modifications (PTMs) of mitochondrial fission and fusion proteins. Schematic model of the three main regulators (protein expression, localization at the mitochondria, and overall GTPase activity) of mitochondrial fission and fusion, which interdependently modulate the balance of fission and fusion.
Fig. 2.

Subpopulations of mitochondria in adult cardiomyocytes. A: illustration of a single adult cardiomyocyte and diagram of different subpopulations of mitochondria. B: illustration of longitudinal section of the area indicated in A including subsarcolemmal mitochondria. C: illustration of longitudinal section of the area indicated in A including perinuclear mitochondria. D: illustration of longitudinal section of the area indicated in A including interfibrillar mitochondria. Note that interfibrillar mitochondria face the sarcoplasmic reticulum (SR). T tubule, transverse tubule. E: transmission electron microscope image of the longitudinal section of a papillary muscle obtained from an adult rabbit ventricle. Arrows show the location of subsarcolemmal mitochondria. Scale bars, 5 µm. F: transmission electron microscope image of the short axis of a papillary muscle obtained from an adult rabbit ventricle. Blue area indicates location of the nucleus. Red areas indicate perinuclear mitochondria. Yellow areas indicate subsarcolemmal mitochondria. Scale bar, 5 µm.
On the basis of studies using cell culture systems, mitochondrial dynamics are suggested as an important modulator for bioenergetics, reactive oxygen species (ROS) generation, spatiotemporal dynamics of Ca2+ signaling, cell growth, and cell death (54, 109, 197, 202). Since mammalian hearts, which are enriched in mitochondria (≅30% of total volume) mostly from CMs, have a high demand for ATP to constantly pump blood through the body (128), it is reasonable to suggest that mitochondrial fission and fusion are important for regulating ATP production in CMs so that they may acquire the energy necessary for performing their function. Transmission electron microscope images show that adult CMs have highly organized and largely static mitochondria that are densely packed 1) into the intermyofibrillar spaces with one or more mitochondria surrounding each sarcomere (i.e., interfibrillar mitochondria) (Fig. 2, D and E), 2) just beneath the plasma membrane (i.e., subsarcolemmal mitochondria) (Fig. 2, B, E, and F), and 3) at the perinuclear region (i.e., perinuclear mitochondria) (Fig. 2, C and F), which likely restricts mitochondrial movement and formation of networks (8, 133, 152, 190). Thus, the activity of mitochondrial dynamics and their physiological relevance to mitochondrial and cellular function in CMs are still under debate. Emerging evidence using state-of-the-art imaging techniques suggests that cardiac mitochondria (especially interfibrillar mitochondria) form mitochondrial networks inside the myofibrils and may possess at least slow fusion activity under physiological conditions. For instance, several groups, using confocal microscopy and a mitochondrial matrix-targeted photoactivable green fluorescent protein, reported that interfibrillar mitochondria form local networks and undergo OPA1- and Mfn1-dependent fusion events to share mitochondrial matrix content with neighboring mitochondria in both adult skeletal and cardiac muscles (52, 53, 151). However, the green fluorescent protein diffusion/sharing events were slower (≥ 3 min) and less frequent in adult CMs compared with nondifferentiated myoblasts or neonatal CMs (52, 53). Using three-dimensional electron microscopy, Balaban’s group also proposed that interfibrillar mitochondria in both skeletal and cardiac muscles communicate through junctions (68, 69, 148), which allows for rapid electrical coupling though mitochondrial networks (103). However, it is still unknown whether there is continuity in the matrix along all of the conducting elements (68, 69, 148). Moreover, the frequency and timescale of fission events in adult CMs remain unclear since the techniques for detecting mitochondrial fission events in live adult CMs are still experimentally challenging. Importantly, abnormal mitochondrial morphologies, especially small, disorganized interfibrillar mitochondria, are frequently observed in the myocardium of both human patients (43, 95, 154, 169) and animal models of cardiac diseases, such as heart failure (HF; 11, 13, 25, 36, 87, 167, 198). These observations indicate that mitochondrial fragmentation in CMs frequently occurs under chronic pathological conditions as has been observed in other human diseases including neurodegenerative diseases, diabetes, and cancer (5, 26). This suggests a link between mitochondrial fragmentation and cardiac pathologies, although the physiological relevance of mitochondrial fission and fusion in adult CMs is still unclear. Consequently, multiple questions still remain: 1) Is this mitochondrial fragmentation caused by the enhancement of mitochondrial fission activity and/or the inhibition of fusion activity? 2) How are the PTMs of mitochondrial fission and fusion proteins involved in these fission and fusion processes? 3) Is the abnormality of cardiac mitochondrial morphology involved in the development of cardiac pathology or just an adaptation/maladaptation to pathological changes in the heart?
It is conceivable that mitochondrial fusion and fission impact all cell types in the heart, including cardiac fibroblasts and vascular endothelial and smooth muscle cells in addition to CMs. Indeed, the roles and functional relevance of mitochondrial fission and fusion proteins in fibroblasts [e.g., embryonic fibroblasts (179)], vascular smooth muscle cells [e.g., pulmonary artery smooth muscle cells (116)], and endothelial cells [e.g., kidney microvascular endothelial cells (194)] in other organs/tissues have emerged. However, the number of publications investigating these cell types in the heart (i.e., cardiac fibroblasts and smooth muscle and endothelial cells from coronary arteries) is extremely limited compared with those focusing on CMs [see review (189)]. Therefore, in this review, we summarize present knowledge regarding the function and regulation of mitochondrial fission and fusion only in CMs, with a focus on the PTMs of each mitochondrial fission/fusion protein. We also discuss the molecular mechanisms underlying abnormal mitochondrial morphology in CMs and their contribution to the development of cardiac diseases, highlighting the crucial role of PTMs of mitochondrial fission and fusion proteins in CMs.
OVERVIEW: MITOCHONDRIAL MORPHOLOGY AND FISSION AND FUSION PROTEINS IN CARDIOMYOCYTES
As described in the introduction, unlike noncardiac cells, adult CMs have highly organized and globular-shaped mitochondria, which can be categorized into three distinct subpopulations depending on their location and function [see also review (133); Fig. 2]. The majority of these mitochondria are classified as interfibrillar mitochondria, and a smaller population are categorized as subsarcolemmal or perinuclear mitochondria. The interfibrillar mitochondria are aligned adjacent to the myofibrils, with one or two mitochondria lying along each sarcomere [i.e., between the 2 transverse tubules (T tubules); Fig. 2, A, D, and E]. These mitochondria are involved in ATP generation for contractile function and Ca2+ signaling. The subsarcolemmal mitochondria are arranged just beneath the sarcolemma and may provide ATP required for ion channel function and cell signaling (Fig. 2, A, B, E, and F). The perinuclear mitochondria form clusters around the nucleus and may provide ATP for transcription and other nuclear activities (Fig. 2, A, C, and F). These specific and compact intracellular mitochondrial arrangements may restrict mitochondrial mobility in adult CMs compared with other cell types, although they still maintain their networks and interaction throughout the myofibrils (see introduction). In the heart and CMs, mitochondrial fission and fusion proteins are highly expressed, including Drp1, Mff, Mfn1, Mfn2, and OPA1, although their stoichiometric ratio may differ during development (e.g., neonatal CMs vs. adult CMs) or under various pathological conditions (e.g., HF; 11, 25, 88; Table 1).
Table 1.
Altered expression levels of mitochondrial fission and fusion proteins in cardiac disease animal models
| Reference | Species | Model | Mitochondrial Morphology | Drp1 | Mfn1 | Mfn2 | OPA1 |
|---|---|---|---|---|---|---|---|
| Chen et al. (25) | Rat | 9 wk post-MI-induced HF | Fragmented mitochondria | → | → | → | ↓ |
| Javadov et al. (88) | Rat | 12 wk post-MI-induced HF | Fragmented mitochondria | → | ND | ↓ | → |
| Jiang et al. (92) | Rat | 8 wk post-MI-induced cardiac injury | ND | ↑ | → | ↓ | ↓ |
| Littlejohns et al. (110) | Mouse | Cardiac I/R injury in 20–21-wk high-fat diet model | Fragmented mitochondria | ↑ | → | ↑ | ↓ |
| Cao et al. (15) | Rat | Cardiac injury in 8-wk high-fat diet model | ND | → | ↓ | → | ↓ |
| Maneechote et al. (115) | Rat | Cardiac I/R injury | Swollen mitochondria | ↑ | ND | → | → |
Drp1, dynamin-related protein-1; HF, heart failure; I/R, ischemia-reperfusion; Mfn1 and Mfn2, mitofusin 1 and 2, respectively; MI, myocardial infarction; ND, not determined; OPA1, optic atrophy protein-1; →, remained unchanged; ↑, increased; ↓, decreased.
Initially, the importance of mitochondrial fission and fusion proteins in maintaining cellular functions has been investigated using cultured cell lines and/or nonexcitable cells (54, 90, 107, 150). Recently, the physiological and pathological importance of mitochondrial fission and fusion proteins in humans has emerged from clinical reports showing that individuals who possess a mutation in these proteins exhibit various clinical phenotypes, especially related to excitable cell functions. These include 1) neurological disorders in Drp1 mutations (21, 58, 127, 174, 188), Mfn2 mutations linked to Charcot-Marie-Tooth disease type 2A (61), and OPA1 mutations linked to autosomal dominant optic atrophy (2, 38, 42) and 2) the mixture of neurological and skeletal muscle disorders in OPA1 mutations linked to Behr syndrome (77). Finally, thanks to recent studies using primary adult CMs and knockout (KO) mouse lines (Table 2), many laboratories have started to precisely assess the roles of fission and fusion proteins in cardiac physiology and their contributions to cardiac pathologies. However, there are multiple technical challenges involved in researching mitochondrial morphology and dynamics using adult CMs that still need to be overcome. For instance, the observation of mitochondrial morphology/dynamics (especially fission events) in live CMs under light/confocal microscopy (52, 53) is still challenging, which necessitates the use of electron microscopy under fixed conditions (68, 69, 148). In addition, it is technically difficult to maintain the integrity of cellular membrane structures such as T tubules (Fig. 2D) in cultured adult CMs for the time periods required for genetic manipulations (e.g., 24–48 h; 54, 139, 200). Thus, the utilization of adult CMs/hearts freshly isolated from KO mouse lines may limit the observable information for investigating the roles of PTMs in mitochondrial fission and fusion proteins. Therefore, it is reasonable to extract the information about cardiac-specific PTMs of mitochondrial fission and fusion proteins from various “cell models for adult CMs” that possess a similar structure of signaling pathways, despite the fact that their cellular membrane integrity and mitochondrial morphology/dynamics at resting conditions are different from those in adult CMs. These include H9c2 cardiac myoblasts (25, 91, 177, 200, 204), the HL-1 mouse atrial CM cell line (56, 147, 206), primary neonatal CMs (12, 45, 91, 114, 211), human induced pluripotent stem cell-derived CMs (79), and embryonic stem cells-derived CMs (97; see also the “Notes” columns in Tables 3 and 4).
Table 2.
Effects of in vivo knockout/ablation of the mitochondrial fission and fusion proteins in the heart
| Reference | Targeted Protein | Model | Cardiac Phenotype | Mitochondrial Morphology | Mitochondrial Function | Other Notes |
|---|---|---|---|---|---|---|
| Kageyama et al. (94); Song et al. (179) | Drp1 | CM-specific (Myh6 nuclear-directed Cre) KO after birth | Neonatal cardiomyopathy (94); modestly enlarged hearts (179) | Connected or enlarged mitochondria (94) | Mitochondrial respiration ↓ (94); Parkin-independent mitophagy signaling ↑ (94) | Died at 1–1.5 wk (94); died by 6 wk (179) |
| Song et al. (179) | Drp1 | Inducible CM-specific (Myh6 MER-Cre-MER) KO (132) | At 6–7 wk after Drp1 deletion, DCM, CM necrosis, cardiac fibrosis, and HF | Elongated and enlarged mitochondria | mPTP opening ↑; Parkin-dependent mitophagy signaling ↑ | |
| Ikeda et al. (82) | Drp1 | Inducible CM-specific (α-MHC-MER-Cre-MER) homozygous KO | At 4–8 wk after Drp1 deletion, cardiac hypertrophy, CM apoptosis, cardiac fibrosis, and HF | Elongated mitochondria | ATP ↓; mPTP opening ↑; ROS ↑; autophagic flux ↓ | Died at 8–13 wk after Drp1 deletion |
| Ikeda et al. (82); Shirakabe et al. (176) | Drp1 | CM-specific (α-MHC) heterozygous KO | Cardiac function at 12 wk →; cardiac hypertrophy at 5 days after TAC ↑; HF at ~4 wk after TAC | Elongated mitochondria; after TAC, enlarged mitochondria at ~24 h and ~4 wk and fragmented mitochondria at 3–5 days | Susceptibility to I/R injury ↑; after 3–5 days of TAC, mitophagy ↑, ATP ↓, and mitochondrial respiration ↓ | Homozygous mice: embryonic lethal |
| Ishihara et al. (85) | Drp1 | Muscle-specific (Mck) KO | Neonatal cardiomyopathy | Connected or enlarged mitochondria | Mitochondrial respiration ↓ | Died at 1–1.5 wk |
| Chen et al. (23) | Mff | Conventional KO | At 13–14 wk, DCM, cardiac fibrosis, and HF | Mitochondrial length →; mitochondria number ↓ | Mitochondrial respiration ↓; mitophagy ↑ | Neuromuscular defects; died at ~13 wk |
| Papanicolaou et al. (144) | Mfn1 | CM-specific (Myh6) KO | Cardiac function → | Fragmented mitochondria | Mitochondrial respiration →; ROS-induced mPTP opening and CM death ↓ | |
| Chen et al. (28) | Mfn1 | CM-specific (Myh6 nuclear-directed “turbo” Cre) KO | Cardiac function at 6–8 wk → | Mean distance between SR and mitochondria → | During pacing and β-adrenergic stimulation, mitochondrial Ca2+ uptake → and oxidation of NAD(P)H and FADH2 → | |
| Papanicolaou et al. (142) | Mfn2 | CM-specific (α-MHC) KO | Modest LV hypertrophy; mild LV systolic dysfunction; recovery after I/R ↑ | Pleomorphic and enlarged mitochondria | Mitochondrial respiration →; time to reach Ca2+-induced mPTP opening ↑ | |
| Chen et al. (28) | Mfn2 | CM-specific (Myh6 nuclear-directed “turbo” Cre) KO | Cardiac function at 6–8 wk → | Enlarged mitochondria; mean distance between SR and mitochondria ↑ | During pacing and β-adrenergic stimulation, mitochondrial Ca2+ uptake ↓, oxidation of NAD(P)H and FADH2 ↑, and mitochondrial ROS ↑ | No apoptosis |
| Chen et al. (30) | Mfn1 and Mfn2 | Inducible CM-specific (Myh6 MER-Cre-MER) DKO | DCM during 5 wk; HF after 7–8 wk | Fragmented mitochondria | Mitochondrial respiration ↓ | |
| Papanicolaou et al. (143) | Mfn1 and Mfn2 | CM-specific (Myh6) DKO | At 1 wk, DCM and HF | At 1 wk, spherical and heterogeneously sized mitochondria and swollen mitochondria | ATP →; early mediators of autophagy ↑ | Died at 6–16 days |
| Chen et al. (30); Kasahara et al. (97) | Mfn1 and Mfn2 | CM-specific (Nkx2.5, which induces robust gene recombination before E9.5 in CMs) DKO | CM differentiation at E9.5 ↓ (97); cardiac development at E12.5–13.5 ↓ (97) | Fragmented mitochondria in embryonic heart (97) | Mitochondrial functions were normal (97) | Died after E9.5 (30); died after E13.5 (97) |
| Hall et al. (75) | Mfn1 and Mfn2 | Inducible CM-specific (Myh6 MER-Cre-MER) DKO | At ~4 wk after DKO, cardiac function →, I/R injury ↓, and contractile function ↓ | Fragmented mitochondria; mean distance between SR and mitochondria ↑ | Mitochondrial respiration ↓; time to reach Ca2+-induced mPTP opening ↑; mitochondrial Ca2+ uptake during I/R ↓ | |
| Piquereau et al. (153) | OPA1 | Heterozygous mutation (329–355del, OPA1+/− ) | Cardiac function at 6 mo →; LV hypertrophy after TAC ↑ | Enlarged mitochondria; cristae disorganization | Mitochondrial respiration →; time to reach Ca2+-induced mPTP opening ↑ | |
| Chen et al. (27) | OPA1 | Heterozygous mutation (Q285 Stop, OPA1+/−) | Cardiomyopathy and HF at 12 mo | Disorganized and fragmented mitochondria ↑; cristae structure ↓ | Mitochondrial respiration ↓; ATP ↓; ROS ↑ | No apoptotic CM death; homozygous mice: embryonic lethal |
CM, cardiomyocyte; DCM, dilated cardiomyopathy; DKO, double knockout; Drp1, dynamin-related protein-1; E9.5, embryonic day 9.5; HF, heart failure; I/R, ischemia-reperfusion; KO, knockout; LV, left ventricular; Mck, muscle creatine kinase; MER, modified estrogen receptor; Mff, mitochondrial fission factor; Mfn1 and Mfn2, mitofusin 1 and 2, respectively; α-MHC, α-myosin heavy chain; mPTP, mitochondrial permeability transition pore; Myh6, myosin, heavy polypeptide 6 cardiac muscle-α; Nkx2.5, NK2 homeobox 5; OPA1, optic atrophy protein-1; ROS, reactive oxygen species; SR, sarcoplasmic reticulum; TAC, transverse aortic constriction; →, remained unchanged; ↑, increased; ↓, decreased.
Table 3.
