

Abstract
Tight regulation of K+ balance is fundamental for normal physiology. Reduced dietary K+ intake, which is common in Western diets, often leads to hypokalemia and associated cardiovascular- and kidney-related pathologies. The distal nephron, and, specifically, the collecting duct (CD), is the major site of controlled K+ reabsorption via H+-K+-ATPase in the state of dietary K+ deficiency. We (Mamenko MV, Boukelmoune N, Tomilin VN, Zaika OL, Jensen VB, O'Neil RG, Pochynyuk OM. Kidney Int 91: 1398–1409, 2017) have previously demonstrated that the transient receptor potential vanilloid type 4 (TRPV4) Ca2+ channel, abundantly expressed in the CD, contributes to renal K+ handling by promoting flow-induced K+ secretion. Here, we investigated a potential role of TRPV4 in controlling H+-K+-ATPase-dependent K+ reabsorption in the CD. Treatment with a K+-deficient diet (<0.01% K+) for 7 days reduced serum K+ levels in wild-type (WT) mice from 4.3 ± 0.2 to 3.3 ± 0.2 mM but not in TRPV4−/− mice (4.3 ± 0.1 and 4.2 ± 0.3 mM, respectively). Furthermore, we detected a significant reduction in 24-h urinary K+ levels in TRPV4−/− compared with WT mice upon switching to K+-deficient diet. TRPV4−/− animals also had significantly more acidic urine on a low-K+ diet, but not on a regular (0.9% K+) or high-K+ (5% K+) diet, which is consistent with increased H+-K+-ATPase activity. Moreover, we detected a greatly accelerated H+-K+-ATPase-dependent intracellular pH extrusion in freshly isolated CDs from TRPV4−/− compared with WT mice fed a K+-deficient diet. Overall, our results demonstrate a novel kaliuretic role of TRPV4 by inhibiting H+-K+-ATPase-dependent K+ reabsorption in the CD. We propose that TRPV4 inhibition could be a novel strategy to manage certain hypokalemic states in clinical settings.
Keywords: collecting duct, intracellular pH, intercalated cells, K+ transport, principal cells
INTRODUCTION
K+ is a predominant intracellular cation that is essential for a wide array of cellular processes, such as setting of resting membrane voltage, generation of the action potential, muscle contraction, etc. (31). Since only a tiny fraction of the whole body K+ is present in extracellular compartments, maintaining plasma K+ within a narrow limit of 3.5–5.0 mM is essential for normal physiology during day-to-day variations of dietary K+ intake (52). Both hyperkalemia and hypokalemia have detrimental pathophysiological consequences for human health, including arrhythmias such as ventricular fibrillation, chronic kidney disease, blood pressure abnormalities, and a greatly increased risk of mortality (14, 18, 47).
The kidney is critical for maintaining systemic K+ homeostasis by eliminating the bulk of ingested K+ in the normal K+ replete state (31). The distal segments of renal nephron, including the connecting tubule (CNT) and collecting duct (CD), are the major sites of controlled K+ transport (16, 49). Apically localized renal outer medullary K+ channels and large-conductance (BK) channels mediate K+ secretion in the presence of elevated K+ intake, whereas H+-K+-ATPase is the dominant pathway of K+ reabsorption in the state of systemic K+ deficiency (13, 15). H+-K+-ATPase hydrolyzes ATP to move K+ against the electrochemical gradient at the expense of proton secretion into the tubular lumen. Two H+-K+-ATPase isoforms, having different catalytic α-subunits (α1 or gastric and α2 or nongastric/colonic), are known to be expressed in intercalated (similarly in both A- and B-types) and, to a lesser extent, in principal cells of the CNT and CD (1, 2, 9, 20, 48). H+-K+-ATPase expression is augmented in response to dietary K+ restriction (1, 2, 19). Mice with genetic deletion of both isoforms demonstrate notable disturbances in K+ and acid-base balance (12), suggesting the physiological relevance of H+-K+-ATPase for kidney function. However, signaling mechanisms controlling H+-K+-ATPase expression and activity remain largely unexplored.
Interestingly, both increases and decreases in dietary K+ intake lead to a notable elevation of tubular flow in the distal nephron (36, 51). Changes in tubular flow rate, by raising intracellular Ca2+ concentration ([Ca2+]i), can modulate numerous physiologically relevant processes in epithelial cells, including transport of water and electrolytes, proliferation, polarization, etc. (34, 40). Transient receptor potential vanilloid type 4 (TRPV4) is a mechanosensitive Ca2+-permeable channel activated in response to increased flow and changes in osmolarity (8, 25, 30, 33). TRPV4 is predominantly expressed in the distal nephron, where its activity is indispensable for flow-induced [Ca2+]i responses in freshly isolated split-opened CNTs/CDs of rodents and in primary cultures of human distal nephron cells (3, 46, 53). Our laboratory recently demonstrated that TRPV4 is necessary for stimulation of flow-induced K+ secretion via BK channels with TRPV4 deficiency, leading to renal K+ retention and hyperkalemia in the presence of high K+ intake (27). However, the role of renal TRPV4 in the state of systemic K+ deficiency remains unknown.
In the present study, we demonstrated the functional interplay between TRPV4 and H+-K+-ATPase activity in the distal nephron. We found that TRPV4 deletion increased H+-K+-ATPase-dependent proton extrusion into the tubular lumen in both intercalated and principal cells of the CD. This augmented K+ reabsorption exhibited as an accelerated reduction of urinary K+ excretion at the expense of urinary acidification. At the systemic level, TRPV4−/− mice were able to maintain a normal plasma K+ level in the presence of systemic K+ deficiency, whereas wild-type (WT) mice developed hypokalemia. Overall, our results suggest a novel role of TRPV4 in inhibition of K+ reabsorption via H+-K+-ATPase in the distal nephron.
MATERIALS AND METHODS
Reagents and animals.
All chemicals and materials were from Sigma (St. Louis, MO), VWR (Radnor, PA), Fisher (Waltham, MA), and Tocris (Ellisville, MO), unless noted otherwise, and were at least of reagent grade. Animal use and welfare adhered to the National Institutes of Health Guide for the Care and Use of Laboratory Animals following protocols reviewed and approved by the Animal Care and Use Committees of the University of Texas Health Science Center at Houston. For experiments, 6- to 10-wk-old C57BL/6 (WT) and TRPV4−/− (C57BL/6 background) mice were used. The generation of TRPV4−/− mice was as previously described (21).
Systemic measurements.
