

Abstract
Polymerase-δ-interacting protein 2 (Poldip2) controls a wide variety of cellular functions and vascular pathologies. To mediate these effects, Poldip2 interacts with numerous proteins and generates reactive oxygen species via the enzyme NADPH oxidase 4 (Nox4). We have previously shown that Poldip2 can activate the Rho family GTPase RhoA, another signaling node within the cell. In this study, we aimed to better understand how Poldip2 activates Rho family GTPases and the functions of the involved proteins in vascular smooth muscle cells (VSMCs). RhoA is activated by guanine nucleotide exchange factors. Using nucleotide-free RhoA (isolated from bacteria) to pulldown active RhoGEFs, we found that the RhoGEF epithelial cell transforming sequence 2 (Ect2) is activated by Poldip2. Ect2 is a critical RhoGEF for Poldip2-mediated RhoA activation, because siRNA against Ect2 prevented Poldip2-mediated RhoA activity (measured by rhotekin pulldowns). Surprisingly, we were unable to detect a direct interaction between Poldip2 and Ect2, as they did not coimmunoprecipitate. Nox4 is not required for Poldip2-driven Ect2 activation, as Poldip2 overexpression induced Ect2 activation in Nox4 knockout VSMCs similar to wild-type cells. However, antioxidant treatment blocked Poldip2-induced Ect2 activation. This indicates a novel reactive oxygen species-driven mechanism by which Poldip2 regulates Rho family GTPases. Finally, we examined the function of these proteins in VSMCs, using siRNA against Poldip2 or Ect2 and determined that Poldip2 and Ect2 are both essential for vascular smooth muscle cell cytokinesis and proliferation.
Keywords: cytokinesis, epithelial cell transforming sequence 2, polymerase-δ-interacting protein 2, RhoGEF, vascular smooth muscle
INTRODUCTION
Polymerase-δ-interacting protein 2 (Poldip2) is a critical signaling hub within the cell, regulating DNA repair (18, 40, 55), mitochondria morphology (2), cell morphology (9, 39), cell migration (9, 39), cell proliferation (1, 4, 31), cell polarity (9), cell adhesion (56), and cell secretions (15, 53). The mechanisms by which it controls these diverse functions have been partially described. Poldip2 has over 35 putative binding partners, which enable it to perform many of these tasks (22). For example, Poldip2 was originally identified in a yeast-two-hybrid screen as a binding partner of polymerase-δ (35) and since then has been shown to interact with multiple other polymerases (18, 40, 55). The binding of Poldip2 to Polλ and Polη enhances the performance of both enzymes to promote DNA damage repair (40). Similarly, we previously determined that Poldip2 binds p22phox, a subunit of multiple enzymes in the NADPH oxidase (Nox) family. When Poldip2 is bound to p22phox within Nox4, it enhances enzyme activity to generate reactive oxygen species (ROS) (39). This function of Poldip2 may have numerous effects in the cell, because ROS act as signaling molecules via direct oxidation of proteins (3). Additionally, Poldip2 is expressed as a 42-kDa protein that in some cells is processed in mitochondria to a 37-kDa form (22). While homozygous deletion of Poldip2 is embryonic lethal, Poldip2 heterozygote (+/−) mice show a clear vascular phenotype: disorganized aortic structure in the tunica media and decreased contractility, the primary physiological function of vascular smooth muscle cells (VSMCs) (53). In pathological conditions, Poldip2 contributes to VSMC migration and secretion of matrix proteins (1, 53). Still, it is unclear how the myriad of functions that Poldip2 controls connect to each other.
One common pathway activated by Poldip2 involves the Rho family GTPase, RhoA (39). Rho family GTPases are known to regulate many of the same functions as Poldip2. While they are particularly well studied for their role in the actin cytoskeleton and thus cell morphology, migration, adhesion, polarity, secretion, and proliferation (6, 21, 57), Rho family GTPases have also been implicated in DNA damage repair (14) and mitochondrial morphology (13). Therefore, we sought to better understand the mechanism by which Poldip2 regulates Rho family GTPases.
Rho GTPases act as molecular switches. They are “on” (interacting with downstream effectors) when GTP-bound and “off” while GDP bound (23). Canonically, guanine nucleotide exchange factors (GEFs) turn “on” GTPases by promoting GDP release, which results in binding of the more abundant guanine nucleotide GTP (45). Conversely, GTPase-activating proteins (GAPs) enhance the intrinsic ability of the GTPase to convert GTP to GDP, thus turning them “off.” Additionally, guanine nucleotide dissociation inhibitors (GDIs) strip Rho family GTPases from the plasma membrane and sequester them away from downstream effectors (8).
In this study, we hypothesized that Poldip2 may promote Rho activity by activating a RhoGEF. We specifically investigated the consequences of this signaling pathway in VSMCs, where both Poldip2 and Rho are physiologically relevant. We identified the RhoGEF Ect2 as a critical intermediate in Poldip2-mediated RhoA activation and an important downstream effector of Poldip2 that also regulates VSMC proliferation.
MATERIALS AND METHODS
Tissue culture.
Rat aortic smooth muscle cells (RASMs; passages 6–13) were cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% calf serum, 4.5 g/l glucose, 2–4 mM glutamine, 100 U/ml penicillin, and 100 μg/ml streptomycin. Similarly, HEK293T cells and mouse aortic smooth muscle cells (MASMs; passages 6–13), including Nox4−/− MASMs isolated as previously described (33) from mice generated by Dr. K. H. Krause (7) and Poldip2+/− MASMs (53), were grown in the same media as RASMs but supplemented with 10% fetal bovine serum instead of calf serum. Human aortic smooth muscle cells (HASMs; passages 8–12) were cultured in Medium 231 (Thermo Fisher Scientific) supplemented with smooth muscle growth supplement (ThermoFisher Scientic), 2 mM glutamine, 100 U/ml penicillin, and 100 μg/ml streptomycin. Cells were passaged approximately every 5 days using 0.25% trypsin and fed every 2 to 4 days. All MASM and RASM cell preparations were generated from a single group of 4–12 male ~10-wk-old euthanized animals, except for wild-type (WT) MASMs. WT MASMs were generated from three distinct groups of 4–12 male ~10-wk-old animals. A single batch of HASMs was purchased (Cascade Biologics c-007-5c, lot 200707-934 generated from an 18-yr-old male) and used. MASMs were from C57BL/6 mice and RASMs were isolated from Sprague-Dawley rats. All protocols for isolation of these cells were completed as approved by the Emory University School of Medicine Institutional Animal Care and Use Committee.
Adenovirus transduction.
Adenoviruses coexpressing Poldip2 and green fluorescent protein (GFP) or GFP alone were prepared in HEK293T cells as described previously (39) and purified by ultracentrifugation in a cesium chloride gradient followed by dialysis (32) (ThermoScientific, 3.5K Dialysis Cassettes). Transduction of VSMCs was performed in serum-free DMEM for 2 h at 37°C and 5% CO2. Following adenovirus transduction, cells were serum deprived for 48–72 h before use in experiments. Infection efficiency ranged from 80 to 90% and was visualized by GFP just before experiments.
Immunoblotting.
VSMCs were lysed in the various pulldown buffers described below. Samples were sonicated and a Bradford assay (Bio-Rad), or Precision Red Advanced Protein Reagent (Cytoskeleton) was used to assess protein concentrations. Equal amounts of protein were aliquoted, and samples were brought to equal volume with water and Laemmli buffer. After boiling for 10 min at 100°C, samples were resolved in 7.5–12% acrylamide gels and transferred to PVDF membranes. PVDF membranes were blocked in 5% bovine serum albumin (BSA) or 5% fat-free milk for at least 30 min before incubation with primary antibodies in Tris-buffered saline with 0.1% Tween (TBST) overnight. Primary antibodies used were as follows: anti-RhoGDI polyclonal rabbit Ab (Cell Signaling 2564; 1:1,000), anti-Ect2 polyclonal rabbit Ab (Millipore 07-1364-EMD; 1:1,000), anti-myc monoclonal mouse Ab (Cell Signaling 2276; 1:1,000), anti-myc rabbit Ab (Cell Signaling 2278; 1:1,000), anti-Poldip2 monoclonal rabbit Ab (Abcam Ab181841; 1:1,000), anti-caseinolytic mitochondrial matrix peptidase chaperone subunit (CLPX) rabbit monoclonal Ab (Abcam Ab168338; 1:1,000), anti-p115 RhoGEF rabbit antibody (Cell Signaling 3669; 1:500), anti-tubulin monoclonal mouse Ab (Sigma T4026; 1:10,000), anti-actin (Sigma A5441; 1:5,000), and anti-GAPDH mouse monoclonal Ab (Millipore MAB374; 1:10,000). Next, membranes were rinsed in TBST for 30 min before incubation with secondary antibodies for 1 h. Secondary antibodies were used at twice the dilution of primary antibodies and were as follows: anti-mouse (Amersham NA931) and anti-rabbit (Jackson laboratories 711035152 and Amersham NA934) horseradish peroxidase-conjugated antibodies. Bands were visualized using enhanced chemiluminescence (SuperSignal West Pico Chemiluminescent Substrate; ThermoFisher Scientific).