Posttranslational modifications of Drp1 in cardiomyocytes/hearts
| Position | Type of Modification | Upstream Molecules | Effect on Mitochondrial Morphology | Notes |
|---|---|---|---|---|
| K532, K535, K558, K568, K594, K597, K606, and K608 (59) | SUMOylation | SENP5 (99, 175) | DeSUMOylation: enlarged and swollen mitochondria (99); fragmented mitochondria (175) | Figueroa-Romero et al. (59) detected SUMOylation sites in noncardiac cells |
| T585 and T586 | O-GlcNAcylation | ND (O-GlcNAc transferase?) | O-GlcNAcylation: fragmentation (66) | |
| S616 | Phosphorylation | CDK1/cyclin B (206); PKCδ (113, 206); ERK1/2 (203); CaMKII (200); ROCK (12); RIP1 (164) | Phosphorylation: fragmentation (113, 203, 206) Dephosphorylation: elongation (206) S616A: fragmentation (91); no significant changes (12) | Xu et al. (200) detected Drp1 phosphorylation at S616 and S637 in adult CMs, but the effect of CaMKII on mitochondrial morphology was analyzed in H9c2 cardiac myoblasts; Lu et al. (113) used the neonatal CMs for detecting Drp1 phosphorylation at S616 but did not provide any direct evidence of PKCδ-dependent Drp1 phosphorylation; Yu et al. (203) detected phosphorylation of Drp1 at S616 using an in vitro kinase assay, and mitochondrial morphological changes were analyzed in H9c2 cells; Zaja et al. (206) used the HL-1 cell line derived from the AT-1 mouse atrial myocyte tumor lineage for detecting Drp1 phosphorylation at S616; Jhun et al. (91) detected Drp1 phosphorylation at S616 in H9c2 cells and heart trusses, but the effect of S616 phosphorylation on mitochondrial morphology was analyzed in H9c2 cells |
| S637 | Phosphorylation | PKA (35); PKD (91); Pim-1 (45); CaN (35, 149, 173, 206) | Phosphorylation: fragmentation (91) Dephosphorylation: fragmentation (149, 173, 206) S637A: no significant changes (91) | Zaja et al. (206) used the HL-1 CM line for detecting Drp1 phosphorylation at S637; Jhun et al. (91) detected Drp1 phosphorylation at S637 in H9c2 cells, neonatal CMs, and heart trusses, but the effect of S637 phosphorylation on mitochondrial morphology was analyzed in H9c2 cells and neonatal CMs; Cribbs and Strack (35) detected S637 phosphorylation in the heart by in vivo isoproterenol injection and exercise, but they did not observe mitochondrial morphology in CMs; Din et al. (45) showed that Pim-1 decreases Drp1 expression and increases S637 phosphorylation, which results in mitochondrial elongation |
Information about sites of conjugation of small ubiquitin-like modifier (SUMO) proteins (SUMOylation) was collected from PhosphoSitePlus (80). CaN, Ca2+-dependent phosphatase calcineurin; CDK1, cyclin-dependent kinase; CM, cardiomyocyte; Drp1, dynamin-related protein-1; ND, not determined; O-GlcNAc, O-linked-N-acetyl-glucosamine; O-GlycNAcylation, O-linked-N-acetyl-glucosamine glycosylation; Pim-1, proto-oncogene serine/threonine-protein kinase; RIP1, receptor-interacting protein-1; ROCK, Rho-associated protein kinase; SENP5, sentrin/SUMO-specific protease 5.
Table 4.
Posttranslational modifications of mitochondrial fusion proteins
| Protein | Type of Modification (Position) | Upstream Molecules | Effect on Mitochondrial Morphology | Detected/Tested in CMs? | Notes |
|---|---|---|---|---|---|
| Mfn1 | Phosphorylation (T562) | ERK (156) | Phosphorylation: fragmentation (156) | No | |
| Mfn1 | Acetylation (K491) | ND (145) | Acetylation-deficient K491R: aggregation (145) Acetylation-mimetic K491Q: elongation (145) | No | |
| Mfn1 | Ubiquitination (ND) | MARCH5 (145) | No | Mfn1 levels are maintained by MARCH5-mediated quality control on acetylated Mfn1 (145) | |
| Mfn2 | Phosphorylation (S27) | JNK (105) | S27D: fragmentation (105) | No | S27 phosphorylation of Mfn2 increased ubiquitination and degradation of Mfn2 by the E3 ubiquitin ligase Huwe1 (105) |
| Mfn2 | Phosphorylation (S442) | PKA (212) | S442A/S442D: no change (212) | No | |
| Mfn2 | Phosphorylation (T111 and S442) | PINK1 (29) | ND | Yes (29) | PINK1-dependent phosphorylation of Mfn2 induces Parkin translocation to the OMM upon membrane depolarization, which subsequently promotes Parkin-mediated ubiquitination of Mfn2 in adult CMs (29) |
| Mfn1/Mfn2 | Ubiquitination (In Mfn2 sequence: K406, K416, K420, K719, K720, K730, and K737) | Parkin (29, 67, 70, 117, 185); USP30 (205) | Ubiquitination: reduced mitochondrial number (185); distance between ER and mitochondria ↑ (117, 185) Nondegradative ubiquitination: restore a normal mitochondrial network (205) | Yes (29) | Parkin-mediated ubiquitination of Mfn2 leads to its degradation, resulting in the selective removal of damaged mitochondria by mitophagy in adult CMs (29) |
| OPA1 | Proteolysis (S1 or S2 cleavage sites; see Fig. 5, A and B, for positions) | Paraplegin (84); PARL (32); YME1L (3, 72, 180, 191); OMA1 (1, 3, 6, 51, 78, 93, 159, 191, 209) | YME1L KO: mitochondrial fragmentation (3, 180, 191) YME1L-OMA1 DKO: maintain L-OPA1 amount, normal mitochondrial morphology (191) OMA1 KO: increased the number of tubular mitochondria (159) | Yes (1, 191) | OMA1 KO alone protects against cardiac dysfunction in various HF mouse models (1); YME1L KO promotes DCM and HF, and YME1L-OMA1 DKO rescues this cardiac phenotype (159, 191) |
| OPA1 | Acetylation (K926 and K931) | SIRT3 (166) | SIRT3 KO: hyperacetylation of OPA1 and mitochondrial fragmentation SIRT3 overexpression: maintain mitochondrial morphology and networks; protect from doxorubicin-mediated cell death | Yes (166) | Acetylation- and deacetylation-mimetic mutants were tested only in OPA1-null MEFs, not in CMs (166) |
Information about ubiquitination sites was collected from Sarraf et al. (168) and Chen and Dorn (29). CMs, cardiomyocytes; DCM, dilated cardiomyopathy; DKO, double knockout; ER, endoplasmic reticulum; HF, heart failure; Huwe1, E3 ubiquitin-protein ligase HUWE1; KO, knockout; L-OPA1, membrane-anchored long form of optic atrophy protein-1; MARCH5, E3 ubiquitin-protein ligase MARCH5; MEFs, mouse embryo fibroblasts; Mfn1 and Mfn2, mitofusin 1 and 2, respectively; ND, not determined; OMA1, overlapping with the m-AAA protease 1 homolog (a zinc metalloprotease); OMM, outer mitochondrial membrane; OPA1, optic atrophy protein-1; paraplegin, an m-AAA metalloprotease; PARL, presenilins-associated rhomboid-like protease; PINK1, phosphatase and tensin homolog (PTEN)-induced putative kinase 1; S1 and S2, sites 1 and 2, respectively; SIRT3, NAD-dependent deacetylase sirtuin-3; USP30, ubiquitin COOH-terminal hydrolase 30; YME1L, ATP-dependent zinc metalloprotease YME1L1.
POSTTRANSLATIONAL MODIFICATIONS OF MITOCHONDRIAL FISSION PROTEINS IN THE HEART
Overview of the Structure of Mitochondrial Fission Proteins and Their Posttranslational Modifications
Mitochondrial fission in mammalian cells (including CMs) is mediated by 1) an ~80-kDa cytosolic soluble protein, Drp1 (also called DLP1; Fig. 3), which was originally discovered as a member of the dynamin family of GTPases that drive cellular membrane remodeling, and 2) its receptors at the OMM [e.g., Mff (65, 138), MiD49, and MiD51, respectively (141, 210); see also introduction]. Human fission factor 1 (hFis1, 17 kDa) is a small OMM protein with a COOH-terminal single transmembrane (TM) domain, which was also proposed to act as a Drp1 receptor in mammalian cells on the basis of initial studies using yeast (123). Indeed, earlier reports showed that overexpression of hFis1 induces mitochondrial fragmentation in mammalian cells (86, 182, 201), but subsequent studies have claimed that KO or knockdown of hFis1 in mammalian cells does not affect Drp1 translocation/association to mitochondria (101, 112, 135, 137, 140). Although these studies do not exclude a possible role for hFis1 in mitochondrial fission independent of the recruitment of Drp1 to the OMM, we mainly focus on the function of the recently discovered Drp1 receptors (i.e., Mff, MiD49, and MiD51) rather than hFis1 in this review. It has been suggested that the relative contributions of each Drp1 receptor for overall fission activity may differ across cell types and tissues (112, 140). Mff is likely the primary Drp1 receptor at the OMM in the heart, since loss of Mff in vivo causes a significant decrease in mitochondrial fission and exacerbates cardiomyopathy (23; Table 2). Structurally, Drp1 is composed of a highly conserved NH2-terminal GTPase domain that provides mechanical force, a middle domain involved in higher-order self-assembly, a variable domain (also known as B insert) that functions in protein-protein interactions, and a GTPase effector domain (GED) that interacts with the NH2-terminal domains to regulate GTPase activity (64, 90, 196; Fig. 3). Importantly, B insert normally blocks the Mff binding sites on Drp1 and retains this protein as a dimer at the cytosol (64, 196). Once B insert binds to cardiolipin at the OMM, this allows for the exposure of the Mff binding site of Drp1 for Drp1-Mff interaction (111, 135). The binding of Drp1 dimers to Mff allows Drp1 assemblies to form functionally active Drp1 oligomers on the OMM as a helical ring structure, which promotes the GTP-mediated constriction of mitochondrial tubules (33, 163).
Fig. 3.

Schematic diagram of dynamin-related protein-1 (Drp1) structure and distribution of posttranslational modifications in cardiomyocytes and hearts. Structure-based domain architecture of human Drp1 isoform 1 is shown on the basis of the published data of Drp1 isoform 2 given by Fröhlich et al. (64). All the posttranslational modifications listed in this diagram are based on reports using native cardiomyocytes or heart tissues (see also Table 3). Information on sites of conjugation of small ubiquitin-like modifier (SUMO) proteins (SUMOylation) was collected from PhosphoSitePlus (80). BSE, bundle signaling element; CaN, Ca2+-dependent phosphatase calcineurin; GED, GTPase effector domain; O-GlcNAc, O-linked-N-acetyl-glucosamine; O-GlycNAcylation, O-linked-N-acetyl-glucosamine glycosylation; Pim-1, proto-oncogene serine/threonine-protein kinase; RIP1, receptor-interacting protein-1; ROCK, Rho-associated protein kinase; SENP5, sentrin/SUMO-specific protease 5; VD, variable domain.
Multiple PTMs of Drp1 have been detected by mass spectrometry-based proteomics, and some of them have been further investigated in cell systems to understand their roles in mitochondrial and cellular functions under physiological and pathophysiological conditions [see our recent review (90)]. These PTMs include phosphorylation, ubiquitination, conjugation of small ubiquitin-like modifier (SUMO) proteins (SUMOylation), S-nitrosylation, and O-linked-N-acetyl-glucosamine glycosylation (O-GlcNAcylation), which can modulate functional properties of Drp1 such as its subcellular localizations, protein-protein interactions (e.g., Drp1-Mff binding), protein stability, and GTPase activity (see also Fig. 1). Among these PTMs, the phosphorylation, SUMOylation, and O-GlcNAcylation of Drp1 have been reported in CMs and hearts (90; Table 3 and Fig. 3). Importantly, since the B insert region contains multiple sites for phosphorylation (Fig. 3), SUMOylation, and O-GlcNAcylation, these PTMs of Drp1 in particular may induce a conformational change of the B insert that controls the binding affinity of Drp1 with Mff and ultimately the mitochondrial fission activity in CMs. In addition to Drp1, there are several reports elucidating the PTMs of Drp1 receptors [e.g., AMP-activated protein kinase (AMPK)-dependent phosphorylation of Mff (49, 187) and E3 ubiquitin-protein ligase MARCH5 (MARCH5)-dependent ubiquitination of MiD49 (199)] and their influence on mitochondrial fission activity in noncardiac cells. Recently, Qi’s group reported the concomitant phosphorylation of Mff and Drp1, possibly by ERK1/2 and PKCδ, respectively, during succinate stimulation in neonatal CMs, suggesting that the functional consequences of increased fission in CMs may be derived from the combination of the effects of phosphorylation of both Drp1 and Mff (113). Future studies are required to precisely delineate the effects of PTMs of Drp1 and Mff separately under cardiac stress signaling.
Phosphorylation and O-GlcNAcylation of Drp1 in Cardiomyocytes and Cardiac Pathology
In this section, we introduce data regarding the function and regulation of mitochondrial fission proteins from adult CMs and cell models for adult CMs (see overview: mitochondrial morphology and fission and fusion proteins in cardiomyocytes). Putative Drp1 inhibitors including mitochondrial division inhibitor 1 (Mdivi-1) have been widely applied to inhibit the GTPase activity of Drp1 in cells and in vivo, which frequently exhibits protective effects from mitochondrial injury and cell death [see review (162)]. However, these beneficial effects of these drugs are likely via nonspecific inhibition of complex I (10), and thus the data obtained by the use of Mdivi-1 may not be suitable for understanding Drp1 function or the roles of PTMs of Drp1. Therefore, to precisely understand the roles of PTMs of Drp1 in CMs, we present our discussion here based on the accumulating data using the mutants of Drp1 that mimic or diminish PTMs of Drp1 as well as the experimental results from genetic modification of upstream signaling pathways, rather than the reports using pharmacological inhibition of Drp1 activity and upstream signaling. A full list of PTMs of Drp1 currently reported from all cell types and tissues is given in our recent review (90). The PTMs of Drp1 specifically tested or detected in CMs/hearts are summarized in Table 3.
Two phosphorylation sites on Drp1, Ser616 and Ser637 within the B insert, are well reported in both noncardiac and cardiac cells [see also our recent review (90)]. Ser616 has been shown to be phosphorylated by cyclin-dependent kinase 1 (CDK1/cyclin B; 116, 183, 184, 206), ERK1/2 (98, 160, 172, 203), PKCδ (47, 113, 157, 206), CaMKII (200), and Rho-associated protein kinase (ROCK; 12). On the other hand, the phosphorylation levels of Ser637 are regulated by PKA (20, 35, 71, 100), CaMKIα (76), PKD (91), Ca2+-dependent phosphatase calcineurin (CaN; 17, 149, 173), and protein phosphatase 2 (PP2A; 44, 119). Additionally, Ser637 is located within a consensus motif for ROCK and has been found to be phosphorylated by ROCK in endothelial cells (194).
Ser616 phosphorylation promotes mitochondrial fission, which is consistent with reports from multiple groups using various noncardiac cells (90). The next question is whether Ser616 phosphorylation of Drp1 under pathological conditions modulates mitochondrial morphology and function in CMs and hearts. Wang and Sheu’s group have recently proposed that CaMKII-dependent phosphorylation of Drp1 at Ser616 activates noncanonical functions of Drp1 [e.g., regulation of transient mitochondrial permeability transition pore (mPTP) opening and oxidative phosphorylation (208)] in adult CMs (200). However, the possibility remains that these effects can be mediated via the canonical function of Drp1 (i.e., increased mitochondrial fission via Ser616 phosphorylation) since the effect of Ser616 phosphorylation on mitochondrial morphology has only been tested in H9c2 cardiac myoblasts (91, 200) and neonatal CMs (12, 113) and has not yet been investigated in adult CMs. Further studies are required to quantitatively assess whether the canonical (i.e., mitochondrial fission) and noncanonical functions of Drp1 are linked to or occur independently of each other and how Ser616 phosphorylation regulates both functions in CMs under pathological conditions such as HF.
Unlike Ser616 phosphorylation, the impact of Ser637 phosphorylation on mitochondrial morphology is highly controversial in both noncardiac and cardiac cells (81). Overexpression of phosphomimetic or dephosphomimetic mutants of Drp1 (Ser637Asp and Ser637Ala, respectively) is frequently utilized to understand the functions of Ser637 sites in various cell systems but has resulted in conflicting outcomes for mitochondrial morphology: expression of Ser637Asp promotes mitochondrial elongation (17, 20, 35) or fragmentation (76), and Ser637Ala promotes fragmentation (17, 35), elongation (76), or no significant changes in mitochondrial morphology (20, 91; Table 3). Moreover, another PTM of Drp1, O-GlcNAcylation at Thr585 and Thr586, has been shown to result in dephosphorylation of Drp1 at Ser637 via an unknown mechanism, which promotes Drp1 translocation to the OMM and mitochondrial fragmentation under hyperglycemic conditions in neonatal CMs (66; Table 3). We also showed that PKD-dependent phosphorylation of Ser637 induces Drp1 association to the OMM, resulting in mitochondrial fragmentation under Gq protein-coupled receptor stimulations including α1-adrenoceptor (α1-AR) stimulation (10 μM phenylephrine for 30 min to 1 h) in neonatal CMs, which initially increases mitochondrial respiration but later promotes mPTP opening and apoptotic signaling activation (91). There are several possible causes for this discrepancy in the field. First, the effects of Ser637 phosphorylation and dephosphorylation have been tested in various cell types that have distinct mitochondrial shapes/networks at resting conditions and possess different sets of upstream signaling molecules (i.e., kinases and phosphatases) for regulating the phosphorylation state of Ser637. Second, because of variability of signaling cascades between different cell types, we cannot exclude the possibility of the coexistence of additional PTMs within Drp1 caused by different upstream molecules in addition to Ser637 phosphorylation. Here, we discuss further the discrepancy of the functions of Ser637 sites in CMs. Cribbs and Strack first showed that acute β-AR stimulation by an injection of the β-AR agonist isoproterenol in animals and exercise (forced swimming) promotes Ser637 phosphorylation in Drp1 in the heart in vivo, under the assumption that this phosphorylation occurs via PKA based on observations from noncardiac cells (35; Table 3). Indeed, the sequences around Ser637 match the consensus motif of PKA. Next, Wang’s group, using isolated adult CMs, confirmed that 12-h incubation with isoproterenol exhibits increased Ser637 phosphorylation by PKA (but not via CaMKII; 200). However, they could not detect increased Ser637 phosphorylation after 2-wk isoproterenol infusion in vivo, and the functional consequences of the Ser637 phosphorylation were not investigated both in CMs and in vivo in this publication (200). These observations can be partly attributed to 1) the activation of the Ca2+-dependent phosphatase CaN in the later phase of chronic β-AR stimulation (146) and/or 2) the activation of PP2A via the β2-AR-Gi protein pathway (108), which may dephosphorylate Drp1 at Ser637, similar to observations in noncardiac cells and neonatal CMs (35, 44, 119, 149). Thus, the phosphorylation levels of Ser637 by PKA/CaN pathways may vary time dependently during chronic β-AR stimulation (i.e., the time course of HF development) in vivo. We also reported that PKD-mediated phosphorylation at Ser637 under Gq protein-coupled receptor stimulation promotes mitochondrial fragmentation (91). Our observation is consistent with the previous results of CaMKIα- and ROCK1-dependent Drp1 phosphorylation at Ser637 in noncardiac cells (76, 194). However, these results show the opposite effect of what PKA does via Ser637. This discrepancy may be partly due to the involvement of different upstream molecules in different AR subtype stimulations in CMs (β-AR vs. α1-AR) that may not only regulate the phosphorylation levels of Ser637 in Drp1 but also modulate the phosphorylation levels of other site(s) within Drp1 or other mitochondrial fission/fusion machinery proteins. In line with this idea, we also tested β-AR- and PKA-mediated changes in Drp1 phosphorylation, its translocation to the OMM, and mitochondrial morphology (91). Indeed, β-AR stimulation activates PKA and increases Drp1 phosphorylation at Ser637, which promotes Drp1 translocation to mitochondria in H9c2 cells, similar to the case of PKD-dependent Drp1 phosphorylation at Ser637 (91). However, β-AR stimulation did not show any significant changes in mitochondrial morphology. To further investigate the molecular mechanisms producing this discrepancy between the effects of PKA and PKD on mitochondrial fission in H9c2 cells, we assessed the changes in phosphorylation levels of Drp1 at Ser616 before and after α1-AR stimulation using a dephosphomimetic mutant of Drp1, Drp1-Ser637Ala. Importantly, α1-AR stimulation significantly decreases Ser616 phosphorylation in cells overexpressing a Drp1-Ser637Ala mutant. This result indicates that phosphorylation at one site within Drp1 may prevent or initiate the phosphorylation or dephosphorylation at the other sites by changing the access of other kinases or phosphatases, since Ser616 and Ser637 may be proximal within the three-dimensional structure of Drp1 (64). Another speculation may be the requirement of basal Ser637 phosphorylation for maintaining Ser616 basal phosphorylation. Further experiments using Drp1 mutants with single or double mutations at Ser616 and Ser637 (17) in CMs may clarify the relative roles of these two sites in mitochondrial fission more precisely. In summary, it is likely that the phosphorylation status of Ser637 under physiological and/or pathological conditions participates in the regulation of mitochondrial fission in CMs as seen in Ser616 phosphorylation.
