

Abstract
Deranged histone deacetylase (HDAC) activity causes uncontrolled proliferation, inflammation, fibrosis, and organ damage. It is unclear whether deranged HDAC activity results in acute kidney injury in the renal hypoperfusion model of bilateral ischemia-reperfusion injury (IRI) and whether in vivo inhibition is an appropriate therapeutic approach to limit injury. Male mice were implanted with intraperitoneal osmotic minipumps containing vehicle, the class I HDAC inhibitor, MS275, or the pan-HDAC inhibitor, trichostatin A (TSA), 3 days before sham/bilateral IRI surgery. Kidney cortical samples were analyzed using histological, immunohistochemical, and Western blotting techniques. HDAC-dependent proliferation rate was measured in immortalized rat epithelial cells and primary mouse or human proximal tubule (PT) cells. There were dynamic changes in cortical HDAC localization and abundance following IRI including a fourfold increase in HDAC4 in the PT. HDAC inhibition resulted in a significantly higher plasma creatinine, increased kidney damage, but reduced interstitial fibrosis compared with vehicle-treated IRI mice. HDAC-inhibited mice had reduced interstitial α-smooth muscle actin, fibronectin expression, and Sirius red-positive area, suggesting that IRI activates HDAC-mediated fibrotic pathways. In vivo proliferation of the kidney epithelium was significantly reduced in TSA-treated, but not MS275-treated, IRI mice, suggesting class II HDACs mediate proliferation. Furthermore, HDAC4 activation increased proliferation of human and mouse PTs. Kidney HDACs are activated during IRI with isoform-specific expression patterns. Our data point to mechanisms whereby IRI activates HDACs resulting in fibrotic pathways but also activation of PT proliferation and repair pathways. This study demonstrates the need to develop isoform-selective HDAC inhibitors for the treatment of renal hypoperfusion-induced injury.
Keywords: fibrosis, histone deacetylase, ischemia-reperfusion injury, kidney cortex, proliferation, proximal tubule
INTRODUCTION
Acute kidney injury (AKI) is defined as a sudden loss of kidney function over the course of hours to days. Although kidney function may return to a normal range, epidemiological data suggest that the incidence of AKI is associated with an increased risk of developing chronic kidney disease (13). From 2000 to 2014, the rate of hospitalizations due to AKI has risen significantly (139–230%) in the United States of America (44). There are many different causes and/or risk factors for AKI, including trauma, hypertension, and diabetes (44). Another prevalent cause of AKI is reduced renal perfusion during cardiac, vascular, or cardiopulmonary surgeries (14) that results in ischemia-reperfusion injury (IRI) and accounts for 23–50% of postsurgery incidences of AKI (14, 16, 25, 41). Furthermore, even mild changes in plasma creatinine are associated with a threefold increase in 30-day mortality (9, 26, 27). Thus there is a significant need to determine the mechanisms involved in IRI that can be potentially targeted to reduce or even prevent IRI in patients.
Histone deacetylases (HDACs) are an attractive target as aberrant HDAC activity is associated with increased fibrosis and inflammation of multiple organs, including kidney (11). HDACs are a large family of enzymes composed of four classes: class I (HDAC1, -2, -3, and -8), II (HDAC4, -5, -6, -7, -9, and -10), and IV (HDAC11) are the zinc-dependent HDACs, and class III (sirtuins) are the NAD+-dependent enzymes. They are epigenetic regulators of chromatin structure and thereby regulate transcription. The current paradigm is that excess HDAC activity results in the silencing of tumor suppressor (e.g., p53; Ref. 21) or antifibrotic (e.g., BMP-7; Ref. 35) genes. However, HDACs also deacetylate nonhistone proteins and regulate protein function through posttranslational modification of lysine residues. Increased HDAC activity deacetylates NF-κB p65 lysine residues, resulting in enhanced transcription of NF-κB-mediated inflammatory species (29). Currently, there are four Food and Drug Administration-approved HDAC inhibitors to treat T cell lymphoma, and perhaps these could be repurposed to prevent fibrosis and inflammation in kidney disease (11). However, not enough data are available with respect to the expression, abundance, and localization of the HDACs in both physiological and pathophysiological contexts.
From RNA-Seq (8, 28) and microarray (61) studies, all of the HDACs (1–11) are expressed in the nephron. However, other kidney cell types, like the vasculature, and their specific subcellular distribution are undetermined. Previous studies are indecisive whether HDACs cause injury or are critical for repair. For example, class I/II/IV, considered pan-HDAC inhibition (HDACi) with valproic acid, reduced inflammation and injury in the cecal ligation-puncture model of sepsis-induced AKI (62). Both pan- and class I-selective HDACi reduced fibrosis and maintained kidney function in the unilateral IRI and cold ischemia transplant models (30). Also, pan- and class I-selective HDACi significantly reduced injury and improved kidney function in the cisplatin-induced AKI model (32, 47). Conversely, in the folic acid and rhabdomyolysis models of AKI, class I-selective HDACi resulted in a worsening of renal function, reduced cell proliferation, and enhanced apoptosis (57). Whether the functional consequences of HDAC activation are model-specific, isoform-specific, or some other factor remains to be determined. Whereas these studies demonstrate that HDACi can affect outcomes in AKI, they also highlight the gap in our understanding of the physiological and pathophysiological role of kidney HDACs.
The purpose of this study was to determine the abundance and localization of the HDACs in the kidney and examine whether bilateral IRI affects this distribution. Next, we sought to determine the effect of HDACi on kidney function and injury in a bilateral IRI model. We hypothesized that HDACi would be protective by limiting renal injury and preserving renal function in the bilateral mouse IRI model.
MATERIALS AND METHODS
Animals.
All animal use and welfare adhered to the NIH Guide for the Care and Use of Laboratory Animals following a protocol reviewed and approved by the Institutional Laboratory Animal Care and Use Committee of the University of Alabama at Birmingham. Ten-week-old male C57BL/6J mice from Jackson (Bar Harbor, ME) were used in all studies because it is well-established that females are protected from IRI (2, 3). Mice were maintained on a standard chow diet (Teklad 7917; Envigo, Madison, WI) and water ad libitum in a room with 12:12-h light-dark cycle.
Bilateral IRI surgery.
All animal surgeries and biological sampling occurred between 8 AM and 12 PM. Mice were anesthetized with 2.5% isoflurane for induction and maintained on 1.5% isoflurane during surgery. Bilateral flank incisions were made, and the kidneys were exposed for atraumatic vascular clamps (cat. no. 18055-05; Fine Science Tools, Foster City, CA) to be placed on both pedicles for 27–30 min (see protocols 1 and 2 below). Body temperature was maintained at 37°C. Sham control mice went through the same procedures except for clamping of the renal pedicles. Mice were provided gel diet (TestDiet, Richmond, IN) for 24 h as an additional source of fluids during recovery.
Protocol 1: reperfusion time course study.
Mice were randomly assigned to the following groups: sham, 1-, 24-, 48-, or 72-h reperfusion; or IRI, 1, 24, 48, or 72 h. Because of 3 deaths in the 72-h IRI group, 4 additional mice (1 sham and 3 72-h IRI) underwent surgeries, resulting in n = 3 per group except for sham and 72-h IRI, n = 4. Atraumatic vascular clamps were placed on both pedicles for 30 min, and reperfusion was allowed for 1–72 h.
Protocol 2: HDACi intervention.
In a separate group of mice, we intervened with two HDAC inhibitors (HDACi) or vehicle before and after IRI. Seven-day osmotic minipumps (1007D; Alzet, Cupertino, CA) were filled with vehicle (acetic acid pH 5.0), a selective class I inhibitor, MS275 (20 mg·kg−1·day−1; entinostat; Cayman Chemical, Ann Arbor, MI; Ref. 57), or a pan-HDACi, trichostatin A (TSA; 1 mg·kg−1·day−1; Cayman Chemical; Ref. 47) and implanted intraperitoneally (i.p.). Three days after implantation, mice were randomly assigned to either sham or bilateral IRI surgery (renal pedicles were clamped for 27 min). Because of some deaths, final sample sizes were sham vehicle, n = 5; IRI vehicle, n = 6; sham MS275, n = 4; IRI MS275, n = 5; sham TSA, n = 6; and IRI TSA, n = 7. Samples were taken at 72 h of reperfusion.
Biological samples.
Blood was taken by cardiac puncture with heparinized syringes, and plasma was collected and stored at −80°C until analysis. Plasma creatinine was measure by liquid chromatography coupled to tandem mass spectrometry (53). The left kidney was processed for histological analysis. The right kidney was decapsulated, and cortical samples were snap-frozen and stored at −80°C.
Histology, immunohistochemistry, and Western blotting.
For histology, kidneys were fixed in 10% formalin, embedded in paraffin, cut into 5-μm-thick sections, and analyzed with Gömöri trichrome stain following manufacturer’s instruction (Richard-Allan Scientific, San Diego, CA). Picrosirius red staining was performed by deparaffinizing the samples, hydrating from 100% ethanol to water, and staining nuclei with Weigert hematoxylin for 8 min, followed by a wash in running tap water for 10 min. The slides were placed in 0.1% Direct Red 80 in saturated picric acid for 1 h at room temperature, then washed with acidified water (0.5% acetic acid) twice, then three changes of 100% ethanol, cleared in xylenes, and mounted. Primary antibody immunoreactivity was visualized on the slides with the ImmPRESS anti-rabbit IgG or anti-mouse IgG (peroxidase) polymer detection kits (cat. no. MP-6401-15 or MP-6402-15; Vector) and ImmPACT DAB Peroxidase (HRP) Substrate (cat. no. SK-4105; Vector), following manufacturer’s instructions. Our Western blot procedure was recently published (18). Antibodies and their concentrations are listed in Table 1.
Table 1.