Mice were acclimated for few days in metabolic cages (3600M021, Techniplast, West Chester, PA) with free access to water and a standard (regular) rodent chow (0.9% K+, TD.7012, Envigo, Madison, WI). After acclimation, baseline measurements of food/water intake and 24-h urinary volume were performed, as previously described by our laboratory (5, 27). Mice were challenged further with a K+-deficient diet (<0.01% K+, TD.88238, Envigo) or a high-K+ diet (5% K+, TD 150699, Envigo) for 2 consecutive days or longer, depending on the experimental design. Urinary K+ concentration was measured using Jenway PFP7 Flame photometer (Bibby Scientific, Burlington, NJ). To estimate the amount of renal K+ excretion, urinary K+ concentration was normalized on the 24-h urine volume. Urinary pH was measured in fresh spot urine samples or after 24-h urine collections in metabolic cages using a MI-410 pH microelectrode (Microelectrodes, Bedford, NH). To minimize contribution of the circadian rhythms, spot urine collections for measuring pH were conducted around 11 AM. As necessary for the experimental design, the TRPV4 antagonist HC-067047 was added to the drinking water at a concentration of 0.075 mg/kg body wt, starting from day 0 (regular K+ diet). Blood samples (~500 μl) were taken by terminal cardiac puncture in anesthetized animals. Serum was separated by centrifugation at 1,300 g in Vacutainer Plus SST plastic tubes with clot activator and gel for serum separation (product no. 367988, BD, Franklin Lakes, NJ).
Isolation of individual CDs.
The procedure for the isolation of CDs from WT and TRPV4−/− mice suitable for fluorescent-based intracellular pH (pHi) measurements closely followed the protocols previously published by our group for patch clamping and intracellular Ca2+ imaging (26, 28). Kidneys were cut into thin slices (<1 mm) with slices placed into an ice-cold bath solution containing (in mM) 150 NaCl, 5 KCl, 1 CaCl2, 2 MgCl2, 5 glucose, and 10 HEPES (pH 7.35). CDs were visually identified by their morphological features (pale color, coarse surface, and, in some cases, bifurcations) and were mechanically isolated from kidney slices by microdissection using watchmaker forceps under a stereomicroscope. Isolated CDs were attached to 5 × 5-mm coverglasses coated with poly-l-lysine. A coverglass containing a CD was placed in a perfusion chamber mounted on an inverted Nikon Eclipse Ti microscope and perfused with the aforementioned bath solution at room temperature. CDs were split opened with two sharpened micropipettes, controlled with different micromanipulators, to gain access to the apical membrane. CDs were used within 2 h of isolation.
pHi measurements.
Split-opened CDs were loaded with 2′,7′-bis-(2-carboxyethyl)-5-(6)-carboxyfluorescein (BCECF) by incubation with 15 μM BCECF-AM in the bath solution for 40 min at room temperature followed by a washout with the bath solution for an additional 10 min. CDs were placed in an open-top imaging study chamber (RC-26GLP, Warner Instruments, Hamden, CT) with a bottom coverslip viewing window, and the chamber was attached to the microscope stage of a Nikon Ti-S Wide-Field Fluorescence Imaging System (Nikon Instruments, Melville, NY) integrated with Lambda XL light source (Sutter Instrument, Novato, CA) and QIClick 1.4 megapixel monochrome charge-coupled device camera (QImaging, Surrey, BC, Canada) via NIS Elements 4.3 imaging software (Nikon Instruments). Cells were imaged with a ×40 Nikon Super Fluor objective, and regions of interest were drawn for individual cells. The BCECF fluorescence intensity ratio was determined by excitation at 495 and 440 nm and calculating the ratio of the emission intensities at 520 nm every 5 s. Experiments were performed under permanent perfusion of a solution containing (in mM) 150 NaCl, 5 KCl, 1 CaCl2, 2 MgCl2, 5 glucose, and 10 HEPES at rate of 1.5 ml/min. Bafilomycin (100 nM, Alfa Aesar, Ward Hill, MA) and 5-(N-ethyl-N-isopropyl)amiloride (EIPA; 10 µM) were added throughout the experiments to block the activity of H+-ATPase and Na+/H+ exchangers, respectively. No significant BCECF bleaching was detected during the timeline of experiments. The changes in the ratio were converted into changes in pHi by performing a calibration in high-K+ solutions (145 mM KCl) with predefined pH (6.0, 7.0, and 8.0, adjusted by HCl and KOH, respectively) in the presence of 15 µM nigericin, as originally described (44). At least six individual CDs from at least three mice were used for each experimental set. For intracellular acidification, a solution containing (in mM) 40 NH4Cl, 110 NaCl, 5 KCl, 1 CaCl2, 2 MgCl2, 5 glucose, and 10 HEPES was applied to the recording chamber. H+-K+-ATPase activity was calculated as a linear slope of the initial pHi recovery rate from the lowest pHi values for each cell after removal of the acidification pulse, similarly to previously described procedures (22).
Immunofluorescent microscopy.
After pHi measurements, split-opened CDs were fixed with 10% neutral buffer formalin for 20 min at room temperature. After fixation, samples were permeabilized by the addition of 0.1% Triton in PBS for 5 min and washed in PBS three times for 5 min. Nonspecific staining was blocked with 10% normal goat serum (Jackson Immunoresearch) in PBS for 30 min at room temperature. After being washed with PBS (3 times for 5 min), samples were incubated for 1.5 h at room temperature in the dark with anti-aquaporin 2 (AQP2) labeled with ATTO-550 (1:100 dilution, Alomone Laboratories) in 1% serum + 0.1% Triton in PBS. After being washed with PBS (3 times for 5 min), samples were stained with 4′,6-diamidino-2-phenylindole (300 nM concentration, Calbiochem, San Diego, CA) to visualize nuclei. Samples were dehydrated and mounted with a permanent mounting media (Thermo Scientific, Pittsburgh, PA). Labeled tubules were examined with an inverted Nikon Eclipse Ti fluorescent microscope using a ×40 Plan-Fluor oil-immersion (1.3 numerical aperture) objective. Samples were excited with 405- and 561-nm laser diodes, and emission was captured with a 16-bit Cool SNAP HQ2 camera (Photometrics) interfaced to a PC running NIS Elements software.
Data analysis.
All summarized data are reported as means ± SE. Statistical comparisons were made using one-way ANOVA with a post hoc Tukey test or one-way repeated-measures ANOVA with a post hoc Bonferroni test (for paired experiments within the same group). P values of <0.05 were considered significant.
