

Abstract
Long-chain acyl-CoA synthetase 4 (ACSL4) has a unique substrate specificity for arachidonic acid. Hepatic ACSL4 is coregulated with the phospholipid (PL)-remodeling enzyme lysophosphatidylcholine (LPC) acyltransferase 3 by peroxisome proliferator-activated receptor δ to modulate the plasma triglyceride (TG) metabolism. In this study, we investigated the acute effects of hepatic ACSL4 deficiency on lipid metabolism in adult mice fed a high-fat diet (HFD). Adenovirus-mediated expression of a mouse ACSL4 shRNA (Ad-shAcsl4) in the liver of HFD-fed mice led to a 43% reduction of hepatic arachidonoyl-CoA synthetase activity and a 53% decrease in ACSL4 protein levels compared with mice receiving control adenovirus (Ad-shLacZ). Attenuated ACSL4 expression resulted in a substantial decrease in circulating VLDL-TG levels without affecting plasma cholesterol. Lipidomics profiling revealed that knocking down ACSL4 altered liver PL compositions, with the greatest impact on accumulation of abundant LPC species (LPC 16:0 and LPC 18:0) and lysophosphatidylethanolamine (LPE) species (LPE 16:0 and LPE 18:0). In addition, fasting glucose and insulin levels were higher in Ad-shAcsl4-transduced mice versus control (Ad-shLacZ). Glucose tolerance testing further indicated an insulin-resistant phenotype upon knockdown of ACSL4. These results provide the first in vivo evidence that ACSL4 plays a role in plasma TG and glucose metabolism and hepatic PL synthesis of hyperlipidemic mice.
Keywords: ACSL4, arachidonic acid metabolism, insulin resistance, phospholipid synthesis, VLDL
INTRODUCTION
The long-chain acyl-CoA synthetase (ACSL) family consists of five enzymes (ACSL1, ACSL3, ACSL4, ACSL5, and ACSL6) that catalyze the formation of fatty acyl-CoAs from ATP, CoA, and long-chain fatty acids (LCFAs) with certain degrees of substrate specificity (10–12, 35, 48, 49). As LCFAs must be esterified to fatty acid (FA)-CoA for entry into various metabolic pathways, including the cellular β-oxidation system responsible for FA oxidation (catabolism) and anabolic pathways for the synthesis of phospholipids (PLs), cholesterol esters, and triglycerides (TGs), ACSLs are considered rate-determining enzymes in the FA metabolism (14). It has been well recognized that even though ACSL enzymes catalyze similar enzymatic reactions to esterify free FA (FFA) to acyl-CoA, they exhibit variable cellular functions and generate distinct metabolic outcomes in an isoform-specific and tissue/cell type-specific manner (2, 5, 28, 29, 35, 57). The current thinking is that substrate specificity, interacting proteins, subcellular localization, tissue-specific expression, upstream signaling regulatory pathways, and nutritional factors all contribute to the unique functions of individual ACSLs.
Within the ACSL family, ACSL4 is unique in that it has a marked substrate preference for polyunsaturated FA (PUFA), including arachidonic acid (AA), and plays an important role in AA cellular metabolism (18, 21, 23). In a feed-forward fashion, AA regulates ACSL4 protein stability (17). ACSL4 is expressed in various tissues of humans and rodents, with the highest levels in brain, placenta, testis, ovary, spleen, and adrenal cortex; low levels are detected in the liver (3, 4). The expression of ACSL4 in normal liver tissue is relatively lower than that of ACSL1, the major ACSL isoform, but its expression is highly elevated in hepatocarcinoma (52) and nonalcoholic fatty liver disease (NAFLD) (22, 51, 56). Several in vitro studies have reported various functions of ACSL4 in the channeling of FA into different metabolic pathways. In rat smooth muscle cells, altered ACSL4 expression was linked to PGE2 secretion (13). In pancreatic cell lines, ACSL4 participates in insulin secretion (1), and diminished ACSL4 expression was associated with reduced glucose-stimulated insulin secretion (20). Another study demonstrated the requirement of ACSL4 in TG-PUFA formation in activated hepatic stellate cells (55). In addition, studies in rat fibroblasts (26) and COS-7 cells (23) showed that exogenous overexpression of ACSL4 promotes AA incorporation into phosphatidylinositol (PI). A recent study, using a novel mouse model of adipocyte-specific ablation of ACSL4 [ACSL4 adipocyte knockout (Ad-KO)], also observed reduced incorporation of AA into PLs of adipocytes derived from Ad-KO mice (19). These studies suggest that ACSL4 may be functionally involved in the channeling of PUFA into PL synthesis.
It has been demonstrated that the PL remodeling enzyme lysophosphatidylcholine (LPC) acyltransferase 3 (LPCAT3) is a critical determinant of TG secretion due to its unique ability to catalyze the incorporation of arachidonate into membranes, which impacts membrane lipid mobility in living cells (31, 39, 43, 54). LPCAT3 catalyzes the formation of phosphatidylcholines (PCs) from saturated LPC, inserting PUFAs at the sn-2 position (40, 42). Mice lacking LPCAT3 in the liver showed reduced plasma VLDL-TG levels and hepatosteatosis (31, 43). Interestingly, our previous studies have shown that ACSL4 and LPCAT3 are coordinately upregulated by the FA-activated nuclear receptor peroxisome proliferator-activated receptor δ (PPARδ) in liver cells (47). We demonstrated that the treatment of mice with the PPARδ agonist L165041 increased hepatic ACSL4 and LPCAT3 expression, which was associated with increased incorporation of arachidonate into liver PC and phosphatidylethanolamine (PE). Collectively, it becomes clear that despite all of these previous reports, the key metabolic function of ACSL4 in liver tissue under normal or disease conditions remains to be elucidated.
In the present study, we took the approach of adenovirus-mediated gene knockdown (KD) to examine the acute effects of hepatic ACSL4 deficiency on plasma and hepatic lipid metabolism in adult mice under a hyperlipidemic state. In view of the prominently altered metabolic features of ACSL4 KD mice, combined with lipidomic analysis, our studies, for the first time, reveal requirements of hepatic ACSL4 in support of circulating TG levels and glucose metabolism and in hepatic PL synthesis. Whole genome transcriptomic analysis of liver tissues further identified p53 signaling pathways and genes involved in apoptosis being affected by ACSL4 deficiency.
MATERIALS AND METHODS
Mice and diets.
All animal studies were approved by the Institutional Animal Care and Use Committee at Veterans Affairs Palo Alto Health Care System (Palo Alto, CA) and were consistent with the National Institutes of Health Guide for the Care and Use of Laboratory Animals. C57BL/6j male mice were purchased from The Jackson Laboratory (Bar Harbor, ME) and were fed a high-fat diet [HFD; No. TD.88137 from Harlan (Envigo, Indianapolis, IN); ~42% of total calories from fat; 0.15% cholesterol] for 8 weeks before adenoviral injections.
Construction of Ad-shAcsl4 adenoviral vector for ACSL4 KD.
A U6 promoter-based vector (pSH-ACSL4) that expresses an shRNA targeting mouse ACSL4 coding region (5′-GCTGCAAATGCCATGAAATTG-3′) was generated using the BLOCK-iT U6 RNAi Entry Vector Kit (Invitrogen; Thermo Fisher Scientific, Waltham, MA), following the manufacturer’s instructions. The sequence identity and orientation of ACSL4 shRNA in pSH-ACSL4 were confirmed by sequencing. The pSH-ACSL4 plasmid was then recombined with the pAD/BLOCK-iT DEST vector to generate adenovirus-mediated expression of a mouse ACSL4 shRNA (Ad-shAcsl4) viral vector and transduced into human embryonic kidney 293A cells. The control adenovirus (Ad-shLacZ) targeting β-galactosidase coding region was constructed with the same procedure of Ad-shAcsl4. Both crude viral stocks were further amplified and purified by Vector Biolabs (Malvern, PA).
Adenoviral transduction in mouse primary hepatocytes.
Mouse primary hepatocytes (MPHs), seeded at 1 × 106 cells/well in 12-well plates, were transduced with Ad-shAcsl4 or the control virus (Ad-shLacZ) at various multiplicities of infection (MOIs) in 1 ml medium containing 0.5% FBS. After 5 h of infection, medium was replaced with fresh medium containing 10% FBS, and cells were harvested 72 h postinfection to isolate total RNA or cell lysates.
Induction of hyperlipidemia in mice fed an HFD.
In one pilot study, 8- to 10-week-old male mice were fed a normal chow diet (NCD; n = 4) or an HFD (n = 4) for 8 weeks. Fasting serum and liver tissues were collected for lipid analysis. In addition, routine hematoxylin and eosin staining was performed on formalin-fixed, paraffin-embedded sections and Oil Red O staining on cryosections of liver tissue from NCD and HFD groups.
Adenoviral infection in mice.
In another study, male mice were fed an HFD for 8 weeks. Continuing on the HFD, 12 mice were divided into two groups with similar total cholesterol (TC) and TG levels. At day 0, mice were fully anesthetized and injected with Ad-shAcsl4 (6 × 109 plaque-forming units/mouse) or an equal viral dose of a control adenovirus (Ad-shLacZ) via the retro-orbital sinus, as previously described (44, 47). Body weight and food intake were monitored throughout the duration of the experiment. After 11 days of infection, mice were unfed for 4 h (10 AM–2 PM) before euthanasia for serum and liver-tissue collections.