Antibodies used in the study
| Concentration or Dilution |
||||||
|---|---|---|---|---|---|---|
| Target | Type | Company | Cat. no. | Lot no. | Western | IHC |
| HDAC1 | Rabbit monoclonal | Abcam | AB109411 | GR53419-13 | 0.77 μg/ml | 7.7 μg/ml |
| HDAC2 | Rabbit monoclonal | Abcam | AB32117 | GR112991-15 | 0.008 μg/ml | 0.008 μg/ml |
| HDAC3 | Rabbit monoclonal | Abcam | SB32369 | GR145298-2 | ND | 1.64 μg/ml |
| HDAC4 | Rabbit polyclonal | Abcam | AB111318 | GR100372-2 | 1 μg/ml | 1 μg/ml |
| HDAC5 | Rabbit monoclonal | Abcam | AB1440 | GR185618-1 | ND | 5 μg/ml |
| HDAC6 | Rabbit monoclonal | Abcam | AB1440 | GR263286-6 | ND | 0.25 μg/ml |
| HDAC7 | Rabbit monoclonal | Abcam | AB166911 | GR119402-10 | ND | 0.24 μg/ml |
| HDAC8 | Rabbit monoclonal | Abcam | AB187139 | GR265652-1 | ND | 0.41 μg/ml |
| HDAC9 | Rabbit monoclonal | Abcam | AB109446 | GR48716-12 | ND | 0.04 μg/ml |
| HDAC10 | Rabbit monoclonal | Abcam | AB108934 | YK022001PC3 | ND | 1:10,000 |
| HDAC11 | Rabbit monoclonal | Abcam | AB18973 | GR254922-8 | ND | 0.1 μg/ml |
| Actin | Mouse monoclonal | Sigma | A1978 | 087M4850V | 1:100K | ND |
| α-Smooth muscle actin | Mouse monoclonal | Sigma | A5228 | 037M4805V | ND | 1/1,000 |
| Acetylated histone H3 | Rabbit polyclonal | Millipore | 06-599 | 2724352 | 0.1 μg/ml | 2 μg/ml |
| Histone H3 unmodified | Rabbit monoclonal | Millipore | 05-928 | 2603378 | 1:1,000 | ND |
| Fibronectin | Rabbit polyclonal | Sigma | F3648 | 047M4809V | ND | 1:1,000 |
| Ki-67 | Rabbit monoclonal | Abcam | AB16667 | GR3185488-1 | ND | 1:100 |
| Kim-1 | Goat polyclonal | R&D Systems | AF1817 | KCA0316011 | 0.25 μg/ml | ND |
| NGAL | Goat polyclonal | R&D Systems | AF1857 | JZP0316051 | 0.25 μg/ml | ND |
HDAC, histone deacetylase; K, thousand; Kim-1, kidney injury marker-1; ND, not determined: Westerns or immunohistochemistry (IHC) were not performed with these antibodies; NGAL, neutrophil gelatinase-associated lipocalin.
RNA extraction, cDNA, and quantitative real-time PCR.
RNA was extracted from kidney cortex samples as previously reported (18). cDNA was reverse-transcribed from 2.5 µg using a SuperScript IV VILO Master Mix kit with ezDNase Enzyme (Thermo Fisher Scientific). SYBR Green-based quantitative real-time PCR was run using Bio-Rad SsoAdvanced Universal master mix and primers. Duplicate reactions were 15 µl of SYBR master mix, 30 pmol of primer pairs, and 12 µl of sterile water, vortexed, split into 2 15-µl reactions in a 96-well plate, and 1 µl of 1:10 diluted cDNA added to each well. Thermal cycler (Bio-Rad CFX96) conditions were polymerase activation, 95°C, 30 s; denature, 95°C, 15 s; anneal and extension, 60°C, 30 s; for 40 cycles. Cycle thresholds were analyzed using the comparative cycle threshold method where each sample was subtracted from the housekeeping gene, Gapdh (Qiagen QuantiTect primer pair, QT01658692), and then subtracted from a vehicle, sham sample. All samples were normalized to vehicle sham group average and then to their respective sham groups to represent fold change. Primer sequences for all other targets were obtained from PrimerBank (59), purchased from Integrated DNA Technologies, and are reported in Table 2.
Table 2.
Quantitative real-time PCR primers used in the study
| Protein | Gene Name | Forward Primer Sequence 5′-3′ | Reverse Primer Sequence 5′-3′ |
|---|---|---|---|
| LC3 | Map1lc3a | GACCGCTGTAAGGAGGTGC | CTTGACCAACTCGCTCATGTTA |
| Beclin-1 | Becn1 | ATGGAGGGGTCTAAGGCGTC | TCCTCTCCTGAGTTAGCCTCT |
| ATG5 | Atg5 | AGCCAGGTGATGATTCACGG | GGCTGGGGGACAATGCTAA |
| ATG7 | Atg7 | GTTCGCCCCCTTTAATAGTGC | TGAACTCCAACGTCAAGCGG |
| PCNA | Pcna | TTTGAGGCACGCCTGATCC | GGAGACGTGAGACGAGTCCAT |
ATG5, autophagy-related 5; ATG7, autophagy-related 7; LC3, light chain 3; PCNA, proliferating cell nuclear antigen.
Cell culture: proliferation assay with WST-1, and scratch tests.
Immortalized rat epithelial cells (NRK-52E; American Type Culture Collection, Manassas, VA) and primary mouse or human proximal tubule (PT) cells (Cell Biologics) were cultured following manufacturer’s instructions. Only passages 4–7 were used in experiments. Cells were grown to confluency in 100-mm plates, trypsinized (0.25% trypsin- 0.1% EDTA; Corning), and resuspended in 25 ml of media. One milliliter of cells in media were mixed with vehicle or HDAC inhibitors listed in Table 5 (Cayman Chemical) and seeded in triplicate for each treatment in 96-well plates. To determine whether these inhibitors significantly affect proliferation rate, 2-(4-iodophenyl)-3-(4-nitrophenyl)-5-(2,4-disulfophenyl)-2H-tetrazolium, monosodium salt (WST-1) assays (ab65475; Abcam, Cambridge, MA) were performed. The WST-1 assay is similar to the traditional 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-2H-tetrazolium bromide assay for measuring proliferation rate, but the WST-1 is more stable and sensitive than 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-2H-tetrazolium bromide. Twenty-four hours after seeding, 10 μl of WST-1 proliferation reagent was added to each well, and the absorbance was measured at 420 nm at 0 and 24 h.
Table 5.
WST-1 proliferation assay of immortalized rat kidney epithelial cells (NRK-52E) or primary mouse proximal tubules
| Condition/Drug Name | Concentration | Description | Immortalized Rat Epithelial | Primary Mouse Proximal Tubules |
|---|---|---|---|---|
| Media | Negative control | 1.49 ± 0.09 | 2.02 ± 0.12 | |
| DMSO | 0.10% | Vehicle control | 1.56 ± 0.09 | 1.89 ± 0.07 |
| MS275 | 8 µM | Class I inhibition | 1.67 ± 0.2 | 1.73 ± 0.02 |
| TSA | 500 nM | Class I/II inhibition | 1.07 ± 0.2* | 1.53 ± 0.09* |
| MS275 | 300 nM | HDAC1 inhibition | 1.61 ± 0.07 | 1.78 ± 0.01 |
| Mocetinostat | 300 nM | HDAC1 inhibition | 1.39 ± 0.2 | 1.79 ± 0.03 |
| MI-192 | 100 nM | HDAC2/HDAC3 inhibition | 1.41 ± 0.3 | 1.78 ± 0.06 |
Values are means of optical density measured at 420 nm ± SE. n = 4 independent cell experiments. ANOVA with post hoc Dunnett test,
P < 0.05 compared with DMSO group.
HDAC, histone deacetylase; TSA, trichostatin A; WST-1, 2-(4-iodophenyl)-3-(4-nitrophenyl)-5-(2,4-disulfophenyl)-2H-tetrazolium, monosodium salt.
For HDAC overexpression studies, cells were resuspended in Opti-MEM (Thermo Fisher Scientific) and seeded in 96 wells with linear polyethyleneimine transfection agent (25,000 mol wt; 33.75 mg/100 ml sterile water; Polysciences) and 125 ng of DNA [empty vector pcDNA3.1, flag-tagged human HDAC1 (#13820), or flag-tagged human HDAC4 (#30485); Addgene]. Four hours after transfection, 100 μl of complete media were added to the wells, and the next morning the media were replaced and WST-1 was added as described above. To a subset of samples, after 24 h of transfection, 500 nM TSA or 0.1% DMSO vehicle was added to the wells along with WST-1 and absorbance measured.
Scratch tests to measure proliferation/migration were performed in 70–80% confluent cells that were randomly assigned to the vehicle (0.1% DMSO), 300 nM MS275, or 500 nM TSA groups. Cells were grown in a 12-well plate, and using a 1-ml pipette tip, a scratch through the middle of each well was made. The media were replaced and included the vehicles or HDACi. Each well was then imaged with a CKX41 inverted light microscope affixed with a SC100 camera (Olympus, Tokyo, Japan) and, 24 h later, imaged again. The average width of each scratch was measured with ImageJ (the average of 10 measures across the scratch).
For all cell culture experiments, technical replicates were averaged per plate, per day. Biological replicates were repeated on different days with different passages (n = 3–5).
Analyses and statistics.
Kidney sections were scored by two blinded investigators. Interstitial fibrosis and brush border integrity were scored on a four-point scale as previously described (22), and the total number of dilated tubules and protein casts were counted per kidney section from each animal. To test for significance of the kidney scoring, acetylated histone H3, or quantitative real-time PCR, Kruskal-Wallis tests were performed because either the data were ordinal or variances were not equal among the groups. Because of two comparisons made in these data sets, α = 0.025, thus statistical significance was ascertained as P < 0.025. α-Smooth muscle actin (α-SMA), fibronectin, Sirius red-positive area, the number of Ki-67-positive nuclei, and integrated density of lysine-acetylated histone H3 nuclei were calculated using ImageJ (Fiji version 2.0.0-rc-69/1.52i). The proportion of HDAC4-positive PTs was counted by two blinded investigators independently, and statistical significance was determined by Kruskal-Wallis ANOVA followed by Dunn post hoc comparison. To test whether there was a significant effect of treatment and/or surgery, a two-way ANOVA with Tukey or Šídák multiple-comparison post hoc was performed. To test for a significant effect of HDACi either in vivo or in vitro, a one-way ANOVA with Dunnett post hoc was performed. In the aforementioned analyses, α = 0.05.