RESULTS
Previous experimental evidence demonstrated that the mechanosensitive TRPV4 channel is abundantly expressed in the distal renal nephron, where it serves as a critical determinant of flow-induced [Ca2+]i elevations as well as basal [Ca2+]i levels (3, 27). TRPV4-mediated Ca2+ influx promotes K+ secretion during high K+ intake, with its disruption leading to a disturbed systemic K+ balance (27). In the present study, we investigated the significance of TRPV4 for renal K+ excretion in the state of K+ deficiency, which is known to increase tubular flow in the distal nephron (7, 36).
We found no differences in serum K+ levels between age-matched WT and TRPV4−/− mice fed a regular K+ diet (0.9% K+). Mean values were 4.3 ± 0.2 and 4.3 ± 0.1 mM, respectively (Fig. 1). In contrast, 1-wk treatment with a K+-deficient (<0.01% K+) diet resulted in hypokalemia of 3.3 ± 0.2 mM in WT mice, whereas TRPV4−/− mice managed to remain normokalemic at 4.2 ± 0.3 mM (Fig. 1). In the state of K+ deficiency, the kidney plays a central role in limiting K+ excretion in urine to maintain systemic K+ balance. Thus, we next used metabolic cage experiment to compare urinary K+ levels in WT and TRPV4−/− mice (Fig. 2). There were no significant difference in 24-h urinary K+ levels between WT and TRPV4−/− mice and between male and female knockout mice after acclimation on a regular K+ regimen. Urinary K+ fell progressively during the tested first and second days when animals were fed a K+-deficient (<0.01% K+) diet. However, the rate of adaptation to the state of K+ deficiency was much accelerated similarly in both male and female TRPV4−/− mice compared with WT mice, suggestive of augmented renal K+ conservation (Fig. 2). Concomitant treatment of WT mice with a TRPV4 antagonist, HC-067047 (0.075 mg/kg body wt, estimated final concentration of 4 µM), also decreased urinary K+ levels, although the magnitude of the effect was considerably smaller compared with that observed in knockout mice (Fig. 2). Food and water intake were similar between WT and TRPV4−/− mice during all tested conditions (Fig. 3, A and B). There were also no differences in whole body weight in the experimental animals (Fig. 3C). As expected, urine production was notably increased in response to a K+-deficient diet, although we did not detect any significant differences between the respective groups (Fig. 3D). Altogether, these results suggest that the observed renal K+ retention in TRPV4−/− mice is most likely a manifestation of the augmented renal tubule K+ reabsorption. Since we found no significant differences between male and female TRPV4−/− mice, we did not discriminate between animal sexes in the following experiments.
Fig. 1.
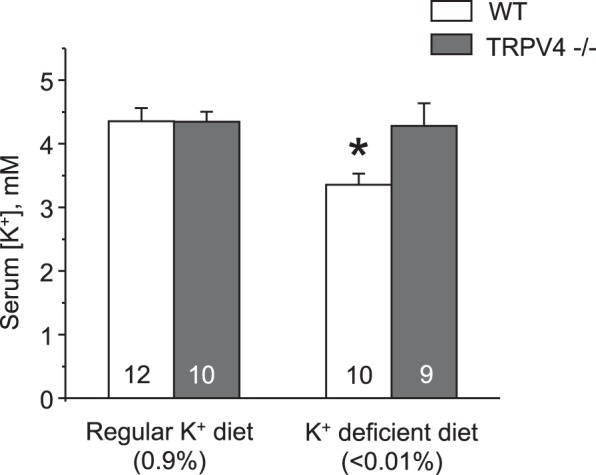
Transient receptor potential vanilloid type 4 (TRPV4)−/− mice are resistant to hypokalemia during systemic K+ deficiency. The summary graph shows serum K+ levels in wild-type (WT) and TRPV4−/− mice kept on regular (0.9% K+) and K+-deficient (<0.01% K+) diets for 7 days. Values are means ± SE. The number of experimental animals for each group is shown on the respective bars. *Significant decrease vs. WT mice on a regular K+ diet.
Fig. 2.
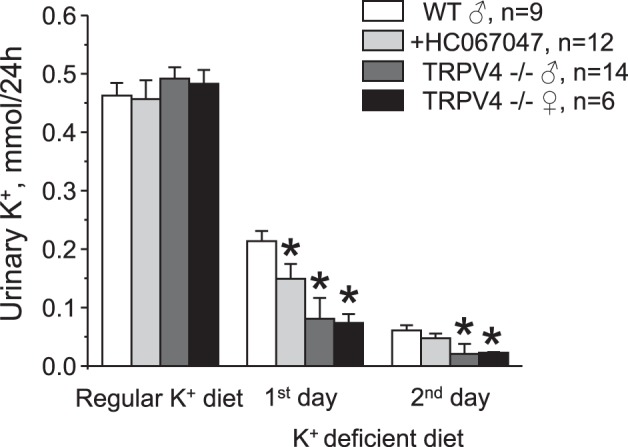
Augmented renal K+ retention in transient receptor potential vanilloid type 4 (TRPV4)−/− mice during a K+-deficient diet. The summary graph shows a comparison of 24-h urinary K+ levels in male wild-type (WT) mice, male WT mice treated with the TRPV4 inhibitor HC-067047 (0.075 mg/kg body wt in drinking water starting from the beginning of the experiment, estimated final concentration of 4 µM), male TRPV4−/− mice, and female TRPV4−/− mice maintained on a regular K+ diet (0.9% K+) and in response to a K+-deficient diet (<0.01% K+) for 1 or 2 days. Values are means ± SE; n, number of experimental animals for each group. *Significant decrease versus the respective value in WT mice.
Fig. 3.

Similar intake and urinary production in wild-type (WT) and transient receptor potential vanilloid type 4 (TRPV4)−/− mice during acute K+ deficiency. A and B: summary graphs showing a comparison of 24-h food (A) and water (B) intake in male WT, male TRPV4−/−, and female TRPV4−/− mice maintained on a regular K+ diet (0.9% K+) and in response to a K+-deficient diet (<0.01% K+) for 1 or 2 days. C: summary graph showing the comparison of whole body animal weights of male WT, male TRPV4−/−, and female TRPV4−/− mice used for balance experiments. D: summary graph showing a comparison of 24-h urinary output in male WT, male TRPV4−/−, and female TRPV4−/− mice maintained on a regular K+ diet (0.9% K+) and in response to a K+-deficient diet (<0.01% K+) for 1 or 2 days. Values are means ± SE; n, number of experimental animals for each group. *Significant increase versus the respective values on the regular K+ diet.