Oral glucose tolerance test.
An oral glucose tolerance test (GTT) was performed after 9 days of adenovirus injection. Mice were unfed overnight (~16 h; 5 PM–9 AM) and were gavaged with glucose at a dose of 2 g/kg body weight. Blood glucose concentrations were measured using a glucometer at 0, 15, 30, 60, 90, and 120 min postglucose administration. The area under the glucose concentration time curve (0–120 min) was calculated using GraphPad Prism 7 (GraphPad Software, La Jolla, CA).
Measurement of serum lipids.
Serum was isolated at room temperature and stored at −80°C. Standard enzymatic methods were used to determine TC, TG, FFA, and PL levels of serum using commercially available kits purchased from Stanbio Laboratory (Boerne, TX). Each sample was assayed in duplicate.
Measurement of serum LTB4.
Mouse serum leukotriene B4 (LTB4) concentration was determined by using the LTB4 enzyme immunoassay kit [Catalog #520111; Cayman Chemical (Ann Arbor, MI)], according to the manufacturer’s protocol.
HPLC separation of serum lipoprotein cholesterol and TGs.
Serum samples (50 μl), obtained on day 11 after adenovirus injection from two animals of the same treatment group, were pooled together, and a total of three pooled serum samples from the Ad-shAcsl4 or Ad-shLacZ group was analyzed for cholesterol and TG levels of each of the major lipoprotein classes, including chylomicron (>80 nm), VLDL (30–80 nm), LDL (16–29 nm), and HDL (8–15 nm), with a dual-detection HPLC system consisting of two tandem-connected TSKgel Lipopropak XL columns (300 × 7.8 mm; Tosoh, Tokyo, Japan) at Skylight Biotech, Inc. (Tokyo, Japan), as described (9).
Measurement of hepatic lipids.
Frozen liver tissue (30 mg) was homogenized in 1 ml chloroform/methanol (2:1) for lipid extraction (35). TC and TGs were measured using kits from Stanbio Laboratory.
Lipidomics.
Individual frozen liver samples (50 mg) from Ad-shAcsl4-infected mice (n = 6) or control mice injected with Ad-shLacZ (n = 6) were subjected to lipidomic analysis at the West Coast Metabolic Center at the University of California, Davis. Six replicas of extracted lipids from each treatment group were applied in a lipidomics study using an automated electrospray ionization-tandem mass spectrometry approach with a group of internal lipid standards. The liquid chromatography/tandem mass spectrometry analyses were carried out on an Agilent 1200 SL ultra-HPLC system (Santa Clara, CA) coupled with an AB Sciex 4000 QTRAP system (Foster City, CA) under a negative multiple-reaction monitoring mode. The detailed methods were described previously (47). The specific peaks on the spectra are normalized and are relative semiquantifications. All data acquisition, analysis, acyl group identification, and normalization were conducted by the West Coast Metabolic Center at the University of California, Davis.
Real-time PCR.
Total RNA isolation, generation of cDNA, and real-time quantitative RT-PCR (qRT-PCR) were conducted as previously reported (8). Each cDNA sample was assayed in duplicate. The correct size of the PCR product and the specificity of each primer pair were validated by examination of PCR products on an agarose gel. Primer sequences, used in real-time PCR, are listed in Table 1. Target mRNA expression in each sample was normalized to the housekeeping gene GAPDH. The comparative threshold cycle (or 2−ΔΔCt) method was used to calculate relative mRNA expression levels.
Table 1.
List of mouse quantitative PCR primer sequences
| Gene | Forward Primer | Reverse Primer |
|---|---|---|
| Acsl1 | ATCTGGTGGAACGAGGCAAG | TCCTTTGGGGTTGCCTGTAG |
| Acsl3 | TCTTGCAAACAAAGCTGAAGGA | GGTTGGAGGCTTCCCATCAA |
| Acsl4 | CTTCCTCTTAAGGCCGGGAC | TGCCATAGCGTTTTTCTTAGATTT |
| Acsl5 | GGCCAAACAGAATGCACAGG | GATGCAGATCTCGCCTTCGT |
| Lpcat3 | GGCCTCTCAATTGCTTATTTCA | AGCACGACACATAGCAAGGA |
| Pparα | AGACACCCTCTCTCCAGCTT | TTCGCCGAAAGAAGCCCTTA |
| Pparδ | CGGACCTGGGGATTAATGGG | ATGGACTGCCTTTACCGTGG |
| Pparγ | TGTGAGACCAACAGCCTGAC | CCGCTTCTTTCAAATCTTGTCTGT |
| Cpt1α | GGCCATCTGTGGGAGTATGT | ACTGTAGCCTGGTGGGTTTG |
| Acox1 | GGGAGTGCTACGGGTTACATG | CCGATATCCCCAACAGTGATG |
| Apaf1 | GGAATCAGCTTGAAGCATGGC | CCCGCTTATGTCCTGCTCATTA |
| Bax | TGAAGACAGGGGCCTTTTTG | AATTCGCCGGAGACACTCG |
| Casp3 | ATGGAGAACAACAAAACCTCAGT | TTGCTCCCATGTATGGTCTTTAC |
| P21 | CCTGGTGATGTCCGACCTG | CCATGAGCGCATCGCAATC |
| Cyclin g1 | ACAACTGACTCTCAGAAACTGC | CATTATCATGGGCCGACTCAAT |
| Il1β | TGCCACCTTTTGACAGTGATG | TGATGTGCTGCTGCGAGATT |
| Tnfα | ACTGAACTTCGGGGTGATCG | CTTGGTGGTTTGCTACGACG |
| Il6 | CACTTCACAAGTCGGAGGCT | CTGCAAGTGCATCATCGTTGT |
| Atf4 | TGGCGAGTGTAAGGAGCTAGAAA | TCTTCCCCCTTGCCTTACG |
| Chop | CTGGAAGCCTGGTATGAGGAT | CAGGGTCAAGAGTAGTGAAGGT |
| Xbp1 | GAATGGACACGCTGGATCCT | GCCACCAGCCTTACTCCACTC |
| Ccl2 | TTAAAAACCTGGATCGGAACCAA | GCATTAGCTTCAGATTTACGGGT |
| Itgax | CTGGATAGCCTTTCTTCTGCTG | GCACACTGTGTCCGAACTCA |
| Chrebp | TGGGTGTTCAGCATCCTCATC | CAGCCAGGCCAGTGAGGTCT |
| Fasn | GTGATAGCCGGTATGTCGGG | TAGAGCCCAGCCTTCCATCT |
| Scd1 | CTGCAGGTTGTGCTAGATGGGATGG | GCCTGGGGTCTTTGGTAAGTAGGC |
| Dgat2 | GGCTACGTTGGCTGGTAACT | TCTTCAGGGTGACTGCGTTC |
| Srebp1c | CAAGGCCATCGACTACATCCG | CACCACTTCGGGTTTCATGC |
| α-SMA | GTCCCAGACATCAGGGAGTAA | TCGGATACTTCAGCGTCAGGA |
| Col1a1 | GCTCCTCTTAGGGGCCACT | CCACGTCTCACCATTGGGG |
| Gapdh | ATGGTGAAGGTCGGTGTGAA | ACTGGAACATGTAGACCATGTAGT |
| Tgf-β | GGTTCATGTCATGGATGGTGC | TGACGTCACTGGAGTTGTACGG |
| Mmp-2 | TTCCCCCGCAAGCCCAAGTG | GAGAAAAGCGCAGCGGAGTGACG |
| Timp-1 | GCATCTCTGGCATCTGGCATC | GCGGTTCTGGGACTTGTGGGC |
Western blot analysis.
Frozen liver (50 mg) was homogenized in radioimmunoprecipitation assay buffer containing 1 mM PMSF and protease inhibitor cocktail (Roche, Indianapolis, IN) and phosphatase inhibitor cocktail (Sigma, St. Louis, MO). After protein quantitation using bicinchoninic acid protein assay reagent (Pierce; Thermo Fisher Scientific), 50 μg homogenate proteins from individual samples were used in SDS-PAGE and Western blot analysis to determine relative protein levels of target proteins, as described (45, 46). Table 2 lists all antibody technical information. Primary antibodies were used as 1:1,000 dilution, and secondary antibodies were used at 1:10,000 dilution. Immunoreactive bands of predicted molecular mass were visualized using a SuperSignal West Femto Maximum Sensitivity Substrate enhanced chemiluminescent kit from Thermo Fisher Scientific and quantified with AlphaView software with normalization by signals of β-actin.
Table 2.
List of antibodies
| Antibody | Species | Catalog No. | Vendor |
|---|---|---|---|
| ACSL4* | Rabbit | ||
| FASN | Mouse | Sc-55580 | Santa Cruz Biotechnology |
| SCD | Mouse | Sc-14720 | Santa Cruz Biotechnology |
| P-JNK | Rabbit | 4668 | Cell Signaling Technology |
| T-JNK | Rabbit | 9252 | Cell Signaling Technology |
| LDLR | Rabbit | 3839–100 | BioVision |
| GAPDH | Mouse | AM4300 | Invitrogen |
| β-Actin | Mouse | A1978 | Sigma |
| p21 | Mouse | sc-817 | Santa Cruz Biotechnology |
| p53 | Mouse | sc-126 | Santa Cruz Biotechnology |
| Mttp | Mouse | ab20737 | Abcam |
| Cpt1α | Mouse | Ab128568 | Abcam |
| Anti-rabbit HRP conjugate | Rabbit | 7074P2 | Cell Signaling Technology |
| Anti-mouse HRP conjugate | Mouse | 7076P2 | Cell Signaling Technology |
HRP, horseradish peroxidase.