RESULTS
IRI affects HDAC localization and abundance in the kidney cortex.
As PT injury is a hallmark of AKI (54), we first performed immunohistochemical analyses determining the localization of the class I, II, and IV HDACs in the kidney cortex of male sham and bilateral IRI mice 72 h after ischemia. A summary of the localization of all of the cortical kidney HDACs is reported in Table 3, and representative images are in Figs. 1–4. The most significant changes observed were with HDAC1 (class I) and HDAC4 (class II). HDAC1 was expressed in the nucleus of every cell type in the cortex, and following IRI there was a significant doubling in HDAC1 abundance by 72 h of reperfusion, which exceeded sham levels (Fig. 1). Although HDAC2 was also expressed in the nuclei, no significant change in abundance was observed compared with sham at any time point (Fig. 2). HDAC4, although expressed at very low levels in the cortical tubules, immediately 1 h after IRI there was expression in the nuclei of the tubules and increased cytoplasmic expression (no longer evident in the nucleus) 24–72 h after IRI compared with sham levels (Fig. 3). Figure 4 is representative images of the other eight HDACs from sham and IRI mice after 72 h of reperfusion (Table 3). Thus, in the mouse kidney, there is expression of HDACs that is significantly affected by bilateral IRI, both in localization and abundance in the renal cortex.
Table 3.
Summary of the localization of the histone deacetylases in the cortex of the sham or IRI mice from Fig. 4
| Sham | 72-h IRI | |
|---|---|---|
| HDAC1 | Nucleus | Increased expression in nucleus |
| HDAC2 | Nucleus | Nucleus |
| HDAC3 | Nucleus, macula densa | Nucleus, macula densa |
| HDAC4 | Low expression | Increased in proximal tubules |
| HDAC5 | Low expression | Nuclei of glomeruli and tubules |
| HDAC6 | Low expression, some nuclei | Highly expressed in distal nephrons |
| HDAC7 | Proximal tubules | Proximal tubules |
| HDAC8 | Low expression | Intrarenal vessels |
| HDAC9 | Interstitial expression | Nuclei of glomeruli and tubules |
| HDAC10 | Low expression | Highly expressed in dilated tubules |
| HDAC11 | Low expression | Interstitial, near glomeruli |
HDAC, histone deacetylase; IRI, ischemia-reperfusion injury.
Fig. 1.

Time course in changes of histone deacetylase-1 (HDAC1) expression and abundance in the male mouse kidney cortex after 30 min of bilateral ischemia followed by 1–72 h of reperfusion (IRI) or sham surgery. A: HDAC1 was immunolocalized to the nuclei of the kidney cortex. Scale bar = 40 μm. B: representative Western blots of cortical lysates and HDAC1 abundance with β-actin and total protein (pr.; Coomassie blue-stained membranes) to demonstrate equal loading. C: relative quantification of HDAC1 cortical abundance normalized to 1-h sham samples. n = 3–4 Animals per time point. Two-factor ANOVA, *P < 0.05 compared with sham from Šídák multiple-comparison post hoc analysis. 50, 50-kDa molecular mass marker; PSxT, PSurgery×Time.
Fig. 4.
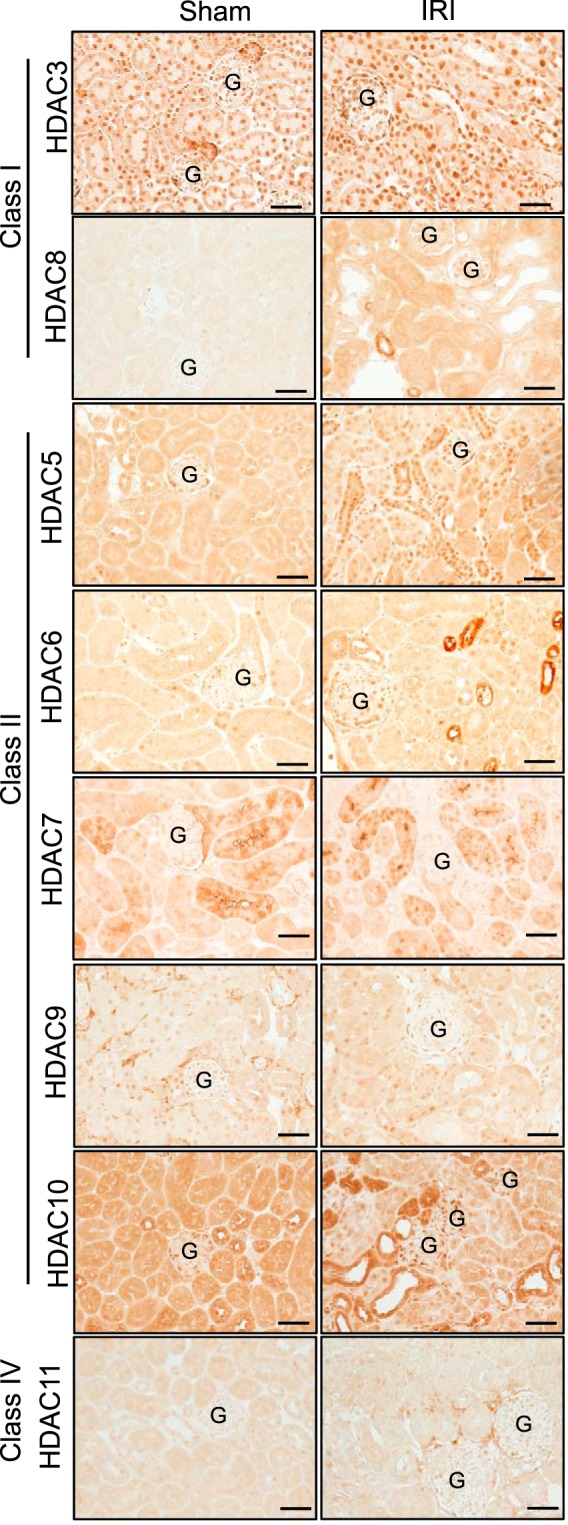
Male mouse kidney cortical expression of class I, II, and IV histone deacetylases (HDACs) 72 h after sham surgery or 30 min of bilateral ischemia (ischemia-reperfusion injury, IRI). G, glomerulus. Scale bar = 40 μm.
Fig. 2.

Time course in changes in histone deacetylase-2 (HDAC2) expression and abundance in the male mouse kidney cortex after 30 min of bilateral ischemia followed by 1–72 h of reperfusion (IRI) or sham surgery. A: HDAC2 was immunolocalized to the nuclei of the kidney cortex. Scale bar = 40 μm. B: representative Western blots of cortical lysates and HDAC2 abundance with β-actin and total protein (pr.; Coomassie blue-stained membranes) to demonstrate equal loading. C: relative quantification of HDAC2 cortical abundance normalized to 1-h sham samples. n = 3–4 Animals per time point. Two-factor ANOVA was reported. 50, 50-kDa molecular mass marker; PSxT, PSurgery×Time.
Fig. 3.

Time course in changes in histone deacetylase-4 (HDAC4) expression and abundance in the male mouse kidney cortex after 30 min of bilateral ischemia followed by 1–72 h of reperfusion (IRI) or sham surgery. A: HDAC4 was immunolocalized to the proximal tubule in the kidney cortex. Scale bar = 40 μm. B: representative Western blots of cortical lysates and HDAC4 abundance with β-actin and total protein (pr.; Coomassie blue-stained membranes) to demonstrate equal loading. C: relative quantification of HDAC4 cortical abundance normalized to 1-h sham samples. n = 3–4 Animals per time point. Two-factor ANOVA, *P < 0.05 compared with sham from Šídák multiple-comparison post hoc analysis. 50, 100, and 150, 50-, 100-, and 150-kDa molecular mass markers; PSxT, PSurgery×Time.
Confirmation of HDAC activation from IRI and in vivo HDAC blockade.
To determine whether these changes in HDAC expression and abundance resulted in HDAC activation following IRI, we determined histone H3 lysine acetylation intensity. A decrease in H3 acetylation represents HDAC activation, whereas an increase in H3 acetylation represents HDACi (38). Histone H3 lysine acetylation was globally reduced in the cortex of vehicle-treated mice, 72 h after IRI, compared with sham mice (P = 0.028; Fig. 5, A and B). Next, to determine whether HDAC activation contributes to IRI, we intervened by delivering in vivo the class I HDACi, MS275, the pan-HDACi, TSA, or vehicle 3 days before IRI surgery using i.p. osmotic minipump. MS275 or TSA significantly augmented histone H3 lysine acetylation compared with IRI vehicle (P = 0.035; Fig. 5, A and B). Furthermore, semiquantitative Western blot confirmed the immunohistochemical findings that in vivo HDACi of IRI mice resulted in higher acetylated histone H3 abundance compared with vehicle-treated IRI mice (P = 0.009; Fig. 5, C and D). However, HDACi treatment to sham mice did not further enhance the abundance of acetylated histone H3 (P = 0.5).
Fig. 5.

Male mouse kidney cortical and nuclear histone deacetylase (HDAC) activity as determined by histone H3 lysine acetylation. Mice were implanted with intraperitoneal osmotic minipumps to deliver continuously vehicle (acetic acid, pH > 5), MS275, the class I HDAC inhibitor (20 mg·kg−1·day−1), or trichostatin A (TSA; 1 mg·kg−1·day−1), the pan-HDAC inhibitor, 3 days before sham surgery or 27 min of bilateral ischemia (ischemia-reperfusion injury, IRI). Samples were obtained at 72 h of reperfusion. A: representative images of lysine-acetylated histone H3 expression in the kidney cortex from the IRI groups. B: average nuclear integrated density of lysine-acetylated histone H3 expression in the kidney cortex. IRI vehicle mice had significantly reduced H3 acetylation, suggesting activation of HDACs. In vivo treatment with MS275 or TSA maintained H3 acetylation similar to sham, suggesting inhibition of HDAC activity. C: representative Western blots of cortical lysates immunoreactive to lysine-acetylated histone H3 (Ac-H3) and total histone H3, with β-actin and total protein (Pr.; Coomassie blue-stained membranes) to demonstrate equal loading. D: summary of the densitometry of the level of acetylated histone H3. n = 4–7 Animals; See materials and methods for exact sample size per group. Kruskal-Wallis ANOVA, *P < 0.025 compared with sham from Dunn multiple-comparison post hoc analysis. G, glomerulus; K, thousand; MW, molecular weight (molecular mass in kilodaltons). Scale bar = 40 μm.