Preparation of beads for pulldowns.
Glutathione S-transferase (GST) tagged nucleotide-free RhoA [RhoA(17A)] or the Rho-binding domain (RBD) of rhotekin (plasmids kindly donated by Adrienne Cox, University of North Carolina-Chapel Hill; Ref. 24) were transformed into Rosetta bacteria and cultured in ~1 liter LB at 37°C. Once an optical density of 0.5–0.6 at a wavelength of 600 nm was achieved, expression of GST-RhoA(17A) or GST-Rhotekin(RBD) was induced with 0.1 mM or 0.3 mM IPTG, respectively, for 24 h at room temperature. Bacteria were spun down, and supernatant was removed and divided into five to six aliquots that were stored at −20°C for up to a few months. When ready to make beads, bacteria were lysed by sonication in bead preparation buffer [20 mM HEPES pH 7.5, 150 mM NaCl, 5 mM MgCl, 1% Triton X-100, 1 mM dithiothreitol (DTT), 10 µg/ml aprotinin, 10 µg/ml leupeptin, and 1 mM PMSF] for GST-RhoA(17A) or in bead lysis buffer (20 mM HEPES pH 7.5, 300 mM NaCl, 10% glycerol, 2 mM EDTA, 1% Triton X-100, 2 mM DTT, 10 μg/ml aprotinin, 10 μg/ml leupeptin, and 1 mM PMSF) for GST-Rhotekin(RBD). Debris were spun down, and the supernatant was incubated with 100–500 µl [for RhoA(17A)] or 500–1000 µl [for Rhotekin(RBD)] of 50% (vol/vol) glutathione (GSH) agarose bead slurry for 1 h at 4°C and rinsed in bead preparation or bead lysis buffer three times. GST-Rhotekin(RBD) was then measured for protein concentration, aliquoted, and stored at −80°C until use. GST-RhoA(17A)-bound GSH agarose beads were blocked in 5% BSA for 1 h and rinsed three more times in bead preparation buffer. Finished GST-RhoA(17A) beads were stored at 4°C and used within 24 h. GST-only control beads were made using the same method as GST-RhoA(17A) beads.
Pulldown assays.
To pulldown active GEFs, GST-RhoA(17A) pulldowns were performed as described previously (19, 24). VSMCs were lysed in bead preparation buffer. After Precision Red Advanced Protein Reagent (Cytoskeleton) was used to determine protein concentration, an equal amount of protein for each condition (~1,000 μg) was combined with an equal amount of GST-RhoA(17A)-bound GSH agarose beads (~80 μg, prepared from bacteria as described above) or GST-bound GSH agarose beads and rocked for 1 h at 4°C. These beads were rinsed three times with bead preparation buffer. Rinsed GST-RhoA(17A) (or GST)-bound GSH agarose beads were then taken to mass spectrometry or incubated with 3× Laemmli buffer and 50 mM DTT to elute proteins for immunoblotting. Total cell lysate was also examined by Western blotting to confirm equal loading of pulldown lysate.
To pulldown active RhoA, GST-Rhotekin(RBD) pulldowns were performed (24). RASMs were lysed in 500 μl (volume for 100 mm dish) pulldown buffer [50 mM Tris pH 7.6, 500 mM NaCl, 10 mM MgCl2 (added just before lysis), 1% Triton X-100, 0.1% SDS, 0.5% deoxycholate, 10 μg/ml aprotinin, 10 μg/ml leupeptin, and 1 mM PMSF], and lysates were clarified by centrifugation (1 min, 12,000 rpm, 4°C). Clarified lysates were then incubated with 60 μg of agarose bead-bound GST-Rhotekin(RBD) (prepared from bacteria, as described above) for 30 min at 4°C with rocking. Beads were then spun down (30 s, 12,000 rpm, 4°C) and washed in washing buffer [50 mM Tris pH 7.6, 150 mM NaCl, 10 mM MgCl2 (added just prior), 1% Triton X-100, 10 μg/ml aprotinin, 10 μg/ml leupeptin, and 1 mM PMSF] three times. After the final wash, 20 μl of 6× SDS sample buffer was added to each sample. Samples were boiled for 3 min, spun down, and stored at −20°C until used for immunoblotting. Total cell lysate was also examined by Western blotting to confirm equal loading of pulldown lysate.
Liquid chromatography–tandem mass spectrometry analysis.
Overnight bead digestion was performed as previously described (36). Briefly, the residual wash buffer was removed and two times bead volume of 50 mM ammonium bicarbonate buffer (ABC buffer) was added. Reduction was performed with 1 mM DTT for 30 min and then samples were alkylated with 5 mM iodoacetamide for 30 min in the dark. The samples were incubated on a rotator at low speed for both steps. The samples were then spun down and digested with 1:100 (wt/wt) lysyl endopeptidase (Wako) for 2 h and further digested overnight with 1:50 (wt/wt) trypsin (Promega). Both digestions were performed at room temperature. The digested samples were then acidified with 1/10 volume of 10% formic acid (FA) and 1% trifluoroacetic acid (TFA). Desalting was performed with 50-mg tC18 SepPak columns (Waters, Milford, MA), and eluents were dried under vacuum. The liquid chromatography-tandem mass spectrometry analysis (LC-MS/MS) analysis was performed according to previously described protocols (50). Loading buffer (10 µl of 0.1% FA, 0.03% TFA, 1% acetonitrile) was added to the dry sample, and 3.5 μl were separated on a 25-cm 1.9-μm C18 (Dr. Maisch, Germany) self-packed column (New Objective, Woburn, MA) by a NanoAcquity UHPLC (Waters) and monitored on an Q-Exactive Plus mass spectrometer (ThermoFisher Scientific, San Jose, CA). Elution was performed over a 120-min gradient at a rate of 400 nl/min with buffer B ranging from 3 to 80% (buffer A: 0.1% formic acid and 5% DMSO in water; buffer B: 0.1% formic and 5% DMSO in acetonitrile). The mass spectrometer cycle was programmed to collect one full MS scan followed by 10 data-dependent MS/MS scans. The MS scans were collected at a resolution of 70,000 (300- to 1,800-m/z range, 1,000,000 AGC, 100-ms maximum ion time), and the MS/MS spectra were acquired at a resolution of 17,500 (2-m/z isolation width, 0.5-m/z isolation offset, 25% collision energy, 100,000 AGC target, and 50-ms maximum ion time). Dynamic exclusion was set to exclude previous sequenced peaks for 30 s within a 10-ppm window. The SageN Sorcerer SEQUEST 4.3 algorithm was used to search and match MS/MS spectra to a Rattus norvegicus database with 29,688 target entries (REFSEQ Version 62). The target database was concatenated to an equal number of pseudo-reversed decoy sequences (12, 60). Searching parameters included mass tolerance of precursor ions (±20 ppm), fully tryptic restriction, dynamic modifications for oxidized Met (+15.9949 Da), four maximal modification sites, and a maximum of four missed cleavages. Only b and y ions were considered for scoring (Xcorr), and Xcorr along with ΔCn were dynamically increased for groups of peptides organized by a combination of fully tryptic and precursor ion charge state to remove false positive hits along with decoys until achieving a false discovery rate (FDR) of < 1%. The FDR was estimated by the number of decoy matches (nd) and total number of assigned matches (nt). FDR = 2 × nd/nt, assuming mismatches in the original database were the same as in the decoy database (51).
Coimmunoprecipitation.