The anti-ACSL4 antibody was provided by Dr. Diana M. Stafforini (Huntsman Cancer Institute, University of Utah, Salt Lake City, UT).
Measurement of liver acyl-CoA synthetase activity.
Approximately 50 mg frozen liver tissue was dounce homogenized, 15 times, in a buffer containing 250 mM sucrose, 10 mM Tris, pH 7.4, 1 mM EDTA, 1 mM DTT, protease inhibitor cocktail (Roche), and phosphatase inhibitor cocktail (Sigma). Homogenates were centrifuged at 16,000 g for 30 min at 4°C. Protein concentrations in supernatants were determined by bicinchoninic acid assay, and aliquots were stored at −80°C. Initial rates of total ACSL activity in liver homogenates were measured using 4 µg liver homogenates at 37°C in the presence of 175 mM Tris (pH 7.4), 8 mM MgCl2, 5 mM DTT, 10 mM ATP, 250 µM CoA, 50 µM [3H]AA, [3H]palmitic acid (PA), or [3H]oleic acid (OA) in 0.5 mM Triton X-100 and 10 μM EDTA in a total volume of 0.1 ml. The reaction was initiated by the addition of the homogenized sample and terminated by the addition of 1 ml Dole’s reagent, as previously described (7). Generated [3H]AA-CoA, [3H]PA-CoA, and [3H]OA-CoA were extracted, and the radioactivity was determined in a scintillation counter. The radioactivity in a reaction that contained all components but omitted homogenate was included as a negative control.
Microarray analysis.
Microarray chip hybridization and data analysis were performed on individual livers of mice fed an HFD (n = 4 per treatment group) at Boston University Microarray and Sequencing Resource Core Facility. Mouse Gene 2.0 ST CEL files were normalized to produce gene-level expression values using the implementation of the Robust Multi-Array Average in the affy package (version 1.36.1) included in the Bioconductor software suite (version 2.11) and an Entrez Gene-specific probe-set mapping (17.0.0) from The Molecular & Behavioral Neuroscience Institute (Brainarray) at the University of Michigan. Differential expression was assessed using the moderated (empirical Bayesian) t-test implemented in the limma package (version 3.14.4). Correction for multiple hypothesis testing was accomplished using the Benjamini-Hochberg false discovery rate. All microarray analyses were performed using the R environment for statistical computing (version 2.15.1). For comparative analysis, general linear models for microarray data were performed for probe sets present on the microarray to identify probe sets that were differentially expressed between Ad-shAcsl4 and Ad-shLacZ groups, based on moderated t-statistics. Probe sets with a 1.5-fold change and a P value <0.05 were considered biologically significant.
Gene Set Enrichment Analysis.
Gene Set Enrichment Analysis (GSEA; version 2.2.1) was used to identify biological terms, pathways, and processes that are coordinately up- or downregulated within each pairwise comparison. The Entrez Gene identifiers of the human homologs of the genes interrogated by the array were ranked according to the moderated t-statistic computed between Ad-shAcsl4 and Ad-shLacZ. Mouse genes with multiple human homologs (or vice versa) were removed before ranking so that the ranked list represents only those human genes that match exactly one mouse gene. This ranked list was then used to perform pre-ranked GSEA analyses using the Entrez Gene versions of the Hallmark, Biocarta, Kyoto Encyclopedia of Genes and Genomes (KEGG), Reactome, Gene Ontology, and transcription factor and microRNA motif gene sets obtained from the Molecular Signatures Database (or MSigDB), version 6.0.
Statistical analysis.
MS Excel and GraphPad Prism 7 were used to calculate averages and SEs, generate graphs, and perform statistical tests. All values are presented as means ± SE. Unpaired Student’s two-tailed t-test was used to compare two groups. Statistical significance is displayed as P < 0.05, P < 0.01, or P < 0.001.
RESULTS
Knocking down hepatic ACSL4 expression in adult mice by adenovirus infection.
To identify specific functions of hepatic ACSL4 in adult mice, we used shRNA technology to deplete hepatic ACSL4 using Ad-shAcsl4. Efficiency of the viral particle was first examined in MPH that were transduced with different MOIs of Ad-shAcsl4 or an shRNA control virus Ad-shLacZ for 3 days. Expression of Acsl4 shRNA caused a dose-dependent reduction in ACSL4 protein levels (Fig. 1A). Real-time PCR analysis of all hepatic ACSL isoforms demonstrated that Ad-shAcsl4, at the highest MOI, reduced Acsl4 mRNA by 95% without affecting mRNA expression of other ACSL isoforms (Fig. 1B).
Fig. 1.
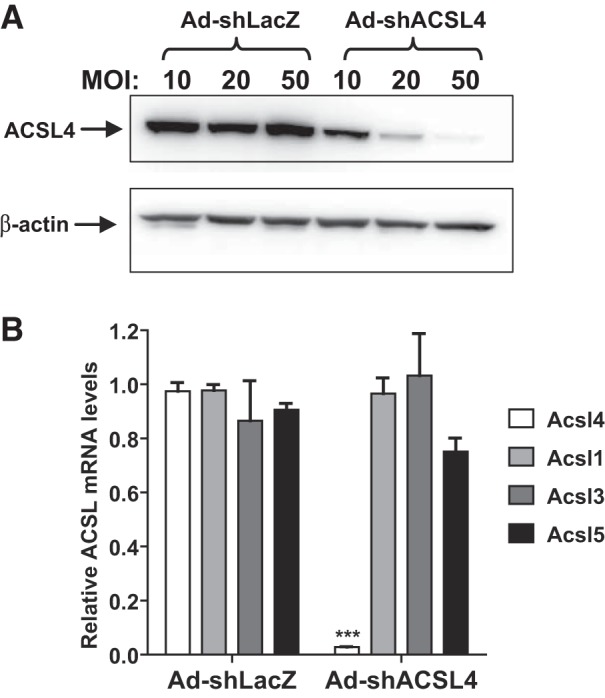
Knockdown of hepatic long-chain acyl-CoA synthetase 4 (ACSL4) in mouse primary hepatocytes (MPHs) by adenovirus-mediated expression of a mouse ACSL4 shRNA (Ad-shAcsl4) transduction. MPHs, seeded in a 12-well cell-culture plate, were transduced with mice receiving control adenovirus (Ad-shLacZ) or Ad-shAcsl4 at an indicated multiplicity of infection (MOI) of adenovirus for 3 days. Total cell lysates and total RNA were separately isolated. A: Western blot analysis of ACSL4 protein using anti-ACSL4 rabbit antibodies. B: real-time PCR analysis of mRNA levels of all ACSL isoforms expressed in mouse liver cells. ***P < 0.001.
To examine the hepatic function of ACSL4 under a hyperlipidemic condition, initially, we conducted a pilot diet study of feeding male C57BL/6J mice either a chow diet (NCD) or an HFD for 8 weeks. Examination of liver tissues showed that compared with chow-fed mice, HFD feeding induced hepatosteatosis, as evidenced by a 6.9-fold increase in liver TG content and a 2.5-fold increase in liver cholesterol content; serum cholesterol levels were increased 1.7-fold by HFD feeding (Fig. 2A). Oil Red O staining of frozen liver sections (Fig. 2B) revealed large lipid droplets in livers of the HFD group that were not detected in livers from the chow diet group. Hepatic gene-expression analysis by qRT-PCR showed that mRNA levels of inflammatory-related gene IL-1β and fibrosis-related genes α-smooth muscle actin and collagen type I mRNA were all elevated in the HFD group compared with the control group (Fig. 2C).
Fig. 2.

High-fat diet exposure increased hepatic steatosis and serum cholesterol and elevated mRNA levels of inflammation and fibrosis-related genes in mice. Male C57BL/6J mice were fed a normal chow diet (NCD; n = 4) or a high-fat diet (HFD; n = 4) for 2 mo. A: hepatic lipid levels [liver triglyceride (TG) and liver total cholesterol (TC)] and serum TC levels were measured. ***P < 0.001 compared with NCD mice. B: Oil Red O staining of representative liver sections of chow (NCD)- and HFD-fed mice. C: quantitative RT-PCR analysis of hepatic gene expression. a-SMA, α-smooth muscle actin. *P < 0.05; ***P < 0.001.
Having established a hyperlipidemic mouse model and confirmed the specificity and efficacy of Ad-shAcsl4 in MPH, Ad-shAcsl4 or control virus (Ad-shLacZ) was injected into male mice fed the HFD for 8 weeks. Analysis of liver tissues of mice euthanized on day 11, after viral injection, showed that transduction of Ad-shAcsl4 resulted in 53% depletion of ACSL4 protein (P < 0.001; Fig. 3A), and Acsl4 mRNA levels were reduced by 64% (P < 0.001). Levels of Acsl5 and Acsl3 mRNAs were unchanged, whereas Acsl1 mRNA levels were increased by 18% (P < 0.05; Fig. 3B). In addition, mRNA levels of Lpcat3 were similar between the two groups. Measurements of total ACSL activity in liver homogenates in the presence of three different 3H-labeled FA substrates (Fig. 3C) demonstrated that the arachidonoyl-CoA synthetase activity in the Ad-shAcsl4 group was markedly reduced by 43% (P < 0.001) compared with the control group, whereas oleoyl-CoA synthetase activities did not differ between the two groups. Interestingly, the palmitoyl-CoA synthetase activity in the Ad-shAcsl4 group was increased by 15% (P < 0.01) compared with the control group, which is consistent with the increased Acsl1 mRNA expression in the ACSL4 KD group. Altogether, these results demonstrate the high efficacy and specificity of Ad-shAcsl4 in the depletion of ACSL4 in mouse liver cells.