In vivo HDAC blockade during IRI resulted in reduced kidney function and damage.
In vivo HDACi had a small but statistically significant effect on plasma creatinine in sham-operated mice at 72-h sample time (sham vehicle, 0.078 ± 0.004 mg/dl; sham MS275, 0.11 ± 0.013 mg/dl; sham TSA, 0.12 ± 0.01 mg/dl; P = 0.02; Fig. 6). IRI of vehicle-treated mice had a significantly higher plasma creatinine compared with vehicle sham mice (IRI vehicle, 0.75 ± 0.29 mg/dl). Both HDACis resulted in a two to three times higher plasma creatinine compared with vehicle-treated mice [PSurgery < 0.01, PTreatment < 0.001, PSurgery×Treatment (PSxT) = 0.007; Fig. 6].
Fig. 6.
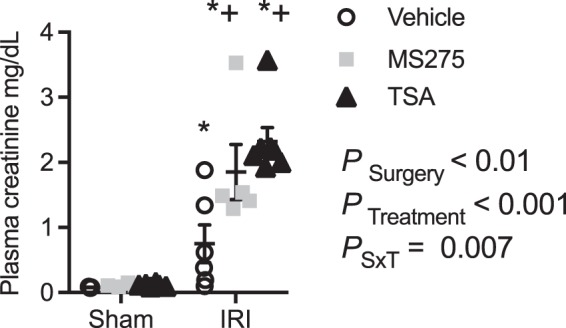
Effect of histone deacetylase inhibitors and ischemia-reperfusion injury (IRI) on plasma creatinine. Mice were implanted with intraperitoneal osmotic minipumps to deliver continuously vehicle (acetic acid, pH > 5), MS275, the class I histone deacetylase (HDAC) inhibitor (20 mg·kg−1·day−1), or trichostatin A (TSA; 1 mg·kg−1·day−1), the pan-HDAC inhibitor, 3 days before sham surgery or 27 min of bilateral ischemia (IRI). Samples were obtained at 72 h of reperfusion. n = 6 Animals. Two-factor ANOVA, *P < 0.05 compared with sham, +P < 0.05 compared with IRI vehicle from Šídák multiple-comparison post hoc analysis. PSxT, PSurgery×Treatment.
Histological analysis determined that in all sham treatments, regardless of intervention, there were minimal interstitial fibrosis, intact brush borders, and no dilated tubules or protein casts (Fig. 7). In IRI vehicle-treated mice, there was a small but significant increase in the interstitial fibrosis score compared with sham (Fig. 7B). This modest increase was significantly reduced by TSA, although it was still statistically significantly higher compared with sham (P = 0.043; Fig. 7B). In all IRI treatments, there was a significant reduction in brush border score compared with shams suggesting a loss of brush border integrity (Fig. 7C). IRI also resulted in a significant number of dilated tubules; however, it was not significantly different among the treatment groups (Fig. 7D). Protein casts were few in the IRI vehicle-treated mice, but there were significantly more in both the TSA and MS275 groups (P = 0.02; Fig. 7E). Kidney injury marker-1 (Kim-1; Refs. 15, 20) and neutrophil gelatinase-associated lipocalin (NGAL; Ref. 37) are markers of proximal tubule injury. As representative images show in Fig. 7F and quantified in Fig. 7, G and H, there was significant increase in abundance of Kim-1 and NGAL in the cortex of IRI mice. However, Kim-1 abundance was further significantly augmented in the MS275- and TSA-treated IRI mice (Fig. 7, F–H). NGAL abundance in HDACi-treated IRI mice was not statistically different from vehicle (P = 0.08).
Fig. 7.

Assessment of kidney damage from mice implanted with intraperitoneal osmotic minipumps to deliver continuously vehicle (acetic acid, pH > 5), MS275, the class I histone deacetylase (HDAC) inhibitor (20 mg·kg−1·day−1), or trichostatin A (TSA; 1 mg·kg−1·day−1), the pan-HDAC inhibitor, 3 days before sham surgery or 27 min of bilateral ischemia (ischemia-reperfusion injury, IRI). Samples were obtained at 72 h of reperfusion. A: representative histological images of cortical kidney sections stained with Gömöri trichrome. Shown are interstitial fibrosis score (B; 0 = none to 4 = 100%), brush border integrity score (C; 0 = 100% intact, 4 = 0% of proximal tubules have a brush border), number of dilated tubules per field (D), and kidney casts per field (E). No dilated tubules or protein casts were observed in any sham kidney cortex. Kruskal-Wallis ANOVA, *P < 0.05 compared with sham, +P < 0.05 compared with IRI vehicle from Dunn multiple-comparison post hoc analysis. F: representative Western blots for neutrophil gelatinase-associated lipocalin (NGAL) and kidney injury marker-1 (Kim-1) cortical abundance. Actin and total protein (Pr.; Coomassie blue-stained membranes) are provided to demonstrate equal loading. Summarized densitometries for cortical NGAL (G) and Kim-1 (H) abundance are shown. Box plots ± minimum and maximum are reported. n = 4–7 Animals; see materials and methods for exact sample size per group. Two-factor ANOVA, *P < 0.05 compared with sham, +P < 0.05 compared with IRI vehicle from Tukey post hoc analysis. AU, arbitrary units; MW, molecular weight (molecular mass in kilodaltons); PSxT, PSurgery×Treatment. Scale bar = 40 μm.
To investigate further the effect of IRI and HDACi on interstitial fibrosis of the kidney cortex, Sirius red staining of collagen and immunohistochemical localization of extracellular matrix proteins α-SMA and fibronectin were determined. There was a significant increase in the Sirius red interstitial staining of collagen in the IRI vehicle-treated mice compared with shams or both HDACi-treated groups (Fig. 8, A and B). There was minimal α-SMA (Fig. 8, C and D) or fibronectin (Fig. 8, E and F) expression in all sham groups of mice. IRI vehicle-treated mice had a significant increase in α-SMA or fibronectin expression in the tubulointerstitial area that was significantly prevented by both HDACi treatments (Fig. 8).
Fig. 8.

Kidney cortical interstitial collagen and extracellular matrix proteins from mice implanted with intraperitoneal osmotic minipumps to deliver continuously vehicle (acetic acid, pH > 5), MS275, the class I histone deacetylase (HDAC) inhibitor (20 mg·kg−1·day−1), or trichostatin A (TSA; 1 mg·kg−1·day−1), the pan-HDAC inhibitor, 3 days before sham surgery or 27 min of bilateral ischemia (ischemia-reperfusion injury, IRI). Samples were obtained at 72 h of reperfusion. Shown are Sirius red staining in the interstitium (A) and percentage of area positive for Sirius red (B). Also shown are expression of α-smooth muscle actin (α-SMA; C) and percentage of area positive for α-SMA (D). Expression of fibronectin (E) and percentage of area positive for fibronectin (F) are shown as well. Box plots ± minimum and maximum are reported. n = 4–7 Animals; see materials and methods for exact sample size per group. Two-factor ANOVA, *P < 0.05 compared with sham, +P < 0.05 compared with IRI vehicle from Tukey post hoc analysis. G, glomerulus; PSxT, PSurgery×Treatment. Scale bar = 40 μm.
In vivo HDACi effects on autophagy in the cortex.
Recent studies suggested that HDAC blockade induced autophagy and prevented kidney injury in the AKI model of cisplatin nephrotoxicity (32). To determine whether autophagy pathways were also significantly affected by continuous HDACi infusion, we measured autophagy marker expression in the cortex. Bilateral IRI led to a general 20–70% increase in autophagy marker expression in the cortex of vehicle-treated mice (Table 4, values are normalized to sham vehicle and reported as fold change of respective sham group). However, in the MS275-treated IRI mice, there was significantly lower expression of Becn1 (beclin-1) but significantly onefold higher expression of Atg7 compared with either vehicle or TSA-treated IRI mice. However, mRNA of other autophagy markers, Lc3 and Atg5, were similar among the IRI groups (Table 4).
Table 4.
Kidney cortex cDNA expression from ischemia-reperfusion injury mice relative to their sham controls
| Target | Vehicle | MS275 | TSA | ANOVA P Value |
|---|---|---|---|---|
| Lc3 | 0.9 (0.1) | 1.2 (0.4) | 1.0 (0.2) | 0.3 |
| Becn1 | 1.4 (0.2) | 1.0 (0.1*) | 1.3 (0.3) | 0.02 |
| Atg5 | 1.2 (0.2) | 1.3 (0.1) | 1.2 (0.1) | 0.5 |
| Atg7 | 1.7 (0.2) | 2.8 (0.6*) | 1.8 (0.4) | 0.01 |
Means (SD) are reported. ANOVA with post hoc Dunnett test,
P < 0.05 compared with vehicle group.
TSA, trichostatin A.
In vivo pan-HDACi, but not class I, significantly reduces proliferation.