Cells were lysed in buffer containing: 50 mM HEPES, 50 mM NaCl, 5 mM EDTA, 10 mM napyrophosphate, 50 mM NaF, 1 mM Na orthovanadate, 1% Triton-x-100, 10 µg/ml aprotinin, 10 µg/ml leupeptin, and 1 mM PMSF and briefly sonicated. To preclear, 500 μg of protein lysate were nutated for 30 min at 4°C with 20 μl protein G PLUS-agarose beads (Santa Cruz Biotechnology). Beads were spun down, and supernatant was added to 2 μg of antibody (anti-Myc monoclonal mouse from Cell Signaling 2276 or normal mouse IgG control from Santa Cruz Biotechnology sc-2025) and 20 μl of protein G PLUS-agarose beads and nutated for 2 h at 4°C. Beads were rinsed three times with PBS to remove nonbound proteins and proteins were eluted with 4× Laemmli buffer and 0.06 mM DTT before immunoblotting.
Small interfering RNA.
Transfection was performed in Opti-MEM reduced serum media. Cells were incubated in a solution containing 25 nM siRNA against the message of interest (from a 20 μM stock) and Lipofectamine RNAiMAX Reagent (volume equal to double the volume of siRNA; Invitrogen) for 5–12 h. Cells were then rinsed in supplemented DMEM (same as described above). siRNA sequences were as follows: Poldip2 (Poldip2-A: sense 5′-CUAUUUCUCCAUAGUGUUU[dT][dT]-3′ antisense: 5′-AAACACUAUGGAGAAAUAG[dT][dT]-3′, Sigma; Poldip2-B: sense 5′-GUCUAUUGGUGGCGAUACU[dT][dT]-3′ antisense 5′-AGUAUCGCCACCAAUAGAC[dT][dT]-3′, Sigma); Ect2 (Ect2-A: sense 5′-CUGACUUACAUGGUACUUU[dT][dT]-3′ antisense 5′-AAAGUACCAUGUAAGUCAG[dT][dT]-3′, Sigma; Ect2-B: sense 5′-GUAACACUAACCAACAGUU[dT][dT]-3′ antisense 5′-AACUGUUGGUUAGUGUUAC[dT][dT]-3′, Sigma); and control (siCtl-1: MISSION siRNA Universal Negative Control #1, Sigma; and siCtl-2: AllStars negative control sense 5′- GGGUAUCGACGAUUACAAAUU-3′ antisense 5′-UUUGUAAUCGUCGAUACCCUG-3′, Qiagen).
Multinucleation analysis.
VSMCs were seeded and grown to 10% confluence on collagen-coated glass coverslips before being treated with siRNA, as described above. Seventy-two hours later, all samples were fixed in 4% paraformaldehyde and subsequently permeabilized with 0.1% Triton X-100 for 10 min. Samples were then incubated with Alexa Fluor568-conjugated phalloidin (ThermoFisher Scientific A12380; 1:100) or with Alexa Fluor488-conjugated phalloidin (ThermoFisher Scientific A12379; 1:200) and TRITC-conjugated wheat germ agglutinin (BioWorld 21761059; 1:400) for 1 h at room temperature and mounted with Vectashield containing DAPI (Vector Laboratories H1200). Slides were imaged by a blinded observer using a Zeiss LSM 510 META Laser Scanning Confocal Microscope System. The number of cells in these blinded images with single or multiple nuclei was counted.
Growth curves.
Prior to the start of the experiment, an equal number of RASMs were plated in quintuplicate. Cells were transfected 24 h later (time 0) with siRNA, as described above and subsequently trypsinized and counted every 24 h using a Scepter Cell Counter (Millipore) for a total of 5 days. Each suspension was counted twice, and the average of two counts was used.
Data analysis.
For all assays concluding in immunoblot, films were scanned and densitometry performed using ImageJ. Values were normalized to a loading control and then expressed as a fold change with respect to the vector-only controls. Significance was determined using a ratio paired t-test, calculated in PRISM (version 7.0b), with a threshold of P < 0.05 considered significant. For multinucleation counts, significance was determined using a Cochran-Mantel-Haenzel test. For proliferation curves, counts were analyzed using a two-way ANOVA with Tukey’s method for correction in PRISM.
RESULTS
Poldip2 enhances activity of the RhoGEF Ect2.
We previously determined that overexpression of Poldip2 enhances activity of RhoA (39). Canonically, this is driven by enhanced activity/expression of RhoGEFs or a decrease in RhoGAPs or RhoGDIs. While there are over 70 RhoGEFs and 70 RhoGAPs (8), only one RhoGDI (RhoGDI-α) is expressed in VSMCs (10). To first determine if Poldip2 regulated RhoGDI expression, we transduced RASMs with viruses expressing both Poldip2 and GFP (AdPoldip2) or GFP alone (AdGFP) and measured RhoGDI-α by Western blot. RhoGDI-α protein levels remained unchanged upon Poldip2 expression (data not shown), suggesting that RhoGDI is unlikely to explain the effect of Poldip2 on RhoA activity, although we did not examine regulation of RhoGDI-α binding to RhoGTPases, which is complex (10, 16).
Instead, we next sought to determine if the activity of any RhoGEFs was affected by Poldip2 expression using a previously validated method of isolating active RhoGEFs from lysate, GST-RhoA(17A) pulldowns (11, 17, 19, 24). When RhoGEFs are activated, they bind with increased affinity to nucleotide-free RhoA [RhoA(17A)] (17); this allows use of purified GST-RhoA(17A) to pull down the pool of active RhoGEFs from lysate (19). Therefore, we used GST-RhoA(17A) (isolated from bacteria) to perform pulldowns for active RhoGEFs in cell lysate from RASMs expressing Poldip2 or a vector control. After elution from pulldown, mass spectrometry was performed to identify and compare the bound proteins in each condition. In addition to performing GST-RhoA(17A) pulldowns, GST only pulldowns were performed with each cell lysate as controls for nonspecific binding.
While no Poldip2-activated GEFs were identified directly by mass spectrometry, we sorted all identified proteins by spectral count (an estimated measure of relative abundance; Refs. 38, 42) and looked for proteins with high spectral counts following GST-RhoA17A pulldown from Poldip2-expressing cells versus lower spectral counts following GST-RhoA17A pulldowns in vector-expressing cells or GST pulldowns. The discovered proteins led us to hypothesize that the RhoGEF Ect2 may be activated by Poldip2.
To explore the possibility that Poldip2 could activate Ect2, we conducted the GST-RhoA(17A) pulldown assay on RASMs overexpressing Poldip2 or GFP alone followed by immunoblot for Ect2 and found that Poldip2 overexpression increased the amount of active Ect2 and slightly increased the amount of total Ect2 (Fig. 1A); however, only the increase in amount of Ect2 pulled down by GST-RhoA(17A) (not total Ect2) was statistically significant upon replication (Fig. 1B). As a negative control, we also examined the effect of Poldip2 expression on the amount of p115-RhoGEF pulled down by GST-RhoA(17A) (Fig. 1A). Poldip2 had no significant effect on the amount of pulled down p115-RhoGEF (Fig. 1C).
Fig. 1.

Polymerase-δ-interacting protein 2 (Poldip2) enhances activity of the RhoGEF epithelial cell transforming sequence 2 (Ect2). A: rat aortic smooth muscle cells were transduced with Myc-Poldip2 (AdPoldip2) or vector-only (AdGFP) adenovirus, and GST-RhoA(17A) pulldowns for active guanine nucleotide exchange factors or GST control pulldowns were performed and immunoblotted for Ect2. Pulldowns were also blotted for p115-RhoGEF to show specificity of RhoGEF activation. Prior to pulldown, some lysate was retained to examine total Ect2 and p115 and to confirm even loading and overexpression (right). The top arrow identifies exogenous Poldip2, and bottom arrow identifies endogenous Poldip2 on the Poldip2 immunoblot. B: the average amount of pulled down or total Ect2 normalized to tubulin is shown (n = 6; *P < 0.05 ratio paired t-test; N.S., not significant; error bars represent SE). C: the average amount of pulled down p115-RhoGEF normalized to tubulin as a fold change above AdGFP is shown (n = 3; N.S., not significant ratio paired t-test; error bars represent SE). D: wild-type (Poldip2+/+) mouse aortic smooth muscle cells (MASMs) and MASMs with heterozygous deletion of Poldip2 (Poldip2+/−) stimulated with serum (5 min, 10% fetal bovine serum) were assessed for active and total Ect2 as above. E: results were quantified (n = 5; *P < 0.05 ratio paired t-test; N.S., not significant; error bars represent SE).