Reduced dietary K+ intake is known to cause an upregulation of H+-K+-ATPase expression in the CD to promote K+ uptake at the expense of proton secretion into the tubular lumen (15). The comparison of urinary pH in metabolic cages did not detect significant differences between WT and TRPV4−/− mice on a regular K+ diet (Fig. 4A). In contrast, urinary pH became significantly lower (i.e., more acidic) in TRPV4−/− mice compared with WT mice starting from the first day on a K+-deficient diet (Fig. 4A). We also monitored pH in freshly collected urinary spot samples from WT and TRPV4−/− mice kept subsequently on K+-deficient, regular K+, and high-K+ (5% K+) diets for 6 days (Fig. 4B). Consistently, pH values were lower in TRPV4−/− mice during the treatment with a K+-deficient diet, whereas no major differences in urinary pH were detected between WT and TRPV4−/− mice on regular and high-K+ diets. These results support the view that TRPV4 deficiency leads to increased H+-K+-ATPase-dependent K+ reabsorption in the CD during a low-K+ diet, which is reflected as a decrease in urinary pH.
Fig. 4.

Transient receptor potential vanilloid type 4 (TRPV4)−/− mice develop urinary acidification when fed a K+-deficient diet. A: summary graph showing the comparison of urinary pH in wild-type (WT) and TRPV4−/− mice in metabolic cages maintained on a regular K+ diet (0.9% K+) and in response to a K+-deficient diet (<0.01% K+) for 1 or 2 days. B: summary graph of time course changes in spot urinary pH for WT and TRPV4−/− mice kept on K+-deficient (<0.01% K+), regular K+ (0.9% K+), and high-K+ (5% K+) diets, as indicated by the respective bars at the top. Values are means ± SE; n, number of experimental animals for each group. *Significant decrease versus the respective value in WT mice.
In the next set of experiments, we quantified the activity of H+-K+-ATPase in freshly isolated CDs from WT and TRPV4−/− mice kept on a K+-deficient diet. To do this, we monitored changes in pHi in split-opened CDs loaded with a pH-sensitive dye, BCECF, using the standard ammonium pulse protocol (22). As expected, isotonic application of 40 mM NH4Cl for 3 min induced a rapid alkalization followed by an acidification on NH4Cl removal, thereby creating a maximal driving force for proton extrusion (Fig. 5A). Thus, the rate of subsequent pHi recovery toward the baseline value represents the activity of proton pumps, such as H+-K+-ATPase. To dissect the H+-K+-ATPase-dependent component, experiments were performed in the presence of bafilomycin (100 nM) and EIPA (10 µM) to block the activity of H+-ATPase and Na+/H+ exchangers, respectively, as was used previously (23). Since H+-K+-ATPase activity and expression are much higher in intercalated cells than in principal cells (22, 23), the cell types were analyzed individually by postexperimental staining of the respective CDs with a principal cell marker, AQP2 (Fig. 5A). As shown by the averaged time courses of pHi changes, the recovery after NH4Cl pulse was notably faster in both intercalated (Fig. 5B) and principal (Fig. 5C) cells in the CDs from TRPV4−/− mice compared with the respective values in WT mice. Furthermore, basal pHi levels were mildly but significantly higher in intercalated cells from TRPV4−/− mice (6.98 ± 0.03) compared with WT mice (6.88 ± 0.03), which would also be indicative of increased H+-K+-ATPase-dependent proton extrusion (Fig. 6A). In contrast, basal pHi levels were similar in principal cells of WT and TRPV4−/− mice (6.82 ± 0.82 and 6.84 ± 0.02, respectively; Fig. 6A), potentially reflecting lower levels of H+-K+-ATPase in this cell type. To quantify H+-K+-ATPase activity, we next fitted the initial pHi recovery after the ammonium pulse with a linear function for each individual cell. As anticipated, H+-K+-ATPase activity was roughly 10 times higher in intercalated cells than in principal cells of WT animals (0.356 ± 0.085 and 0.034 ± 0.003 ΔpHi/min, respectively; Fig. 6B). Importantly, both values were significantly greater in TRPV4−/− mice (0.582 ± 0.100 and 0.049 ± 0.005 ΔpHi/min), strongly suggesting augmented H+-K+-ATPase activity on TRPV4 deletion.
Fig. 5.

H+-K+-ATPase-dependent proton extrusion in split-opened collecting ducts (CDs) from wild-type (WT) and transient receptor potential vanilloid type 4 (TRPV4)−/− mice. A: representative pseudocolor images (blue: acidic; red: alkali) of intracellular pH (pHi) in a split-opened CD loaded with the pH-sensitive dye 2′,7′-bis-(2-carboxyethyl)-5-(6)-carboxyfluorescein (BCECF) at baseline (1), on application of 40 mM NH4Cl (2), immediately after NH4Cl removal (3), and recovery toward baseline pHi values (4). A confocal micrograph of the same split-opened CD probed with anti-aquaporin 2 (AQP2; pseudocolor red) is shown on the right. AQP2-negative intercalated cells are shown by white arrows. Nuclear 4,6-diamidino-2-phenylindole staining is shown in pseudocolor blue. B and C: summary graphs showing the time course of pHi changes in intercalated (B) and principal (C) cells from split-opened CDs of WT and TRPV4−/− mice kept on a K+-deficient diet (<0.01% K+) for 1 wk. Application of the NH4Cl pulse is shown by the black horizontal bars at the top. The time points shown in A are marked as 1–4. Experiments were performed in the continuous presence of bafilomycin (100 nM) and 5-(N-ethyl-N-isopropyl)amiloride (EIPA; 10 µM) to dissect H+-K+-ATPase-dependent pHi recovery. n, number of individual tested cells for each group.
Fig. 6.

Increased H+-K+-ATPase activity in both intercalated and principal cells of the collecting duct (CD) from transient receptor potential vanilloid type 4 (TRPV4)−/− mice on a K+-deficient diet. A: summary graph showing the comparison of basal intracellular pH (pHi) levels in principal and intercalated cells of CDs from wild-type (WT) and TRPV4−/− mice kept on a K+-deficient diet (<0.01% K+) for 1 wk. B: summary graphs showing the comparison of H+-K+-ATPase activity in intercalated (left) and principal (right) cells from WT and TRPV4−/− mice. Activity was calculated as a linear slope of pHi recovery after 40 mM NH4Cl application, as shown in Fig. 5. Values are means ± SE. The number of experimental animals for each group is shown on the respective bars. *Significant change versus the respective value in WT mice.