To determine whether HDACi inhibit proliferation in the injured kidney, we determined the number of proliferating nuclei with anti-Ki-67 immunoreactivity. In all sham samples, there were 45 ± 3 Ki-67-positive nuclei per field (Fig. 9, A and B). In contrast, in the IRI vehicle group, we found on average 825 ± 49 Ki-67-positive nuclei per field. Likewise, in the MS275 group, there were 650 ± 48 Ki-67-positive nuclei per field (Fig. 9, A and B). However, the mice treated with TSA had significantly fewer Ki-67-positive nuclei, although still significantly more than sham controls (545 ± 88 positive nuclei per field; PSurgery < 0.001, PTreatment = 0.02, PSxT = 0.05; Fig. 9). In agreement with these findings, mRNA expression of the proliferating cell nuclear antigen was significantly increased in all IRI groups; however, it was significantly attenuated in the TSA-treated mice [fold change from respective sham: IRI, 2.9 ± 0.7 arbitrary units (AU); MS275, 2.7 ± 0.3 AU; TSA, 2.2 ± 0.2 AU; P = 0.02].
Fig. 9.

Assessment of putative regenerative pathways in the kidney cortex from mice implanted with intraperitoneal osmotic minipumps to deliver continuously vehicle (acetic acid, pH > 5), MS275, the class I histone deacetylase (HDAC) inhibitor (20 mg·kg−1·day−1), or trichostatin A (TSA; 1 mg·kg−1·day−1), the pan-HDAC inhibitor, 3 days before sham surgery or 27 min of bilateral ischemia (ischemia-reperfusion injury, IRI). Samples were obtained at 72 h of reperfusion. A: representative images of kidney cortex immunoreactive to anti-Ki-67, a marker of cells active in the cell cycle that may be proliferating. B: quantification of the number of (#) Ki-67-positive cells in the cortex. C: representative Westerns of the abundance of protein kinase B (Akt) and phosphorylated S473 Akt (pAKT) from cortical lysates. Actin and total protein (pr.; Coomassie blue-stained membranes) are provided to demonstrate equal loading. Densitometries of phosphorylated Akt (pAKT; D) and total Akt (E) are shown. F: ratio of phosphorylated to total Akt among the groups. Box plots ± minimum and maximum are reported. n = 4–7 Animals; see materials and methods for exact sample size per group. Two-factor ANOVA, *P < 0.05 compared with sham, +P < 0.05 compared with IRI vehicle from Tukey post hoc analysis. AU, arbitrary units; G, glomerulus; MW, molecular weight (molecular mass in kilodaltons); PSxT, PSurgery×Treatment. Scale bar = 40 μm.
Others have reported that the proportion of activated Akt (protein kinase B) is critical for kidney repair after AKI (60) and that class I HDACi prevents this regenerative pathway (57). To determine whether that was also true in our model, we measured total Akt, phosphorylated S473 Akt, and the proportion of phosphorylated/total Akt from renal cortical lysates. Seventy-two hours after bilateral ischemia, there was a significant increase in phosphorylated and total Akt in all treatment groups (PSurgery < 0.001; Fig. 9, C–F). In TSA-treated mice, both sham and IRI, there was a significant increase in phosphorylated and total Akt compared with vehicle or MS275-treated mice. However, the ratio of phosphorylated to total Akt was significantly different among the IRI groups; vehicle-treated mice had a greater proportion of phosphorylated Akt than HDACi IRI mice. Furthermore, MS275-treated mice had a significantly lower proportion of phosphorylated Akt compared with sham, whereas TSA-treated IRI mice were not significantly different from TSA sham mice (PSurgery = 0.6, PTreatment = 0.1, PSxT < 0.01; Fig. 9, C–F).
To determine whether HDACi reduce proliferation of the kidney epithelium, we performed a series of in vitro proliferation and migration experiments with immortalized renal epithelial or primary PT cells. As displayed in Table 5, only the pan-HDACi, TSA, significantly reduced proliferation of both rat renal epithelial cells (NRK-52E) and primary mouse PT. Moreover, 24 h after scratch test with rat renal epithelia cells, 92.3 ± 3% of the scratch had been repopulated by vehicle-treated cells (Fig. 10A). Likewise, in the MS275-treated cells, 80 ± 9% of the scratch was filled. However, in the TSA-treated cells, the scratch was only reduced by 19.7 ± 12%.
Fig. 10.

Proliferation and migration assays. A: representative proliferation/migration assays using the scratch test with immortalized rat kidney epithelial cells (NRK-52E) 24 h after original scratch. Thin lines outline the scratch. %Change from original scratch width is reported ±SE. n = 5 independent cell experiments. B: proliferation rate was determined by 2-(4-iodophenyl)-3-(4-nitrophenyl)-5-(2,4-disulfophenyl)-2H-tetrazolium, monosodium salt (WST-1) assay of cell overexpression of empty vector control, histone deacetylase-1 (HDAC1), or HDAC4 in rat kidney epithelial cells for 24 h. HDAC4 resulted in an increase in proliferation rate as determined by the WST-1 microplate assay. n = 3 independent cell experiments. ANOVA with post hoc Tukey test, *P < 0.05 compared with vector group. TSA, trichostatin A. Scale bar = 40 μm.
Because we observed a fourfold increase in PT HDAC4 expression after IRI (Fig. 3), we further wanted to determine whether specifically HDAC4 activity regulates PT proliferation. To determine whether MS275 or TSA significantly reduced the number of HDAC4-positive PTs, PTs were counted in a blinded fashion and the percentage of HDAC4-positive PTs was determined. PT HDAC4 abundance was not significantly reduced by in vivo MS275 or TSA administration (vehicle IRI, 60 ± 3.8% PTs per field; MS275 IRI, 62 ± 2.2%; TSA, 71 ± 2.4%). In fact, there was a small but significant increase in the proportion of HDAC4-positive PTs in the TSA-treated mice (Kruskal-Wallis ANOVA, P = 0.028; Dunn multiple-comparisons, TSA vs. vehicle or MS275, P = 0.1). Next, we transfected immortalized rat renal epithelial cells and primary human PT cells with empty vector control, HDAC1 (which was significantly increased 2-fold by IRI; Fig. 1), or HDAC4. In rat renal epithelial cells (Fig. 10B) and primary human PT (Fig. 11), overexpression of HDAC4 significantly increased proliferation compared with both control and HDAC1-expressing cells. Moreover, treatment of primary human PT with TSA for 24 h prevented the HDAC4-mediated increase in proliferation (PTransfection = 0.08, PTreatment = 0.4, PTransfection×Treatment = 0.004; Fig. 11).
Fig. 11.
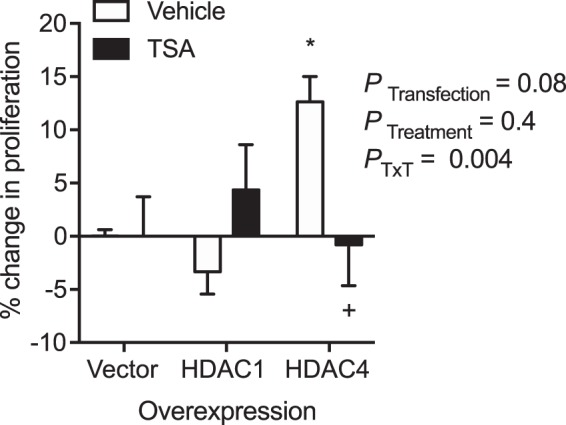
Proliferation of primary human proximal tubules. Cells were transfected with empty vector control, histone deacetylase-1 (HDAC1), or HDAC4 plasmids 24 h before incubation with vehicle (0.1% DMSO) or the pan-HDAC inhibitor, trichostatin A (TSA), and the 2-(4-iodophenyl)-3-(4-nitrophenyl)-5-(2,4-disulfophenyl)-2H-tetrazolium, monosodium salt proliferation reagent for 24 h. %Change from vector control is reported ±SE. n = 5 independent cell experiments for vehicle groups, n = 3 independent cell experiments for TSA groups. Two-factor ANOVA, *P < 0.05 compared with vector vehicle group. +P < 0.05 compared with HDAC4 vehicle group from Tukey post hoc analysis. PTxT, PTransfection×Treatment.
DISCUSSION
The zinc-dependent HDACs are a large family of enzymes expressed throughout the body, including the kidney. Although normally they function in the regulation of gene transcription and deacetylate lysine residues, resulting in changes in protein function, in disease states they can be deranged. The classic example is in T cell lymphoma where deranged HDAC activity is causative in this cancer (31). As such, there are four Food and Drug Administration-approved HDACi as treatment options (31). The beneficial effects of HDACi result in activation of anti-inflammatory, antifibrotic, and antiproliferative pathways, thus suggesting that these drugs may be repurposed for other diseases, including kidney diseases (11). Unfortunately, the adverse events reported with HDACi, such as hyponatremia, hypokalemia, edema, and changes in blood pressure (45, 46, 48), may be problematic for people with reduced kidney function. In fact, to date, studies testing HDACi in models of acute and chronic kidney disease have been conflicting, showing either a benefit (30, 32, 43, 47, 62) or potentiating damage (57). Thus, to understand better whether HDACi are appropriate for treating and/or even preventing kidney disease, we need to understand HDAC expression, distribution, and functions in the kidney.
Nuclear expression of HDAC1–3 in the kidney cortex confirmed pervious reports (43, 57). However, our study also identified nuclear expression of HDAC9 in uninjured kidneys and HDAC1–5 and -10 after kidney injury. These HDACs were activated in the nucleus because there was a significant reduction of histone H3 lysine acetylation in the IRI mice compared with both sham and in vivo HDACi groups. Thus bilateral IRI results in nuclear activation of class I and II HDACs.
Additional roles of HDACs in the regulation of lysine acetylation of nonhistone proteins are emerging. Acetylome studies from the rat kidney determined that >2,000 proteins are lysine-acetylated with significant expression in the cytosol and nonnuclear organelles (19, 34). These acetylated proteins are involved in biological pathways such as metabolism, actin cytoskeleton, regulation of apoptosis, oxidation-reduction status, and ion regulation (19, 34). We confirmed in this study nonnuclear expression of HDAC3 in the macula densa and HDAC7 in PTs of the uninjured kidney. In the male rat kidney, HDAC7 was reported in the cortical collecting duct, where it functions to deacetylate the epithelial sodium channel α-subunit and thereby promotes degradation of the channel via lysine ubiquitination pathways (5). Our reports of HDAC7 also in PTs, in both sham and injured kidneys, suggest it may affect nonnuclear protein lysine acetylation. Determination of the function of HDAC3 and HDAC7 in the uninjured/injured kidney requires further investigation.