Fig. 3.

Depletion of long-chain acyl-CoA synthetase 4 (ACSL4) in the liver of high-fat diet (HFD)-fed mice. Male C57BL/6J mice, fed an HFD for 8 weeks, were injected with adenovirus-mediated expression of a mouse ACSL4 shRNA (Ad-shAcsl4; n = 6) or mice receiving control adenovirus (Ad-shLacZ; n = 6). Eleven days after adenoviral injection, mice were euthanized, and serum samples and livers were isolated at the termination of the experiment. All data are means ± SE of 6 liver samples per group. A: Western blot analysis of hepatic ACSL4 protein and β-actin levels in individual livers of Ad-shAcsl4- or Ad-shLacZ-injected mice. The protein abundance of ACSL4 was quantified with normalization with β-actin using AlphaView software. Significance was determined by unpaired Student’s t-test; n = 6 mice per group; **P < 0.01. B: real-time PCR was conducted to determine relative expression levels of ACSL isoform mRNAs and lysophosphatidylcholine acyltransferase 3 (Lpcat3) after normalization with GAPDH mRNA levels. Significance was determined by unpaired Student’s t-test; n = 6 mice per group; **P < 0.01; *P < 0.05. C: total ACSL enzymatic activity in liver-tissue homogenates was measured using 4 µg homogenate proteins at 37°C in the presence of [3H]-labeled arachidonoyl-CoA (arachidonic acid), [3H]-labeled oleoyl-CoA (oleic acid), or [3H]-labeled palmitoyl-CoA (palmitic acid). Significance was determined by unpaired Student’s t-test; n = 6 mice per group; ***P < 0.001; **P < 0.01.
Ad-shAcsl4 mice fed an HFD had altered VLDL-TG and FFA metabolism.
The body weight, liver weight, epididymal white adipose tissue mass, and food intake did not differ between the two groups of mice injected with Ad-shAcsl4 or Ad-shLacZ (Fig. 4). Measurements of serum lipid levels showed that the transient KD of liver ACSL4 did not affect circulating levels of TC or PL but significantly lowered serum TG levels by 36% (P < 0.01) and reduced serum FFA abundance by 31% (P < 0.001) compared with the control group (Fig. 5, A–D). HPLC analysis of lipoprotein TG and cholesterol profiles of serum samples from the Ad-shAcsl4 and control groups revealed that the reduction of TG was primarily caused by a substantial decrease in the VLDL fraction (Fig. 5, E and F), whereas lipoprotein-cholesterol fractions (Fig. 5, G and H) only showed a small decrease in VLDL-cholesterol in hepatic ACSL4-deficient mice, which is consistent with an overall lack of effects of ACSL4 KD on serum cholesterol levels in hyperlipidemic mice.
Fig. 4.

Effects of adenoviral injection on mouse body weight, food intake, liver weight (wt.), epididymal white adipose tissue (EpiWAT) mass, and the Epi WAT index. Male mice fed a high-fat diet for 8 weeks were injected with adenovirus-mediated expression of a mouse long-chain acyl-CoA synthetase 4 shRNA (Ad-shAcsl4) or mice receiving control adenovirus (Ad-shLacZ). Body weight and food intake were recorded throughout the treatment duration. After 11 days of adenoviral injection, mice were euthanized, and 4-h fasting serum and liver samples were collected. All data are means ± SE with n = 6 per group. A: body weight measurement. B: food intake. C: liver weight. D: EpiWAT mass. E: EpiWAT index. n.s, not significant.
Fig. 5.

Hepatic long-chain acyl-CoA synthetase 4 (ACSL4) deficiency reduced plasma VLDL- triglyceride (TG) and free fatty acid (FFA). Eleven days after adenoviral injection, mice were unfed for 4 h and then euthanized for serum and liver sample collections. TG, FFA, total cholesterol (TC), and phospholipid (PL) levels were measured. A–D: significance was determined by unpaired Student’s t-test; n = 6 mice per group; **P < 0.01; ***P < 0.001. A: serum TG. B: serum FFA [nonesterified fatty acid (NEFA)]. C: serum TC. D: serum PL. E–H: serum samples from 2 animals of the same treatment group were pooled together, and a total of 3 pooled serum samples from each group were analyzed for TG (E and F) and TC (G and H) distribution in HPLC-separated lipoprotein factions; n = 3; *P < 0.05. Ad-shAcsl4, mouse ACSL4 shRNA; Ad-shLacZ, mice receiving control adenovirus; CM, chylomicron; n.s, not significant.
Next, we extracted hepatic lipids from livers of mice infected with Ad-shAcsl4 or Ad-shLacZ. ACSL4 depletion led to a 20% increase in hepatic nonesterified FA (FFA) levels without effects on hepatic cholesterol, TG, or PL (Fig. 6, A–D). Altogether, these results reveal a function of hepatic ACSL4 in the modulation of plasma VLDL-TG metabolism in mice fed an HFD.
Fig. 6.

Analysis of hepatic lipid contents in high-fat diet-fed mice injected with mouse long-chain acyl-CoA synthetase 4 shRNA (Ad-shAcsl4) or mice receiving control adenovirus (Ad-shLacZ). Lipids were extracted from individual livers of Ad-shAcsl4- and Ad-shLacZ-injected mice and measured for nonesterified fatty acid (NEFA; A), triglyceride (TG; B), total cholesterol (TC; C), and phospholipid (PL; D). All data are means ± SE of 6 liver samples per group. n.s, not significant. *P < 0.05.
Knocking down ACSL4 generated an insulin-resistant phenotype.
In addition to changes in serum TG and FFA levels, we observed that ACSL4 KD mice, fed an HFD, exhibited modest elevations in fasting serum glucose and insulin levels compared with HFD-fed control mice (Fig. 7, A and B). GTTs further indicated a greater degree of glucose intolerance, with a 26% increase in the area under the curve of serum glucose during the GTT, thereby leading to a higher score of homeostatic model assessment to determine insulin resistance in hepatic ACSL4 KD mice compared with control mice (Fig. 7, C–E).
Fig. 7.

Effects of hepatic long-chain acyl-CoA synthetase 4 (ACSL4) deficiency on serum glucose and insulin level. A and B: 11 days after adenoviral injection, 4 h fasting serum samples of individual mice were measured for glucose and insulin levels. C–E: glucose tolerance test (GTT) was performed at 9 days after injection of mouse ACSL4 shRNA (Ad-shAcsl4) or mice receiving control adenovirus (Ad-shLacZ). Mice were unfed overnight before administration of glucose (2 g/kg body weight). A–E: significance was determined by unpaired Student’s t-test; n = 6 mice per group; *P < 0.05; **P < 0.01. AUC, area under the curve; HOMA-IR, homeostatic model assessment-insulin resistance.
Saturated LPC and LPE species selectively accumulated in ACSL4-depleted liver.
Previous studies have shown that reduced incorporation of arachidonate at the sn-2 position of PC, due to deficiency of LPCAT3, resulted in reduced plasma VLDL-TG concentrations (43), an observation similar to the reduced VLDL-TG phenotype in ACSL4 KD mice. Thus we performed a lipidomics analysis to profile PL molecular species in liver tissues of ACSL4 KD mice (Ad-shAcsl4) and control mice (Ad-shLacZ). Figure 8A shows that LPC (16:0) was the most abundant LPC species in liver tissue, and its level was increased by ~45% (P < 0.001) in Ad-shAcsl4-transduced mice. We also detected significant enrichment of three other LPC species. Furthermore, levels of lyso-PE (LPE) species 18:0 and 16:0 were increased 80% (P < 0.001) and 70% (P < 0.001), respectively, of control by KD of ACSL4 (Fig. 8B). Accumulations of saturated LPC and LPE are in line with reduced AA-CoA synthetase activity in ACSL4-depleted liver tissue (Fig. 3C) and support the role of ACSL4 in hepatic PL synthesis. Ablation of LPCAT3 in the liver of mice fed a Western diet was previously shown to increase PC (34:1) abundance significantly (43). Interestingly, we detected a 34% increase (P < 0.001) of this PC species in ACSL4-depleted liver compared with control, despite unchanged levels of abundant PC and PE species containing arachidonates (Fig. 8, C and D). In addition, the ceramide species (d42:1) was increased by 36% by ACSL4 depletion (Fig. 8E), and no significant changes were observed in PI species between the two groups (Fig. 8F). Altogether, these results indicate that attenuated ACSL4 activity in the liver of HFD-fed mice resulted in accumulation of saturated LPC and LPE, which could account, at least in part, for the reduced plasma VLDL abundance, owing to the insufficient supply of arachidonate substrates to LPCAT3.
Fig. 8.