DISCUSSION
In the present study, we provided multicomponent experimental evidence in support of inhibitory actions of the Ca2+-permeable TRPV4 channel on H+-K+-ATPase-dependent K+reabsorption in intercalated and principal cells of the CD during dietary K+ restriction (Fig. 7). We further showed that disruption of TRPV4 activity augments renal K+ conservation to preclude the development of hypokalemia during low-K+ diet challenge.
Fig. 7.

Proposed role of transient receptor potential vanilloid type 4 (TRPV4) in the regulation of H+-K+-ATPase-dependent K+ reabsorption in the collecting duct (CD) during regular and K+-deficient states. Larger icon sizes reflect higher expression levels of transport proteins. During the regular K+ diet (left), TRPV4 has no appreciable role in the regulation of CD K+ reabsorption. Tubular flow is not elevated, and the activity of TRPV4 channels is at a baseline. H+-K+-ATPase expression is also low. The CD secretes K+ due to the activity of the Ca2+-independent renal outer medullary K+(ROMK) channel in principal cells. In the K+-deficient state (right), there is virtually no ROMK expression, whereas H+-K+-ATPase levels are upregulated. Activation of TRPV4 by high tubular flow inhibits H+-K+-ATPase activity in both principal and intercalated cells to decrease/shape K+ reabsorption in the CD. While intercalated cells account for only ~25% of total CD cells, the activity of H+-K+-ATPase is nearly 10-fold higher in intercalated cells compared with principal cells (see Fig. 6B). Overall, the total contribution of H+-K+-ATPase activity would be still roughly three times higher in intercalated cells, even after correction for cell numbers. [Ca2+]i, intracellular Ca2+ concentration.
Even an intermediate dietary K+ restriction is detrimental to cardiovascular health, leading to disturbances in K+ homeostasis, with serum K+ falling to ∼3.5 mM, accompanied with renal Na+ retention and hypertension (10, 11, 50). This occurs despite notable renal K+ conservation with urinary K+ excretion of only <1% of the filtered load (4). The major finding of the present study is that ablation of the mechanosensitive TRPV4 channel facilitates renal K+ conservation in response to a K+-deficient diet (Fig. 2) to mitigate the development of hypokalemia after at least 1 wk (Fig. 1). Of interest, hypokalemia leads to polyuria, in part because of reduced expression of distal tubule Na+ transporters: Na+-K+-2Cl− cotransporter 2 and Na+-Cl− cotransporter (NCC) (7). Thus, it is conceivable to suggest that a low-K+ diet increases fluid delivery to the CD, resulting in TRPV4 activation, as previously reported by our laboratory (3, 27). We also provided a proof of principle that systemic blockade of TRPV4 activity with HC-067047 decreases urinary K+ levels during a low-K+ diet (Fig. 2), arguing for its pharmacological potential in treatment of hypokalemic states in the clinical setting. The considerably smaller antikaliuretic effect observed with HC-067047 treatment might be due to a suboptimal dosage and/or a limited bioavailability of the TRPV4 antagonist. Other factors related to developmental alterations of TRPV4 deletion versus short-term administration of the antagonist might also contribute. Future translational studies are necessary to carefully evaluate the efficiency and safety of TRPV4 blockade in correcting hypokalemia. It is important to stress that global deletion of the channel in TRPV4−/− mice does not allow one to sort out a contribution of extra-CD and extrarenal factors in the observed phenotype during the state of K+ deficiency. Indeed, a previous study (54) demonstrated a role of TRPV4 in endothelium-dependent relaxation in small mesenteric arteries, potentially indicating disturbed hemodynamics in knockout mice. On the contrary, TRPV4−/− mice are normotensive and have a similarity to WT urinary volume at baseline (17) and during a low-K+ diet (Fig. 3). Thus, it is most likely that putative changes in renal hemodynamics play a minor role in the observed K+ retention on TRPV4 deletion. Convincing evidence identified a critical role of the distal convoluted tubule and specifically the NCC-mediated Na+ reabsorption in controlling systemic K+ homeostasis via control of fluid delivery to the CD (43). However, it has been recently reported that WT and TRPV4−/− mice have similar urinary Na+ levels at baseline and in response to dietary Na+ restriction, which is associated with augmented NCC activity (17). TRPV4 knockout mice also had a similarity to WT AQP2 levels, urinary volume at baseline, and after water deprivation, arguing against alterations in fluid delivery to the CDs. While investigation of the interaction between TRPV4 and NCC is beyond the scope of the present study, it is highly unlikely that NCC expression/activity was significantly affected in TRPV4−/− mice.
Activation of TRPV4 by mechanical forces has been reported to modulate K+ flux across the plasma membrane in different cell types. Thus, flow-induced TRPV4 Ca2+ influx stimulates Ca2+-activated K+ channels, such as small conductance-3 and BK channels, in endothelial and smooth muscle cells, leading to vasodilation (24, 41, 42). In choroid plexus epithelial cells, TRPV4 activity is similarly crucial for activation of small conductance-2 and intermediate-conductance K+ channels to control the production of cerebrospinal fluid (35). Moreover, we recently reported that TRPV4 regulates flow-induced K+ secretion via BK channels in the distal nephron, and this is an essential component for adaptation to dietary K+ load (27). In the present study, we discovered that TRPV4 also inhibits H+-K+-ATPase-dependent K+ reabsorption in the CD during the state of K+ deficiency. It is interesting that, in all of the aforementioned cases, TRPV4 favors K+ redistribution out of cells directly (by stimulating K+ secretion) or indirectly (by inhibiting K+ absorption), pointing to a fundamental physiological role of the channel in setting/driving transmembrane ion gradients in response to exogenous mechanical inputs. The counterintuitive relevance of TRPV4-mediated inhibition of K+ reabsorption on a low-K+ diet deserves an additional explanation. Evolutionarily, it is much more important to eliminate K+ in urine than to conserve it, since ancient diets were rich in K+ (45). The proposed role of TRPV4 in the kidney to augment excretion of K+ aligns well with this concept. On the contrary, kaliuretic actions of TRPV4 interfere with renal K+ conservation during dietary K+ restriction, thereby contributing to hypokalemia.