Nonnuclear expression of HDAC6 in the distal tubules was found in our study. HDAC6 is predominantly cytosolic, and it is the key deacetylase to regulate α-tubulin acetylation (17) and binds misfolded proteins, facilitating their transport via the microtubules to the aggresome-autophagy pathway (7, 23, 42). Moreover, HDAC6 deacetylates the transcription factor EB, which in an acetylated state accumulates in the nucleus and leads to an upregulation of autophagy genes (4). Taken together, these data suggest that an upregulation of HDAC6 in the distal nephrons following IRI may inhibit autophagy, resulting in excess cell injury. In support of this hypothesis, previous studies have determined that in vivo inhibition of HDAC6 can attenuate kidney injury in 5/6 nephrectomy chronic kidney disease model (4) and rhabdomyolysis- (52) or cisplatin- (55) induced models of AKI.
Given these dynamic changes in HDAC abundance and activity in the renal cortex following severe bilateral IRI, we wanted to intervene with two different HDACi before injury, to determine whether they can provide any benefit to limit renal damage and/or maintain renal function. Unfortunately, in our study, mice treated with HDACi before bilateral ischemia and during reperfusion had a higher plasma creatinine and more kidney damage at our 72-h time end point. This is similar to findings reported in the folic acid and rhabdomyolysis models of AKI (57) but contradicts findings in cisplatin-induced nephrotoxicity (47), unilateral ureteral obstruction (UUO) models (24, 33, 39, 43), and unilateral IR (12, 30). It is important to note that the pharmacokinetics of HDACi are vastly different between humans and rodents. MS275 and TSA are quickly metabolized in rodents (t1/2 = 1 h; Refs. 1, 50) compared with humans (t1/2 = 50 h; Ref. 49). Often in rodent model studies, HDACi are delivered by single or daily i.p. injections (30, 32, 47, 62). Our study used i.p. osmotic minipumps to deliver a constant concentration of HDACi, to mimic pharmacokinetics of humans better, and this may partially explain the difference in findings among these studies. For example, in the cisplatin model of nephrotoxicity, when TSA was given via i.p. daily, there was a significant enhancement of tubular autophagy and renoprotection (32). In our study of bilateral IRI, we found increased autophagy marker expression in the cortex, but continuous HDACi did not further enhance autophagy, and in some cases the autophagy markers (e.g., beclin) were reduced compared with the vehicle-treated mice. Although beyond the scope of this study, comparisons among intermittent and continuous delivery of HDACi and understanding pharmacokinetics of HDACi will be critical in determining appropriate therapeutic dosing in various etiologies of AKI.
After injury, regenerative and repair intracellular signaling mechanisms are critical for healing. One example is activation of the kinase Akt (protein kinase B). Activation of Akt through phosphorylation results in further signaling cascades leading to regulation of cell cycle and cell survival (6). Akt phosphorylation is critical for renal repair in models of AKI, including IRI (51, 60). In our bilateral IRI study, continuous infusion of HDACi failed to increase the proportion of cortical phosphorylated Akt, similar to what was reported in the folic acid and rhabdomyolysis models (57). Furthermore, we determined that cells actively in the cell cycle and likely proliferating were significantly reduced in the TSA-treated mice. Given that we observed a significant increase in PT HDAC4 following IRI, and TSA inhibits HDAC4, we hypothesized that HDAC4 may regulate PT proliferation. Data with respect to HDAC4 in kidney physiology and disease are limited. In a mouse UUO model, HDAC1, -4, -5, -6, and -10 were upregulated in the kidney, and inhibition with a resveratrol metabolite (piceatannol) attenuated only HDAC4 and -5 upregulation (10). This attenuation of HDAC4 and -5 was associated with a decrease in renal fibrosis and decreased MAPK phosphorylation, but epithelial proliferation was not an end point in this study (10). From our study, within 1 h after ischemia, HDAC4 was highly expressed in the tubular epithelial nuclei followed by enhanced expression in the cytosol of PT. Manipulation of HDAC4 levels and activity in primary human PT resulted in alterations in proliferation rate. In our study, in vivo HDACi did not significantly affect PT HDAC4 expression but did significantly reduce proliferation. Thus our data point to a mechanism whereby bilateral IRI activates HDAC4, resulting in PT proliferation and perhaps repair.
Previously, it was reported that 48-h inhibition of class I HDACs with MS275 or knockdown of HDAC1, -3, or -8, but not HDAC2, from immortalized mouse renal PT cells resulted in reduced proliferation (56). In our cell culture studies with immortalized rat kidney epithelial cells and primary mouse PTs treated with a variety of class I HDAC inhibitors for 24 h, we did not observe a significant change in proliferation as determined by the WST-1 assay or scratch wound healing tests. In agreement with this finding, overexpression of HDAC1 failed to affect proliferation rate significantly in either rat or human PTs. These conflicting results again highlight that HDACi delivery and exposure time can significantly affect the end points measured in a study. A thorough investigation into this is warranted.
Although our HDACi mice had more kidney damage and higher plasma creatinine at 72 h, we did detect improvement in one injury marker, interstitial fibrosis. Although our bilateral model of IRI is not a robust fibrosis model, we did detect a reduction in interstitial fibrosis in HDACi-treated mice as determined by Gömöri trichrome staining scoring, Sirius red-positive area, and extracellular matrix protein expression of α-SMA and fibronectin. This is similar to results reported in a unilateral model, which presented reduced fibrosis with both TSA and MS275 even 30 days after injury (30). Reduced fibrosis with HDACi was also reported in the aristolochic acid (40), UUO (10, 24, 33, 35, 36), and Adriamycin (58) models of kidney injury. In the cortical interstitium after severe IRI, there was increased expression of HDAC1 and -11, suggesting that these HDACs may promote profibrotic pathways early after injury. Although the mechanisms of HDAC-mediated kidney interstitial fibrosis are still emerging, data by multiple investigators demonstrate two pathways: 1) HDACi-mediated reduced phosphorylation of Smad3 (33, 39, 43) and 2) HDACi-mediated reduction in proinflammatory cytokines (24, 33, 36, 39, 40). Inhibition of either or both of these pathways is associated with reduced interstitial fibrosis, α-SMA, fibronectin, and collagen expression and decreased transforming growth factor-β, monocyte chemoattractant protein-1, and colony-stimulating factor-1 (24, 33, 36, 39, 40, 43). Whether these are direct effects of deacetylase activity or indirect signaling pathways remains to be determined.
Although our study highlights mechanisms of HDAC1- and HDAC4-mediated injury and repair, we cannot exclude significant contribution to other mechanisms involving the other nine HDACs, as the majority of them had a significant change in expression following bilateral IRI. This is a clinically relevant finding given that reduced renal perfusion occurs during cardiac, vascular, and cardiopulmonary surgeries (14) and is associated with a postsurgery AKI incidence of 23–50% of patients (14, 16, 25, 41). Thus, to prevent inhibition of critical HDAC-mediated repair pathways best, isoform-selective HDACi may be beneficial and are currently an active area of drug development (63). Ensuring they are selective in vivo will be key to determine whether HDACi are a viable option for preventing or treating kidney diseases.
GRANTS
Research reported in this publication was supported by National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) under award no. K01-DK-105038 to K. A. Hyndman [a Pittman Scholar at University of Alabama at Birmingham (UAB)] and American Society of Nephrology Joseph A. Carlucci Research Fellowship to M. Kasztan. We acknowledge support from the UAB-University of California at San Diego O’Brien Core Center for Acute Kidney Injury Research (NIDDK Grant P30-DK-079337) for this project.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
K.A.H. conceived and designed research; K.A.H., M.K., L.D.M., and S.M.-P. performed experiments; K.A.H., M.K., and L.D.M. analyzed data; K.A.H. and M.K. interpreted results of experiments; K.A.H. and M.K. prepared figures; K.A.H. drafted manuscript; K.A.H., M.K., L.D.M., and S.M.-P. edited and revised manuscript; K.A.H., M.K., L.D.M., and S.M.-P. approved final version of manuscript.