To confirm that this effect was not an artifact of overexpression, we also compared Ect2 pulled down by GST-RhoA(17A) upon Poldip2 depletion. Homozygous deletion of Poldip2 is embryonic lethal (4, 53); therefore, we compared MASMs from Poldip2+/+ and Poldip2+/− mice. After serum stimulation (5 min, 10% serum), Ect2 activity was significantly higher in Poldip2+/+ cells compared with Poldip2+/− cells, but total Ect2 levels were unchanged (Fig. 1, D and E).
Ect2 is required for Poldip2-mediated RhoA activation.
Poldip2 overexpression causes activation of both RhoA (39) and the RhoGEF Ect2 (Fig. 1). To determine if Ect2 is the critical RhoGEF mediating Poldip2-induced activation of RhoA, we examined the ability of Poldip2 to activate RhoA upon Ect2 depletion. Relative RhoA activity was examined using rhotekin pulldown assays. The rho binding domain (RBD) of rhotekin preferentially binds GTP-bound RhoA and thus is routinely used as method to pull down active RhoA from solution and compare relative RhoA activity (19). As expected (39) and shown in Fig. 2, A and B, overexpression of Poldip2 significantly enhanced RhoA activity compared with cells expressing GFP alone in the presence of control siRNA. However, in RASMs with Ect2 depleted via transfection with Ect2 siRNA (Fig. 2A), overexpression of Poldip2 did not significantly increase RhoA activity above that seen in cells expressing the vector control (Fig. 2B). This demonstrates that Ect2 is required for Poldip2-mediated RhoA activation.
Fig. 2.
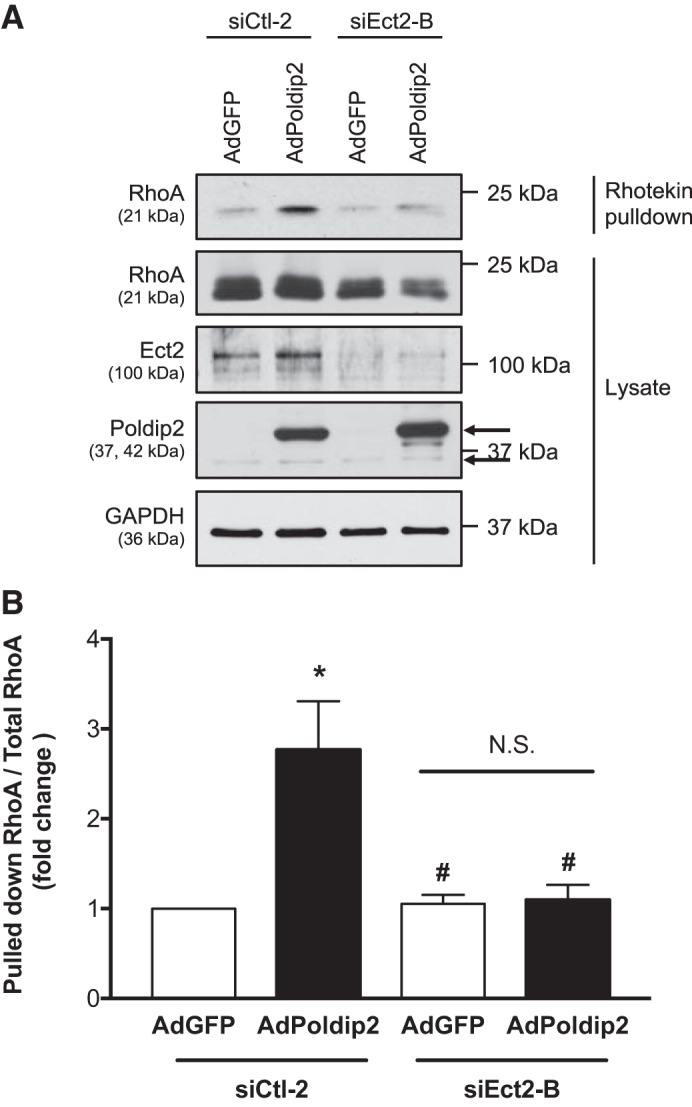
Epithelial cell transforming sequence 2 (Ect2) is required for polymerase-δ-interacting protein 2 (Poldip2)-mediated RhoA activation. A: rat aortic smooth muscle cells were transfected with control or Ect2 siRNA and 24 h later transduced with Myc-Poldip2 (AdPoldip2) or vector-only (AdGFP) adenovirus. GST-Rhotekin(RBD) pulldowns for active RhoA were performed and immunoblotted for RhoA. Prior to pulldown, some lysate was retained to examine total RhoA and to confirm even loading, overexpression, and knockdown. The top arrow identifies exogenous Poldip2, and bottom arrow identifies endogenous Poldip2 on the Poldip2 immunoblot. B: the average amount of active RhoA normalized to total RhoA is shown (n = 5; *P < 0.05 vs. siCtl-2 AdGFP,; #P < 0.05 vs. siCtl-2 AdPoldip2 ratio paired t-test; N.S., not significant; error bars represent SE).
ROS, but not from Nox4, are required for Poldip2-mediated Ect2 activity.
We have previously determined that ROS are required for Poldip2-induced RhoA activation (39). Because our data show that the Rho GEF Ect2 is an intermediate in this pathway, we hypothesized that Poldip2-induced Ect2 activation would similarly require ROS. To examine this possibility, we compared the effect of Poldip2 overexpression on Ect2 activity in the presence or absence of the antioxidant N-acetyl-cysteine (NAC). In the presence of NAC, Poldip2 overexpression had no significant effect on the amount of Ect2 pulled down by GST-Rho(A17A) compared with the vector-only control (Fig. 3, A and B), suggesting that Poldip2-induced Ect2 activation requires ROS. However, NAC treatment also significantly reduced total levels of Ect2 compared with untreated cells (Fig. 3B). Poldip2 interacts with Nox4 to generate ROS (39); therefore, we predicted that Nox4 was the relevant source of ROS. To determine if Nox4 is required for Poldip2 to activate Ect2, we overexpressed Poldip2 in Nox4−/− MASMs and examined Ect2 activity via GST-RhoA(17A) pulldown. Poldip2 increased the amount of Ect2 pulled down by GST-RhoA(17A) in both WT and Nox4−/− MASMs (Fig. 3C), indicating that Nox4 is not required for Poldip2 to enhance Ect2 activity. In fact, on average, Poldip2 enhanced Ect2 activity sixfold in Nox4−/− MASMs compared with twofold in WT MASMs, suggesting that the interaction of Poldip2 with Nox4 may actually inhibit/compete with the ability of Poldip2 to activate Ect2.
Fig. 3.

Reactive oxygen species, but not Nox4, are required for polymerase-δ-interacting protein 2 (Poldip2)-mediated epithelial cell transforming sequence 2 (Ect2) activity. A: rat aortic smooth muscle cells (RASMs) were transduced with Myc-Poldip2 (AdPoldip2) or vector-only (AdGFP) adenovirus and the following day treated with 10–20 mM N-acetyl-cysteine (NAC). Twenty-four hours later, GST-RhoA(17A) pulldowns for active guanine nucleotide exchange factors (GEFs) were performed and immunoblotted for Ect2. Prior to pulldown, some lysate was retained to examine total Ect2 and confirm even loading and overexpression. The top arrow identifies exogenous Poldip2, and bottom arrow identifies endogenous Poldip2 on the Poldip2 immunoblot. Representative immunoblots are from the same film and exposure for each blotted protein. The vertical black line between lanes indicates cropping of extraneous lanes. B: the average amount of pulled down and total Ect2 normalized to tubulin is shown (n = 3–6; *P < 0.05 vs. AdGFP; #P < 0.05 vs. no NAC ratio paired t-test; N.S., not significant; error bars represent SE). C: Similarly, wild-type (WT) and Nox4 knockout (Nox4−/−) mouse aortic smooth muscle cells (MASMs) were transduced with Myc-Poldip2 (AdPoldip2) or vector-only (AdGFP) adenovirus and GST-RhoA(17A) pulldowns for active GEFs were performed and immunoblotted for Ect2. The average amount of pulled down Ect2 normalized to tubulin is shown as a fold change above AdGFP (n = 3–7; *P < 0.05; **P < 0.01 ratio paired t-test; error bars represent SE).
Poldip2 does not appear to interact with Ect2 in basal conditions.