SUMOylation of Drp1 in Cardiomyocytes and Cardiac Pathology
Drp1 is also shown to be modified by the conjugation of SUMO proteins (i.e., SUMOylation), which occurs at multiple nonconsensus lysine sites within its B insert including Lys594, Lys597, Lys606, and Lys608 (59; Fig. 3 and Table 3). SUMOylation of Drp1 has been extensively studied in noncardiac cells, but its physiological and pathological relevance to mitochondrial and cellular functions is still controversial. For instance, McBride’s group showed that SUMOylation of Drp1 by mitochondrial-anchored protein/SUMO ligase (MAPL) stabilizes its oligomeric form on the OMM resulting in increased mitochondrial fission during apoptosis (155, 195), whereas deSUMOylation of Drp1 by sentrin/SUMO-specific protease 5 (SENP5) reduces mitochondrial fission assessed by knockdown of SENP5 (214). Later, they also reported that SENP5 translocation from the nucleus to the mitochondria and deSUMOylation of Drp1 at the onset of mitosis also resulted in increased Drp1 oligomerization, followed by mitochondrial fragmentation (213). On the other hand, Henley’s group showed that SENP3 (a SUMO-2/3-specific protease)-mediated deSUMOylation of Drp1 plays a major role in regulating mitochondrial fission (73). Currently, there are only few reports proposing significant roles of SUMOylation and deSUMOylation of Drp1 in CMs/hearts (99, 175), especially focusing on SENP5. Importantly, SENP5 was reported as only SENP that is overexpressed in human HF (99). In transgenic mice with CM-specific overexpression of SENP5 (99), decreased SUMO-2/3-modified Drp1 induces enlarged (or swollen) mitochondria and CM apoptosis, leading to cardiomyopathy and HF (99). Moreover, Calvert’s group found that the expression of SENP5 increases in conventional DJ-1 [also known as Parkinson’s disease autosomal recessive, early onset 7 (Park7)] KO mice, and DJ-1 deficiency exacerbates acute ischemia-reperfusion (I/R) injury partly via decreased SUMO-2/3-modified Drp1 (175). Importantly, decreased SUMO-2/3-modified Drp1 is preferentially localized in the mitochondrial fraction without changing the whole cell expression level of Drp1, suggesting that the SUMOylation state of Drp1 regulated by SENP5 likely influences its subcellular localization (i.e., mitochondrial translocation) rather than Drp1 protein expression in CMs, consistent with the results from one of the reports from McBride’s group using noncardiac cells (213). However, SENP5 may target mitochondrial fission/fusion proteins other than Drp1 in these in vivo models (99, 175), which could cause the unbalance of SUMOylation-deSUMOylation in other fission/fusion proteins that may ultimately impact mitochondrial morphology and/or function in vivo. Further studies are required to understand the relative contributions of SUMOylationof Drp1 to the regulation of mitochondrial morphology and function compared with other PTMs of Drp1 under HF. Since the SUMO conjugation pathways are likely to be potential targets in the prevention and treatment of HF (113, 118), the physiological and pathological relevance of SUMOylation with regard to mitochondrial fission and fusion proteins (see also roles of posttranslational modifications of mitochondrial fission and fusion proteins in mitochondrial fragmentation under heart failure) in CMs is a topic to explore in the future.
POSTTRANSLATIONAL MODIFICATIONS OF MITOCHONDRIAL FUSION PROTEINS IN THE HEART
Overview of the Structure of Mitochondrial Fusion Proteins and Their Posttranslational Modifications
Because mitochondria are double-membraned organelles, mitochondrial fusion requires initial fusion of the OMM, mediated by Mfn1 and Mfn2, followed by fusion of the inner mitochondrial membrane (IMM), which occurs mainly via OPA1 (18, 150, 192). The precise mechanisms underlying the coupling of OMM and IMM fusion machinery remain unclear, but the main driving force for mitochondrial fusion is GTP hydrolysis catalyzed by the appropriate GTPase. Mfn1 and Mfn2 are highly homologous dynamin-related GTPases (~80% similarity in humans) and share a similar molecular structure, consisting of an NH2-terminal GTPase domain, a heptad-repeat 1 domain (HR1), two TM domains, and a COOH-terminal heptad-repeat 2 domain (HR2; 161; Fig. 4A). Mfn1 and Mfn2 also possess a unique topology by spanning the OMM twice with their TM domains, which results in a short loop within the intermembrane space (IMS) and leaves major parts of the protein, including the GTPase domain, HR1, and HR2, exposed to the cytosol (Fig. 4B). Between HR1 and the TM domains, Mfn2 (but not Mfn1) possesses a proline-rich domain, which is likely responsible for Mfn2-specific protein-protein interactions (Fig. 4, A and B). Based on recent reports of mammalian Mfn1 structure (16, 158), the proposed model for the OMM fusion step is as follows (Fig. 4C): 1) HR2s initiate the tethering of adjacent mitochondria by forming HR2-HR2 dimers; 2) GTPase domains of Mfn proteins may contact each other across OMM tubules, suggesting that GTP-dependent oligomerization is also involved in the initial tethering in addition to the HR2-HR2 interaction; and 3) the membranes are pulled together by a power stroke dependent on GTP hydrolysis, allowing for the fusion of the two OMMs [see also review (61)]. Although this model was developed on the basis of structural information about Mfn1, it is likely to be applicable to Mfn2. Indeed, several groups have shown that the HR2 of Mfn interacts with the HR2 of other Mfn molecules to form homodimers (Mfn1-Mfn1 or Mfn2-Mfn2) and/or heterodimers (Mfn1-Mfn2) to initiate mitochondrial fusion (22, 102, 161). These results suggest that Mfn1 and Mfn2 are partially redundant in their function, as either homodimer can suffice to initiate mitochondrial fusion (61). However, several functions that differ between Mfn1 and Mfn2 have been reported. This may be partly due to the difference in 1) GTPase activities between Mfn1 and Mfn2 (recombinant Mfn1 exhibits an approximately eightfold higher GTPase activity than Mfn2; 83) and 2) subcellular expression patterns [e.g., in addition to the OMM, Mfn2 (but not Mfn1) is localized in the endoplasmic reticulum (ER)/sarcoplasmic reticulum (SR) and tethers ER/SR to the mitochondria; 28, 39]. On the basis of KO studies of Mfn2 in cell systems and in vivo (Table 2), in addition to its unquestioned role in OMM fusion, it has been proposed that Mfn2 serves as a key regulator of 1) ER-mitochondrial Ca2+ coupling (60), 2) ER stress response machinery at ER-mitochondria contact sites (124, 129, 171), 3) mitochondrial bioenergetics (170), and 4) axonal transport of mitochondria (120). Moreover, in addition to the interaction of HR2-HR2 and GTPase dimerization described above, Dorn’s group reported the importance of intramolecular binding of HR1 and HR2 in Mfn2 (62). They proposed that there are two states of Mfn2 structure: 1) at resting conditions, HR2 is restrained by antiparallel intramolecular binding to HR1, which forms the nonpermissive state; and 2) in the tethering-permissive state, destabilization of HR1-HR2 binding unfolds and extends HR2 into the cytosol, followed by the classic model of the HR2-HR2 interaction.
Fig. 4.

Schematic diagram of structure and distribution of posttranslational modifications of mitofusin 1 and 2 (Mfn1 and Mfn2, respectively), and mechanism of outer mitochondrial membrane (OMM) fusion by Mfn proteins. A: structure-based domain architectures of human Mfn1 and Mfn2 are depicted on the basis of the published data given by Franco et al. (62; see also Table 4). TM, transmembrane domain; HR, heptad-repeat domain. B: Mfn2 topology at the OMM, with two TM domains, a small bridge structure at the intermembrane space, and large structures at the cytosolic space. C: representative cartoon shows Mfn2-Mfn2 interaction. Initial tethering is mediated between heptad-repeat 2 domains (HR2s) from the opposite OMM (step 1). Next, dimerization of GTPase domains occurs (step 2), and power stroke from GTPase hydrolysis promotes OMM fusion (step 3). CMs, cardiomyocytes; IMM, inner mitochondrial membrane; PINK1, phosphatase and tensin homolog (PTEN)-induced putative kinase 1; PR, proline-rich domain.
IMM fusion is regulated by the IMM-associated GTPase OPA1 in mammalian systems (7, 104, 107). OPA1 consists of an NH2-terminal mitochondrial-targeting sequence (MTS), a TM, an HR, a GTPase domain, a middle domain, and a COOH-terminal GED (Fig. 5A). The NH2-terminal TM of OPA1 is embedded in the IMM, and the main part of the protein is located in the IMS, allowing it to interact with the loop structure of Mfn proteins for successful coordination to complete mitochondrial fusion (31, 181). The exceptional complexity of the regulation of this protein compared with other mitochondrial fission and fusion proteins is due to the existence of multiple alternative splicing variants [8 variants in humans involving exons 4, 4b, and 5b (40)] and proteolytic processing in the mitochondria (Fig. 5, B and C). Since all OPA1 proteins have an MTS at the NH2 terminus, all OPA1 proteins can be imported into mitochondria via the OMM and IMM translocases. Subsequently, all OPA1 proteins can be proteolytically cleaved in the mitochondria at the NH2-terminal side, which forms combinations of membrane-anchored long forms (termed L-OPA1) and soluble short forms of OPA1 (termed S-OPA1; 7, 107; Fig. 5B). Historically, protease presenilins-associated rhomboid-like protease (PARL; 32) and the m-AAA metalloprotease paraplegin (84) were proposed as the main molecules responsible for OPA1 processing (Table 4). However, later studies found that cells lacking PARL or paraplegin maintain normal OPA1 processing (50, 72). The present established model for OPA1 processing is mediated via the function of two mitochondrial proteases, the i-AAA metalloprotease ATP-dependent zinc metalloprotease YME1L1 (YME1L; 3, 72, 180, 191) and the zinc metalloprotease, overlapping with the m-AAA protease 1 homolog (OMA1; 1, 3, 51, 78, 191; Table 4 and Fig. 5, A and B). All variants of OPA1 contain exon 5 with the site 1 (S1) cleavage site targeted by OMA1, whereas half of variants contain the site 2 (S2) cleavage site processed by another peptidase, YME1L (Fig. 5, B and C). Since cleavage at S1 is incomplete without the exon 4b-encoded sequence, some of the variants result in both uncleaved L-OPA1 and cleaved S-OPA1 (Fig. 5C). S-OPA1 is distributed in the IMS because of the lack of a membrane-anchoring TM, but it still retains its GTPase domain (Fig. 5B). S-OPA1 may associate with the IMM via interactions with L-OPA1 integrated in the IMM. The next question is whether there are different roles between L-OPA1 and S-OPA1. Regarding the disputed function of OPA1 (i.e., IMM fusion), the relative contributions of OPA1 isoforms are still controversial. Originally, Chan’s group reported that both L-OPA1 and S-OPA1 are required for successful fusion between mitochondria (180). However, other groups have shown that L-OPA1 alone is sufficient for mitochondrial fusion, whereas S-OPA1 alone is not capable of forming fusion machinery or has a minor role in mitochondrial fusion (3, 84, 106, 186). On the basis of KO studies in cell systems, OPA1 is also proposed to have other functions in addition to mitochondrial fusion. These include 1) maintenance of mtDNA content, 2) regulation of mitochondrial bioenergetics, and 3) cristae organization [see reviews (40, 107)]. Originally, the proposed mechanism underlying the regulation of cristae tightness (i.e., the narrowness of the width of the intracristal space formed between 2 cristae membranes) and bioenergetics by OPA1 is via 1) trans-interaction of L-OPA1 or 2) L-OPA1 oligomerization facilitated by S-OPA1 as a molecular staple (40, 107), which regulates the assembly and stability of the respiratory chain supercomplexes as well as respiratory efficiency (34). However, these models may require significant revision based on recent findings, using an OPA1 KO cell line with selective expression of each OPA1 isoform (41, 106). Using this system, two groups confirmed that S-OPA1 has a minor role or no function in mitochondrial fusion and L-OPA1 is sufficient for recovering mitochondrial fusion in OPA1 KO cell lines (41, 106). Moreover, Yoon’s group showed that expression of OPA1 variant 5, which produces only S-OPA1 (see Fig. 5C), is sufficient to maintain mtDNA, respiratory complexes, and cristae structure, indistinguishable from the functions of L-OPA1 (106). Zanna’s group tested all eight variants and found no differences in the effect of bioenergetics between each variant (41). These results indicate that S-OPA1 alone is sufficient for the maintenance of mitochondrial bioenergetics, which supports the original idea from Scorrano’s group that the mitochondrial fusion function of OPA1 is molecularly distinct and independent from cristae maintenance by OPA1 (63).
Fig. 5.

Schematic diagram of optic atrophy protein-1 (OPA1) structure and variant-dependent proteolysis. A: structure-based domain architecture of human OPA1 is depicted. Numbers in the domains indicate the exon number. Red exons are involved in the formation of splice variants. B: variant-dependent proteolysis and the structures of the membrane-anchored long and soluble short forms of OPA1 (L-OPA1 and S-OPA1, respectively). C: summary table of variant-dependent proteolysis. GED, GTPase effector domain; IMM, inner mitochondrial membrane; IMS, intermembrane space; MPP, mitochondrial processing peptidase; MTS, mitochondrial-targeting sequence; OMA1, overlapping with the m-AAA protease 1 homolog; S1 and S2, sites 1 and 2; TM, transmembrane domain; YME1L, i-AAA metalloprotease.
In addition to the KO/ablation studies both in cell systems and in vivo (see Table 2), PTMs of Mfn1, Mfn2, and OPA1 have also been investigated (Table 4). Multiple PTMs of human mitochondrial fusion proteins such as phosphorylation (in Mfn1, Mfn2, and OPA1), acetylation (in Mfn1, Mfn2, and OPA1), methylation (in Mfn1 and OPA1), and ubiquitination (in Mfn1, Mfn2, and OPA1) have been detected by mass spectrometry-based proteomics as shown in online open databases [e.g., PhosphoSitePlus (80)]. However, the regulation of mitochondrial fusion proteins by PTMs remains largely uncharacterized compared with Drp1. For instance, the following PTMs of Mfn1 and Mfn2 have been investigated in noncardiac cells: 1) phosphorylation (in Mfn1 and Mfn2), 2) acetylation (in Mfn1), 3) ubiquitination (in Mfn1 and Mfn2), and 4) cysteine oxidation (in Mfn2; Table 4 and Fig. 4A). Currently, there is a single report of PTMs of Mfn2 (phosphorylation and ubiquitination) in CMs/hearts, which will be discussed in the following sections. Although Mfn1 is highly expressed in the heart compared with Mfn2 (57), there are no reports of PTMs of Mfn1 in CMs/hearts. The investigation of PTMs of OPA1 mainly focuses on proteolytic processes as described above. Most of the prior studies use genetic manipulations of the responsible proteases to modify the L-OPA1-to-S-OPA1 ratio in cells or in vivo (3, 72, 78, 180, 191) or the overexpression of specific OPA1 variants and/or a mutant OPA1 variant 1 that lacks S1 sites, which forms only L-OPA1 in the mitochondria (41, 106). In the following section, we mainly discuss the physiological and pathological relevance of the proteolytic cleavage of OPA1 in CMs and hearts and also introduce PTMs of OPA1 specifically reported in CMs.
Phosphorylation and Ubiquitination of Mfn2 in Cardiomyocytes and Cardiac Pathology
In the heart, Mfn1 and Mfn2 are required for early embryonic (30, 97) and postnatal development (143). Moreover, double KO (DKO) of both Mfn1 and Mfn2 in adult mice showed dramatic changes in mitochondrial morphology, respiration, and contractile function, resulting in HF and CM death (28, 75; see Table 2). Similar results were observed from CM-specific Mfn2 single-KO mice, but their cardiac phenotype was milder than that of the DKO (28, 142). Although Mfn1 expression is higher than that of Mfn2 in the heart (57), CM-specific Mfn1 KO does not cause any significant changes in cardiac function (28, 143). Given that both Mfn1 and Mfn2 are capable of inducing mitochondrial fusion, these observations indicate that 1) Mfn1 cannot take the place of (or substitute for) Mfn2-specific functions such as mitochondria-SR tethering and mitophagy; 2) Mfn2 can compensate mitochondrial fusion activity under Mfn1 deletion; and 3) inhibition of Mfn2-specific functions by Mfn2 KO may contribute to the mechanisms of cardiac pathology observed in the transgenic mice. Indeed, Mfn2 KO increases the distance between mitochondria and SR (28) and delays in Ca2+-induced mPTP opening (142). Moreover, Chen and Dorn showed that CM-specific Mfn2 KO promotes cardiomyopathy via impaired mitophagy with the accumulation of damaged mitochondria (29). They also showed that Mfn2 serves as a Parkin receptor on damaged mitochondria, which promotes mitochondrial recruitment of Parkin; phosphatase and tensin homolog (PTEN)-induced putative kinase 1 (PINK1)-dependent Mfn2 phosphorylation, possibly at Thr111 and Ser442, facilitates Parkin binding to Mfn2, followed by its Parkin-mediated ubiquitination of Mfn2 (Table 4 and Fig. 4A). Thus, ablation of Mfn2 in mouse CMs prevented depolarization-induced translocation of Parkin to the mitochondria and suppressed mitophagy. Historically, Mfn2 was shown to interact with the E3 ubiquitin ligase MARCH5 (96, 125), but evidence for neither direct ubiquitination nor degradation of Mfn2 was observed in these initial publications. Later, Taanman’s group reported the ubiquitination of both Mfn1 and Mfn2 in a PINK1/Parkin-dependent manner upon induction of mitophagy (67), indicating that inhibition of the PINK1/Parkin/Mfn1/2 pathway may contribute to the pathogenesis of Parkinson’s disease. Thus, the report from Chen and Dorn in CMs (29) is indeed well recapitulated by the findings of Taanman’s group in neurons (67). Similarly, on the basis of in silico consensus sequence analysis, several groups showed that Mfn2 can be phosphorylated by PKA at Ser442 in noncardiac cells (24, 212), which is the same site as the predicted PINK1-dependent phosphorylation site on Mfn2 in CMs (Table 4 and Fig. 4A). These results suggest that the function of Mfn2 is posttranslationally regulated in CMs and hearts. However, future studies may be required to further address the following questions: 1) Does the activity of the PINK1/Parkin pathway change under cardiac pathological conditions such as HF, myocardial infarction, and I/R? 2) Since Parkin expression is lower in the heart compared with in neurons (46), what is the relative contribution of Mfn2-mediated mitophagy compared with other mechanisms such as Drp1-mediated mitophagy (82, 164, 176)? 3) Since PKA signaling is one of the predominant cell-signaling pathways in CMs and also important in the development of cardiac pathologies (9), can PKA phosphorylate Mfn2 similar to PINK1? 4) Do PTMs of Mfn2 modulate other Mfn2 functions such as mitochondria-SR interactions under physiological and pathological conditions? Finally, since there are several reports regarding PTMs of Mfn1 (145, 156) and their functions in noncardiac cells (but not in CMs) and Mfn1 is highly expressed in CMs (57), it should be noted that the investigation of PTMs of Mfn1 and their functional relevance in CMs may also be an important topic to explore in the future.