REFERENCES
- 1.Acharya MR, Sparreboom A, Sausville EA, Conley BA, Doroshow JH, Venitz J, Figg WD. Interspecies differences in plasma protein binding of MS-275, a novel histone deacetylase inhibitor. Cancer Chemother Pharmacol 57: 275–281, 2006. doi: 10.1007/s00280-005-0058-8. [DOI] [PubMed] [Google Scholar]
- 2.Aufhauser DD Jr, Wang Z, Murken DR, Bhatti TR, Wang Y, Ge G, Redfield RR 3rd, Abt PL, Wang L, Svoronos N, Thomasson A, Reese PP, Hancock WW, Levine MH. Improved renal ischemia tolerance in females influences kidney transplantation outcomes. J Clin Invest 126: 1968–1977, 2016. doi: 10.1172/JCI84712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Boddu R, Fan C, Rangarajan S, Sunil B, Bolisetty S, Curtis LM. Unique sex- and age-dependent effects in protective pathways in acute kidney injury. Am J Physiol Renal Physiol 313: F740–F755, 2017. doi: 10.1152/ajprenal.00049.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brijmohan AS, Batchu SN, Majumder S, Alghamdi TA, Thieme K, McGaugh S, Liu Y, Advani SL, Bowskill BB, Kabir MG, Geldenhuys L, Siddiqi FS, Advani A. HDAC6 inhibition promotes transcription factor EB activation and is protective in experimental kidney disease. Front Pharmacol 9: 34, 2018. doi: 10.3389/fphar.2018.00034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Butler PL, Staruschenko A, Snyder PM. Acetylation stimulates the epithelial sodium channel by reducing its ubiquitination and degradation. J Biol Chem 290: 12497–12503, 2015. doi: 10.1074/jbc.M114.635540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cantley LC. The phosphoinositide 3-kinase pathway. Science 296: 1655–1657, 2002. doi: 10.1126/science.296.5573.1655. [DOI] [PubMed] [Google Scholar]
- 7.Cebotaru L, Liu Q, Yanda MK, Boinot C, Outeda P, Huso DL, Watnick T, Guggino WB, Cebotaru V. Inhibition of histone deacetylase 6 activity reduces cyst growth in polycystic kidney disease. Kidney Int 90: 90–99, 2016. doi: 10.1016/j.kint.2016.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen L, Lee JW, Chou CL, Nair AV, Battistone MA, Păunescu TG, Merkulova M, Breton S, Verlander JW, Wall SM, Brown D, Burg MB, Knepper MA. Transcriptomes of major renal collecting duct cell types in mouse identified by single-cell RNA-seq. Proc Natl Acad Sci USA 114: E9989–E9998, 2017. doi: 10.1073/pnas.1710964114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chertow GM, Levy EM, Hammermeister KE, Grover F, Daley J. Independent association between acute renal failure and mortality following cardiac surgery. Am J Med 104: 343–348, 1998. doi: 10.1016/S0002-9343(98)00058-8. [DOI] [PubMed] [Google Scholar]
- 10.Choi SY, Piao ZH, Jin L, Kim JH, Kim GR, Ryu Y, Lin MQ, Kim HS, Kee HJ, Jeong MH. Piceatannol attenuates renal fibrosis induced by unilateral ureteral obstruction via downregulation of histone deacetylase 4/5 or p38-MAPK signaling. PLoS One 11: e0167340, 2016. doi: 10.1371/journal.pone.0167340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chun P. Therapeutic effects of histone deacetylase inhibitors on kidney disease. Arch Pharm Res 41: 162–183, 2018. doi: 10.1007/s12272-017-0998-7. [DOI] [PubMed] [Google Scholar]
- 12.Cianciolo Cosentino C, Skrypnyk NI, Brilli LL, Chiba T, Novitskaya T, Woods C, West J, Korotchenko VN, McDermott L, Day BW, Davidson AJ, Harris RC, de Caestecker MP, Hukriede NA. Histone deacetylase inhibitor enhances recovery after AKI. J Am Soc Nephrol 24: 943–953, 2013. doi: 10.1681/ASN.2012111055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Coca SG, Singanamala S, Parikh CR. Chronic kidney disease after acute kidney injury: a systematic review and meta-analysis. Kidney Int 81: 442–448, 2012. doi: 10.1038/ki.2011.379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Evans RG, Lankadeva YR, Cochrane AD, Marino B, Iguchi N, Zhu MZ, Hood SG, Smith JA, Bellomo R, Gardiner BS, Lee CJ, Smith DW, May CN. Renal haemodynamics and oxygenation during and after cardiac surgery and cardiopulmonary bypass. Acta Physiol (Oxf) 222: e12995, 2018. doi: 10.1111/apha.12995. [DOI] [PubMed] [Google Scholar]
- 15.Han WK, Bailly V, Abichandani R, Thadhani R, Bonventre JV. Kidney injury molecule-1 (KIM-1): a novel biomarker for human renal proximal tubule injury. Kidney Int 62: 237–244, 2002. doi: 10.1046/j.1523-1755.2002.00433.x. [DOI] [PubMed] [Google Scholar]
- 16.Hu J, Chen R, Liu S, Yu X, Zou J, Ding X. Global incidence and outcomes of adult patients with acute kidney injury after cardiac surgery: a systematic review and meta-analysis. J Cardiothorac Vasc Anesth 30: 82–89, 2016. doi: 10.1053/j.jvca.2015.06.017. [DOI] [PubMed] [Google Scholar]
- 17.Hubbert C, Guardiola A, Shao R, Kawaguchi Y, Ito A, Nixon A, Yoshida M, Wang XF, Yao TP. HDAC6 is a microtubule-associated deacetylase. Nature 417: 455–458, 2002. doi: 10.1038/417455a. [DOI] [PubMed] [Google Scholar]
- 18.Hyndman KA, Mironova EV, Giani JF, Dugas C, Collins J, McDonough AA, Stockand JD, Pollock JS. Collecting duct nitric oxide synthase 1ß activation maintains sodium homeostasis during high sodium intake through suppression of aldosterone and renal angiotensin II pathways. J Am Heart Assoc 6: e006896, 2017. doi: 10.1161/JAHA.117.006896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hyndman KA, Yang CR, Jung HJ, Umejiego EN, Chou CL, Knepper MA. Proteomic determination of the lysine acetylome and phosphoproteome in the rat native inner medullary collecting duct. Physiol Genomics 50: 669–679, 2018. doi: 10.1152/physiolgenomics.00029.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ichimura T, Bonventre JV, Bailly V, Wei H, Hession CA, Cate RL, Sanicola M. Kidney injury molecule-1 (KIM-1), a putative epithelial cell adhesion molecule containing a novel immunoglobulin domain, is up-regulated in renal cells after injury. J Biol Chem 273: 4135–4142, 1998. doi: 10.1074/jbc.273.7.4135. [DOI] [PubMed] [Google Scholar]
- 21.Juan LJ, Shia WJ, Chen MH, Yang WM, Seto E, Lin YS, Wu CW. Histone deacetylases specifically down-regulate p53-dependent gene activation. J Biol Chem 275: 20436–20443, 2000. doi: 10.1074/jbc.M000202200. [DOI] [PubMed] [Google Scholar]
- 22.Kasztan M, Fox BM, Speed JS, De Miguel C, Gohar EY, Townes TM, Kutlar A, Pollock JS, Pollock DM. Long-term endothelin-A receptor antagonism provides robust renal protection in humanized sickle cell disease mice. J Am Soc Nephrol 28: 2443–2458, 2017. doi: 10.1681/ASN.2016070711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kawaguchi Y, Kovacs JJ, McLaurin A, Vance JM, Ito A, Yao TP. The deacetylase HDAC6 regulates aggresome formation and cell viability in response to misfolded protein stress. Cell 115: 727–738, 2003. doi: 10.1016/S0092-8674(03)00939-5. [DOI] [PubMed] [Google Scholar]
- 24.Kinugasa F, Noto T, Matsuoka H, Urano Y, Sudo Y, Takakura S, Mutoh S. Prevention of renal interstitial fibrosis via histone deacetylase inhibition in rats with unilateral ureteral obstruction. Transpl Immunol 23: 18–23, 2010. doi: 10.1016/j.trim.2010.02.003. [DOI] [PubMed] [Google Scholar]
- 25.Lagny MG, Jouret F, Koch JN, Blaffart F, Donneau AF, Albert A, Roediger L, Krzesinski JM, Defraigne JO. Incidence and outcomes of acute kidney injury after cardiac surgery using either criteria of the RIFLE classification. BMC Nephrol 16: 76, 2015. doi: 10.1186/s12882-015-0066-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lassnigg A, Schmid ER, Hiesmayr M, Falk C, Druml W, Bauer P, Schmidlin D. Impact of minimal increases in serum creatinine on outcome in patients after cardiothoracic surgery: do we have to revise current definitions of acute renal failure? Crit Care Med 36: 1129–1137, 2008. doi: 10.1097/CCM.0b013e318169181a. [DOI] [PubMed] [Google Scholar]
- 27.Lassnigg A, Schmidlin D, Mouhieddine M, Bachmann LM, Druml W, Bauer P, Hiesmayr M. Minimal changes of serum creatinine predict prognosis in patients after cardiothoracic surgery: a prospective cohort study. J Am Soc Nephrol 15: 1597–1605, 2004. doi: 10.1097/01.ASN.0000130340.93930.DD. [DOI] [PubMed] [Google Scholar]
- 28.Lee JW, Chou CL, Knepper MA. Deep sequencing in microdissected renal tubules identifies nephron segment-specific transcriptomes. J Am Soc Nephrol 26: 2669–2677, 2015. doi: 10.1681/ASN.2014111067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Leus NG, van der Wouden PE, van den Bosch T, Hooghiemstra WT, Ourailidou ME, Kistemaker LE, Bischoff R, Gosens R, Haisma HJ, Dekker FJ. HDAC 3-selective inhibitor RGFP966 demonstrates anti-inflammatory properties in RAW 264.7 macrophages and mouse precision-cut lung slices by attenuating NF-κB p65 transcriptional activity. Biochem Pharmacol 108: 58–74, 2016. doi: 10.1016/j.bcp.2016.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Levine MH, Wang Z, Bhatti TR, Wang Y, Aufhauser DD, McNeal S, Liu Y, Cheraghlou S, Han R, Wang L, Hancock WW. Class-specific histone/protein deacetylase inhibition protects against renal ischemia reperfusion injury and fibrosis formation. Am J Transplant 15: 965–973, 2015. doi: 10.1111/ajt.13106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li Y, Seto E. HDACs and HDAC inhibitors in cancer development and therapy. Cold Spring Harb Perspect Med 6: a026831, 2016. doi: 10.1101/cshperspect.a026831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liu J, Livingston MJ, Dong G, Tang C, Su Y, Wu G, Yin XM, Dong Z. Histone deacetylase inhibitors protect against cisplatin-induced acute kidney injury by activating autophagy in proximal tubular cells. Cell Death Dis 9: 322, 2018. doi: 10.1038/s41419-018-0374-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liu N, He S, Ma L, Ponnusamy M, Tang J, Tolbert E, Bayliss G, Zhao TC, Yan H, Zhuang S. Blocking the class I histone deacetylase ameliorates renal fibrosis and inhibits renal fibroblast activation via modulating TGF-beta and EGFR signaling. PLoS One 8: e54001, 2013. doi: 10.1371/journal.pone.0054001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lundby A, Lage K, Weinert BT, Bekker-Jensen DB, Secher A, Skovgaard T, Kelstrup CD, Dmytriyev A, Choudhary C, Lundby C, Olsen JV. Proteomic analysis of lysine acetylation sites in rat tissues reveals organ specificity and subcellular patterns. Cell Rep 2: 419–431, 2012. doi: 10.1016/j.celrep.2012.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Manson SR, Song JB, Hruska KA, Austin PF. HDAC dependent transcriptional repression of Bmp-7 potentiates TGF-β mediated renal fibrosis in obstructive uropathy. J Urol 191: 242–252, 2014. doi: 10.1016/j.juro.2013.06.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Marumo T, Hishikawa K, Yoshikawa M, Hirahashi J, Kawachi S, Fujita T. Histone deacetylase modulates the proinflammatory and -fibrotic changes in tubulointerstitial injury. Am J Physiol Renal Physiol 298: F133–F141, 2010. doi: 10.1152/ajprenal.00400.2009. [DOI] [PubMed] [Google Scholar]