In the present study, we demonstrated feasibility to reliably monitor activity of H+-K+-ATPase activity in split-opened CDs with the precision to discriminate the signals from principal and intercalated cells by combining pHi measurements with BCECF followed by immunofluorescent staining with a principal cell marker, AQP2 (Fig. 5). This offers an alternative and more efficient experimental output compared with studies in perfused CDs, where only a small fraction of cells can be used for analysis (23). We observed that only ∼25% of all tested cells were intercalated cells, whereas the majority of cells show positive AQP2 signal, thus being principal cells (Fig. 5). This is in agreement with the well-defined ratio between the cell types in the CDs (32, 37). Moreover, we found ~10 times higher H+-K+-ATPase activity, measured as pHi recovery after the NH4Cl pulse in the presence of bafilomycin and EIPA, in intercalated cells than in principal cells (Fig. 6). This resonates nicely with the previously reported predominant expression of the pump in intercalated cells, pointing to a central role in mediating K+ reabsorption, despite their fewer numbers in the CDs (15). In our study, we found that H+-K+-ATPase-dependent pHi extrusion in split-opened CDs from mice kept on a K+-deficient diet was roughly twice higher compared with the values reported in perfused tubules from mice on a regular K+ regimen (22). Again, this is consistent with the documented upregulation of H+-K+-ATPase expression by dietary K+ restriction (13). Although we did not discriminate between the A- and B-types of intercalated cells, both subtypes exhibit similar activity of the enzyme (22). Moreover, statistical analysis of H+-K+-ATPase activity demonstrated a bell-shaped distribution, arguing against different expression of the pump in A- and B-cell types (not shown). We did observe that H+-K+-ATPase-dependent pHi recovery was significantly accelerated in both principal and intercalated cells of split-opened CDs from TRPV4−/− mice (Figs. 5 and 6). This directly demonstrates an inhibitory action of TRPV4 expressed in both principal and intercalated cells (3) on K+ reabsorption in the CD. Identification of the signaling pathway of this regulation is beyond the scope of the present study, but it is feasible to speculate that it involves elevations of [Ca2+]i due to TRPV4 Ca2+ influx.
In addition to governing distal nephron K+ reabsorption, H+-K+-ATPases also play a notable role in the regulation of acid-base balance (15). Metabolic acidosis greatly increases H+-K+-ATPase expression and activity (6, 38, 39). Mice lacking both α1- and α2-isoforms have lower blood concentration levels, which is indicative of impaired distal nephron H+ secretion in the presence of mineralocorticoid excess (12). Consistently, we found a significantly lower urinary pH in TRPV4−/− mice fed a K+-deficient diet to increase H+-K+-ATPase expression (Fig. 4). While global TRPV4 deletion does not allow us to rule out the potential contribution of extrarenal factors in urinary acidification, this process was clearly dependent on K+ intake, since no major differences were found between WT and TRPV4−/− mice on regular and high-K+ diets (Fig. 4). Regardless, our results indicate that pharmacological intervention targeting TRPV4 not only might be instrumental for correction K+ homeostasis but also could be used to affect systemic acid-base balance, at least when K+ intake is reduced. Of interest, numerous very specific mutations in TRPV4 (leading to both decreased and increased channel activity) have been associated with a puzzling set of skeletal diseases and neuropathies in humans (for a review, see Ref. 29). Thus, it is possible that alterations in systemic pH balance, particularly during dietary K+ restriction, which is common in Western diets (14), may underlie or serve as a driving force for the development of these pathologies by affecting bone growth/resorption and cartilage formation in patients bearing such TRPV4 polymorphisms.
In conclusion, this study unraveled a previously unrecognized role of TRPV4 in the regulation of K+ reabsorption in the CD. We showed that TRPV4 ablation increases H+-K+-ATPase activity to facilitate renal K+ conservation in the state of K+ deficiency. Finally, we demonstrated that TRPV4 blockade is beneficial in maintaining systemic K+ homeostasis and in precluding a drop in plasma K+ levels. Thus, this study offers a novel way to protect against hypokalemia by inhibiting TRPV4, which could potentially improve cardiovascular function in patients with electrolyte imbalances.
GRANTS
This work was supported by National Institute of Diabetes and Digestive and Kidney Diseases Grant DK-119170 (to O. Pochynyuk), American Heart Association Grant 17GRNT33660488 (to O. Pochynyuk), an American Society of Nephrology Ben J. Lipps Research Fellowship (to V. Tomilin), and American Heart Association Grant 15SDG25550150 (to M. Mamenko)
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
V.T., M.M., O.Z., and O.P. performed experiments; V.T., M.M., O.Z., and O.P. analyzed data; V.T., M.M., O.Z., and O.P. prepared figures; V.T., M.M., O.Z., C.S.W., and O.P. edited and revised manuscript; V.T., M.M., O.Z., C.S.W., and O.P. approved final version of manuscript; M.M., C.S.W., and O.P. conceived and designed research; M.M., O.Z., C.S.W., and O.P. interpreted results of experiments; O.P. drafted manuscript.
REFERENCES
- 1.Ahn KY, Park KY, Kim KK, Kone BC. Chronic hypokalemia enhances expression of the H+-K+-ATPase α2-subunit gene in renal medulla. Am J Physiol Renal Physiol 271: F314–F321, 1996. doi: 10.1152/ajprenal.1996.271.2.F314. [DOI] [PubMed] [Google Scholar]
- 2.Ahn KY, Turner PB, Madsen KM, Kone BC. Effects of chronic hypokalemia on renal expression of the “gastric” H+-K+-ATPase α-subunit gene. Am J Physiol Renal Physiol 270: F557–F566, 1996. doi: 10.1152/ajprenal.1996.270.4.F557. [DOI] [PubMed] [Google Scholar]