Proteolytic Cleavage of OPA1 in Cardiomyocytes and Cardiac Pathology
As described above, proteolytic cleavage of OPA1 is regulated by YME1L and OMA1 (see posttranslational modifications of mitochondrial fusion proteins in the heart, Overview of the Structure of Mitochondrial Fusion Proteins and Their Posttranslational Modifications). In noncardiac cells, it has been shown that cellular stress, mitochondrial membrane depolarization, and KO of YME1L can activate the other protease, OMA1, which promotes the conversion of L-OPA1 into S-OPA1 and subsequent mitochondrial fragmentation (3, 6, 93, 159, 209; Table 4). Later, Langer’s group investigated the role of OPA1 processing on mitochondrial morphology in the CMs and cardiac function in the heart by generating CM-specific YME1L and OMA1 KO mouse lines (191). First, they confirmed via conventional KO that YME1L, but not OMA1, is essential for embryogenesis. Next, using cardiac-specific YME1L KO mice, they found that the ablation of YME1L activates OMA1 and accelerated OPA1 proteolysis (see also Fig. 5, B and C), which promotes mitochondrial fragmentation and altered cardiac metabolism, followed by dilated cardiomyopathy and HF. Importantly, these cardiac and mitochondrial dysfunctions can be rescued by OMA1 KO because of the prevention of excessive OPA1 cleavage. The authors conclude that L-OPA1 is sufficient to maintain heart function, and OMA1 is a critical regulator of CM survival and the maintenance of mitochondrial function via regulating proteolytic cleavage of OPA1. Finally, Enríquez’s group also reported that OMA1 KO alone protects against cardiac dysfunction in various HF mouse models (1). These reports raise the intriguing possibility that the proteolytic cleavage of OPA1 is critical for the maintenance of mitochondrial morphology and function in the heart and dysregulation of this process contributes to the pathogenesis of HF. However, since proteases such as YME1L and OMA1 likely have multiple target proteins in CMs, it is difficult to completely exclude the possibility that the phenotypes observed in these mouse lines are mediated by the alterations in the proteolysis of these other proteins in CMs, but not solely via OPA1 cleavage. Future studies are required to delineate the differential roles of L-OPA1 and S-OPA-1 in CMs by reexpressing specific OPA1 variants and/or a mutant OPA1 variant 1 (41, 106; see Fig. 5C) in the OPA1-ablated mouse lines (see Table 2).
Acetylation and O-GlcNAcylation of OPA1 in Cardiomyocytes and Cardiac Pathology
In addition to proteolysis, several other PTMs of OPA1 are reported such as acetylation and O-GlcNAcylation (Table 4). Gupta’s group first found the hyperacetylation of OPA1 in cardiac pressure overload animal models (166). In addition, they found that a mitochondrial deacetylase, NAD-dependent deacetylase sirtuin-3 (SIRT3), is capable of directly deacetylating OPA1 at its GED (Lys926 and Lys931; see Fig. 5A), which increases its GTPase activity. Moreover, they showed that CM-specific SIRT3 KO promotes hyperacetylation of OPA1 and mitochondrial fragmentation in the heart. Finally, they confirmed that SIRT3-dependent activation of OPA1 protects CMs from doxorubicin-mediated cell death by maintaining mitochondrial morphology and networks. Later, using H9c2 cells, De Rasmo’s group proposed that PKA signaling, which is one of the major cell-signaling pathways in CMs (9), can modify SIRT3 activity and regulate OPA1 activity (177). The reduction of the mitochondrial cAMP pool promotes SIRT3 protein degradation, followed by hyperacetylation of OPA1, and also triggers the proteolytic cleavage of L-OPA1 and subsequent CM apoptosis. These observations suggest that cardiac stress such as pressure overload or chronic AR stimulation induces OPA1 hyperacetylation, which may contribute to the reduction of the activity of mitochondrial fusion machinery in CMs under HF in vivo. Since SIRT3 KO or overexpression changes the acetylation levels of multiple proteins in the CMs, experimental designs such as reexpressing deacetylation- or acetylation-mimetic OPA1 mutants in the OPA1 ablation animal models (see Table 2) may be appropriate to further elucidate the role of acetylation on OPA1 at Lys926 and Lys931 in CMs and hearts. Similarly, although O-GlcNAcylation of OPA1 was detected under hyperglycemic conditions in neonatal CMs (114), further studies using O-GlcNAcylation site mutants of OPA1 may be necessary to precisely dissect the role of this PTM for OPA1 function in CMs.
ROLES OF POSTTRANSLATIONAL MODIFICATIONS OF MITOCHONDRIAL FISSION AND FUSION PROTEINS IN MITOCHONDRIAL FRAGMENTATION UNDER HEART FAILURE
As described in the introduction, abnormal mitochondrial morphologies, especially small, disorganized interfibrillar mitochondria, are a hallmark feature of failing hearts in both human patients (43, 95, 154, 169) and animal HF models (11, 13, 25, 36, 87, 167, 198). As summarized in this review, mitochondrial dynamics have been widely studied using KO/overexpression systems (Table 2) and/or manipulation of PTMs of mitochondrial fission and fusion proteins (Tables 3 and 4), which has helped advance the understanding of the pathogenesis and molecular mechanisms of HF. In this section, we integrate the information listed from overview: mitochondrial morphology and fission and fusion proteins in cardiomyocytes, posttranslational modifications of mitochondrial fission proteins in the heart, and posttranslational modifications of mitochondrial fusion proteins in the heart and comprehensively discuss how PTMs of mitochondrial fission and fusion proteins affect mitochondrial morphology and function under HF (Fig. 6).
Fig. 6.

Posttranslational modifications (PTMs) of mitochondrial fission and fusion proteins by cardiac pathological signaling promote heart failure but also serve as compensatory mechanisms for protecting cardiomyocytes (CMs). DLP1, dynamin-related protein-1; Mfn2, mitofusin 2; mPTP, mitochondrial permeability transition pore; OPA1, optic atrophy protein-1; ROS, reactive oxygen species.
Enhancement of Mitochondrial Fission Activity under Heart Failure
As described in the introduction, overall fission activity is determined by at least the following three factors: 1) the total expression levels of fission proteins in the cell; 2) the expression levels of these proteins at the mitochondria; and 3) the activities of GTPases (see also Fig. 1). First, increased Drp1 protein expression is observed in several animal models of HF (but not all models), which may promote mitochondrial fission (Table 1). Second, as shown in posttranslational modifications of mitochondrial fission proteins in the heart, Overview of the Structure of Mitochondrial Fission Proteins and Their Posttranslational Modifications, the phosphorylation status of Drp1 regulates the translocation of Drp1 from the cytosol to the OMM (or the binding affinity to the Drp1 receptor Mff), which increases the amount of Drp1 at the OMM. AR signaling, hypoxia, and hyperglycemic signaling in the CMs, which are critical in HF pathogenesis, myocardial infarction, and diabetic cardiomyopathy, respectively, and oxidative stress-sensitive signaling (e.g., CaMKII, PKCδ, and PKD activation) are likely involved in this mechanism (Table 3 and Fig. 6). For instance, Shirakabe and colleagues reported increased Drp1 translocation to mitochondria in a mouse model of nonischemic HF generated by transverse aortic constriction (176). Since these Drp1 phosphorylations likely do not modulate the GTPase activity of Drp1 in vitro or in situ (35), increased mitochondrial fission activity under chronic HF is mainly a result of increased levels of Drp1 at the OMM (see also Fig. 1). Moreover, SENP5 is overexpressed in human HF (99), and SENP5-dependent deSUMOylation of Drp1 may also promote the translocation of Drp1 to the mitochondria under pathological conditions in the heart, followed by mitochondrial fragmentation and CM apoptosis (175; see also posttranslational modifications of mitochondrial fission proteins in the heart, SUMOylation of Drp1 in Cardiomyocytes and Cardiac Pathology). Finally, we also need to take into consideration that Drp1 receptors at the OMM (e.g., Mff) also receive PTMs and may contribute to Drp1 retention at the OMM, although there is currently less information regarding the PTMs of Drp1 receptors compared with Drp1 (see also posttranslational modifications of mitochondrial fission proteins in the heart, Overview of the Structure of Mitochondrial Fission Proteins and Their Posttranslational Modifications).
The last question is whether GTPase activity of Drp1 itself changes under HF. There are numerous reports that genetic and pharmacological inhibition of GTPase activity of Drp1 (see also posttranslational modifications of mitochondrial fission proteins in the heart, Phosphorylation and O-GlcNAcylation of Drp1 in Cardiomyocytes and Cardiac Pathology) is beneficial for protecting CMs and hearts from mitochondrial dysfunction [e.g., I/R injury (134, 207)]. However, these observations do not directly indicate that the GTPase activity of Drp1 itself increases under cardiac pathological conditions. Further studies are needed to directly measure the GTPase activity of Drp1 in CMs/hearts under various pathological conditions or in animal models of HF. For instance, Nishimura and colleagues recently found that the GTP-binding activity of Drp1 is significantly increased in a mouse model of myocardial infarction (131), indicating that the molecular mechanisms underlying enhanced mitochondrial fission activity during HF may differ in various types of cardiac stress (e.g., ischemic vs. nonischemic).
In summary, future experiments should be designed to quantitatively address the relative contributions of 1) Drp1 expression levels, 2) Drp1 translocation to the OMM, and 3) GTPase activity of Drp1 to the overall activity of fission machinery in CMs under HF.
Inhibition of Mitochondrial Fusion Activity under Heart Failure
As shown in posttranslational modifications of mitochondrial fusion proteins in the heart, mitochondrial fusion is mediated via Mfn1, Mfn2, and OPA1. Although Mfn1 is highly expressed in the CMs, PTMs of Mfn1 have not been well investigated. Parkin-mediated ubiquitination of Mfn2 leads to its degradation, resulting in decreased fusion activity, and promotes mitochondrial fragmentation, which leads to the selective removal of damaged mitochondria by mitophagy in adult CMs (29; Fig. 6; see also roles of posttranslational modifications of mitochondrial fission and fusion proteins in mitochondrial fragmentation under heart failure, Mitochondrial Fragmentation and Mitochondrial Dysfunction under Heart Failure). Parkin-dependent ubiquitination of Mfn2 is likely involved in the decreased Mfn2 expression observed in some HF animal models (Table 1), but further studies are required to understand how Parkin/PINK1/Mfn2 can be activated under HF.
Fusion activity of OPA1 is mainly regulated by the amount of L-OPA1, although the role of S-OPA1 in CMs is still under debate. Since the stress response protease OMA1 regulates OPA1 proteolysis and subsequently modulates the amount of L-OPA1 in the heart (1, 191), activation of OPA1 proteolysis is a possible mechanism for decreasing fusion activity in HF (Fig. 6). Moreover, hyperacetylation of OPA1 possibly via decreased activity of PKA and SIRT3 may also be involved in the activation of OPA1 proteolysis under HF (177).
Mitochondrial Fragmentation and Mitochondrial Dysfunction under Heart Failure
Overall, the mitochondrial fragmentation frequently observed in HF is likely attributed to the following two aspects that simultaneously occur via multiple signaling pathways activated under HF (Fig. 6): 1) increased mitochondrial fission activity (see roles of posttranslational modifications of mitochondrial fission and fusion proteins in mitochondrial fragmentation under heart failure, Enhancement of Mitochondrial Fission Activity under Heart Failure) and 2) decreased fusion activity (see roles of posttranslational modifications of mitochondrial fission and fusion proteins in mitochondrial fragmentation under heart failure, Inhibition of Mitochondrial Fusion Activity under Heart Failure). The next question is how mitochondrial fragmentation and/or these two aspects via PTMs of mitochondrial fission and fusion proteins mentioned above cause cardiac mitochondrial dysfunction under HF.
It is still largely unknown whether mitochondrial fission and fusion events influence the beat-to-beat-based regulation of physiological excitation-contraction/metabolism coupling in CMs. On the other hand, it is well documented and demonstrated that mitochondrial fragmentation occurs under both acute and chronic cardiac stress (see introduction) and pharmacological inhibition of the GTPase activity of Drp1 protects CMs and hearts from mPTP opening (82, 115, 134, 173, 200, 207). Although it is still unclear how Drp1 is involved in mPTP opening, several possible molecular mechanisms have been proposed: 1) direct association of Drp1 to the mPTP structure at the OMM triggers mPTP opening; 2) indirect association of Drp1 with other OMM proteins indirectly facilitates mPTP opening (208); and/or 3) there is a link between mitochondrial morphology including mitochondrial cristae structures and contact site stabilization [155; see also reviews (74, 178)]. Recent studies strongly suggest that mitochondrial ATP synthase (FoF1 ATP synthase) is a critical component of mPTP and the c-subunit of mitochondrial ATP synthase serves as a pore-forming subunit of mPTP (74, 122). However, the molecular identity of the OMM-spanning component of mPTP is still unknown. Therefore, it is currently very challenging to investigate the possibility of a direct association of Drp1 with the OMM-spanning component of mPTP. Second, since Drp1 can associate with other OMM-bound proteins and also increase mPTP-independent OMM permeability, it is plausible that Drp1 indirectly facilitates mPTP activity via its association with other OMM proteins (121). However, a link between macrostructural and microstructural changes in mitochondrial morphology is the most reasonable of these three possibilities since inhibition of GTPase activity of Drp1 efficiently blocks mPTP opening. Mitochondrial fragmentation by Drp1 may initially increase the efficiency of electron transport at the electron transport chain (ETC) by affecting the ultrastructural and spatial organization of the respiratory chain and ATP synthase (i.e., cristae remodeling), thus stimulating forward electron flow through the ETC and increasing ATP production (34). Indeed, several groups including ours have shown that respiration increases under short-term α1-AR stimulation or high-glucose treatment (89, 91, 204). Moreover, Wang and Sheu’s group showed that pharmacological and genetic inhibition of the GTPase activity of Drp1 inhibits the ETC activity and the frequency of transient mPTP opening (208). Under well-coupled conditions, stimulation of forward flow by itself would oxidize the ETC and thereby lower ROS levels (130). However, increased electron flow at the ETC may oxidize NADH and NADPH and thus deplete the antioxidative capacity (4). Another possibility is that mitochondrial fragmentation may structurally limit the size of the matrix cavity and mitochondrial network (see also introduction), which contributes to the decrease in the amount of antioxidative enzymes in the matrix. Thus, chronic mitochondrial fragmentation serves as one of the upstream mechanisms for increased mitochondrial ROS, mitochondrial membrane depolarization, and enhanced long-lasting mPTP opening under HF (Fig. 6).
Changes in the levels of PTMs of mitochondrial fission and fusion proteins also alter mitochondrial function in addition to maintenance of mitochondrial morphology. First, dysregulation of mitochondrial cristae formation (or loss of its structure) under HF is well reported in addition to the alteration of global mitochondrial morphology (i.e., mitochondrial fragmentation), which also contributes to inefficient energy production in cardiac mitochondria under HF (13, 25, 36). As discussed in posttranslational modifications of mitochondrial fusion proteins in the heart, Proteolytic Cleavage of OPA1 in Cardiomyocytes and Cardiac Pathology and Acetylation and O-GlcNAcylation of OPA1 in Cardiomyocytes and Cardiac Pathology, this is partly due to the dysregulation of OPA1 activity via proteolysis and alteration of the L-OPA1-to-S-OPA1 ratio. However, it is still unclear why the existence of S-OPA1 is insufficient for maintaining the cristae structures in cardiac mitochondria under HF (1, 191), which is clearly different from observations in noncardiac cells (41, 106). Further studies are needed to address whether other IMM proteins that stabilize cristae morphology [e.g., coiled-coil-helix-coiled-helix domain containing 3 (CHCHD3) and mitofilin (37)] also alter its function during HF in addition to OPA1. Although OMM association of Drp1 is also proposed as a direct regulator of mitochondrial bioenergetics in adult CMs (193), further studies are required to identify the mechanisms of how events in the OMM (i.e., Drp1 translocation) are transmitted to the ETC (which is located at the IMM) and how PTMs of Drp1, especially its phosphorylation, contribute to this noncanonical function of Drp1. Finally, mitochondria-SR interaction may be altered during HF, which contributes to the alterations of Ca2+ and ROS cross talk between these two organelles and the regulation of mitochondrial bioenergetics in CMs (14; see also posttranslational modifications of mitochondrial fission proteins in the heart, Overview of the Structure of Mitochondrial Fission Proteins and Their Posttranslational Modifications, and Fig. 2D). Since the importance of Mfn2 tethering functions in CMs has been tested only under Mfn2 KO conditions (28), next steps include exploring whether PTMs of Mfn2 influence mitochondria-SR tethering by changing the three-dimensional structure of Mfn2 or the expression levels of Mfn2 at the contact sites.