Poldip2 is a multifunctional protein with many binding partners (22), and Ect2 often directly interacts with its activators (26, 27, 61). To determine if Poldip2 and Ect2 interact directly, we immunoprecipitated Myc-Poldip2 and probed for Ect2. Even using mild wash conditions, with which we were able to immunoprecipitate Myc-Poldip2 and other known binding partners [such as caseinolytic mitochondrial matrix peptidase chaperone subunit (CLPX) (44), shown here], we were unable to detect Ect2 coimmunoprecipitating with Myc-Poldip2 (Fig. 4).
Fig. 4.
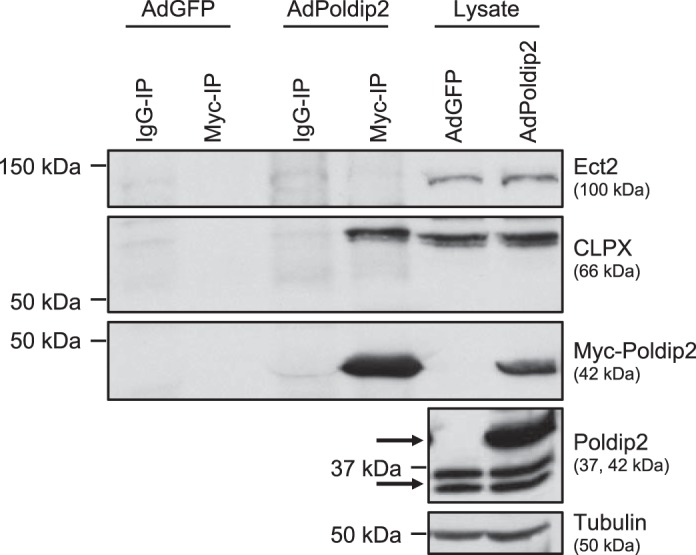
Epithelial cell transforming sequence 2 (Ect2) and polymerase-δ-interacting protein 2 (Poldip2) do not appear to interact in basal conditions. Myc immunoprecipitation was performed on rat aortic smooth muscle cells (RASMs) (shown here) or human aortic smooth muscle cells transduced with vector-only (AdGFP) or Myc-Poldip2 (AdPoldip2) and immunoblotted for Ect2, Myc, and the positive control caseinolytic mitochondrial matrix peptidase chaperone subunit (CLPX) (n = 4). Tubulin was used as a loading control and endogenous Poldip2 shows relative levels of overexpression in lysates. The top arrow identifies exogenous Poldip2, and bottom arrow identifies endogenous Poldip2 on the Poldip2 immunoblot.
Poldip2 and Ect2 both regulate VSMC cytokinesis and proliferation.
Ect2-mediated Rho activation has been implicated in cytokinesis (30, 54), yet some cell types (for example, various tumor cell lines) are able to complete cytokinesis in the absence of Ect2 (24, 25, 28). The potential role of Ect2 in cytokinesis of vascular cells has not been tested. To determine if Poldip2 and Ect2 regulate VSMC cytokinesis, we used two independent siRNA sequences directed to each mRNA to transiently knockdown either Poldip2 or Ect2 and examined the number of multinucleated cells. An increase in the number of multinucleated cells indicates completion of nuclear division but failure to complete cytokinesis at the end of mitosis. Knockdown of either Poldip2 or Ect2 (Fig. 5A) significantly increased the number of multinucleated RASMs (Fig. 5, B and C). Consistent with this observation, knockdown of Poldip2 or Ect2 also significantly decreased cell growth 72, 96, and 120 h after transfection with siRNA (Fig. 5D).
Fig. 5.

Polymerase-δ-interacting protein 2 (Poldip2) and epithelial cell transforming sequence 2 (Ect2) both regulate VSMC cytokinesis and proliferation. A–C: rat aortic smooth muscle cells (RASMs) were plated on collagen-coated coverslips and transfected with either of 2 different siRNA sequences against the same target: control (siCtl-1 and 2), Poldip2 (siPoldip2-A and B), or Ect2 (siEct2-A and B). 72 h after siRNA transfection, the cells were lysed to confirm knockdown by immunoblot (A) or stained with phalloidin (green), wheat germ agglutinin (red), and DAPI (blue) (B). The number of multinucleated cells (examples indicated by white arrow) in each condition was counted by a blinded observer and the average percentage of multinucleated cells graphed (n = 4–7; ****P < 0.0001 vs. siCtl-1; ####P < 0.0001 vs. siCtl-2 Cochran-Mantel-Haenzel test; error bars = SE) (C). D: a separate group of RASMs was plated in 6-well plates in equal numbers. The following day, the cells were treated in quintuplicate with control (siCtl-2), Poldip2, or Ect2 siRNA. Knockdown was confirmed by immunoblot at 24 h after transfection and the number of RASMs in each condition was counted every 24 h for 5 days following transfection at time 0 (n = 3; ####P < 0.0001 vs. siCtl-2 at 72-, 96-, and 120-h two-way ANOVA with Tukey’s method for correction; error bars represent SE).
DISCUSSION
Poldip2 is a multifunctional protein, regulating DNA polymerase activity (18, 40, 55), mitochondrial morphology (2), cell migration (9, 39), extracellular matrix secretion (15, 53), cell adhesion (56), and cell proliferation (1, 4, 31). While the molecular mechanisms by which it controls such a variety of tasks are unclear, there have been two previous studies dissecting the role of Poldip2 in cell proliferation. Klaile and colleagues (31) determined that Poldip2 regulates mitotic spindle formation and chromosome segregation, and we have previously reported that Poldip2 regulates cell cycle check point progression (4). Here we provide further insight into the role of Poldip2 during cell proliferation, showing that Poldip2 regulates cytokinesis. We also show that Poldip2 promotes activation of the RhoGEF Ect2 to activate RhoA (the small GTPase responsible for cleavage furrow ingression during cytokinesis; Ref. 61) and that Ect2 regulates VSMC cytokinesis.
This function of Poldip2 is likely separate from our previous finding relating Poldip2 and control of the cell cycle. We previously proposed that Poldip2 influences cell cycle check points via the transcriptional regulators E2F and p53 (4), both of which have been reported to control Ect2 expression (48, 49). However, in this study, we did not observe a significant increase in Ect2 expression upon overexpression of Poldip2 (Fig. 1B), suggesting the existence of an additional mechanism. Furthermore, knockout of Poldip2 caused growth arrest of mouse embryonic fibroblasts in the G2/M phase of the cell cycle (4), when Ect2 expression (34, 47) (or possibly phosphorylation-regulated activity; Ref. 54) is highest. If Ect2 regulation by Poldip2 is simply a consequence of the role of Poldip2 and Ect2 in the cell cycle, then we would predict that depletion of Poldip2 would increase both Ect2 activation and expression by increasing the number of cells in G2/M. Instead, we observed that Ect2 activity was decreased and Ect2 expression unchanged upon Poldip2 depletion (Fig. 1, D and E). Thus it appears that Poldip2 has multiple functions in cell division.
It is less clear if Ect2 activation is discrete from the role of Poldip2 at the mitotic spindle. Both Poldip2 (31) and Ect2 (54) have been shown to localize to the mitotic spindle during mitosis, hinting at a possible point of interaction. However, we were not able to coimmunoprecipitate the two proteins, suggesting that they may not directly interact at the spindle, although we cannot rule out an interaction below the level of detection. It may be necessary to synchronize the cells before coimmunoprecipitation or otherwise modify our coimmunoprecipitation protocol to detect any interaction. Consistent with a separate mechanism from the effect of Poldip2 on the mitotic spindle, the observed defects in spindle assembly and chromosome segregation upon Poldip2 knockdown (31) have not been described for Ect2. Our data are consistent with the alternative possibility that Poldip2 depletion reduces Ect2 activity at the cleavage furrow, leading to reduced RhoA activity and no cleavage furrow ingression because RhoA no longer activates myosin II and actin polymerization (61).