Knocking down hepatic long-chain acyl-CoA synthetase 4 (ACSL4) increases saturated lysophosphatidylcholine (LPC) and lysophosphatidylethanolamine (LPE) species in mouse liver samples. Charged surface hybrid column-electrospray ionization quadrupole time-of-flight tandem mass spectrometry analysis of the abundance of LPC species (A), LPE species (B), phosphatidylcholine (PC) species (C), phosphatidylethanolamine (PE) species (D), ceramide species (E), and phosphatidylinositol (PI) species (F) in livers of high-fat diet-fed mice injected with mouse ACSL4 shRNA (Ad-shAcsl4) or mice receiving control adenovirus (Ad-shLacZ). Values are the means ± SE of 6 liver samples per group; *P < 0.05; ***P < 0.001 compared with the Ad-shLacZ group.
Changes of lipogenic and oxidative pathways by ACSL4 depletion.
To understand further the above-observed complex changes in plasma TG, glucose, FFA, and hepatic lipid levels, we examined the expression of genes involved in FA synthesis and β-oxidation. qRT-PCR analysis revealed that hepatic expression of SREBP1c and its target genes encoding lipogenic enzymes Fas, Scd1, and Dgat2 was increased in ACSL4-deficient livers (Fig. 9A). Western blot analysis, using anti-FASN and anti-SCD1 antibodies, further demonstrated the increased protein expression in liver homogenates of Ad-shAcsl4 mice (Fig. 9B). Measurement of a panel of FA-oxidative genes revealed increased expression of PPARα and its target genes CPT1α and AcoX1, whereas PPARδ and PPARγ mRNA levels were unchanged (Fig. 9C). Increased expression of CPT1α protein in ACSL4 KD liver was also confirmed by Western blotting (Fig. 9D). Thus these data suggest that ACSL4 deficiency increased both the synthesis and degradation of FAs, thus resulting in a slightly increased hepatic FFA content without substantial changes in hepatic TG abundance in mice in the context of an HFD feeding. As ACSL1 is a direct target gene of PPARα (53), increases in ACSL1 mRNA level and palmitoyl-CoA activity are likely the result of increased PPARα activity in ACSL4 KD mice. We also noticed that PPARδ expression is unchanged, which is consistent with the lack of effect of ACSL4 KD on Lpcat3 mRNA levels.
Fig. 9.

Effects of long-chain acyl-CoA synthetase 4 (ACSL4) knockdown on the fatty acid (FA) lipogenic and oxidative pathways. A and C: quantitative RT-PCR was conducted to determine relative expression levels of mRNAs of lipogenic genes (A) and genes involved in FA β-oxidation (C) after normalization with GAPDH mRNA levels. Significance was determined by unpaired Student’s t-test; n = 6 mice per group; *P < 0.05; **P < 0.01; ***P < 0.001. B and D: Western blot analysis of FASN and SCD1 (B) and CPT1α (D) and β-actin levels in individual livers of mouse ACSL4 shRNA (Ad-shAcsl4)- or mice receiving control adenovirus (Ad-shLacZ)-injected mice. The protein abundance of target protein was quantified with normalization with β-actin using AlphaView software. Significance was determined by unpaired Student’s t-test; n = 6 mice per group; ***P < 0.001. n.s, not significant.
ACSL4 KD protects from HFD-associated inflammatory mediator production and p53 activation.
It is known that free AA can be metabolized by cyclooxygenases, lipoxygenase, and cytochrome p450 enzyme systems into lipid mediators, producing inflammatory eicosanoids (PGE2, PGF2α, thromboxane B2, and LTB4) (38, 41, 50). Furthermore, studies conducted in extrahepatic cell types have shown that suppression of ACSL4 activity or siRNA-mediated KD of ACSL4 expression enhanced IL-1β-dependent production of PG (13, 25, 26, 34). Thus we initially postulated that hepatic ACSL4 KD would increase liver inflammatory mediator production, due to potentially altered AA metabolite profiles in the liver. With the use of qRT-PCR, we measured mRNA levels of a panel of inflammatory genes TNF-α, IL-1β, IL-6, chemokine (C-C motif) ligand (CCL2), and integrin alpha X (Itgax) and surprisingly, found that their mRNA levels were significantly lower in ACSL4 KD mice than those of control mice on an HFD (Fig. 10A). In addition, ACSL4 KD attenuated the expression of stress-marker genes activating transcription factor 4 (Atf4), C/EBP homolog protein (Chop), and Xbp1 (Fig. 10A) and a panel of fibrotic genes (Fig. 10A). Analysis of liver homogenates for stress-induced JNK kinase further showed that the HFD-induced phosphorylation and activation of JNK were greatly attenuated by knocking down hepatic ACSL4 (Fig. 10B). Moreover, measurement of serum LTB4 showed a 51.6% (P < 0.01) reduction of this inflammatory eicosanoid in ACSL4 KD mice on an HFD (Fig. 10C). Further analysis of liver histology and Oil Red O staining did not reveal overall histological differences in either fatty change or inflammation between liver-tissue samples of six ACSL4 KD and six LacZ control animals fed an HFD, possibly because changes in gene expression occurred earlier than histological changes of fatty liver (data not shown).
Fig. 10.

Long-chain acyl-CoA synthetase 4 (ACSL4) knockdown attenuated high-fat diet (HFD)-induced hepatic inflammatory and stress-induced gene mRNA levels. A: real-time PCR was conducted to determine relative expression levels of hepatic mRNAs of proinflammatory genes, stress-induced genes [endoplasmic reticulum (ER)], and fibrotic genes after normalization with GAPDH mRNA levels. Significance was determined by Student’s t-test; n = 6 mice per group; *P < 0.05; **P < 0.01; ***P < 0.001. B: Western blot analysis of phosphorylated (p)-JNK and total JNK in liver homogenates of HFD-fed mice with mouse ACSL4 shRNA (Ad-shAcsl4) or mice receiving control adenovirus (Ad-shLacZ) infection. C: serum leukotriene B4 (LTB4) concentrations were measured in serum samples of mice fed an HFD. Significance was determined by unpaired Student’s t-test; n = 6 mice per group; **P < 0.01.
It was recently reported that ablation of ACSL4 in adipose tissue of HFD-fed mice is associated with protection against HFD-induced p53 activation and its downstream effects on inflammation (19). To determine whether the reduced inflammatory mediator production observed in Ad-shAcsl4 mice is associated with diminished p53 activation, we performed Western blot analysis to examine p53 protein expression in liver samples of Ad-shAcsl4 and Ad-shLacZ mice. The results showed a substantial reduction of p53 protein levels in the ACSL4 KD group (Fig. 11A). By contrast, LDL receptor (LDLR) protein levels were similar between the two groups, which correlates with the lack of effect of ACSL4 KD on plasma and hepatic cholesterol levels.
Fig. 11.

Repression of high-fat diet (HFD)-induced p53 activation and downstream events. A: Western blot analysis of p53, LDL receptor (LDLR), p21, and GAPDH in liver homogenates of HFD-fed mice with mouse long-chain acyl-CoA synthetase 4 shRNA (Ad-shAcsl4) or mice receiving control adenovirus (Ad-shLacZ) infection. B: heatmap illustration of changes in gene expression of the Kyoto Encyclopedia of Genes and Genomes (KEGG) p53 signaling pathway in livers of Ad-shAcsl4-injected mice (n = 6) compared with Ad-shLacZ mice (n = 6). Graph shows fold changes in all genes within the indicated pathway that have a P value <0.05 and a fold change >1.5. C: heatmap illustration of changes in expression of apoptotic genes. Graph shows fold changes in all genes within this gene set that have a P value <0.05 and a fold change >1.5. D: real-time quantitative RT-PCR was performed to determine the relative mRNA expression levels of genes in the p53 signaling pathway after normalization with GAPDH mRNA levels to validate the gene array results. Significance was determined by Student’s t-test; n = 6 mice per group; *P < 0.05; ***P < 0.001.
To understand further the impact of ACSL4 KD on p53, as well as p53-mediated downstream events, we carried out gene-expression profiling of individual livers of HFD-fed mice infected with Ad-shAcsl4 or control Ad-shLacZ. With the use of a cutoff criterion of 1.5-fold change and a P value of <0.05, we identified 1,164 genes that were downregulated and 416 genes that were upregulated by ACSL4 deficiency. Importantly, GSEA KEGG pathway analysis revealed that the p53 signaling pathway was the third-most enriched biological pathway downregulated following depletion of ACSL4. Eighteen genes within this gene set were significantly repressed (Fig. 11B). The well-known p53 target genes p21 (cyclin-dependent kinase inhibitor 1A) and cyclin G1 were reduced by 9.3-fold (P < 0.0001) and 3.5-fold (P < 0.0001), respectively. Reduction of p21 protein levels in Ad-shAcsl4 mice was further shown by Western blotting with an anti-p21 antibody (Fig. 11A). Consequentially, the p53-activated apoptosis signaling pathway was attenuated in the liver of the ACSL4 KD group compared with control mice on an HFD (Fig. 11C). These microarray results were further validated by qRT-PCR analysis (Fig. 11D). Taken together, these results suggest that in the context of HFD-fed mice, similar to ablation of adipocyte ACSL4, knocking down hepatic ACSL4 results in protection of HFD-induced p53 activation and its downstream proinflammatory response.