- 3.Berrout J, Jin M, Mamenko M, Zaika O, Pochynyuk O, O’Neil RG. Function of transient receptor potential cation channel subfamily V member 4 (TRPV4) as a mechanical transducer in flow-sensitive segments of the renal collecting duct system. J Biol Chem 287: 8782–8791, 2012. doi: 10.1074/jbc.M111.308411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chen P, Guzman JP, Leong PK, Yang LE, Perianayagam A, Babilonia E, Ho JS, Youn JH, Wang WH, McDonough AA. Modest dietary K+ restriction provokes insulin resistance of cellular K+ uptake and phosphorylation of renal outer medulla K+ channel without fall in plasma K+ concentration. Am J Physiol Cell Physiol 290: C1355–C1363, 2006. doi: 10.1152/ajpcell.00501.2005. [DOI] [PubMed] [Google Scholar]
- 5.Cherezova A, Tomilin V, Buncha V, Zaika O, Ortiz PA, Mei F, Cheng X, Mamenko M, Pochynyuk O. Urinary concentrating defect in mice lacking Epac1 or Epac2. FASEB J 33: 2156–2170, 2019. doi: 10.1096/fj.201800435R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cheval L, Morla L, Elalouf JM, Doucet A. Kidney collecting duct acid-base “regulon”. Physiol Genomics 27: 271–281, 2006. doi: 10.1152/physiolgenomics.00069.2006. [DOI] [PubMed] [Google Scholar]
- 7.Elkjaer ML, Kwon TH, Wang W, Nielsen J, Knepper MA, Frøkiaer J, Nielsen S. Altered expression of renal NHE3, TSC, BSC-1, and ENaC subunits in potassium-depleted rats. Am J Physiol Renal Physiol 283: F1376–F1388, 2002. doi: 10.1152/ajprenal.00186.2002. [DOI] [PubMed] [Google Scholar]
- 8.Everaerts W, Nilius B, Owsianik G. The vanilloid transient receptor potential channel TRPV4: from structure to disease. Prog Biophys Mol Biol 103: 2–17, 2010. doi: 10.1016/j.pbiomolbio.2009.10.002. [DOI] [PubMed] [Google Scholar]
- 9.Fejes-Tóth G, Náray-Fejes-Tóth A. Immunohistochemical localization of colonic H-K-ATPase to the apical membrane of connecting tubule cells. Am J Physiol Renal Physiol 281: F318–F325, 2001. doi: 10.1152/ajprenal.2001.281.2.F318. [DOI] [PubMed] [Google Scholar]
- 10.Gallen IW, Rosa RM, Esparaz DY, Young JB, Robertson GL, Batlle D, Epstein FH, Landsberg L. On the mechanism of the effects of potassium restriction on blood pressure and renal sodium retention. Am J Kidney Dis 31: 19–27, 1998. doi: 10.1053/ajkd.1998.v31.pm9428447. [DOI] [PubMed] [Google Scholar]
- 11.Greenlee M, Wingo CS, McDonough AA, Youn JH, Kone BC. Narrative review: evolving concepts in potassium homeostasis and hypokalemia. Ann Intern Med 150: 619–625, 2009. doi: 10.7326/0003-4819-150-9-200905050-00008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Greenlee MM, Lynch IJ, Gumz ML, Cain BD, Wingo CS. Mineralocorticoids stimulate the activity and expression of renal H+,K+-ATPases. J Am Soc Nephrol 22: 49–58, 2011. doi: 10.1681/ASN.2010030311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Greenlee MM, Lynch IJ, Gumz ML, Cain BD, Wingo CS. The renal H,K-ATPases. Curr Opin Nephrol Hypertens 19: 478–482, 2010. doi: 10.1097/MNH.0b013e32833ce65f. [DOI] [PubMed] [Google Scholar]
- 14.Gritter M, Rotmans JI, Hoorn EJ. Role of dietary K+ in natriuresis, blood pressure reduction, cardiovascular protection, and renoprotection. Hypertension 73: 15–23, 2019. doi: 10.1161/HYPERTENSIONAHA.118.11209. [DOI] [PubMed] [Google Scholar]
- 15.Gumz ML, Lynch IJ, Greenlee MM, Cain BD, Wingo CS. The renal H+-K+-ATPases: physiology, regulation, and structure. Am J Physiol Renal Physiol 298: F12–F21, 2010. doi: 10.1152/ajprenal.90723.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hebert SC, Desir G, Giebisch G, Wang W. Molecular diversity and regulation of renal potassium channels. Physiol Rev 85: 319–371, 2005. doi: 10.1152/physrev.00051.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Janas S, Seghers F, Schakman O, Alsady M, Deen P, Vriens J, Tissir F, Nilius B, Loffing J, Gailly P, Devuyst O. TRPV4 is associated with central rather than nephrogenic osmoregulation. Pflugers Arch 468: 1595–1607, 2016. doi: 10.1007/s00424-016-1850-5. [DOI] [PubMed] [Google Scholar]
- 18.Kovesdy CP. Management of hyperkalaemia in chronic kidney disease. Nat Rev Nephrol 10: 653–662, 2014. doi: 10.1038/nrneph.2014.168. [DOI] [PubMed] [Google Scholar]
- 19.Kraut JA, Hiura J, Besancon M, Smolka A, Sachs G, Scott D. Effect of hypokalemia on the abundance of HK alpha 1 and HK alpha 2 protein in the rat kidney. Am J Physiol Renal Physiol 272: F744–F750, 1997. doi: 10.1152/ajprenal.1997.272.6.F744. [DOI] [PubMed] [Google Scholar]
- 20.Kraut JA, Starr F, Sachs G, Reuben M. Expression of gastric and colonic H+-K+-ATPase in the rat kidney. Am J Physiol Renal Physiol 268: F581–F587, 1995. doi: 10.1152/ajprenal.1995.268.4.F581. [DOI] [PubMed] [Google Scholar]
- 21.Liedtke W, Friedman JM. Abnormal osmotic regulation in trpv4−/− mice. Proc Natl Acad Sci USA 100: 13698–13703, 2003. doi: 10.1073/pnas.1735416100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lynch IJ, Greenlee MM, Gumz ML, Rudin A, Xia SL, Wingo CS. Heterogeneity of H-K-ATPase-mediated acid secretion along the mouse collecting duct. Am J Physiol Renal Physiol 298: F408–F415, 2010. doi: 10.1152/ajprenal.00333.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lynch IJ, Rudin A, Xia SL, Stow LR, Shull GE, Weiner ID, Cain BD, Wingo CS. Impaired acid secretion in cortical collecting duct intercalated cells from H-K-ATPase-deficient mice: role of HKα isoforms. Am J Physiol Renal Physiol 294: F621–F627, 2008. doi: 10.1152/ajprenal.00412.2007. [DOI] [PubMed] [Google Scholar]
- 24.Ma X, Du J, Zhang P, Deng J, Liu J, Lam FF, Li RA, Huang Y, Jin J, Yao X. Functional role of TRPV4-KCa2.3 signaling in vascular endothelial cells in normal and streptozotocin-induced diabetic rats. Hypertension 62: 134–139, 2013. doi: 10.1161/HYPERTENSIONAHA.113.01500. [DOI] [PubMed] [Google Scholar]
- 25.Mamenko M, Zaika O, Boukelmoune N, O’Neil RG, Pochynyuk O. Deciphering physiological role of the mechanosensitive TRPV4 channel in the distal nephron. Am J Physiol Renal Physiol 308: F275–F286, 2015. doi: 10.1152/ajprenal.00485.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mamenko M, Zaika O, O’Neil RG, Pochynyuk O. Ca2+ imaging as a tool to assess TRP channel function in murine distal nephrons. Methods Mol Biol 998: 371–384, 2013. doi: 10.1007/978-1-62703-351-0_29. [DOI] [PubMed] [Google Scholar]