Our novel observation that Poldip2 activation of RhoA is mediated by the RhoGEF Ect2 raises the additional question of how Poldip2 enhances Ect2 activity. In general, Ect2 is autoinhibited by its phosphoserine-binding BRCT domains folding over its catalytic DH domain (20, 29, 46), yet there are multiple mechanisms described for release of this autoinhibition. During cytokinesis, a central spindle associated protein, MgcRacGAP, is phosphorylated by Plk-1 (5, 58) to create a docking site for the BRCT domains of Ect2 (5, 58, 61). This recruits Ect2 to the central spindle and prevents autoinhibition of Ect2 by the BRCT domains, holding it in an active conformation and allowing for activation of RhoA in a narrow area of the cell cortex above the central spindle (61). Additionally, studies using in vitro phosphatase treatment (54) and phosphodeficient mutants of specific residues on Ect2 (20, 26, 43) suggest that phosphorylation activates Ect2. Consistent with an important role for phosphorylation, the COOH terminus of Ect2 (a.a. 775–882), which does not contain any known functional domains but is ~30% composed of phosphorylatable residues, is required for optimal Ect2 activity (52). Therefore, it is most likely that Poldip2 regulates phosphorylation of Ect2 or protein:protein interactions with the BRCT domains of Ect2 to enhance its activity.
Our initial mass spectrometry screen identified PHGDH as a potential Poldip2-regulated protein: higher spectral counts of PHGDH were observed in the proteins bound by GST-RhoA(17A) when Poldip2 was overexpressed. One explanation for this increased binding to GST-RhoA(17A) is increased binding of PHGDH to the active RhoGEFs bound to GST-RhoA(17A). Since PHGDH has been described to bind the BRCT domains of Ect2 (59), it is possible that Poldip2 enhances PHGDH binding to Ect2 to activate Ect2 [increasing binding of both proteins to GST-RhoA(17A)]. This would be an extremely interesting mechanism of activation, yet it is untested and how ROS and Nox fit into the signaling pathway is currently unclear. PHGDH is essential for proliferation and functions as the enzyme responsible for the committed step in l-serine biogenesis (41). This has led to the conclusion that serine biogenesis is required for proliferation of certain cell types. However, the effect of PHGDH depletion on proliferation cannot be rescued by exogenous serine, indicating another essential role of PHGDH in proliferation (41). Possibly Ect2 activation and control of cytokinesis is the missing function.
Alternatively, Poldip2 may regulate ROS-sensitive kinases/phosphatases (3) to enhance Ect2 activation. We previously determined that Poldip2-induced activation of RhoA was inhibited by the antioxidant NAC (39), suggesting a ROS-dependent mechanism. When we examined the effect of NAC on Poldip2-induced Ect2 activity we also observed that NAC inhibited the ability of Poldip2 to activate Ect2 (Fig. 3, A and B). However, NAC treatment also caused a dramatic decrease in total Ect2 (Fig. 3, A and B), making these experiments difficult to interpret. Moreover, Poldip2 was able to activate Ect2 even in the absence of Nox4 (Fig. 3C), suggesting that if ROS are involved, they are derived from another source. One possibility is Nox1, although Ect2 activity is highly variable in conditions required for Nox1−/− cell growth (data not shown). Another possibility is mitochondrial ROS, although the ability of Poldip2 to regulate mitochondrial ROS has not been reported yet.
In summary, Poldip2 is a novel regulator of Ect2, and both proteins play a critical role in VSMC proliferation. Excessive VSMC proliferation is a hallmark of atherosclerosis and restenosis (37). Therefore, understanding this role of Poldip2 in VSMCs is an essential step towards potential therapeutic targeting of Poldip2 in these diseases.
GRANTS
This work was supported by American Heart Association Grant 16POST30400020 (to L. P. Huff), National Heart, Lung, and Blood Institute Grants T32-HL-007745 (to L. P. Huff) and P01-HL-095070 (to K. K. Griendling), and Emory University’s SURE and SIRE programs (to D. S. Kikuchi). Additionally, this study was supported in part by the Emory Integrated Proteomics Core (EIPC), which is subsidized by the Emory University School of Medicine and is one of the Emory Integrated Core Facilities. Additional support was provided by the Georgia Clinical & Translational Science Alliance of the National Institutes of Health under Award Number UL1TR002378.
DISCLAIMERS
The content is solely the responsibility of the authors and does not necessarily reflect the official views of the National Institutes of Health or the American Heart Association.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
L.P.H., D.S.K., B.L., and K.K.G. conceived and designed research; L.P.H., D.S.K., E.F., S.J.F., and M.Z.T. performed experiments; L.P.H., D.S.K., S.J.F., and B.L. analyzed data; L.P.H., D.S.K., B.L., and K.K.G. interpreted results of experiments; L.P.H., D.S.K., E.F., and S.J.F. prepared figures; L.P.H. and D.S.K. drafted manuscript; L.P.H., D.S.K., B.L., and K.K.G. edited and revised manuscript; L.P.H., D.S.K., E.F., S.J.F., M.Z.T., B.L., and K.K.G. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Dr. Duc Duong of the Emory Integrated Proteomics Core for help with the mass spectrometry experiments, Dr. Eugene Huang and Megan Warnock of the Emory Data Analysis and Biostatistics Core for biostatistical guidance, and Dr. Lula Hilenski of the Emory Microscopy in Medicine Core. We also appreciate the gift of plasmids from Dr. Adrienne Cox (University of North Carolina at Chapel Hill).
REFERENCES
- 1.Amanso AM, Lassègue B, Joseph G, Landázuri N, Long JS, Weiss D, Taylor WR, Griendling KK. Polymerase δ-interacting protein 2 promotes postischemic neovascularization of the mouse hindlimb. Arterioscler Thromb Vasc Biol 34: 1548–1555, 2014. doi: 10.1161/ATVBAHA.114.303873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Arakaki N, Nishihama T, Kohda A, Owaki H, Kuramoto Y, Abe R, Kita T, Suenaga M, Himeda T, Kuwajima M, Shibata H, Higuti T. Regulation of mitochondrial morphology and cell survival by Mitogenin I and mitochondrial single-stranded DNA binding protein. Biochim Biophys Acta 1760: 1364–1372, 2006. doi: 10.1016/j.bbagen.2006.05.012. [DOI] [PubMed] [Google Scholar]
- 3.Brown DI, Griendling KK. Regulation of signal transduction by reactive oxygen species in the cardiovascular system. Circ Res 116: 531–549, 2015. doi: 10.1161/CIRCRESAHA.116.303584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brown DI, Lassègue B, Lee M, Zafari R, Long JS, Saavedra HI, Griendling KK. Poldip2 knockout results in perinatal lethality, reduced cellular growth and increased autophagy of mouse embryonic fibroblasts. PLoS One 9: e96657, 2014. doi: 10.1371/journal.pone.0096657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Burkard ME, Maciejowski J, Rodriguez-Bravo V, Repka M, Lowery DM, Clauser KR, Zhang C, Shokat KM, Carr SA, Yaffe MB, Jallepalli PV. Plk1 self-organization and priming phosphorylation of HsCYK-4 at the spindle midzone regulate the onset of division in human cells. PLoS Biol 7: e1000111, 2009. doi: 10.1371/journal.pbio.1000111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Burridge K, Wennerberg K. Rho and Rac take center stage. Cell 116: 167–179, 2004. doi: 10.1016/S0092-8674(04)00003-0. [DOI] [PubMed] [Google Scholar]