DISCUSSION
ACSL4 is an ACSL family member that has a key role in AA metabolism. Within this family, ACSL4 is the prominent isoform showing elevated expression in hepatocarcinoma (52) and NAFLD (22, 51, 56), both of which exhibit abnormal hepatic lipid metabolism. Our previous studies showed that ACSL4 expression and AA-CoA synthetase activity were upregulated in liver tissue of hyperlipidemic hamsters via a PPARδ-mediated mechanism (16). We also demonstrated that ACSL4 was coregulated with Lpcat3 in the liver of mice by activation of PPARδ to modulate plasma TG metabolism (47). However, until the present study, no reports in the literature have addressed the specific roles played by hepatic ACSL4 in lipid metabolism in any live animal models. By use of a KD approach, the aim of this current study was to assess directly the function of hepatic ACSL4 in hyperlipidemic adult mice, which display the pathological condition of NAFLD. Our study results revealed for the first time that hepatic ACSL4 is required for metabolism of plasma VLDL-TG and glucose and for PL synthesis in the liver of mice in the context of HFD feeding.
We first demonstrated that adenovirus-mediated expression of Acsl4 shRNA led to efficient and specific depletion of Acsl4 mRNA and protein in cultured MPH in vitro and in the liver of mice fed an HFD. With the use of three different 3H-labeled FA substrates, we further showed that ACSL4 KD in mice reduced AA-CoA synthetase activity to a level nearly one-half of control without any attenuation of fatty-CoA synthetase activities using saturated (PA) or monounsaturated (OA) LCFA as substrates in liver tissue, which provided in vivo evidence for the preference of ACSL4 for arachidonate as substrate.
One important finding of this current study is that hepatic ACSL4 KD reduced serum VLDL-TG without any effects on plasma and hepatic cholesterol levels, thereby suggesting that ACSL4 may direct PUFA into cellular TG pathways. The similar LDLR expression in liver tissues of Ad-shLacZ and Ad-shAcsl4 also suggests that cholesterol homeostasis is undisturbed by ACSL4 KD in HFD-fed mice.
One potential mechanism underlying the reduction of VLDL-TG may be related to the attenuation of hepatic LPCAT3 activity. Previous studies by other investigators have demonstrated that Lpcat3-dependent production of arachidonoyl PLs is a key determinant of TG secretion (43, 54). Genetic deletion of the Lpcat3 gene in the liver led to reduced plasma VLDL-TG and increased liver fat content. In our ACSL4 KD mice, the reduction of AA-CoA activity and substantial increases in saturated LPC and LPE species are indicative of reduced Lpcat3 activity. However, as ACSL4 KD did not alter Lpcat3 mRNA expression, it is probable that decreased activity was due to diminished precursor substrates in the setting of ACSL4 KD. This was accompanied by lower plasma TG in the absence of notable changes in liver TG content. It is plausible that the partial depletion of hepatic ACSL4 and a relatively short period of KD diminished the impact of deficiency in the pool of hepatic arachidonate-CoA substrates on LPCAT3 activity. The lack of accumulation of hepatic TG could also be explained by the observation of an increased FA β-oxidation pathway, as well as an elevated lipogenic pathway, that perhaps counteracted their individual effects on TG content in the ACSL4-depleted liver of HFD-fed mice.
The anti-inflammatory effect of hepatic ACSL4 depletion in mice on an HFD diet is an interesting, new finding. We observed reduced expression of proinflammatory genes, including TNF-α and IL-1β, and stress-inducible genes, such as Chop and ATF4, and fibrotic genes in ACSL4 KD mice compared with control mice receiving Ad-shLacZ. These changes in gene expression are corroborated by the marked reduction of phosphorylated JNK abundance in liver homogenates and the lower blood concentration of LTB4, a biomarker of inflammation. These observations seemingly contradict our previous thinking about the relationship between ACSL4 and inflammation status. In smooth muscle cells, siRNA-mediated depletion of ACSL4 led to increases in PGE2 production (13); in fibroblast 3Y1 cells, suppression of ACSL4 enzymatic activity and reduction of ACSL4 protein expression both resulted in an enhanced cyclooxygenase 2 pathway (26). However, recent studies in breast cancer cells suggested that ACSL4 is involved in ferroptosis, since depletion of ACSL4 strongly protected against ferroptosis by blocking PUFA incorporation into PLs (6, 58). Thus in the case of ferroptosis and our study, depletion of ACSL4 has a consistent protective effect.
Regarding anti-inflammation, a recent study of ACSL4 ablation in adipose tissue by Killion et al. (19) provided a clue that pointed to the reduction of HFD-induced p53 activation as a possible underlying mechanism for the reduced inflammation. These authors further reported that the reduction of p53 in ACSL4-ablated adipocytes derived from Ad-KO mice fed HFD was associated with increased expression of glutathione-mediated detoxification gene expression. Similar to the findings of ACSL4 deficiency in adipocytes of mice fed an HFD, in our study, hepatic depletion of ACSL4 lowered the p53 protein abundance in liver homogenates of HFD-fed mice compared with that of HFD-fed control mice. This was accompanied by a marked reduction of the p53 signaling pathway, which was revealed by whole genome transcriptomic analysis. However, the gene array data did not show significant changes in expression patterns of genes involved in detoxification or lipid peroxidation between Ad-shLacZ and Ad-shAcsl4 groups. Nevertheless, the suppression of p53 activation likely accounts for the anti-inflammatory effect of hepatic ACSL4 KD in HFD-fed mice.
Lastly, the apparent insulin-resistant phenotype observed in ACSL4 KD mice, which exhibited lower plasma TG and reduced hepatic inflammatory mediator production, is a puzzle. We observed modest increases in blood glucose and insulin levels in Ad-shAcsl4 mice compared with control mice. Our analysis of hepatic expression of genes involved in glucose metabolism by qRT-PCR and gene-array pathway analysis did not identify obvious candidate genes responsible for the elevation of plasma glucose and insulin levels. However, ceramide and LPCs have been linked to insulin resistance (15, 24, 30, 32, 33, 37), and their accumulations in the liver of Ad-shAcsl4 mice could provide a connection to the insulin-resistant phenotype. Previous studies have shown that certain PC species produced by the liver are signaling molecules that can affect systemic insulin sensitivity (27). Thus it is possible that the insulin resistance in hepatic ACSL4 KD mice stems from the response of extrahepatic metabolic tissues, such as skeletal muscle or adipocytes, to altered lipid signaling molecules secreted from the liver. Further investigations are needed to identify the causal factor or specific lipid mediators responsible for the insulin resistance in ACSL4 KD mice on an HFD.
In summary, with the use of an adenoviral-mediated gene KD approach, we have identified new functions of hepatic ACSL4 in plasma TG metabolism and glucose metabolism, as well as in liver PL synthesis. Furthermore, we demonstrated an unexpected, inverse relationship between p53 activation and ACSL4 expression in the liver of mice on an HFD. Altogether, our new findings from this first in vivo study of hepatic function of ACSL4 warrant further investigations to define clearly all ACSL4-channeled PUFA metabolic pathways in the liver under normal physiologic and pathological conditions.
GRANTS
Support for this study was provided by the Department of Veterans Affairs (Office of Research and Development, Medical Research Service) Grants I01 BX001419 (to J. Liu) and I01 BX000398 (to F. B. Kraemer) and by the National Center for Complementary and Integrative Health Grant R01AT006336-01A1 (to J. Liu) and National Institute of Diabetes and Digestive and Kidney Diseases Grant P30DK116074 (to F. B. Kraemer).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
J.L. conceived and designed research; A.B.S. and C.F.K.K. performed experiments; A.B.S., C.F.K.K., R.A.S., and J.L. analyzed data; A.B.S., F.B.K., R.A.S., and J.L. interpreted results of experiments; A.B.S., R.A.S., and J.L. prepared figures; J.L. drafted manuscript; A.B.S., F.B.K., R.A.S., and J.L. edited and revised manuscript; A.B.S., C.F.K.K., F.B.K., R.A.S., and J.L. approved final version of manuscript.