The last question that needs to be addressed is whether PTMs of mitochondrial fission and fusion proteins are involved in the development of cardiac pathology or just an adaptation/maladaptation to pathological changes in the heart. As described above, mitochondrial fragmentation may initially work as a compensatory mechanism for matching up with the increased energetic demands in failing hearts due to energy depletion or the inefficient energy production (e.g., increased glycolysis) during HF. However, this may soon turn into a detrimental pathway that increases ROS and mPTP opening, thus activating apoptotic signaling. In addition, mitochondrial fragmentation and the PTMs of fission and fusion proteins may act as an important compensatory mechanism, eliminating damaged mitochondria (i.e., mitophagy, see also posttranslational modifications of mitochondrial fusion proteins in the heart, Overview of the Structure of Mitochondrial Fusion Proteins and Their Posttranslational Modifications and Phosphorylation and Ubiquitination of Mfn2 in Cardiomyocytes and Cardiac Pathology; Fig. 6). Indeed, Sadoshima’s group has recently proposed that Ser616 phosphorylation of Drp1 and subsequent mitochondrial fragmentation are involved in Ras-related protein Rab-9A (Rab9)-associated mitophagy after ischemic stress (164; Table 3). Although multiple technologies have recently been developed, especially the use of fluorescent-labeling techniques, quantitative measurements of mitophagy activity in cells and tissues are still challenging. This includes visualization of 1) colocalization and recruitment of accessory factors [e.g., microtubule-associated protein 1A/1B-light chain 3 (LC3; 82, 94, 176) and Rab9 (164)] by immunostaining and/or overexpression of the fluorescent protein-fused factors and 2) the changes in pH in mitochondria when they are incorporated into lysosomes by overexpressing mitochondria-targeted (mt) acid-stable fluorescent protein [e.g., mt-Keima; see also reviews (48, 165)]. Importantly, it should be noted that PTMs of mitochondrial fission and fusion proteins change the mitochondrial morphology/network and the mitochondrial membrane potential, which directly or indirectly affects these fluorescence-based measurements for mitophagy. For instance, mt-Keima concentration in the mitochondrial matrix may be increased simply by mitochondrial fission events that limit the space available for the diffusion of mt-Keima in the mitochondrial network. This would result in changes of its fluorescence value independent of pH change. The other thing to consider is how to titrate the fluorescence value of mt-Keima in mitochondria with different membrane potentials, since mt-Keima may change its fluorescence value in depolarized mitochondria (but not in the process of mitophagy) independent of lysosomal association. The development of new technologies/tools is crucial to dissect the events that alter mitochondrial morphology, membrane potential, and pH to more quantitatively measure the mitophagic flux. This will help advance our understanding of the molecular mechanisms linking PTMs of fission and fusion proteins and mitophagy in CMs under cardiac stress.
In summary, PTMs of mitochondrial fission and fusion proteins by cardiac pathological signaling may serve as a cause of dysregulated mitophagy and mitochondrial dysfunction and simultaneously work as a compensatory mechanism for mitochondrial and cellular function in CMs and hearts (Fig. 6).
SUMMARY AND CONCLUDING REMARKS
As summarized in this review, there has been substantial progress in understanding the detailed molecular mechanisms of how PTMs of mitochondrial fission and fusion proteins modulate important mitochondrial functions such as mitochondrial dynamics, bioenergetics, Ca2+ transport, ROS generation, mPTP activity, and mitophagy in noncardiac cells. Moreover, thanks to recent studies using cultured CM model cells, adult CMs, and cardiac-specific KO mouse lines, the biological importance of mitochondrial fission and fusion proteins in cardiac physiology and pathologies has been gaining recognition. Changes in the activity of mitochondrial fission and fusion proteins via their PTMs in CMs likely contribute to the development of mitochondrial and cellular dysfunctions and also serve as compensatory mechanisms to protect the CMs from cardiac stress. Therefore, modulations of these PTMs of fission and fusion proteins may have promise as potential therapeutic targets for preventing and/or treating cardiac disease. However, it is still up for debate whether altered levels of PTMs of mitochondrial fission and fusion proteins are simply the reflection of overall impairment of cellular function at the end stage of these diseases; some types of PTMs of mitochondrial fission and fusion proteins may be the consequence of the activation of multiple cell-signaling pathways under HF and may not have functional relevance. Approaches such as quantitative measurements of mitochondrial morphology/dynamics, mitochondrial Ca2+ handling, bioenergetics, mitochondrial ROS levels, and mPTP activity in cultured CM model cells and adult CMs, combined with the expression of mutant proteins that prevent or enhance PTM effects, may provide clear answers as to whether these PTMs contribute to the regulation of mitochondrial morphology and functions. Finally, in addition to cardiac-specific KO/ablation animals, the development of knock-in mouse lines carrying PTM-mimetic or PTM-removal mutants of fission and fusion proteins may further address the physiological and pathological importance of these modifications in vivo.
GRANTS
This work was supported by Brown University Karen T. Romer Undergraduate Teaching and Research Award (to S. M. Adaniya), American Physiological Society (APS) 2017 Shih-Chun Wang Young Investigator Award (to J. O-Uchi), National Heart, Lung, and Blood Institute Grant R01-HL-136757 (to J. O-Uchi), American Heart Association (AHA) Grant 16SDG27260248 (to J. O-Uchi), National Institute of General Medical Sciences Grant U54-GM-115677 (to B. S. Jhun), and AHA Grant 18CDA34110091 (to B. S. Jhun). B. S. Jhun is a recipient of the 2018 New Investigator Award from the APS Cell and Molecular Physiology Section.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
S.M.A., J.O-U., Y.K., and B.S.J. prepared figures; S.M.A., J.O-U., and B.S.J. drafted manuscript; S.M.A., J.O-U., M.W.C., Y.K., and B.S.J. edited and revised manuscript; S.M.A., J.O-U., M.W.C., Y.K., and B.S.J. approved final version of manuscript.
REFERENCES
- 1.Acin-Perez R, Lechuga-Vieco AV, Del Mar Muñoz M, Nieto-Arellano R, Torroja C, Sánchez-Cabo F, Jiménez C, González-Guerra A, Carrascoso I, Benincá C, Quiros PM, López-Otín C, Castellano JM, Ruíz-Cabello J, Jiménez-Borreguero LJ, Enríquez JA. Ablation of the stress protease OMA1 protects against heart failure in mice. Sci Transl Med 10: eaan4935, 2018. doi: 10.1126/scitranslmed.aan4935. [DOI] [PubMed] [Google Scholar]
- 2.Alexander C, Votruba M, Pesch UE, Thiselton DL, Mayer S, Moore A, Rodriguez M, Kellner U, Leo-Kottler B, Auburger G, Bhattacharya SS, Wissinger B. OPA1, encoding a dynamin-related GTPase, is mutated in autosomal dominant optic atrophy linked to chromosome 3q28. Nat Genet 26: 211–215, 2000. doi: 10.1038/79944. [DOI] [PubMed] [Google Scholar]
- 3.Anand R, Wai T, Baker MJ, Kladt N, Schauss AC, Rugarli E, Langer T. The i-AAA protease YME1L and OMA1 cleave OPA1 to balance mitochondrial fusion and fission. J Cell Biol 204: 919–929, 2014. doi: 10.1083/jcb.201308006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Aon MA, Cortassa S, O’Rourke B. Redox-optimized ROS balance: a unifying hypothesis. Biochim Biophys Acta 1797: 865–877, 2010. doi: 10.1016/j.bbabio.2010.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Archer SL. Mitochondrial fission and fusion in human diseases. N Engl J Med 370: 1074, 2014. doi: 10.1056/NEJMc1316254. [DOI] [PubMed] [Google Scholar]
- 6.Baker MJ, Lampe PA, Stojanovski D, Korwitz A, Anand R, Tatsuta T, Langer T. Stress-induced OMA1 activation and autocatalytic turnover regulate OPA1-dependent mitochondrial dynamics. EMBO J 33: 578–593, 2014. doi: 10.1002/embj.201386474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Belenguer P, Pellegrini L. The dynamin GTPase OPA1: more than mitochondria? Biochim Biophys Acta 1833: 176–183, 2013. doi: 10.1016/j.bbamcr.2012.08.004. [DOI] [PubMed] [Google Scholar]
- 8.Beraud N, Pelloux S, Usson Y, Kuznetsov AV, Ronot X, Tourneur Y, Saks V. Mitochondrial dynamics in heart cells: very low amplitude high frequency fluctuations in adult cardiomyocytes and flow motion in non beating Hl-1 cells. J Bioenerg Biomembr 41: 195–214, 2009. doi: 10.1007/s10863-009-9214-x. [DOI] [PubMed] [Google Scholar]
- 9.Bers DM. Calcium cycling and signaling in cardiac myocytes. Annu Rev Physiol 70: 23–49, 2008. doi: 10.1146/annurev.physiol.70.113006.100455. [DOI] [PubMed] [Google Scholar]
- 10.Bordt EA, Clerc P, Roelofs BA, Saladino AJ, Tretter L, Adam-Vizi V, Cherok E, Khalil A, Yadava N, Ge SX, Francis TC, Kennedy NW, Picton LK, Kumar T, Uppuluri S, Miller AM, Itoh K, Karbowski M, Sesaki H, Hill RB, Polster BM. The putative Drp1 inhibitor mdivi-1 is a reversible mitochondrial complex I inhibitor that modulates reactive oxygen species. Dev Cell 40: 583–594.e6, 2017. doi: 10.1016/j.devcel.2017.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Boudina S, Sena S, Theobald H, Sheng X, Wright JJ, Hu XX, Aziz S, Johnson JI, Bugger H, Zaha VG, Abel ED. Mitochondrial energetics in the heart in obesity-related diabetes: direct evidence for increased uncoupled respiration and activation of uncoupling proteins. Diabetes 56: 2457–2466, 2007. doi: 10.2337/db07-0481. [DOI] [PubMed] [Google Scholar]
- 12.Brand CS, Tan VP, Brown JH, Miyamoto S. RhoA regulates Drp1 mediated mitochondrial fission through ROCK to protect cardiomyocytes. Cell Signal 50: 48–57, 2018. doi: 10.1016/j.cellsig.2018.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bugger H, Schwarzer M, Chen D, Schrepper A, Amorim PA, Schoepe M, Nguyen TD, Mohr FW, Khalimonchuk O, Weimer BC, Doenst T. Proteomic remodelling of mitochondrial oxidative pathways in pressure overload-induced heart failure. Cardiovasc Res 85: 376–384, 2010. doi: 10.1093/cvr/cvp344. [DOI] [PubMed] [Google Scholar]
- 14.Cao JL, Adaniya SM, Cypress MW, Suzuki Y, Kusakari Y, Jhun BS, O-Uchi J. Role of mitochondrial Ca2+ homeostasis in cardiac muscles. Arch Biochem Biophys 663: 276–287, 2019. doi: 10.1016/j.abb.2019.01.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cao K, Xu J, Pu W, Dong Z, Sun L, Zang W, Gao F, Zhang Y, Feng Z, Liu J. Punicalagin, an active component in pomegranate, ameliorates cardiac mitochondrial impairment in obese rats via AMPK activation. Sci Rep 5: 14014, 2015. doi: 10.1038/srep14014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cao YL, Meng S, Chen Y, Feng JX, Gu DD, Yu B, Li YJ, Yang JY, Liao S, Chan DC, Gao S. MFN1 structures reveal nucleotide-triggered dimerization critical for mitochondrial fusion. Nature 542: 372–376, 2017. doi: 10.1038/nature21077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cereghetti GM, Stangherlin A, Martins de Brito O, Chang CR, Blackstone C, Bernardi P, Scorrano L. Dephosphorylation by calcineurin regulates translocation of Drp1 to mitochondria. Proc Natl Acad Sci USA 105: 15803–15808, 2008. doi: 10.1073/pnas.0808249105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chan DC. Fusion and fission: interlinked processes critical for mitochondrial health. Annu Rev Genet 46: 265–287, 2012. doi: 10.1146/annurev-genet-110410-132529. [DOI] [PubMed] [Google Scholar]
- 19.Chang CR, Blackstone C. Dynamic regulation of mitochondrial fission through modification of the dynamin-related protein Drp1. Ann NY Acad Sci 1201: 34–39, 2010. doi: 10.1111/j.1749-6632.2010.05629.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chang CR, Blackstone C. Cyclic AMP-dependent protein kinase phosphorylation of Drp1 regulates its GTPase activity and mitochondrial morphology. J Biol Chem 282: 21583–21587, 2007. doi: 10.1074/jbc.C700083200. [DOI] [PubMed] [Google Scholar]
- 21.Chao YH, Robak LA, Xia F, Koenig MK, Adesina A, Bacino CA, Scaglia F, Bellen HJ, Wangler MF. Missense variants in the middle domain of DNM1L in cases of infantile encephalopathy alter peroxisomes and mitochondria when assayed in Drosophila. Hum Mol Genet 25: 1846–1856, 2016. doi: 10.1093/hmg/ddw059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen H, Detmer SA, Ewald AJ, Griffin EE, Fraser SE, Chan DC. Mitofusins Mfn1 and Mfn2 coordinately regulate mitochondrial fusion and are essential for embryonic development. J Cell Biol 160: 189–200, 2003. doi: 10.1083/jcb.200211046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen H, Ren S, Clish C, Jain M, Mootha V, McCaffery JM, Chan DC. Titration of mitochondrial fusion rescues Mff-deficient cardiomyopathy. J Cell Biol 211: 795–805, 2015. doi: 10.1083/jcb.201507035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen KH, Guo X, Ma D, Guo Y, Li Q, Yang D, Li P, Qiu X, Wen S, Xiao RP, Tang J. Dysregulation of HSG triggers vascular proliferative disorders. Nat Cell Biol 6: 872–883, 2004. doi: 10.1038/ncb1161. [DOI] [PubMed] [Google Scholar]
- 25.Chen L, Gong Q, Stice JP, Knowlton AA. Mitochondrial OPA1, apoptosis, and heart failure. Cardiovasc Res 84: 91–99, 2009. doi: 10.1093/cvr/cvp181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chen L, Knowlton AA. Mitochondria and heart failure: new insights into an energetic problem. Minerva Cardioangiol 58: 213–229, 2010. [PMC free article] [PubMed] [Google Scholar]
- 27.Chen L, Liu T, Tran A, Lu X, Tomilov AA, Davies V, Cortopassi G, Chiamvimonvat N, Bers DM, Votruba M, Knowlton AA. OPA1 mutation and late-onset cardiomyopathy: mitochondrial dysfunction and mtDNA instability. J Am Heart Assoc 1: e003012, 2012. doi: 10.1161/JAHA.112.003012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chen Y, Csordás G, Jowdy C, Schneider TG, Csordás N, Wang W, Liu Y, Kohlhaas M, Meiser M, Bergem S, Nerbonne JM, Dorn GW II, Maack C. Mitofusin 2-containing mitochondrial-reticular microdomains direct rapid cardiomyocyte bioenergetic responses via interorganelle Ca2+ crosstalk. Circ Res 111: 863–875, 2012. doi: 10.1161/CIRCRESAHA.112.266585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chen Y, Dorn GW II. PINK1-phosphorylated mitofusin 2 is a Parkin receptor for culling damaged mitochondria. Science 340: 471–475, 2013. doi: 10.1126/science.1231031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chen Y, Liu Y, Dorn GW II. Mitochondrial fusion is essential for organelle function and cardiac homeostasis. Circ Res 109: 1327–1331, 2011. doi: 10.1161/CIRCRESAHA.111.258723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cipolat S, Martins de Brito O, Dal Zilio B, Scorrano L. OPA1 requires mitofusin 1 to promote mitochondrial fusion. Proc Natl Acad Sci USA 101: 15927–15932, 2004. doi: 10.1073/pnas.0407043101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cipolat S, Rudka T, Hartmann D, Costa V, Serneels L, Craessaerts K, Metzger K, Frezza C, Annaert W, D’Adamio L, Derks C, Dejaegere T, Pellegrini L, D’Hooge R, Scorrano L, De Strooper B. Mitochondrial rhomboid PARL regulates cytochrome c release during apoptosis via OPA1-dependent cristae remodeling. Cell 126: 163–175, 2006. doi: 10.1016/j.cell.2006.06.021. [DOI] [PubMed] [Google Scholar]
- 33.Clinton RW, Francy CA, Ramachandran R, Qi X, Mears JA. Dynamin-related protein 1 oligomerization in solution impairs functional interactions with membrane-anchored mitochondrial fission factor. J Biol Chem 291: 478–492, 2016. doi: 10.1074/jbc.M115.680025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cogliati S, Frezza C, Soriano ME, Varanita T, Quintana-Cabrera R, Corrado M, Cipolat S, Costa V, Casarin A, Gomes LC, Perales-Clemente E, Salviati L, Fernandez-Silva P, Enriquez JA, Scorrano L. Mitochondrial cristae shape determines respiratory chain supercomplexes assembly and respiratory efficiency. Cell 155: 160–171, 2013. doi: 10.1016/j.cell.2013.08.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cribbs JT, Strack S. Reversible phosphorylation of Drp1 by cyclic AMP-dependent protein kinase and calcineurin regulates mitochondrial fission and cell death. EMBO Rep 8: 939–944, 2007. doi: 10.1038/sj.embor.7401062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dai DF, Hsieh EJ, Chen T, Menendez LG, Basisty NB, Tsai L, Beyer RP, Crispin DA, Shulman NJ, Szeto HH, Tian R, MacCoss MJ, Rabinovitch PS. Global proteomics and pathway analysis of pressure-overload-induced heart failure and its attenuation by mitochondrial-targeted peptides. Circ Heart Fail 6: 1067–1076, 2013. doi: 10.1161/CIRCHEARTFAILURE.113.000406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Darshi M, Mendiola VL, Mackey MR, Murphy AN, Koller A, Perkins GA, Ellisman MH, Taylor SS. ChChd3, an inner mitochondrial membrane protein, is essential for maintaining crista integrity and mitochondrial function. J Biol Chem 286: 2918–2932, 2011. doi: 10.1074/jbc.M110.171975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Davies VJ, Hollins AJ, Piechota MJ, Yip W, Davies JR, White KE, Nicols PP, Boulton ME, Votruba M. Opa1 deficiency in a mouse model of autosomal dominant optic atrophy impairs mitochondrial morphology, optic nerve structure and visual function. Hum Mol Genet 16: 1307–1318, 2007. doi: 10.1093/hmg/ddm079. [DOI] [PubMed] [Google Scholar]