- 7.Carnesecchi S, Deffert C, Donati Y, Basset O, Hinz B, Preynat-Seauve O, Guichard C, Arbiser JL, Banfi B, Pache JC, Barazzone-Argiroffo C, Krause KH. A key role for NOX4 in epithelial cell death during development of lung fibrosis. Antioxid Redox Signal 15: 607–619, 2011. doi: 10.1089/ars.2010.3829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cherfils J, Zeghouf M. Regulation of small GTPases by GEFs, GAPs, and GDIs. Physiol Rev 93: 269–309, 2013. doi: 10.1152/physrev.00003.2012. [DOI] [PubMed] [Google Scholar]
- 9.Datla SR, McGrail DJ, Vukelic S, Huff LP, Lyle AN, Pounkova L, Lee M, Seidel-Rogol B, Khalil MK, Hilenski LL, Terada LS, Dawson MR, Lassègue B, Griendling KK. Poldip2 controls vascular smooth muscle cell migration by regulating focal adhesion turnover and force polarization. Am J Physiol Heart Circ Physiol 307: H945–H957, 2014. doi: 10.1152/ajpheart.00918.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dovas A, Couchman JR. RhoGDI: multiple functions in the regulation of Rho family GTPase activities. Biochem J 390: 1–9, 2005. doi: 10.1042/BJ20050104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dubash AD, Wennerberg K, García-Mata R, Menold MM, Arthur WT, Burridge K. A novel role for Lsc/p115 RhoGEF and LARG in regulating RhoA activity downstream of adhesion to fibronectin. J Cell Sci 120: 3989–3998, 2007. doi: 10.1242/jcs.003806. [DOI] [PubMed] [Google Scholar]
- 12.Elias JE, Gygi SP. Target-decoy search strategy for increased confidence in large-scale protein identifications by mass spectrometry. Nat Methods 4: 207–214, 2007. doi: 10.1038/nmeth1019. [DOI] [PubMed] [Google Scholar]
- 13.Fransson A, Ruusala A, Aspenström P. Atypical Rho GTPases have roles in mitochondrial homeostasis and apoptosis. J Biol Chem 278: 6495–6502, 2003. doi: 10.1074/jbc.M208609200. [DOI] [PubMed] [Google Scholar]
- 14.Fritz G, Henninger C. Rho GTPases: novel players in the regulation of the dna damage response? Biomolecules 5: 2417–2434, 2015. doi: 10.3390/biom5042417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fujii M, Amanso A, Abrahão TB, Lassègue B, Griendling KK. Polymerase delta-interacting protein 2 regulates collagen accumulation via activation of the Akt/mTOR pathway in vascular smooth muscle cells. J Mol Cell Cardiol 92: 21–29, 2016. doi: 10.1016/j.yjmcc.2016.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Garcia-Mata R, Boulter E, Burridge K. The ‘invisible hand’: regulation of RHO GTPases by RHOGDIs. Nat Rev Mol Cell Biol 12: 493–504, 2011. doi: 10.1038/nrm3153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.García-Mata R, Wennerberg K, Arthur WT, Noren NK, Ellerbroek SM, Burridge K. Analysis of activated GAPs and GEFs in cell lysates. Methods Enzymol 406: 425–437, 2006. doi: 10.1016/S0076-6879(06)06031-9. [DOI] [PubMed] [Google Scholar]
- 18.Guilliam TA, Bailey LJ, Brissett NC, Doherty AJ. PolDIP2 interacts with human PrimPol and enhances its DNA polymerase activities. Nucleic Acids Res 44: 3317–3329, 2016. doi: 10.1093/nar/gkw175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Guilluy C, Dubash AD, García-Mata R. Analysis of RhoA and Rho GEF activity in whole cells and the cell nucleus. Nat Protoc 6: 2050–2060, 2011. doi: 10.1038/nprot.2011.411. [DOI] [PubMed] [Google Scholar]
- 20.Hara T, Abe M, Inoue H, Yu LR, Veenstra TD, Kang YH, Lee KS, Miki T. Cytokinesis regulator ECT2 changes its conformation through phosphorylation at Thr-341 in G2/M phase. Oncogene 25: 566–578, 2006. doi: 10.1038/sj.onc.1209078. [DOI] [PubMed] [Google Scholar]
- 21.Heasman SJ, Ridley AJ. Mammalian Rho GTPases: new insights into their functions from in vivo studies. Nat Rev Mol Cell Biol 9: 690–701, 2008. doi: 10.1038/nrm2476. [DOI] [PubMed] [Google Scholar]
- 22.Hernandes MS, Lassègue B, Griendling KK. Polymerase δ-interacting protein 2: a multifunctional protein. J Cardiovasc Pharmacol 69: 335–342, 2017. doi: 10.1097/FJC.0000000000000465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hodge RG, Ridley AJ. Regulating Rho GTPases and their regulators. Nat Rev Mol Cell Biol 17: 496–510, 2016. doi: 10.1038/nrm.2016.67. [DOI] [PubMed] [Google Scholar]
- 24.Huff LP, Decristo MJ, Trembath D, Kuan PF, Yim M, Liu J, Cook DR, Miller CR, Der CJ, Cox AD. The role of Ect2 nuclear RhoGEF activity in ovarian cancer cell transformation. Genes Cancer 4: 460–475, 2013. doi: 10.1177/1947601913514851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Justilien V, Ali SA, Jamieson L, Yin N, Cox AD, Der CJ, Murray NR, Fields AP. Ect2-Dependent rRNA Synthesis Is Required for KRAS-TRP53-Driven Lung Adenocarcinoma. Cancer Cell 31: 256–269, 2017. doi: 10.1016/j.ccell.2016.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Justilien V, Jameison L, Der CJ, Rossman KL, Fields AP. Oncogenic activity of Ect2 is regulated through protein kinase C iota-mediated phosphorylation. J Biol Chem 286: 8149–8157, 2011. doi: 10.1074/jbc.M110.196113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kamijo K, Ohara N, Abe M, Uchimura T, Hosoya H, Lee JS, Miki T. Dissecting the role of Rho-mediated signaling in contractile ring formation. Mol Biol Cell 17: 43–55, 2006. doi: 10.1091/mbc.e05-06-0569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kanada M, Nagasaki A, Uyeda TQ. Novel functions of Ect2 in polar lamellipodia formation and polarity maintenance during “contractile ring-independent” cytokinesis in adherent cells. Mol Biol Cell 19: 8–16, 2008. doi: 10.1091/mbc.e07-04-0370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kim JE, Billadeau DD, Chen J. The tandem BRCT domains of Ect2 are required for both negative and positive regulation of Ect2 in cytokinesis. J Biol Chem 280: 5733–5739, 2005. doi: 10.1074/jbc.M409298200. [DOI] [PubMed] [Google Scholar]
- 30.Kimura K, Tsuji T, Takada Y, Miki T, Narumiya S. Accumulation of GTP-bound RhoA during cytokinesis and a critical role of ECT2 in this accumulation. J Biol Chem 275: 17233–17236, 2000. doi: 10.1074/jbc.C000212200. [DOI] [PubMed] [Google Scholar]
- 31.Klaile E, Kukalev A, Obrink B, Müller MM. PDIP38 is a novel mitotic spindle-associated protein that affects spindle organization and chromosome segregation. Cell Cycle 7: 3180–3186, 2008. doi: 10.4161/cc.7.20.6813. [DOI] [PubMed] [Google Scholar]
- 32.Lassègue B, Sorescu D, Szöcs K, Yin Q, Akers M, Zhang Y, Grant SL, Lambeth JD, Griendling KK. Novel gp91(phox) homologues in vascular smooth muscle cells : nox1 mediates angiotensin II-induced superoxide formation and redox-sensitive signaling pathways. Circ Res 88: 888–894, 2001. doi: 10.1161/hh0901.090299. [DOI] [PubMed] [Google Scholar]
- 33.Lee MY, San Martin A, Mehta PK, Dikalova AE, Garrido AM, Datla SR, Lyons E, Krause KH, Banfi B, Lambeth JD, Lassègue B, Griendling KK. Mechanisms of vascular smooth muscle NADPH oxidase 1 (Nox1) contribution to injury-induced neointimal formation. Arterioscler Thromb Vasc Biol 29: 480–487, 2009. doi: 10.1161/ATVBAHA.108.181925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liot C, Seguin L, Siret A, Crouin C, Schmidt S, Bertoglio J. APC(cdh1) mediates degradation of the oncogenic Rho-GEF Ect2 after mitosis. PLoS One 6: e23676, 2011. doi: 10.1371/journal.pone.0023676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liu L, Rodriguez-Belmonte EM, Mazloum N, Xie B, Lee MY. Identification of a novel protein, PDIP38, that interacts with the p50 subunit of DNA polymerase delta and proliferating cell nuclear antigen. J Biol Chem 278: 10041–10047, 2003. doi: 10.1074/jbc.M208694200. [DOI] [PubMed] [Google Scholar]