REFERENCES
- 1.Ansari IH, Longacre MJ, Stoker SW, Kendrick MA, O’Neill LM, Zitur LJ, Fernandez LA, Ntambi JM, MacDonald MJ. Characterization of Acyl-CoA synthetase isoforms in pancreatic beta cells: gene silencing shows participation of ACSL3 and ACSL4 in insulin secretion. Arch Biochem Biophys 618: 32–43, 2017. doi: 10.1016/j.abb.2017.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Askari B, Kanter JE, Sherrid AM, Golej DL, Bender AT, Liu J, Hsueh WA, Beavo JA, Coleman RA, Bornfeldt KE. Rosiglitazone inhibits acyl-CoA synthetase activity and fatty acid partitioning to diacylglycerol and triacylglycerol via a peroxisome proliferator-activated receptor-gamma-independent mechanism in human arterial smooth muscle cells and macrophages. Diabetes 56: 1143–1152, 2007. doi: 10.2337/db06-0267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cao Y, Murphy KJ, McIntyre TM, Zimmerman GA, Prescott SM. Expression of fatty acid-CoA ligase 4 during development and in brain. FEBS Lett 467: 263–267, 2000. doi: 10.1016/S0014-5793(00)01159-5. [DOI] [PubMed] [Google Scholar]
- 4.Cao Y, Traer E, Zimmerman GA, McIntyre TM, Prescott SM. Cloning, expression, and chromosomal localization of human long-chain fatty acid-CoA ligase 4 (FACL4). Genomics 49: 327–330, 1998. doi: 10.1006/geno.1998.5268. [DOI] [PubMed] [Google Scholar]
- 5.Coleman RA, Lewin TM, Van Horn CG, Gonzalez-Baró MR. Do long-chain acyl-CoA synthetases regulate fatty acid entry into synthetic versus degradative pathways? J Nutr 132: 2123–2126, 2002. doi: 10.1093/jn/132.8.2123. [DOI] [PubMed] [Google Scholar]
- 6.Doll S, Proneth B, Tyurina YY, Panzilius E, Kobayashi S, Ingold I, Irmler M, Beckers J, Aichler M, Walch A, Prokisch H, Trümbach D, Mao G, Qu F, Bayir H, Füllekrug J, Scheel CH, Wurst W, Schick JA, Kagan VE, Angeli JP, Conrad M. ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat Chem Biol 13: 91–98, 2017. doi: 10.1038/nchembio.2239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dong B, Kan CF, Singh AB, Liu J. High-fructose diet downregulates long-chain acyl-CoA synthetase 3 expression in liver of hamsters via impairing LXR/RXR signaling pathway. J Lipid Res 54: 1241–1254, 2013. doi: 10.1194/jlr.M032599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dong B, Wu M, Li H, Kraemer FB, Adeli K, Seidah NG, Park SW, Liu J. Strong induction of PCSK9 gene expression through HNF1alpha and SREBP2: mechanism for the resistance to LDL-cholesterol lowering effect of statins in dyslipidemic hamsters. J Lipid Res 51: 1486–1495, 2010. doi: 10.1194/jlr.M003566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dong B, Young M, Liu X, Singh AB, Liu J. Regulation of lipid metabolism by obeticholic acid in hyperlipidemic hamsters. J Lipid Res 58: 350–363, 2017. doi: 10.1194/jlr.M070888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ellis JM, Frahm JL, Li LO, Coleman RA. Acyl-coenzyme A synthetases in metabolic control. Curr Opin Lipidol 21: 212–217, 2010. doi: 10.1097/MOL.0b013e32833884bb. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ellis JM, Li LO, Wu PC, Koves TR, Ilkayeva O, Stevens RD, Watkins SM, Muoio DM, Coleman RA. Adipose acyl-CoA synthetase-1 directs fatty acids toward beta-oxidation and is required for cold thermogenesis. Cell Metab 12: 53–64, 2010. doi: 10.1016/j.cmet.2010.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ellis JM, Mentock SM, Depetrillo MA, Koves TR, Sen S, Watkins SM, Muoio DM, Cline GW, Taegtmeyer H, Shulman GI, Willis MS, Coleman RA. Mouse cardiac acyl coenzyme a synthetase 1 deficiency impairs Fatty Acid oxidation and induces cardiac hypertrophy. Mol Cell Biol 31: 1252–1262, 2011. doi: 10.1128/MCB.01085-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Golej DL, Askari B, Kramer F, Barnhart S, Vivekanandan-Giri A, Pennathur S, Bornfeldt KE. Long-chain acyl-CoA synthetase 4 modulates prostaglandin E2 release from human arterial smooth muscle cells. J Lipid Res 52: 782–793, 2011. doi: 10.1194/jlr.M013292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Grevengoed TJ, Klett EL, Coleman RA. Acyl-CoA metabolism and partitioning. Annu Rev Nutr 34: 1–30, 2014. doi: 10.1146/annurev-nutr-071813-105541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ha CY, Kim JY, Paik JK, Kim OY, Paik YH, Lee EJ, Lee JH. The association of specific metabolites of lipid metabolism with markers of oxidative stress, inflammation and arterial stiffness in men with newly diagnosed type 2 diabetes. Clin Endocrinol (Oxf) 76: 674–682, 2012. doi: 10.1111/j.1365-2265.2011.04244.x. [DOI] [PubMed] [Google Scholar]
- 16.Kan CF, Singh AB, Dong B, Shende VR, Liu J. PPARδ activation induces hepatic long-chain acyl-CoA synthetase 4 expression in vivo and in vitro. Biochim Biophys Acta 1851: 577–587, 2015. doi: 10.1016/j.bbalip.2015.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kan CF, Singh AB, Stafforini DM, Azhar S, Liu J. Arachidonic acid downregulates acyl-CoA synthetase 4 expression by promoting its ubiquitination and proteasomal degradation. J Lipid Res 55: 1657–1667, 2014. doi: 10.1194/jlr.M045971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kang MJ, Fujino T, Sasano H, Minekura H, Yabuki N, Nagura H, Iijima H, Yamamoto TT. A novel arachidonate-preferring acyl-CoA synthetase is present in steroidogenic cells of the rat adrenal, ovary, and testis. Proc Natl Acad Sci USA 94: 2880–2884, 1997. doi: 10.1073/pnas.94.7.2880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Killion EA, Reeves AR, El Azzouny MA, Yan QW, Surujon D, Griffin JD, Bowman TA, Wang C, Matthan NR, Klett EL, Kong D, Newman JW, Han X, Lee MJ, Coleman RA, Greenberg AS. A role for long-chain acyl-CoA synthetase-4 (ACSL4) in diet-induced phospholipid remodeling and obesity-associated adipocyte dysfunction. Mol Metab 9: 43–56, 2018. doi: 10.1016/j.molmet.2018.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Klett EL, Chen S, Edin ML, Li LO, Ilkayeva O, Zeldin DC, Newgard CB, Coleman RA. Diminished acyl-CoA synthetase isoform 4 activity in INS 832/13 cells reduces cellular epoxyeicosatrienoic acid levels and results in impaired glucose-stimulated insulin secretion. J Biol Chem 288: 21618–21629, 2013. doi: 10.1074/jbc.M113.481077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Klett EL, Chen S, Yechoor A, Lih FB, Coleman RA. Long-chain acyl-CoA synthetase isoforms differ in preferences for eicosanoid species and long-chain fatty acids. J Lipid Res 58: 884–894, 2017. [Erratum in: J Lipid Res 58: 2365, 2017.] doi: 10.1194/jlr.M072512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kotronen A, Yki-Järvinen H, Aminoff A, Bergholm R, Pietiläinen KH, Westerbacka J, Talmud PJ, Humphries SE, Hamsten A, Isomaa B, Groop L, Orho-Melander M, Ehrenborg E, Fisher RM. Genetic variation in the ADIPOR2 gene is associated with liver fat content and its surrogate markers in three independent cohorts. Eur J Endocrinol 160: 593–602, 2009. doi: 10.1530/EJE-08-0900. [DOI] [PubMed] [Google Scholar]
- 23.Küch EM, Vellaramkalayil R, Zhang I, Lehnen D, Brügger B, Stremmel W, Ehehalt R, Poppelreuther M, Füllekrug J. Differentially localized acyl-CoA synthetase 4 isoenzymes mediate the metabolic channeling of fatty acids towards phosphatidylinositol. Biochim Biophys Acta 1841: 227–239, 2014. doi: 10.1016/j.bbalip.2013.10.018. [DOI] [PubMed] [Google Scholar]
- 24.Kurek K, Piotrowska DM, Wiesiołek-Kurek P, Łukaszuk B, Chabowski A, Górski J, Zendzian-Piotrowska M. Inhibition of ceramide de novo synthesis reduces liver lipid accumulation in rats with nonalcoholic fatty liver disease. Liver Int 34: 1074–1083, 2014. doi: 10.1111/liv.12331. [DOI] [PubMed] [Google Scholar]
- 25.Kuwata H, Hara S. Inhibition of long-chain acyl-CoA synthetase 4 facilitates production of 5, 11-dihydroxyeicosatetraenoic acid via the cyclooxygenase-2 pathway. Biochem Biophys Res Commun 465: 528–533, 2015. doi: 10.1016/j.bbrc.2015.08.054. [DOI] [PubMed] [Google Scholar]
- 26.Kuwata H, Yoshimura M, Sasaki Y, Yoda E, Nakatani Y, Kudo I, Hara S. Role of long-chain acyl-coenzyme A synthetases in the regulation of arachidonic acid metabolism in interleukin 1β-stimulated rat fibroblasts. Biochim Biophys Acta 1841: 44–53, 2014. doi: 10.1016/j.bbalip.2013.09.015. [DOI] [PubMed] [Google Scholar]
- 27.Lee JM, Lee YK, Mamrosh JL, Busby SA, Griffin PR, Pathak MC, Ortlund EA, Moore DD. A nuclear-receptor-dependent phosphatidylcholine pathway with antidiabetic effects. Nature 474: 506–510, 2011. doi: 10.1038/nature10111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lewin TM, Kim JH, Granger DA, Vance JE, Coleman RA. Acyl-CoA synthetase isoforms 1, 4, and 5 are present in different subcellular membranes in rat liver and can be inhibited independently. J Biol Chem 276: 24674–24679, 2001. doi: 10.1074/jbc.M102036200. [DOI] [PubMed] [Google Scholar]