- 39.de Brito OM, Scorrano L. Mitofusin 2 tethers endoplasmic reticulum to mitochondria. Nature 456: 605–610, 2008. doi: 10.1038/nature07534. [DOI] [PubMed] [Google Scholar]
- 40.Del Dotto V, Fogazza M, Carelli V, Rugolo M, Zanna C. Eight human OPA1 isoforms, long and short: what are they for? Biochim Biophys Acta Bioenerg 1859: 263–269, 2018. doi: 10.1016/j.bbabio.2018.01.005. [DOI] [PubMed] [Google Scholar]
- 41.Del Dotto V, Mishra P, Vidoni S, Fogazza M, Maresca A, Caporali L, McCaffery JM, Cappelletti M, Baruffini E, Lenaers G, Chan D, Rugolo M, Carelli V, Zanna C. OPA1 isoforms in the hierarchical organization of mitochondrial functions. Cell Rep 19: 2557–2571, 2017. doi: 10.1016/j.celrep.2017.05.073. [DOI] [PubMed] [Google Scholar]
- 42.Delettre C, Lenaers G, Griffoin JM, Gigarel N, Lorenzo C, Belenguer P, Pelloquin L, Grosgeorge J, Turc-Carel C, Perret E, Astarie-Dequeker C, Lasquellec L, Arnaud B, Ducommun B, Kaplan J, Hamel CP. Nuclear gene OPA1, encoding a mitochondrial dynamin-related protein, is mutated in dominant optic atrophy. Nat Genet 26: 207–210, 2000. doi: 10.1038/79936. [DOI] [PubMed] [Google Scholar]
- 43.Diakos NA, Navankasattusas S, Abel ED, Rutter J, McCreath L, Ferrin P, McKellar SH, Miller DV, Park SY, Richardson RS, Deberardinis R, Cox JE, Kfoury AG, Selzman CH, Stehlik J, Fang JC, Li DY, Drakos SG. Evidence of glycolysis up-regulation and pyruvate mitochondrial oxidation mismatch during mechanical unloading of the failing human heart: implications for cardiac reloading and conditioning. JACC Basic Transl Sci 1: 432–444, 2016. doi: 10.1016/j.jacbts.2016.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dickey AS, Strack S. PKA/AKAP1 and PP2A/Bβ2 regulate neuronal morphogenesis via Drp1 phosphorylation and mitochondrial bioenergetics. J Neurosci 31: 15716–15726, 2011. doi: 10.1523/JNEUROSCI.3159-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Din S, Mason M, Völkers M, Johnson B, Cottage CT, Wang Z, Joyo AY, Quijada P, Erhardt P, Magnuson NS, Konstandin MH, Sussman MA. Pim-1 preserves mitochondrial morphology by inhibiting dynamin-related protein 1 translocation. Proc Natl Acad Sci USA 110: 5969–5974, 2013. doi: 10.1073/pnas.1213294110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ding WX, Yin XM. Mitophagy: mechanisms, pathophysiological roles, and analysis. Biol Chem 393: 547–564, 2012. doi: 10.1515/hsz-2012-0119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Divakaruni SS, Van Dyke AM, Chandra R, LeGates TA, Contreras M, Dharmasri PA, Higgs HN, Lobo MK, Thompson SM, Blanpied TA. Long-term potentiation requires a rapid burst of dendritic mitochondrial fission during induction. Neuron 100: 860–875.e7, 2018. doi: 10.1016/j.neuron.2018.09.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dolman NJ, Chambers KM, Mandavilli B, Batchelor RH, Janes MS. Tools and techniques to measure mitophagy using fluorescence microscopy. Autophagy 9: 1653–1662, 2013. doi: 10.4161/auto.24001. [DOI] [PubMed] [Google Scholar]
- 49.Ducommun S, Deak M, Sumpton D, Ford RJ, Núñez Galindo A, Kussmann M, Viollet B, Steinberg GR, Foretz M, Dayon L, Morrice NA, Sakamoto K. Motif affinity and mass spectrometry proteomic approach for the discovery of cellular AMPK targets: identification of mitochondrial fission factor as a new AMPK substrate. Cell Signal 27: 978–988, 2015. doi: 10.1016/j.cellsig.2015.02.008. [DOI] [PubMed] [Google Scholar]
- 50.Duvezin-Caubet S, Koppen M, Wagener J, Zick M, Israel L, Bernacchia A, Jagasia R, Rugarli EI, Imhof A, Neupert W, Langer T, Reichert AS. OPA1 processing reconstituted in yeast depends on the subunit composition of the m-AAA protease in mitochondria. Mol Biol Cell 18: 3582–3590, 2007. doi: 10.1091/mbc.e07-02-0164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ehses S, Raschke I, Mancuso G, Bernacchia A, Geimer S, Tondera D, Martinou JC, Westermann B, Rugarli EI, Langer T. Regulation of OPA1 processing and mitochondrial fusion by m-AAA protease isoenzymes and OMA1. J Cell Biol 187: 1023–1036, 2009. doi: 10.1083/jcb.200906084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Eisner V, Cupo RR, Gao E, Csordás G, Slovinsky WS, Paillard M, Cheng L, Ibetti J, Chen SR, Chuprun JK, Hoek JB, Koch WJ, Hajnóczky G. Mitochondrial fusion dynamics is robust in the heart and depends on calcium oscillations and contractile activity. Proc Natl Acad Sci USA 114: E859–E868, 2017. doi: 10.1073/pnas.1617288114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Eisner V, Lenaers G, Hajnóczky G. Mitochondrial fusion is frequent in skeletal muscle and supports excitation-contraction coupling. J Cell Biol 205: 179–195, 2014. doi: 10.1083/jcb.201312066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Eisner V, Picard M, Hajnóczky G. Mitochondrial dynamics in adaptive and maladaptive cellular stress responses. Nat Cell Biol 20: 755–765, 2018. doi: 10.1038/s41556-018-0133-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Elgass K, Pakay J, Ryan MT, Palmer CS. Recent advances into the understanding of mitochondrial fission. Biochim Biophys Acta 1833: 150–161, 2013. doi: 10.1016/j.bbamcr.2012.05.002. [DOI] [PubMed] [Google Scholar]
- 56.El-Sikhry HE, Alsaleh N, Dakarapu R, Falck JR, Seubert JM. Novel roles of epoxyeicosanoids in regulating cardiac mitochondria. PLoS One 11: e0160380, 2016. doi: 10.1371/journal.pone.0160380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Eura Y, Ishihara N, Yokota S, Mihara K. Two mitofusin proteins, mammalian homologues of FZO, with distinct functions are both required for mitochondrial fusion. J Biochem 134: 333–344, 2003. doi: 10.1093/jb/mvg150. [DOI] [PubMed] [Google Scholar]
- 58.Fahrner JA, Liu R, Perry MS, Klein J, Chan DC. A novel de novo dominant negative mutation in DNM1L impairs mitochondrial fission and presents as childhood epileptic encephalopathy. Am J Med Genet A 170: 2002–2011, 2016. doi: 10.1002/ajmg.a.37721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Figueroa-Romero C, Iñiguez-Lluhí JA, Stadler J, Chang CR, Arnoult D, Keller PJ, Hong Y, Blackstone C, Feldman EL. SUMOylation of the mitochondrial fission protein Drp1 occurs at multiple nonconsensus sites within the B domain and is linked to its activity cycle. FASEB J 23: 3917–3927, 2009. doi: 10.1096/fj.09-136630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Filadi R, Greotti E, Turacchio G, Luini A, Pozzan T, Pizzo P. Mitofusin 2 ablation increases endoplasmic reticulum-mitochondria coupling. Proc Natl Acad Sci USA 112: E2174–E2181, 2015. doi: 10.1073/pnas.1504880112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Filadi R, Pendin D, Pizzo P.. Mitofusin 2: from functions to disease. Cell Death Dis 9: 330, 2018. doi: 10.1038/s41419-017-0023-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Franco A, Kitsis RN, Fleischer JA, Gavathiotis E, Kornfeld OS, Gong G, Biris N, Benz A, Qvit N, Donnelly SK, Chen Y, Mennerick S, Hodgson L, Mochly-Rosen D, Dorn GW II. Correcting mitochondrial fusion by manipulating mitofusin conformations. Nature 540: 74–79, 2016. doi: 10.1038/nature20156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Frezza C, Cipolat S, Martins de Brito O, Micaroni M, Beznoussenko GV, Rudka T, Bartoli D, Polishuck RS, Danial NN, De Strooper B, Scorrano L. OPA1 controls apoptotic cristae remodeling independently from mitochondrial fusion. Cell 126: 177–189, 2006. doi: 10.1016/j.cell.2006.06.025. [DOI] [PubMed] [Google Scholar]
- 64.Fröhlich C, Grabiger S, Schwefel D, Faelber K, Rosenbaum E, Mears J, Rocks O, Daumke O. Structural insights into oligomerization and mitochondrial remodelling of dynamin 1-like protein. EMBO J 32: 1280–1292, 2013. doi: 10.1038/emboj.2013.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Gandre-Babbe S, van der Bliek AM. The novel tail-anchored membrane protein Mff controls mitochondrial and peroxisomal fission in mammalian cells. Mol Biol Cell 19: 2402–2412, 2008. doi: 10.1091/mbc.e07-12-1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Gawlowski T, Suarez J, Scott B, Torres-Gonzalez M, Wang H, Schwappacher R, Han X, Yates JR III, Hoshijima M, Dillmann W. Modulation of dynamin-related protein 1 (DRP1) function by increased O-linked-β-N-acetylglucosamine modification (O-GlcNAc) in cardiac myocytes. J Biol Chem 287: 30024–30034, 2012. doi: 10.1074/jbc.M112.390682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Gegg ME, Cooper JM, Chau KY, Rojo M, Schapira AH, Taanman JW. Mitofusin 1 and mitofusin 2 are ubiquitinated in a PINK1/parkin-dependent manner upon induction of mitophagy. Hum Mol Genet 19: 4861–4870, 2010. doi: 10.1093/hmg/ddq419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Glancy B, Hartnell LM, Combs CA, Femnou A, Sun J, Murphy E, Subramaniam S, Balaban RS. Power grid protection of the muscle mitochondrial reticulum. Cell Rep 19: 487–496, 2017. [Erratum in Cell Rep 23: 2832, 2018.] 10.1016/j.celrep.2017.03.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Glancy B, Hartnell LM, Malide D, Yu ZX, Combs CA, Connelly PS, Subramaniam S, Balaban RS. Mitochondrial reticulum for cellular energy distribution in muscle. Nature 523: 617–620, 2015. doi: 10.1038/nature14614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Glauser L, Sonnay S, Stafa K, Moore DJ. Parkin promotes the ubiquitination and degradation of the mitochondrial fusion factor mitofusin 1. J Neurochem 118: 636–645, 2011. doi: 10.1111/j.1471-4159.2011.07318.x. [DOI] [PubMed] [Google Scholar]
- 71.Gomes LC, Di Benedetto G, Scorrano L. During autophagy mitochondria elongate, are spared from degradation and sustain cell viability. Nat Cell Biol 13: 589–598, 2011. doi: 10.1038/ncb2220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Griparic L, Kanazawa T, van der Bliek AM. Regulation of the mitochondrial dynamin-like protein Opa1 by proteolytic cleavage. J Cell Biol 178: 757–764, 2007. doi: 10.1083/jcb.200704112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Guo C, Hildick KL, Luo J, Dearden L, Wilkinson KA, Henley JM. SENP3-mediated deSUMOylation of dynamin-related protein 1 promotes cell death following ischaemia. EMBO J 32: 1514–1528, 2013. doi: 10.1038/emboj.2013.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Halestrap AP, Richardson AP. The mitochondrial permeability transition: a current perspective on its identity and role in ischaemia/reperfusion injury. J Mol Cell Cardiol 78: 129–141, 2015. doi: 10.1016/j.yjmcc.2014.08.018. [DOI] [PubMed] [Google Scholar]
- 75.Hall AR, Burke N, Dongworth RK, Kalkhoran SB, Dyson A, Vicencio JM, Dorn GW II, Yellon DM, Hausenloy DJ. Hearts deficient in both Mfn1 and Mfn2 are protected against acute myocardial infarction. Cell Death Dis 7: e2238, 2016. doi: 10.1038/cddis.2016.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Han XJ, Lu YF, Li SA, Kaitsuka T, Sato Y, Tomizawa K, Nairn AC, Takei K, Matsui H, Matsushita M. CaM kinase Iα-induced phosphorylation of Drp1 regulates mitochondrial morphology. J Cell Biol 182: 573–585, 2008. doi: 10.1083/jcb.200802164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Hauser S, Schuster S, Theurer Y, Synofzik M, Schöls L. Generation of optic atrophy 1 patient-derived induced pluripotent stem cells (iPS-OPA1-BEHR) for disease modeling of complex optic atrophy syndromes (Behr syndrome). Stem Cell Res (Amst) 17: 426–429, 2016. doi: 10.1016/j.scr.2016.09.012. [DOI] [PubMed] [Google Scholar]
- 78.Head B, Griparic L, Amiri M, Gandre-Babbe S, van der Bliek AM. Inducible proteolytic inactivation of OPA1 mediated by the OMA1 protease in mammalian cells. J Cell Biol 187: 959–966, 2009. doi: 10.1083/jcb.200906083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Hoque A, Sivakumaran P, Bond ST, Ling NX, Kong AM, Scott JW, Bandara N, Hernández D, Liu GS, Wong RC, Ryan MT, Hausenloy DJ, Kemp BE, Oakhill JS, Drew BG, Pébay A, Lim SY. Mitochondrial fission protein Drp1 inhibition promotes cardiac mesodermal differentiation of human pluripotent stem cells. Cell Death Discov 4: 39, 2018. doi: 10.1038/s41420-018-0042-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Hornbeck PV, Zhang B, Murray B, Kornhauser JM, Latham V, Skrzypek E. PhosphoSitePlus, 2014: mutations, PTMs and recalibrations. Nucleic Acids Res 43: D512–D520, 2015. doi: 10.1093/nar/gku1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Hu C, Huang Y, Li L. Drp1-dependent mitochondrial fission plays critical roles in physiological and pathological progresses in mammals. Int J Mol Sci 18: E144, 2017. doi: 10.3390/ijms18010144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Ikeda Y, Shirakabe A, Maejima Y, Zhai P, Sciarretta S, Toli J, Nomura M, Mihara K, Egashira K, Ohishi M, Abdellatif M, Sadoshima J. Endogenous Drp1 mediates mitochondrial autophagy and protects the heart against energy stress. Circ Res 116: 264–278, 2015. doi: 10.1161/CIRCRESAHA.116.303356. [DOI] [PubMed] [Google Scholar]
- 83.Ishihara N, Eura Y, Mihara K. Mitofusin 1 and 2 play distinct roles in mitochondrial fusion reactions via GTPase activity. J Cell Sci 117: 6535–6546, 2004. doi: 10.1242/jcs.01565. [DOI] [PubMed] [Google Scholar]
- 84.Ishihara N, Fujita Y, Oka T, Mihara K. Regulation of mitochondrial morphology through proteolytic cleavage of OPA1. EMBO J 25: 2966–2977, 2006. doi: 10.1038/sj.emboj.7601184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Ishihara T, Ban-Ishihara R, Maeda M, Matsunaga Y, Ichimura A, Kyogoku S, Aoki H, Katada S, Nakada K, Nomura M, Mizushima N, Mihara K, Ishihara N. Dynamics of mitochondrial DNA nucleoids regulated by mitochondrial fission is essential for maintenance of homogeneously active mitochondria during neonatal heart development. Mol Cell Biol 35: 211–223, 2015. doi: 10.1128/MCB.01054-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.James DI, Parone PA, Mattenberger Y, Martinou JC. hFis1, a novel component of the mammalian mitochondrial fission machinery. J Biol Chem 278: 36373–36379, 2003. doi: 10.1074/jbc.M303758200. [DOI] [PubMed] [Google Scholar]
- 87.Javadov S, Karmazyn M, Escobales N. Mitochondrial permeability transition pore opening as a promising therapeutic target in cardiac diseases. J Pharmacol Exp Ther 330: 670–678, 2009. doi: 10.1124/jpet.109.153213. [DOI] [PubMed] [Google Scholar]
- 88.Javadov S, Rajapurohitam V, Kilić A, Hunter JC, Zeidan A, Said Faruq N, Escobales N, Karmazyn M. Expression of mitochondrial fusion-fission proteins during post-infarction remodeling: the effect of NHE-1 inhibition. Basic Res Cardiol 106: 99–109, 2011. doi: 10.1007/s00395-010-0122-3. [DOI] [PubMed] [Google Scholar]
- 89.Jhun BS, Lee H, Jin ZG, Yoon Y. Glucose stimulation induces dynamic change of mitochondrial morphology to promote insulin secretion in the insulinoma cell line INS-1E. PLoS One 8: e60810, 2013. doi: 10.1371/journal.pone.0060810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Jhun BS, O-Uchi J, Adaniya SM, Cypress MW, Yoon Y. Adrenergic regulation of Drp1-driven mitochondrial fission in cardiac physio-pathology. Antioxidants 7: 195, 2018. doi: 10.3390/antiox7120195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Jhun BS, O-Uchi J, Adaniya SM, Mancini TJ, Cao JL, King ME, Landi AK, Ma H, Shin M, Yang D, Xu X, Yoon Y, Choudhary G, Clements RT, Mende U, Sheu SS. Protein kinase D activation induces mitochondrial fragmentation and dysfunction in cardiomyocytes. J Physiol 596: 827–855, 2018. doi: 10.1113/JP275418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Jiang HK, Wang YH, Sun L, He X, Zhao M, Feng ZH, Yu XJ, Zang WJ. Aerobic interval training attenuates mitochondrial dysfunction in rats post-myocardial infarction: roles of mitochondrial network dynamics. Int J Mol Sci 15: 5304–5322, 2014. doi: 10.3390/ijms15045304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Jiang X, Jiang H, Shen Z, Wang X. Activation of mitochondrial protease OMA1 by Bax and Bak promotes cytochrome c release during apoptosis. Proc Natl Acad Sci USA 111: 14782–14787, 2014. doi: 10.1073/pnas.1417253111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Kageyama Y, Hoshijima M, Seo K, Bedja D, Sysa-Shah P, Andrabi SA, Chen W, Höke A, Dawson VL, Dawson TM, Gabrielson K, Kass DA, Iijima M, Sesaki H. Parkin-independent mitophagy requires Drp1 and maintains the integrity of mammalian heart and brain. EMBO J 33: 2798–2813, 2014. doi: 10.15252/embj.201488658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Karamanlidis G, Bautista-Hernandez V, Fynn-Thompson F, Del Nido P, Tian R. Impaired mitochondrial biogenesis precedes heart failure in right ventricular hypertrophy in congenital heart disease. Circ Heart Fail 4: 707–713, 2011. doi: 10.1161/CIRCHEARTFAILURE.111.961474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Karbowski M, Neutzner A, Youle RJ. The mitochondrial E3 ubiquitin ligase MARCH5 is required for Drp1 dependent mitochondrial division. J Cell Biol 178: 71–84, 2007. doi: 10.1083/jcb.200611064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Kasahara A, Cipolat S, Chen Y, Dorn GW II, Scorrano L. Mitochondrial fusion directs cardiomyocyte differentiation via calcineurin and Notch signaling. Science 342: 734–737, 2013. doi: 10.1126/science.1241359. [DOI] [PubMed] [Google Scholar]