- 37.Mishra J, Ma Q, Prada A, Mitsnefes M, Zahedi K, Yang J, Barasch J, Devarajan P. Identification of neutrophil gelatinase-associated lipocalin as a novel early urinary biomarker for ischemic renal injury. J Am Soc Nephrol 14: 2534–2543, 2003. doi: 10.1097/01.ASN.0000088027.54400.C6. [DOI] [PubMed] [Google Scholar]
- 38.Morales V, Giamarchi C, Chailleux C, Moro F, Marsaud V, Le Ricousse S, Richard-Foy H. Chromatin structure and dynamics: functional implications. Biochimie 83: 1029–1039, 2001. doi: 10.1016/S0300-9084(01)01347-5. [DOI] [PubMed] [Google Scholar]
- 39.Nguyễn-Thanh T, Kim D, Lee S, Kim W, Park SK, Kang KP. Inhibition of histone deacetylase 1 ameliorates renal tubulointerstitial fibrosis via modulation of inflammation and extracellular matrix gene transcription in mice. Int J Mol Med 41: 95–106, 2018. doi: 10.3892/ijmm.2017.3218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Novitskaya T, McDermott L, Zhang KX, Chiba T, Paueksakon P, Hukriede NA, de Caestecker MP. A PTBA small molecule enhances recovery and reduces postinjury fibrosis after aristolochic acid-induced kidney injury. Am J Physiol Renal Physiol 306: F496–F504, 2014. doi: 10.1152/ajprenal.00534.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.O’Neal JB, Shaw AD, Billings FT 4th. Acute kidney injury following cardiac surgery: current understanding and future directions. Crit Care 20: 187, 2016. doi: 10.1186/s13054-016-1352-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ouyang H, Ali YO, Ravichandran M, Dong A, Qiu W, MacKenzie F, Dhe-Paganon S, Arrowsmith CH, Zhai RG. Protein aggregates are recruited to aggresome by histone deacetylase 6 via unanchored ubiquitin C termini. J Biol Chem 287: 2317–2327, 2012. doi: 10.1074/jbc.M111.273730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pang M, Kothapally J, Mao H, Tolbert E, Ponnusamy M, Chin YE, Zhuang S. Inhibition of histone deacetylase activity attenuates renal fibroblast activation and interstitial fibrosis in obstructive nephropathy. Am J Physiol Renal Physiol 297: F996–F1005, 2009. doi: 10.1152/ajprenal.00282.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pavkov ME, Harding JL, Burrows NR. Trends in hospitalizations for acute kidney injury – United States, 2000–2014. MMWR Morb Mortal Wkly Rep 67: 289–293, 2018. doi: 10.15585/mmwr.mm6710a2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pili R, Salumbides B, Zhao M, Altiok S, Qian D, Zwiebel J, Carducci MA, Rudek MA. Phase I study of the histone deacetylase inhibitor entinostat in combination with 13-cis retinoic acid in patients with solid tumours. Br J Cancer 106: 77–84, 2012. doi: 10.1038/bjc.2011.527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ramalingam SS, Maitland ML, Frankel P, Argiris AE, Koczywas M, Gitlitz B, Thomas S, Espinoza-Delgado I, Vokes EE, Gandara DR, Belani CP. Carboplatin and paclitaxel in combination with either vorinostat or placebo for first-line therapy of advanced non-small-cell lung cancer. J Clin Oncol 28: 56–62, 2010. doi: 10.1200/JCO.2009.24.9094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ranganathan P, Hamad R, Mohamed R, Jayakumar C, Muthusamy T, Ramesh G. Histone deacetylase-mediated silencing of AMWAP expression contributes to cisplatin nephrotoxicity. Kidney Int 89: 317–326, 2016. doi: 10.1038/ki.2015.326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ree AH, Dueland S, Folkvord S, Hole KH, Seierstad T, Johansen M, Abrahamsen TW, Flatmark K. Vorinostat, a histone deacetylase inhibitor, combined with pelvic palliative radiotherapy for gastrointestinal carcinoma: the Pelvic Radiation and Vorinostat (PRAVO) phase 1 study. Lancet Oncol 11: 459–464, 2010. doi: 10.1016/S1470-2045(10)70058-9. [DOI] [PubMed] [Google Scholar]
- 49.Ryan QC, Headlee D, Acharya M, Sparreboom A, Trepel JB, Ye J, Figg WD, Hwang K, Chung EJ, Murgo A, Melillo G, Elsayed Y, Monga M, Kalnitskiy M, Zwiebel J, Sausville EA. Phase I and pharmacokinetic study of MS-275, a histone deacetylase inhibitor, in patients with advanced and refractory solid tumors or lymphoma. J Clin Oncol 23: 3912–3922, 2005. doi: 10.1200/JCO.2005.02.188. [DOI] [PubMed] [Google Scholar]
- 50.Sanderson L, Taylor GW, Aboagye EO, Alao JP, Latigo JR, Coombes RC, Vigushin DM. Plasma pharmacokinetics and metabolism of the histone deacetylase inhibitor trichostatin A after intraperitoneal administration to mice. Drug Metab Dispos 32: 1132–1138, 2004. doi: 10.1124/dmd.104.000638. [DOI] [PubMed] [Google Scholar]
- 51.Sharples EJ, Patel N, Brown P, Stewart K, Mota-Philipe H, Sheaff M, Kieswich J, Allen D, Harwood S, Raftery M, Thiemermann C, Yaqoob MM. Erythropoietin protects the kidney against the injury and dysfunction caused by ischemia-reperfusion. J Am Soc Nephrol 15: 2115–2124, 2004. doi: 10.1097/01.ASN.0000135059.67385.5D. [DOI] [PubMed] [Google Scholar]
- 52.Shi Y, Xu L, Tang J, Fang L, Ma S, Ma X, Nie J, Pi X, Qiu A, Zhuang S, Liu N. Inhibition of HDAC6 protects against rhabdomyolysis-induced acute kidney injury. Am J Physiol Renal Physiol 312: F502–F515, 2017. doi: 10.1152/ajprenal.00546.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Takahashi N, Boysen G, Li F, Li Y, Swenberg JA. Tandem mass spectrometry measurements of creatinine in mouse plasma and urine for determining glomerular filtration rate. Kidney Int 71: 266–271, 2007. doi: 10.1038/sj.ki.5002033. [DOI] [PubMed] [Google Scholar]
- 54.Takaori K, Nakamura J, Yamamoto S, Nakata H, Sato Y, Takase M, Nameta M, Yamamoto T, Economides AN, Kohno K, Haga H, Sharma K, Yanagita M. Severity and frequency of proximal tubule injury determines renal prognosis. J Am Soc Nephrol 27: 2393–2406, 2016. doi: 10.1681/ASN.2015060647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tang J, Shi Y, Liu N, Xu L, Zang X, Li P, Zhang J, Zheng X, Qiu A, Zhuang S. Blockade of histone deacetylase 6 protects against cisplatin-induced acute kidney injury. Clin Sci (Lond) 132: 339–359, 2018. doi: 10.1042/CS20171417. [DOI] [PubMed] [Google Scholar]
- 56.Tang J, Yan Y, Zhao TC, Bayliss G, Yan H, Zhuang S. Class I histone deacetylase activity is required for proliferation of renal epithelial cells. Am J Physiol Renal Physiol 305: F244–F254, 2013. doi: 10.1152/ajprenal.00126.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tang J, Yan Y, Zhao TC, Gong R, Bayliss G, Yan H, Zhuang S. Class I HDAC activity is required for renal protection and regeneration after acute kidney injury. Am J Physiol Renal Physiol 307: F303–F316, 2014. doi: 10.1152/ajprenal.00102.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Van Beneden K, Geers C, Pauwels M, Mannaerts I, Verbeelen D, van Grunsven LA, Van den Branden C. Valproic acid attenuates proteinuria and kidney injury. J Am Soc Nephrol 22: 1863–1875, 2011. doi: 10.1681/ASN.2010111196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wang X, Spandidos A, Wang H, Seed B. PrimerBank: a PCR primer database for quantitative gene expression analysis, 2012 update. Nucleic Acids Res 40: D1144–D1149, 2012. doi: 10.1093/nar/gkr1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Xie L, Zheng X, Qin J, Chen Z, Jin Y, Ding W. Role of PI3-kinase/Akt signalling pathway in renal function and cell proliferation after renal ischaemia/reperfusion injury in mice. Nephrology (Carlton) 11: 207–212, 2006. doi: 10.1111/j.1440-1797.2006.00558.x. [DOI] [PubMed] [Google Scholar]
- 61.Yu MJ, Miller RL, Uawithya P, Rinschen MM, Khositseth S, Braucht DW, Chou CL, Pisitkun T, Nelson RD, Knepper MA. Systems-level analysis of cell-specific AQP2 gene expression in renal collecting duct. Proc Natl Acad Sci USA 106: 2441–2446, 2009. doi: 10.1073/pnas.0813002106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zheng Q, Liu W, Liu Z, Zhao H, Han X, Zhao M. Valproic acid protects septic mice from renal injury by reducing the inflammatory response. J Surg Res 192: 163–169, 2014. doi: 10.1016/j.jss.2014.05.030. [DOI] [PubMed] [Google Scholar]
- 63.Zwergel C, Valente S, Jacob C, Mai A. Emerging approaches for histone deacetylase inhibitor drug discovery. Expert Opin Drug Discov 10: 599–613, 2015. doi: 10.1517/17460441.2015.1038236. [DOI] [PubMed] [Google Scholar]