- 29.Li LO, Grevengoed TJ, Paul DS, Ilkayeva O, Koves TR, Pascual F, Newgard CB, Muoio DM, Coleman RA. Compartmentalized acyl-CoA metabolism in skeletal muscle regulates systemic glucose homeostasis. Diabetes 64: 23–35, 2015. doi: 10.2337/db13-1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li LO, Klett EL, Coleman RA. Acyl-CoA synthesis, lipid metabolism and lipotoxicity. Biochim Biophys Acta 1801: 246–251, 2010. doi: 10.1016/j.bbalip.2009.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li Z, Jiang H, Ding T, Lou C, Bui HH, Kuo MS, Jiang XC. Deficiency in lysophosphatidylcholine acyltransferase 3 reduces plasma levels of lipids by reducing lipid absorption in mice. Gastroenterology 149: 1519–1529, 2015. doi: 10.1053/j.gastro.2015.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Longato L, Tong M, Wands JR, de la Monte SM. High fat diet induced hepatic steatosis and insulin resistance: Role of dysregulated ceramide metabolism. Hepatol Res 42: 412–427, 2012. doi: 10.1111/j.1872-034X.2011.00934.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lyn-Cook LE Jr, Lawton M, Tong M, Silbermann E, Longato L, Jiao P, Mark P, Wands JR, Xu H, de la Monte SM. Hepatic ceramide may mediate brain insulin resistance and neurodegeneration in type 2 diabetes and non-alcoholic steatohepatitis. J Alzheimers Dis 16: 715–729, 2009. doi: 10.3233/JAD-2009-0984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Maloberti PM, Duarte AB, Orlando UD, Pasqualini ME, Solano AR, López-Otín C, Podestá EJ. Functional interaction between acyl-CoA synthetase 4, lipooxygenases and cyclooxygenase-2 in the aggressive phenotype of breast cancer cells. PLoS One 5: e15540, 2010. doi: 10.1371/journal.pone.0015540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mashek DG, Li LO, Coleman RA. Rat long-chain acyl-CoA synthetase mRNA, protein, and activity vary in tissue distribution and in response to diet. J Lipid Res 47: 2004–2010, 2006. doi: 10.1194/jlr.M600150-JLR200. [DOI] [PubMed] [Google Scholar]
- 37.Motley ED, Kabir SM, Gardner CD, Eguchi K, Frank GD, Kuroki T, Ohba M, Yamakawa T, Eguchi S. Lysophosphatidylcholine inhibits insulin-induced Akt activation through protein kinase C-alpha in vascular smooth muscle cells. Hypertension 39: 508–512, 2002. doi: 10.1161/hy02t2.102907. [DOI] [PubMed] [Google Scholar]
- 38.Orlando UD, Garona J, Ripoll GV, Maloberti PM, Solano AR, Avagnina A, Gomez DE, Alonso DF, Podestá EJ. The functional interaction between Acyl-CoA synthetase 4, 5-lipooxygenase and cyclooxygenase-2 controls tumor growth: a novel therapeutic target. PLoS One 7: e40794, 2012. doi: 10.1371/journal.pone.0040794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Paalvast Y, de Boer JF, Groen AK. Developments in intestinal cholesterol transport and triglyceride absorption. Curr Opin Lipidol 28: 248–254, 2017. doi: 10.1097/MOL.0000000000000415. [DOI] [PubMed] [Google Scholar]
- 40.Pan X, Chen G, Kazachkov M, Greer MS, Caldo KM, Zou J, Weselake RJ. In vivo and in vitro evidence for biochemical coupling of reactions catalyzed by lysophosphatidylcholine acyltransferase and diacylglycerol acyltransferase. J Biol Chem 290: 18068–18078, 2015. doi: 10.1074/jbc.M115.654798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pérez-Chacón G, Astudillo AM, Balgoma D, Balboa MA, Balsinde J. Control of free arachidonic acid levels by phospholipases A2 and lysophospholipid acyltransferases. Biochim Biophys Acta 1791: 1103–1113, 2009. doi: 10.1016/j.bbalip.2009.08.007. [DOI] [PubMed] [Google Scholar]
- 42.Pérez-Chacón G, Astudillo AM, Ruipérez V, Balboa MA, Balsinde J. Signaling role for lysophosphatidylcholine acyltransferase 3 in receptor-regulated arachidonic acid reacylation reactions in human monocytes. J Immunol 184: 1071–1078, 2010. doi: 10.4049/jimmunol.0902257. [DOI] [PubMed] [Google Scholar]
- 43.Rong X, Wang B, Dunham MM, Hedde PN, Wong JS, Gratton E, Young SG, Ford DA, Tontonoz P. Lpcat3-dependent production of arachidonoyl phospholipids is a key determinant of triglyceride secretion. eLife 4: e06557, 2015. doi: 10.7554/eLife.06557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shende VR, Wu M, Singh AB, Dong B, Kan CF, Liu J. Reduction of circulating PCSK9 and LDL-C levels by liver-specific knockdown of HNF1α in normolipidemic mice. J Lipid Res 56: 801–809, 2015. doi: 10.1194/jlr.M052969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Singh AB, Kan CF, Dong B, Liu J. SREBP2 activation induces hepatic long-chain acyl-CoA synthetase 1 (ACSL1) expression in vivo and in vitro through a sterol regulatory element (SRE) motif of the ACSL1 C-promoter. J Biol Chem 291: 5373–5384, 2016. doi: 10.1074/jbc.M115.696872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Singh AB, Kan CF, Shende V, Dong B, Liu J. A novel posttranscriptional mechanism for dietary cholesterol-mediated suppression of liver LDL receptor expression. J Lipid Res 55: 1397–1407, 2014. doi: 10.1194/jlr.M049429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Singh AB, Liu J. Identification of hepatic lysophosphatidylcholine acyltransferase 3 as a novel target gene regulated by peroxisome proliferator-activated receptor δ. J Biol Chem 292: 884–897, 2017. doi: 10.1074/jbc.M116.743575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Soupene E, Dinh NP, Siliakus M, Kuypers FA. Activity of the acyl-CoA synthetase ACSL6 isoforms: role of the fatty acid Gate-domains. BMC Biochem 11: 18, 2010. doi: 10.1186/1471-2091-11-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Soupene E, Kuypers FA. Mammalian long-chain acyl-CoA synthetases. Exp Biol Med (Maywood) 233: 507–521, 2008. doi: 10.3181/0710-MR-287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Spector AA. Arachidonic acid cytochrome P450 epoxygenase pathway. J Lipid Res 50, Suppl: S52–S56, 2009. doi: 10.1194/jlr.R800038-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Stepanova M, Hossain N, Afendy A, Perry K, Goodman ZD, Baranova A, Younossi Z. Hepatic gene expression of Caucasian and African-American patients with obesity-related non-alcoholic fatty liver disease. Obes Surg 20: 640–650, 2010. doi: 10.1007/s11695-010-0078-2. [DOI] [PubMed] [Google Scholar]
- 52.Sung YK, Hwang SY, Park MK, Bae HI, Kim WH, Kim JC, Kim M. Fatty acid-CoA ligase 4 is overexpressed in human hepatocellular carcinoma. Cancer Sci 94: 421–424, 2003. doi: 10.1111/j.1349-7006.2003.tb01458.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Suzuki H, Watanabe M, Fujino T, Yamamoto T. Multiple promoters in rat acyl-CoA synthetase gene mediate differential expression of multiple transcripts with 5′-end heterogeneity. J Biol Chem 270: 9676–9682, 1995. doi: 10.1074/jbc.270.16.9676. [DOI] [PubMed] [Google Scholar]
- 54.Taniguchi K, Hikiji H, Okinaga T, Hashidate-Yoshida T, Shindou H, Ariyoshi W, Shimizu T, Tominaga K, Nishihara T. Essential role of lysophosphatidylcholine acyltransferase 3 in the induction of macrophage polarization in PMA-treated U937 cells. J Cell Biochem 116: 2840–2848, 2015. doi: 10.1002/jcb.25230. [DOI] [PubMed] [Google Scholar]
- 55.Tuohetahuntila M, Spee B, Kruitwagen HS, Wubbolts R, Brouwers JF, van de Lest CH, Molenaar MR, Houweling M, Helms JB, Vaandrager AB. Role of long-chain acyl-CoA synthetase 4 in formation of polyunsaturated lipid species in hepatic stellate cells. Biochim Biophys Acta 1851: 220–230, 2015. doi: 10.1016/j.bbalip.2014.12.003. [DOI] [PubMed] [Google Scholar]
- 56.Westerbacka J, Kolak M, Kiviluoto T, Arkkila P, Sirén J, Hamsten A, Fisher RM, Yki-Järvinen H. Genes involved in fatty acid partitioning and binding, lipolysis, monocyte/macrophage recruitment, and inflammation are overexpressed in the human fatty liver of insulin-resistant subjects. Diabetes 56: 2759–2765, 2007. doi: 10.2337/db07-0156. [DOI] [PubMed] [Google Scholar]
- 57.Wu M, Liu H, Chen W, Fujimoto Y, Liu J. Hepatic expression of long-chain acyl-CoA synthetase 3 is upregulated in hyperlipidemic hamsters. Lipids 44: 989–998, 2009. doi: 10.1007/s11745-009-3341-3. [DOI] [PubMed] [Google Scholar]
- 58.Yuan H, Li X, Zhang X, Kang R, Tang D. Identification of ACSL4 as a biomarker and contributor of ferroptosis. Biochem Biophys Res Commun 478: 1338–1343, 2016. doi: 10.1016/j.bbrc.2016.08.124. [DOI] [PubMed] [Google Scholar]