- 36.Liu X, Zhao B, Sun L, Bhuripanyo K, Wang Y, Bi Y, Davuluri RV, Duong DM, Nanavati D, Yin J, Kiyokawa H. Orthogonal ubiquitin transfer identifies ubiquitination substrates under differential control by the two ubiquitin activating enzymes. Nat Commun 8: 14286, 2017. doi: 10.1038/ncomms14286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Louis SF, Zahradka P. Vascular smooth muscle cell motility: From migration to invasion. Exp Clin Cardiol 15: e75–e85, 2010. [PMC free article] [PubMed] [Google Scholar]
- 38.Lundgren DH, Hwang SI, Wu L, Han DK. Role of spectral counting in quantitative proteomics. Expert Rev Proteomics 7: 39–53, 2010. doi: 10.1586/epr.09.69. [DOI] [PubMed] [Google Scholar]
- 39.Lyle AN, Deshpande NN, Taniyama Y, Seidel-Rogol B, Pounkova L, Du P, Papaharalambus C, Lassègue B, Griendling KK. Poldip2, a novel regulator of Nox4 and cytoskeletal integrity in vascular smooth muscle cells. Circ Res 105: 249–259, 2009. doi: 10.1161/CIRCRESAHA.109.193722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Maga G, Crespan E, Markkanen E, Imhof R, Furrer A, Villani G, Hübscher U, van Loon B. DNA polymerase δ-interacting protein 2 is a processivity factor for DNA polymerase λ during 8-oxo-7,8-dihydroguanine bypass. Proc Natl Acad Sci USA 110: 18850–18855, 2013. doi: 10.1073/pnas.1308760110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mattaini KR, Sullivan MR, Vander Heiden MG. The importance of serine metabolism in cancer. J Cell Biol 214: 249–257, 2016. doi: 10.1083/jcb.201604085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Neilson KA, Ali NA, Muralidharan S, Mirzaei M, Mariani M, Assadourian G, Lee A, van Sluyter SC, Haynes PA. Less label, more free: approaches in label-free quantitative mass spectrometry. Proteomics 11: 535–553, 2011. doi: 10.1002/pmic.201000553. [DOI] [PubMed] [Google Scholar]
- 43.Niiya F, Tatsumoto T, Lee KS, Miki T. Phosphorylation of the cytokinesis regulator ECT2 at G2/M phase stimulates association of the mitotic kinase Plk1 and accumulation of GTP-bound RhoA. Oncogene 25: 827–837, 2006. doi: 10.1038/sj.onc.1209124. [DOI] [PubMed] [Google Scholar]
- 44.Paredes F, Sheldon K, Lassègue B, Williams HC, Faidley EA, Benavides GA, Torres G, Sanhueza-Olivares F, Yeligar SM, Griendling KK, Darley-Usmar V, San Martin A. Poldip2 is an oxygen-sensitive protein that controls PDH and αKGDH lipoylation and activation to support metabolic adaptation in hypoxia and cancer. Proc Natl Acad Sci USA 115: 1789–1794, 2018. doi: 10.1073/pnas.1720693115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rossman KL, Der CJ, Sondek J. GEF means go: turning on RHO GTPases with guanine nucleotide-exchange factors. Nat Rev Mol Cell Biol 6: 167–180, 2005. doi: 10.1038/nrm1587. [DOI] [PubMed] [Google Scholar]
- 46.Saito S, Liu XF, Kamijo K, Raziuddin R, Tatsumoto T, Okamoto I, Chen X, Lee CC, Lorenzi MV, Ohara N, Miki T. Deregulation and mislocalization of the cytokinesis regulator ECT2 activate the Rho signaling pathways leading to malignant transformation. J Biol Chem 279: 7169–7179, 2004. doi: 10.1074/jbc.M306725200. [DOI] [PubMed] [Google Scholar]
- 47.Sakata H, Rubin JS, Taylor WG, Miki T. A Rho-specific exchange factor Ect2 is induced from S to M phases in regenerating mouse liver. Hepatology 32: 193–199, 2000. doi: 10.1053/jhep.2000.8271. [DOI] [PubMed] [Google Scholar]
- 48.Scoumanne A, Chen X. The epithelial cell transforming sequence 2, a guanine nucleotide exchange factor for Rho GTPases, is repressed by p53 via protein methyltransferases and is required for G1-S transition. Cancer Res 66: 6271–6279, 2006. doi: 10.1158/0008-5472.CAN-06-0121. [DOI] [PubMed] [Google Scholar]
- 49.Seguin L, Liot C, Mzali R, Harada R, Siret A, Nepveu A, Bertoglio J. CUX1 and E2F1 regulate coordinated expression of the mitotic complex genes Ect2, MgcRacGAP, and MKLP1 in S phase. Mol Cell Biol 29: 570–581, 2009. doi: 10.1128/MCB.01275-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Seyfried NT, Dammer EB, Swarup V, Nandakumar D, Duong DM, Yin L, Deng Q, Nguyen T, Hales CM, Wingo T, Glass J, Gearing M, Thambisetty M, Troncoso JC, Geschwind DH, Lah JJ, Levey AI. A multi-network approach identifies protein-specific co-expression in asymptomatic and symptomatic Alzheimer’s disease. Cell Syst 4: 60–72.e4, 2017. doi: 10.1016/j.cels.2016.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Seyfried NT, Gozal YM, Donovan LE, Herskowitz JH, Dammer EB, Xia Q, Ku L, Chang J, Duong DM, Rees HD, Cooper DS, Glass JD, Gearing M, Tansey MG, Lah JJ, Feng Y, Levey AI, Peng J. Quantitative analysis of the detergent-insoluble brain proteome in frontotemporal lobar degeneration using SILAC internal standards. J Proteome Res 11: 2721–2738, 2012. doi: 10.1021/pr2010814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Solski PA, Wilder RS, Rossman KL, Sondek J, Cox AD, Campbell SL, Der CJ. Requirement for C-terminal sequences in regulation of Ect2 guanine nucleotide exchange specificity and transformation. J Biol Chem 279: 25226–25233, 2004. doi: 10.1074/jbc.M313792200. [DOI] [PubMed] [Google Scholar]
- 53.Sutliff RL, Hilenski LL, Amanso AM, Parastatidis I, Dikalova AE, Hansen L, Datla SR, Long JS, El-Ali AM, Joseph G, Gleason RL Jr, Taylor WR, Hart CM, Griendling KK, Lassègue B. Polymerase delta interacting protein 2 sustains vascular structure and function. Arterioscler Thromb Vasc Biol 33: 2154–2161, 2013. doi: 10.1161/ATVBAHA.113.301913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tatsumoto T, Xie X, Blumenthal R, Okamoto I, Miki T. Human ECT2 is an exchange factor for Rho GTPases, phosphorylated in G2/M phases, and involved in cytokinesis. J Cell Biol 147: 921–928, 1999. doi: 10.1083/jcb.147.5.921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tissier A, Janel-Bintz R, Coulon S, Klaile E, Kannouche P, Fuchs RP, Cordonnier AM. Crosstalk between replicative and translesional DNA polymerases: PDIP38 interacts directly with Poleta. DNA Repair (Amst) 9: 922–928, 2010. doi: 10.1016/j.dnarep.2010.04.010. [DOI] [PubMed] [Google Scholar]
- 56.Vukelic S, Xu Q, Seidel-Rogol B, Faidley EA, Dikalova AE, Hilenski LL, Jorde U, Poole LB, Lassègue B, Zhang G, Griendling KK. NOX4 (NADPH oxidase 4) and Poldip2 (polymerase δ-interacting protein 2) induce filamentous actin oxidation and promote its interaction with vinculin during integrin-mediated cell adhesion. Arterioscler Thromb Vasc Biol 38: 2423–2434, 2018. doi: 10.1161/ATVBAHA.118.311668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wennerberg K, Der CJ. Rho-family GTPases: it’s not only Rac and Rho (and I like it). J Cell Sci 117: 1301–1312, 2004. doi: 10.1242/jcs.01118. [DOI] [PubMed] [Google Scholar]
- 58.Wolfe BA, Takaki T, Petronczki M, Glotzer M. Polo-like kinase 1 directs assembly of the HsCyk-4 RhoGAP/Ect2 RhoGEF complex to initiate cleavage furrow formation. PLoS Biol 7: e1000110, 2009. doi: 10.1371/journal.pbio.1000110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Woods NT, Mesquita RD, Sweet M, Carvalho MA, Li X, Liu Y, Nguyen H, Thomas CE, Iversen ES Jr, Marsillac S, Karchin R, Koomen J, Monteiro AN. Charting the landscape of tandem BRCT domain-mediated protein interactions. Sci Signal 5: rs6, 2012. doi: 10.1126/scisignal.2002255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Xu P, Duong DM, Peng J. Systematical optimization of reverse-phase chromatography for shotgun proteomics. J Proteome Res 8: 3944–3950, 2009. doi: 10.1021/pr900251d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yüce O, Piekny A, Glotzer M. An ECT2-centralspindlin complex regulates the localization and function of RhoA. J Cell Biol 170: 571–582, 2005. doi: 10.1083/jcb.200501097. [DOI] [PMC free article] [PubMed] [Google Scholar]