- 98.Kashatus JA, Nascimento A, Myers LJ, Sher A, Byrne FL, Hoehn KL, Counter CM, Kashatus DF. Erk2 phosphorylation of Drp1 promotes mitochondrial fission and MAPK-driven tumor growth. Mol Cell 57: 537–551, 2015. doi: 10.1016/j.molcel.2015.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Kim EY, Zhang Y, Beketaev I, Segura AM, Yu W, Xi Y, Chang J, Wang J. SENP5, a SUMO isopeptidase, induces apoptosis and cardiomyopathy. J Mol Cell Cardiol 78: 154–164, 2015. doi: 10.1016/j.yjmcc.2014.08.003. [DOI] [PubMed] [Google Scholar]
- 100.Kim H, Scimia MC, Wilkinson D, Trelles RD, Wood MR, Bowtell D, Dillin A, Mercola M, Ronai ZA. Fine-tuning of Drp1/Fis1 availability by AKAP121/Siah2 regulates mitochondrial adaptation to hypoxia. Mol Cell 44: 532–544, 2011. doi: 10.1016/j.molcel.2011.08.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Koirala S, Guo Q, Kalia R, Bui HT, Eckert DM, Frost A, Shaw JM. Interchangeable adaptors regulate mitochondrial dynamin assembly for membrane scission. Proc Natl Acad Sci USA 110: E1342–E1351, 2013. doi: 10.1073/pnas.1300855110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Koshiba T, Detmer SA, Kaiser JT, Chen H, McCaffery JM, Chan DC. Structural basis of mitochondrial tethering by mitofusin complexes. Science 305: 858–862, 2004. doi: 10.1126/science.1099793. [DOI] [PubMed] [Google Scholar]
- 103.Kurz FT, Derungs T, Aon MA, O’Rourke B, Armoundas AA. Mitochondrial networks in cardiac myocytes reveal dynamic coupling behavior. Biophys J 108: 1922–1933, 2015. doi: 10.1016/j.bpj.2015.01.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Landes T, Leroy I, Bertholet A, Diot A, Khosrobakhsh F, Daloyau M, Davezac N, Miquel MC, Courilleau D, Guillou E, Olichon A, Lenaers G, Arnauné-Pelloquin L, Emorine LJ, Belenguer P. OPA1 (dys)functions. Semin Cell Dev Biol 21: 593–598, 2010. doi: 10.1016/j.semcdb.2009.12.012. [DOI] [PubMed] [Google Scholar]
- 105.Leboucher GP, Tsai YC, Yang M, Shaw KC, Zhou M, Veenstra TD, Glickman MH, Weissman AM. Stress-induced phosphorylation and proteasomal degradation of mitofusin 2 facilitates mitochondrial fragmentation and apoptosis. Mol Cell 47: 547–557, 2012. doi: 10.1016/j.molcel.2012.05.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Lee H, Smith SB, Yoon Y. The short variant of the mitochondrial dynamin OPA1 maintains mitochondrial energetics and cristae structure. J Biol Chem 292: 7115–7130, 2017. doi: 10.1074/jbc.M116.762567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Lee H, Yoon Y. Mitochondrial membrane dynamics: functional positioning of OPA1. Antioxidants (Basel) 7: E186, 2018. doi: 10.3390/antiox7120186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Lei M, Wang X, Ke Y, Solaro RJ. Regulation of Ca2+ transient by PP2A in normal and failing heart. Front Physiol 6: 13, 2015. doi: 10.3389/fphys.2015.00013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Liesa M, Palacín M, Zorzano A. Mitochondrial dynamics in mammalian health and disease. Physiol Rev 89: 799–845, 2009. doi: 10.1152/physrev.00030.2008. [DOI] [PubMed] [Google Scholar]
- 110.Littlejohns B, Pasdois P, Duggan S, Bond AR, Heesom K, Jackson CL, Angelini GD, Halestrap AP, Suleiman MS. Hearts from mice fed a non-obesogenic high-fat diet exhibit changes in their oxidative state, calcium and mitochondria in parallel with increased susceptibility to reperfusion injury. PLoS One 9: e100579, 2014. doi: 10.1371/journal.pone.0100579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Liu R, Chan DC. The mitochondrial fission receptor Mff selectively recruits oligomerized Drp1. Mol Biol Cell 26: 4466–4477, 2015. doi: 10.1091/mbc.E15-08-0591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Losón OC, Song Z, Chen H, Chan DC. Fis1, Mff, MiD49, and MiD51 mediate Drp1 recruitment in mitochondrial fission. Mol Biol Cell 24: 659–667, 2013. doi: 10.1091/mbc.e12-10-0721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Lu YT, Li LZ, Yang YL, Yin X, Liu Q, Zhang L, Liu K, Liu B, Li J, Qi LW. Succinate induces aberrant mitochondrial fission in cardiomyocytes through GPR91 signaling. Cell Death Dis 9: 672, 2018. doi: 10.1038/s41419-018-0708-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Makino A, Suarez J, Gawlowski T, Han W, Wang H, Scott BT, Dillmann WH. Regulation of mitochondrial morphology and function by O-GlcNAcylation in neonatal cardiac myocytes. Am J Physiol Regul Integr Comp Physiol 300: R1296–R1302, 2011. doi: 10.1152/ajpregu.00437.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Maneechote C, Palee S, Kerdphoo S, Jaiwongkam T, Chattipakorn SC, Chattipakorn N. Differential temporal inhibition of mitochondrial fission by Mdivi-1 exerts effective cardioprotection in cardiac ischemia/reperfusion injury. Clin Sci (Lond) 132: 1669–1683, 2018. doi: 10.1042/CS20180510. [DOI] [PubMed] [Google Scholar]
- 116.Marsboom G, Toth PT, Ryan JJ, Hong Z, Wu X, Fang YH, Thenappan T, Piao L, Zhang HJ, Pogoriler J, Chen Y, Morrow E, Weir EK, Rehman J, Archer SL. Dynamin-related protein 1-mediated mitochondrial mitotic fission permits hyperproliferation of vascular smooth muscle cells and offers a novel therapeutic target in pulmonary hypertension. Circ Res 110: 1484–1497, 2012. doi: 10.1161/CIRCRESAHA.111.263848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.McLelland GL, Goiran T, Yi W, Dorval G, Chen CX, Lauinger ND, Krahn AI, Valimehr S, Rakovic A, Rouiller I, Durcan TM, Trempe JF, Fon EA. Mfn2 ubiquitination by PINK1/parkin gates the p97-dependent release of ER from mitochondria to drive mitophagy. eLife 7: e32866, 2018. doi: 10.7554/eLife.32866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Mendler L, Braun T, Müller S. The ubiquitin-like SUMO system and heart function: from development to disease. Circ Res 118: 132–144, 2016. doi: 10.1161/CIRCRESAHA.115.307730. [DOI] [PubMed] [Google Scholar]
- 119.Merrill RA, Slupe AM, Strack S. N-terminal phosphorylation of protein phosphatase 2A/Bβ2 regulates translocation to mitochondria, dynamin-related protein 1 dephosphorylation, and neuronal survival. FEBS J 280: 662–673, 2013. doi: 10.1111/j.1742-4658.2012.08631.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Misko A, Jiang S, Wegorzewska I, Milbrandt J, Baloh RH. Mitofusin 2 is necessary for transport of axonal mitochondria and interacts with the Miro/Milton complex. J Neurosci 30: 4232–4240, 2010. doi: 10.1523/JNEUROSCI.6248-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Montessuit S, Somasekharan SP, Terrones O, Lucken-Ardjomande S, Herzig S, Schwarzenbacher R, Manstein DJ, Bossy-Wetzel E, Basañez G, Meda P, Martinou JC. Membrane remodeling induced by the dynamin-related protein Drp1 stimulates Bax oligomerization. Cell 142: 889–901, 2010. doi: 10.1016/j.cell.2010.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Morciano G, Giorgi C, Bonora M, Punzetti S, Pavasini R, Wieckowski MR, Campo G, Pinton P. Molecular identity of the mitochondrial permeability transition pore and its role in ischemia-reperfusion injury. J Mol Cell Cardiol 78: 142–153, 2015. doi: 10.1016/j.yjmcc.2014.08.015. [DOI] [PubMed] [Google Scholar]
- 123.Mozdy AD, McCaffery JM, Shaw JM. Dnm1p GTPase-mediated mitochondrial fission is a multi-step process requiring the novel integral membrane component Fis1p. J Cell Biol 151: 367–380, 2000. doi: 10.1083/jcb.151.2.367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Muñoz JP, Ivanova S, Sánchez-Wandelmer J, Martínez-Cristóbal P, Noguera E, Sancho A, Díaz-Ramos A, Hernández-Alvarez MI, Sebastián D, Mauvezin C, Palacín M, Zorzano A. Mfn2 modulates the UPR and mitochondrial function via repression of PERK. EMBO J 32: 2348–2361, 2013. doi: 10.1038/emboj.2013.168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Nakamura N, Kimura Y, Tokuda M, Honda S, Hirose S. MARCH-V is a novel mitofusin 2- and Drp1-binding protein able to change mitochondrial morphology. EMBO Rep 7: 1019–1022, 2006. doi: 10.1038/sj.embor.7400790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Nan J, Zhu W, Rahman MS, Liu M, Li D, Su S, Zhang N, Hu X, Yu H, Gupta MP, Wang J. Molecular regulation of mitochondrial dynamics in cardiac disease. Biochim Biophys Acta Mol Cell Res 1864: 1260–1273, 2017. doi: 10.1016/j.bbamcr.2017.03.006. [DOI] [PubMed] [Google Scholar]
- 127.Nasca A, Legati A, Baruffini E, Nolli C, Moroni I, Ardissone A, Goffrini P, Ghezzi D. Biallelic mutations in DNM1L are associated with a slowly progressive infantile encephalopathy. Hum Mutat 37: 898–903, 2016. doi: 10.1002/humu.23033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Neubauer S. The failing heart: an engine out of fuel. N Engl J Med 356: 1140–1151, 2007. doi: 10.1056/NEJMra063052. [DOI] [PubMed] [Google Scholar]
- 129.Ngoh GA, Papanicolaou KN, Walsh K. Loss of mitofusin 2 promotes endoplasmic reticulum stress. J Biol Chem 287: 20321–20332, 2012. doi: 10.1074/jbc.M112.359174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Nicholls DG. Mitochondrial membrane potential and aging. Aging Cell 3: 35–40, 2004. doi: 10.1111/j.1474-9728.2003.00079.x. [DOI] [PubMed] [Google Scholar]
- 131.Nishimura A, Shimauchi T, Tanaka T, Shimoda K, Toyama T, Kitajima N, Ishikawa T, Shindo N, Numaga-Tomita T, Yasuda S, Sato Y, Kuwahara K, Kumagai Y, Akaike T, Ide T, Ojida A, Mori Y, Nishida M. Hypoxia-induced interaction of filamin with Drp1 causes mitochondrial hyperfission-associated myocardial senescence. Sci Signal 11: eaat5185, 2018. doi: 10.1126/scisignal.aat5185. [DOI] [PubMed] [Google Scholar]
- 132.Oka T, Maillet M, Watt AJ, Schwartz RJ, Aronow BJ, Duncan SA, Molkentin JD. Cardiac-specific deletion of Gata4 reveals its requirement for hypertrophy, compensation, and myocyte viability. Circ Res 98: 837–845, 2006. doi: 10.1161/01.RES.0000215985.18538.c4. [DOI] [PubMed] [Google Scholar]
- 133.Ong SB, Hall AR, Hausenloy DJ. Mitochondrial dynamics in cardiovascular health and disease. Antioxid Redox Signal 19: 400–414, 2013. doi: 10.1089/ars.2012.4777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Ong SB, Subrayan S, Lim SY, Yellon DM, Davidson SM, Hausenloy DJ. Inhibiting mitochondrial fission protects the heart against ischemia/reperfusion injury. Circulation 121: 2012–2022, 2010. doi: 10.1161/CIRCULATIONAHA.109.906610. [DOI] [PubMed] [Google Scholar]
- 135.Osellame LD, Singh AP, Stroud DA, Palmer CS, Stojanovski D, Ramachandran R, Ryan MT. Cooperative and independent roles of the Drp1 adaptors Mff, MiD49 and MiD51 in mitochondrial fission. J Cell Sci 129: 2170–2181, 2016. doi: 10.1242/jcs.185165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Otera H, Ishihara N, Mihara K. New insights into the function and regulation of mitochondrial fission. Biochim Biophys Acta 1833: 1256–1268, 2013. doi: 10.1016/j.bbamcr.2013.02.002. [DOI] [PubMed] [Google Scholar]
- 137.Otera H, Miyata N, Kuge O, Mihara K. Drp1-dependent mitochondrial fission via MiD49/51 is essential for apoptotic cristae remodeling. J Cell Biol 212: 531–544, 2016. doi: 10.1083/jcb.201508099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Otera H, Wang C, Cleland MM, Setoguchi K, Yokota S, Youle RJ, Mihara K. Mff is an essential factor for mitochondrial recruitment of Drp1 during mitochondrial fission in mammalian cells. J Cell Biol 191: 1141–1158, 2010. doi: 10.1083/jcb.201007152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.O-Uchi J, Jhun BS, Xu S, Hurst S, Raffaello A, Liu X, Yi B, Zhang H, Gross P, Mishra J, Ainbinder A, Kettlewell S, Smith GL, Dirksen RT, Wang W, Rizzuto R, Sheu SS. Adrenergic signaling regulates mitochondrial Ca2+ uptake through Pyk2-dependent tyrosine phosphorylation of the mitochondrial Ca2+ uniporter. Antioxid Redox Signal 21: 863–879, 2014. doi: 10.1089/ars.2013.5394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Palmer CS, Elgass KD, Parton RG, Osellame LD, Stojanovski D, Ryan MT. Adaptor proteins MiD49 and MiD51 can act independently of Mff and Fis1 in Drp1 recruitment and are specific for mitochondrial fission. J Biol Chem 288: 27584–27593, 2013. doi: 10.1074/jbc.M113.479873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Palmer CS, Osellame LD, Laine D, Koutsopoulos OS, Frazier AE, Ryan MT. MiD49 and MiD51, new components of the mitochondrial fission machinery. EMBO Rep 12: 565–573, 2011. doi: 10.1038/embor.2011.54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Papanicolaou KN, Khairallah RJ, Ngoh GA, Chikando A, Luptak I, O’Shea KM, Riley DD, Lugus JJ, Colucci WS, Lederer WJ, Stanley WC, Walsh K. Mitofusin-2 maintains mitochondrial structure and contributes to stress-induced permeability transition in cardiac myocytes. Mol Cell Biol 31: 1309–1328, 2011. doi: 10.1128/MCB.00911-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Papanicolaou KN, Kikuchi R, Ngoh GA, Coughlan KA, Dominguez I, Stanley WC, Walsh K. Mitofusins 1 and 2 are essential for postnatal metabolic remodeling in heart. Circ Res 111: 1012–1026, 2012. doi: 10.1161/CIRCRESAHA.112.274142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Papanicolaou KN, Ngoh GA, Dabkowski ER, O’Connell KA, Ribeiro RF Jr, Stanley WC, Walsh K. Cardiomyocyte deletion of mitofusin-1 leads to mitochondrial fragmentation and improves tolerance to ROS-induced mitochondrial dysfunction and cell death. Am J Physiol Heart Circ Physiol 302: H167–H179, 2012. doi: 10.1152/ajpheart.00833.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Park YY, Nguyen OT, Kang H, Cho H. MARCH5-mediated quality control on acetylated Mfn1 facilitates mitochondrial homeostasis and cell survival. Cell Death Dis 5: e1172, 2014. doi: 10.1038/cddis.2014.142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Parra V, Rothermel BA. Calcineurin signaling in the heart: the importance of time and place. J Mol Cell Cardiol 103: 121–136, 2017. doi: 10.1016/j.yjmcc.2016.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Parra V, Verdejo HE, Iglewski M, Del Campo A, Troncoso R, Jones D, Zhu Y, Kuzmicic J, Pennanen C, Lopez-Crisosto C, Jaña F, Ferreira J, Noguera E, Chiong M, Bernlohr DA, Klip A, Hill JA, Rothermel BA, Abel ED, Zorzano A, Lavandero S. Insulin stimulates mitochondrial fusion and function in cardiomyocytes via the Akt-mTOR-NFκB-Opa-1 signaling pathway. Diabetes 63: 75–88, 2014. doi: 10.2337/db13-0340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Patel KD, Glancy B, Balaban RS. The electrochemical transmission in I-band segments of the mitochondrial reticulum. Biochim Biophys Acta 1857: 1284–1289, 2016. doi: 10.1016/j.bbabio.2016.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Pennanen C, Parra V, López-Crisosto C, Morales PE, Del Campo A, Gutierrez T, Rivera-Mejías P, Kuzmicic J, Chiong M, Zorzano A, Rothermel BA, Lavandero S. Mitochondrial fission is required for cardiomyocyte hypertrophy mediated by a Ca2+-calcineurin signaling pathway. J Cell Sci 127: 2659–2671, 2014. doi: 10.1242/jcs.139394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Pernas L, Scorrano L. Mito-morphosis: mitochondrial fusion, fission, and cristae remodeling as key mediators of cellular function. Annu Rev Physiol 78: 505–531, 2016. doi: 10.1146/annurev-physiol-021115-105011. [DOI] [PubMed] [Google Scholar]
- 151.Pham AH, McCaffery JM, Chan DC. Mouse lines with photo-activatable mitochondria to study mitochondrial dynamics. Genesis 50: 833–843, 2012. doi: 10.1002/dvg.22050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Piquereau J, Caffin F, Novotova M, Lemaire C, Veksler V, Garnier A, Ventura-Clapier R, Joubert F. Mitochondrial dynamics in the adult cardiomyocytes: which roles for a highly specialized cell? Front Physiol 4: 102, 2013. doi: 10.3389/fphys.2013.00102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Piquereau J, Caffin F, Novotova M, Prola A, Garnier A, Mateo P, Fortin D, Huynh H, Nicolas V, Alavi MV, Brenner C, Ventura-Clapier R, Veksler V, Joubert F. Down-regulation of OPA1 alters mouse mitochondrial morphology, PTP function, and cardiac adaptation to pressure overload. Cardiovasc Res 94: 408–417, 2012. doi: 10.1093/cvr/cvs117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Pisano A, Cerbelli B, Perli E, Pelullo M, Bargelli V, Preziuso C, Mancini M, He L, Bates MG, Lucena JR, Della Monica PL, Familiari G, Petrozza V, Nediani C, Taylor RW, d’Amati G, Giordano C. Impaired mitochondrial biogenesis is a common feature to myocardial hypertrophy and end-stage ischemic heart failure. Cardiovasc Pathol 25: 103–112, 2016. doi: 10.1016/j.carpath.2015.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Prudent J, Zunino R, Sugiura A, Mattie S, Shore GC, McBride HM. MAPL SUMOylation of Drp1 stabilizes an ER/mitochondrial platform required for cell death. Mol Cell 59: 941–955, 2015. doi: 10.1016/j.molcel.2015.08.001. [DOI] [PubMed] [Google Scholar]
- 156.Pyakurel A, Savoia C, Hess D, Scorrano L. Extracellular regulated kinase phosphorylates mitofusin 1 to control mitochondrial morphology and apoptosis. Mol Cell 58: 244–254, 2015. doi: 10.1016/j.molcel.2015.02.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Qi X, Disatnik MH, Shen N, Sobel RA, Mochly-Rosen D. Aberrant mitochondrial fission in neurons induced by protein kinase Cδ under oxidative stress conditions in vivo. Mol Biol Cell 22: 256–265, 2011. doi: 10.1091/mbc.e10-06-0551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Qi Y, Yan L, Yu C, Guo X, Zhou X, Hu X, Huang X, Rao Z, Lou Z, Hu J. Structures of human mitofusin 1 provide insight into mitochondrial tethering. J Cell Biol 215: 621–629, 2016. doi: 10.1083/jcb.201609019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Quirós PM, Ramsay AJ, Sala D, Fernández-Vizarra E, Rodríguez F, Peinado JR, Fernández-García MS, Vega JA, Enríquez JA, Zorzano A, López-Otín C. Loss of mitochondrial protease OMA1 alters processing of the GTPase OPA1 and causes obesity and defective thermogenesis in mice. EMBO J 31: 2117–2133, 2012. doi: 10.1038/emboj.2012.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Roe AJ, Qi X. Drp1 phosphorylation by MAPK1 causes mitochondrial dysfunction in cell culture model of Huntington’s disease. Biochem Biophys Res Commun 496: 706–711, 2018. doi: 10.1016/j.bbrc.2018.01.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Rojo M, Legros F, Chateau D, Lombès A. Membrane topology and mitochondrial targeting of mitofusins, ubiquitous mammalian homologs of the transmembrane GTPase Fzo. J Cell Sci 115: 1663–1674, 2002. [DOI] [PubMed] [Google Scholar]
- 162.Rosdah AA, K Holien J, Delbridge LM, Dusting GJ, Lim SY. Mitochondrial fission: a drug target for cytoprotection or cytodestruction? Pharmacol Res Perspect 4: e00235, 2016. doi: 10.1002/prp2.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Rosenbloom AB, Lee SH, To M, Lee A, Shin JY, Bustamante C. Optimized two-color super resolution imaging of Drp1 during mitochondrial fission with a slow-switching Dronpa variant. Proc Natl Acad Sci USA 111: 13093–13098, 2014. doi: 10.1073/pnas.1320044111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Saito T, Nah J, Oka SI, Mukai R, Monden Y, Maejima Y, Ikeda Y, Sciarretta S, Liu T, Li H, Baljinnyam E, Fraidenraich D, Fritzky L, Zhai P, Ichinose S, Isobe M, Hsu CP, Kundu M, Sadoshima J. An alternative mitophagy pathway mediated by Rab9 protects the heart against ischemia. J Clin Invest 129: 802–819, 2019. doi: 10.1172/JCI122035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Saito T, Sadoshima J. Molecular mechanisms of mitochondrial autophagy/mitophagy in the heart. Circ Res 116: 1477–1490, 2015. doi: 10.1161/CIRCRESAHA.116.303790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Samant SA, Zhang HJ, Hong Z, Pillai VB, Sundaresan NR, Wolfgeher D, Archer SL, Chan DC, Gupta MP. SIRT3 deacetylates and activates OPA1 to regulate mitochondrial dynamics during stress. Mol Cell Biol 34: 807–819, 2014. doi: 10.1128/MCB.01483-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Santulli G, Xie W, Reiken SR, Marks AR. Mitochondrial calcium overload is a key determinant in heart failure. Proc Natl Acad Sci USA 112: 11389–11394, 2015. doi: 10.1073/pnas.1513047112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Sarraf SA, Raman M, Guarani-Pereira V, Sowa ME, Huttlin EL, Gygi SP, Harper JW. Landscape of the PARKIN-dependent ubiquitylome in response to mitochondrial depolarization. Nature 496: 372–376, 2013. doi: 10.1038/nature12043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Schaper J, Froede R, Hein S, Buck A, Hashizume H, Speiser B, Friedl A, Bleese N. Impairment of the myocardial ultrastructure and changes of the cytoskeleton in dilated cardiomyopathy. Circulation 83: 504–514, 1991. doi: 10.1161/01.CIR.83.2.504. [DOI] [PubMed] [Google Scholar]
- 170.Schrepfer E, Scorrano L. Mitofusins, from mitochondria to metabolism. Mol Cell 61: 683–694, 2016. doi: 10.1016/j.molcel.2016.02.022. [DOI] [PubMed] [Google Scholar]
- 171.Sebastián D, Hernández-Alvarez MI, Segalés J, Sorianello E, Muñoz JP, Sala D, Waget A, Liesa M, Paz JC, Gopalacharyulu P, Orešič M, Pich S, Burcelin R, Palacín M, Zorzano A. Mitofusin 2 (Mfn2) links mitochondrial and endoplasmic reticulum function with insulin signaling and is essential for normal glucose homeostasis. Proc Natl Acad Sci USA 109: 5523–5528, 2012. doi: 10.1073/pnas.1108220109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Serasinghe MN, Wieder SY, Renault TT, Elkholi R, Asciolla JJ, Yao JL, Jabado O, Hoehn K, Kageyama Y, Sesaki H, Chipuk JE. Mitochondrial division is requisite to RAS-induced transformation and targeted by oncogenic MAPK pathway inhibitors. Mol Cell 57: 521–536, 2015. doi: 10.1016/j.molcel.2015.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Sharp WW, Fang YH, Han M, Zhang HJ, Hong Z, Banathy A, Morrow E, Ryan JJ, Archer SL. Dynamin-related protein 1 (Drp1)-mediated diastolic dysfunction in myocardial ischemia-reperfusion injury: therapeutic benefits of Drp1 inhibition to reduce mitochondrial fission. FASEB J 28: 316–326, 2014. doi: 10.1096/fj.12-226225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Sheffer R, Douiev L, Edvardson S, Shaag A, Tamimi K, Soiferman D, Meiner V, Saada A. Postnatal microcephaly and pain insensitivity due to a de novo heterozygous DNM1L mutation causing impaired mitochondrial fission and function. Am J Med Genet A 170: 1603–1607, 2016. doi: 10.1002/ajmg.a.37624. [DOI] [PubMed] [Google Scholar]
- 175.Shimizu Y, Lambert JP, Nicholson CK, Kim JJ, Wolfson DW, Cho HC, Husain A, Naqvi N, Chin LS, Li L, Calvert JW. DJ-1 protects the heart against ischemia-reperfusion injury by regulating mitochondrial fission. J Mol Cell Cardiol 97: 56–66, 2016. doi: 10.1016/j.yjmcc.2016.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Shirakabe A, Zhai P, Ikeda Y, Saito T, Maejima Y, Hsu CP, Nomura M, Egashira K, Levine B, Sadoshima J. Drp1-dependent mitochondrial autophagy plays a protective role against pressure overload-induced mitochondrial dysfunction and heart failure. Circulation 133: 1249–1263, 2016. doi: 10.1161/CIRCULATIONAHA.115.020502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Signorile A, Santeramo A, Tamma G, Pellegrino T, D’Oria S, Lattanzio P, De Rasmo D. Mitochondrial cAMP prevents apoptosis modulating Sirt3 protein level and OPA1 processing in cardiac myoblast cells. Biochim Biophys Acta Mol Cell Res 1864: 355–366, 2017. doi: 10.1016/j.bbamcr.2016.11.022. [DOI] [PubMed] [Google Scholar]
- 178.Silva FSG, Costa CF, Marques RJ, Oliveira PJ, Pereira GC. Pharmacological targeting of the mitochondrial permeability transition pore for cardioprotection. In: Mitochondrial Biology and Experimental Therapeutics, edited by Oliveira PJ. Cham, Switzerland: Springer, 2018, p. 423–490. [Google Scholar]
- 179.Song M, Mihara K, Chen Y, Scorrano L, Dorn GW II. Mitochondrial fission and fusion factors reciprocally orchestrate mitophagic culling in mouse hearts and cultured fibroblasts. Cell Metab 21: 273–286, 2015. doi: 10.1016/j.cmet.2014.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Song Z, Chen H, Fiket M, Alexander C, Chan DC. OPA1 processing controls mitochondrial fusion and is regulated by mRNA splicing, membrane potential, and Yme1L. J Cell Biol 178: 749–755, 2007. doi: 10.1083/jcb.200704110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Song Z, Ghochani M, McCaffery JM, Frey TG, Chan DC. Mitofusins and OPA1 mediate sequential steps in mitochondrial membrane fusion. Mol Biol Cell 20: 3525–3532, 2009. doi: 10.1091/mbc.e09-03-0252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Stojanovski D, Koutsopoulos OS, Okamoto K, Ryan MT. Levels of human Fis1 at the mitochondrial outer membrane regulate mitochondrial morphology. J Cell Sci 117: 1201–1210, 2004. doi: 10.1242/jcs.01058. [DOI] [PubMed] [Google Scholar]
- 183.Strack S, Wilson TJ, Cribbs JT. Cyclin-dependent kinases regulate splice-specific targeting of dynamin-related protein 1 to microtubules. J Cell Biol 201: 1037–1051, 2013. doi: 10.1083/jcb.201210045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Taguchi N, Ishihara N, Jofuku A, Oka T, Mihara K. Mitotic phosphorylation of dynamin-related GTPase Drp1 participates in mitochondrial fission. J Biol Chem 282: 11521–11529, 2007. doi: 10.1074/jbc.M607279200. [DOI] [PubMed] [Google Scholar]
- 185.Tanaka A, Cleland MM, Xu S, Narendra DP, Suen DF, Karbowski M, Youle RJ. Proteasome and p97 mediate mitophagy and degradation of mitofusins induced by Parkin. J Cell Biol 191: 1367–1380, 2010. doi: 10.1083/jcb.201007013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Tondera D, Grandemange S, Jourdain A, Karbowski M, Mattenberger Y, Herzig S, Da Cruz S, Clerc P, Raschke I, Merkwirth C, Ehses S, Krause F, Chan DC, Alexander C, Bauer C, Youle R, Langer T, Martinou JC. SLP-2 is required for stress-induced mitochondrial hyperfusion. EMBO J 28: 1589–1600, 2009. doi: 10.1038/emboj.2009.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Toyama EQ, Herzig S, Courchet J, Lewis TL Jr, Losón OC, Hellberg K, Young NP, Chen H, Polleux F, Chan DC, Shaw RJ. AMP-activated protein kinase mediates mitochondrial fission in response to energy stress. Science 351: 275–281, 2016. doi: 10.1126/science.aab4138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Vanstone JR, Smith AM, McBride S, Naas T, Holcik M, Antoun G, Harper ME, Michaud J, Sell E, Chakraborty P, Tetreault M, Majewski J, Baird S, Boycott KM, Dyment DA, MacKenzie A, Lines MA; Care4Rare Consortium . DNM1L-related mitochondrial fission defect presenting as refractory epilepsy. Eur J Hum Genet 24: 1084–1088, 2016. doi: 10.1038/ejhg.2015.243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Vásquez-Trincado C, García-Carvajal I, Pennanen C, Parra V, Hill JA, Rothermel BA, Lavandero S. Mitochondrial dynamics, mitophagy and cardiovascular disease. J Physiol 594: 509–525, 2016. doi: 10.1113/JP271301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Vendelin M, Béraud N, Guerrero K, Andrienko T, Kuznetsov AV, Olivares J, Kay L, Saks VA. Mitochondrial regular arrangement in muscle cells: a “crystal-like” pattern. Am J Physiol Cell Physiol 288: C757–C767, 2005. doi: 10.1152/ajpcell.00281.2004. [DOI] [PubMed] [Google Scholar]
- 191.Wai T, García-Prieto J, Baker MJ, Merkwirth C, Benit P, Rustin P, Rupérez FJ, Barbas C, Ibañez B, Langer T. Imbalanced OPA1 processing and mitochondrial fragmentation cause heart failure in mice. Science 350: aad0116, 2015. doi: 10.1126/science.aad0116. [DOI] [PubMed] [Google Scholar]
- 192.Wai T, Langer T. Mitochondrial dynamics and metabolic regulation. Trends Endocrinol Metab 27: 105–117, 2016. doi: 10.1016/j.tem.2015.12.001. [DOI] [PubMed] [Google Scholar]
- 193.Wang W, Fernandez-Sanz C, Sheu SS. Regulation of mitochondrial bioenergetics by the non-canonical roles of mitochondrial dynamics proteins in the heart. Biochim Biophys Acta Mol Basis Dis 1864, Part B: 1991–2001, 2018. doi: 10.1016/j.bbadis.2017.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Wang W, Wang Y, Long J, Wang J, Haudek SB, Overbeek P, Chang BH, Schumacker PT, Danesh FR. Mitochondrial fission triggered by hyperglycemia is mediated by ROCK1 activation in podocytes and endothelial cells. Cell Metab 15: 186–200, 2012. doi: 10.1016/j.cmet.2012.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Wasiak S, Zunino R, McBride HM. Bax/Bak promote sumoylation of DRP1 and its stable association with mitochondria during apoptotic cell death. J Cell Biol 177: 439–450, 2007. doi: 10.1083/jcb.200610042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Wenger J, Klinglmayr E, Fröhlich C, Eibl C, Gimeno A, Hessenberger M, Puehringer S, Daumke O, Goettig P. Functional mapping of human dynamin-1-like GTPase domain based on x-ray structure analyses. PLoS One 8: e71835, 2013. doi: 10.1371/journal.pone.0071835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Westermann B. Bioenergetic role of mitochondrial fusion and fission. Biochim Biophys Acta 1817: 1833–1838, 2012. doi: 10.1016/j.bbabio.2012.02.033. [DOI] [PubMed] [Google Scholar]
- 198.Wüst RC, de Vries HJ, Wintjes LT, Rodenburg RJ, Niessen HW, Stienen GJ. Mitochondrial complex I dysfunction and altered NAD(P)H kinetics in rat myocardium in cardiac right ventricular hypertrophy and failure. Cardiovasc Res 111: 362–372, 2016. doi: 10.1093/cvr/cvw176. [DOI] [PubMed] [Google Scholar]
- 199.Xu S, Cherok E, Das S, Li S, Roelofs BA, Ge SX, Polster BM, Boyman L, Lederer WJ, Wang C, Karbowski M. Mitochondrial E3 ubiquitin ligase MARCH5 controls mitochondrial fission and cell sensitivity to stress-induced apoptosis through regulation of MiD49 protein. Mol Biol Cell 27: 349–359, 2016. doi: 10.1091/mbc.e15-09-0678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Xu S, Wang P, Zhang H, Gong G, Gutierrez Cortes N, Zhu W, Yoon Y, Tian R, Wang W. CaMKII induces permeability transition through Drp1 phosphorylation during chronic β-AR stimulation. Nat Commun 7: 13189, 2016. doi: 10.1038/ncomms13189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Yoon Y, Krueger EW, Oswald BJ, McNiven MA. The mitochondrial protein hFis1 regulates mitochondrial fission in mammalian cells through an interaction with the dynamin-like protein DLP1. Mol Cell Biol 23: 5409–5420, 2003. doi: 10.1128/MCB.23.15.5409-5420.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Youle RJ, van der Bliek AM. Mitochondrial fission, fusion, and stress. Science 337: 1062–1065, 2012. doi: 10.1126/science.1219855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Yu T, Jhun BS, Yoon Y. High-glucose stimulation increases reactive oxygen species production through the calcium and mitogen-activated protein kinase-mediated activation of mitochondrial fission. Antioxid Redox Signal 14: 425–437, 2011. doi: 10.1089/ars.2010.3284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Yu T, Robotham JL, Yoon Y. Increased production of reactive oxygen species in hyperglycemic conditions requires dynamic change of mitochondrial morphology. Proc Natl Acad Sci USA 103: 2653–2658, 2006. doi: 10.1073/pnas.0511154103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Yue W, Chen Z, Liu H, Yan C, Chen M, Feng D, Yan C, Wu H, Du L, Wang Y, Liu J, Huang X, Xia L, Liu L, Wang X, Jin H, Wang J, Song Z, Hao X, Chen Q. A small natural molecule promotes mitochondrial fusion through inhibition of the deubiquitinase USP30. Cell Res 24: 482–496, 2014. doi: 10.1038/cr.2014.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Zaja I, Bai X, Liu Y, Kikuchi C, Dosenovic S, Yan Y, Canfield SG, Bosnjak ZJ. Cdk1, PKCδ and calcineurin-mediated Drp1 pathway contributes to mitochondrial fission-induced cardiomyocyte death. Biochem Biophys Res Commun 453: 710–721, 2014. doi: 10.1016/j.bbrc.2014.09.144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Zepeda R, Kuzmicic J, Parra V, Troncoso R, Pennanen C, Riquelme JA, Pedrozo Z, Chiong M, Sánchez G, Lavandero S. Drp1 loss-of-function reduces cardiomyocyte oxygen dependence protecting the heart from ischemia-reperfusion injury. J Cardiovasc Pharmacol 63: 477–487, 2014. doi: 10.1097/FJC.0000000000000071. [DOI] [PubMed] [Google Scholar]
- 208.Zhang H, Wang P, Bisetto S, Yoon Y, Chen Q, Sheu SS, Wang W. A novel fission-independent role of dynamin-related protein 1 in cardiac mitochondrial respiration. Cardiovasc Res 113: 160–170, 2017. doi: 10.1093/cvr/cvw212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Zhang K, Li H, Song Z. Membrane depolarization activates the mitochondrial protease OMA1 by stimulating self-cleavage. EMBO Rep 15: 576–585, 2014. doi: 10.1002/embr.201338240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Zhao J, Liu T, Jin S, Wang X, Qu M, Uhlén P, Tomilin N, Shupliakov O, Lendahl U, Nistér M. Human MIEF1 recruits Drp1 to mitochondrial outer membranes and promotes mitochondrial fusion rather than fission. EMBO J 30: 2762–2778, 2011. doi: 10.1038/emboj.2011.198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Zhao T, Huang X, Han L, Wang X, Cheng H, Zhao Y, Chen Q, Chen J, Cheng H, Xiao R, Zheng M. Central role of mitofusin 2 in autophagosome-lysosome fusion in cardiomyocytes. J Biol Chem 287: 23615–23625, 2012. doi: 10.1074/jbc.M112.379164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Zhou W, Chen KH, Cao W, Zeng J, Liao H, Zhao L, Guo X. Mutation of the protein kinase A phosphorylation site influences the anti-proliferative activity of mitofusin 2. Atherosclerosis 211: 216–223, 2010. doi: 10.1016/j.atherosclerosis.2010.02.012. [DOI] [PubMed] [Google Scholar]
- 213.Zunino R, Braschi E, Xu L, McBride HM. Translocation of SenP5 from the nucleoli to the mitochondria modulates DRP1-dependent fission during mitosis. J Biol Chem 284: 17783–17795, 2009. doi: 10.1074/jbc.M901902200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Zunino R, Schauss A, Rippstein P, Andrade-Navarro M, McBride HM. The SUMO protease SENP5 is required to maintain mitochondrial morphology and function. J Cell Sci 120: 1178–1188, 2007. doi: 10.1242/jcs.03418. [DOI] [PubMed] [Google Scholar]