- 27.Mamenko MV, Boukelmoune N, Tomilin VN, Zaika OL, Jensen VB, O’Neil RG, Pochynyuk OM. The renal TRPV4 channel is essential for adaptation to increased dietary potassium. Kidney Int 91: 1398–1409, 2017. doi: 10.1016/j.kint.2016.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mironova E, Bugay V, Pochynyuk O, Staruschenko A, Stockand JD. Recording ion channels in isolated, split-opened tubules. Methods Mol Biol 998: 341–353, 2013. doi: 10.1007/978-1-62703-351-0_27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nilius B, Voets T. The puzzle of TRPV4 channelopathies. EMBO Rep 14: 152–163, 2013. doi: 10.1038/embor.2012.219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nilius B, Vriens J, Prenen J, Droogmans G, Voets T. TRPV4 calcium entry channel: a paradigm for gating diversity. Am J Physiol Cell Physiol 286: C195–C205, 2004. doi: 10.1152/ajpcell.00365.2003. [DOI] [PubMed] [Google Scholar]
- 31.Palmer BF. Regulation of potassium homeostasis. Clin J Am Soc Nephrol 10: 1050–1060, 2015. doi: 10.2215/CJN.08580813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pearce D, Soundararajan R, Trimpert C, Kashlan OB, Deen PM, Kohan DE. Collecting duct principal cell transport processes and their regulation. Clin J Am Soc Nephrol 10: 135–146, 2015. doi: 10.2215/CJN.05760513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pochynyuk O, Zaika O, O’Neil RG, Mamenko M. Novel insights into TRPV4 function in the kidney. Pflugers Arch 465: 177–186, 2013. doi: 10.1007/s00424-012-1190-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Praetorius HA, Leipziger J. Intrarenal purinergic signaling in the control of renal tubular transport. Annu Rev Physiol 72: 377–393, 2010. doi: 10.1146/annurev-physiol-021909-135825. [DOI] [PubMed] [Google Scholar]
- 35.Preston D, Simpson S, Halm D, Hochstetler A, Schwerk C, Schroten H, Blazer-Yost BL. Activation of TRPV4 stimulates transepithelial ion flux in a porcine choroid plexus cell line. Am J Physiol Cell Physiol 315: C357–C366, 2018. doi: 10.1152/ajpcell.00312.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rieg T, Vallon V, Sausbier M, Sausbier U, Kaissling B, Ruth P, Osswald H. The role of the BK channel in potassium homeostasis and flow-induced renal potassium excretion. Kidney Int 72: 566–573, 2007. doi: 10.1038/sj.ki.5002369. [DOI] [PubMed] [Google Scholar]
- 37.Roy A, Al-bataineh MM, Pastor-Soler NM. Collecting duct intercalated cell function and regulation. Clin J Am Soc Nephrol 10: 305–324, 2015. doi: 10.2215/CJN.08880914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Silver RB, Frindt G, Mennitt P, Satlin LM. Characterization and regulation of H-K-ATPase in intercalated cells of rabbit cortical collecting duct. J Exp Zool 279: 443–455, 1997. doi: 10.1002/(SICI)1097-010X(19971201)279:5<443:AID-JEZ6>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- 39.Silver RB, Mennitt PA, Satlin LM. Stimulation of apical H-K-ATPase in intercalated cells of cortical collecting duct with chronic metabolic acidosis. Am J Physiol Renal Physiol 270: F539–F547, 1996. doi: 10.1152/ajprenal.1996.270.3.F539. [DOI] [PubMed] [Google Scholar]
- 40.Sipos A, Vargas S, Peti-Peterdi J. Direct demonstration of tubular fluid flow sensing by macula densa cells. Am J Physiol Renal Physiol 299: F1087–F1093, 2010. doi: 10.1152/ajprenal.00469.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sonkusare SK, Bonev AD, Ledoux J, Liedtke W, Kotlikoff MI, Heppner TJ, Hill-Eubanks DC, Nelson MT. Elementary Ca2+ signals through endothelial TRPV4 channels regulate vascular function. Science 336: 597–601, 2012. doi: 10.1126/science.1216283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sullivan MN, Earley S. TRP channel Ca2+ sparklets: fundamental signals underlying endothelium-dependent hyperpolarization. Am J Physiol Cell Physiol 305: C999–C1008, 2013. doi: 10.1152/ajpcell.00273.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Terker AS, Zhang C, McCormick JA, Lazelle RA, Zhang C, Meermeier NP, Siler DA, Park HJ, Fu Y, Cohen DM, Weinstein AM, Wang WH, Yang CL, Ellison DH. Potassium modulates electrolyte balance and blood pressure through effects on distal cell voltage and chloride. Cell Metab 21: 39–50, 2015. doi: 10.1016/j.cmet.2014.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Thomas JA, Buchsbaum RN, Zimniak A, Racker E. Intracellular pH measurements in Ehrlich ascites tumor cells utilizing spectroscopic probes generated in situ. Biochemistry 18: 2210–2218, 1979. doi: 10.1021/bi00578a012. [DOI] [PubMed] [Google Scholar]
- 45.Tobian L. Dietary sodium chloride and potassium have effects on the pathophysiology of hypertension in humans and animals. Am J Clin Nutr 65, Suppl: 606S–611S, 1997. doi: 10.1093/ajcn/65.2.606S. [DOI] [PubMed] [Google Scholar]
- 46.Tomilin V, Reif GA, Zaika O, Wallace DP, Pochynyuk O. Deficient transient receptor potential vanilloid type 4 function contributes to compromised [Ca2+]i homeostasis in human autosomal-dominant polycystic kidney disease cells. FASEB J 32: 4612–4623, 2018. doi: 10.1096/fj.201701535RR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Unwin RJ, Luft FC, Shirley DG. Pathophysiology and management of hypokalemia: a clinical perspective. Nat Rev Nephrol 7: 75–84, 2011. doi: 10.1038/nrneph.2010.175. [DOI] [PubMed] [Google Scholar]
- 48.Verlander JW, Moudy RM, Campbell WG, Cain BD, Wingo CS. Immunohistochemical localization of H-K-ATPase α2c-subunit in rabbit kidney. Am J Physiol Renal Physiol 281: F357–F365, 2001. doi: 10.1152/ajprenal.2001.281.2.F357. [DOI] [PubMed] [Google Scholar]
- 49.Wang WH, Giebisch G. Regulation of potassium (K) handling in the renal collecting duct. Pflugers Arch 458: 157–168, 2009. doi: 10.1007/s00424-008-0593-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Weiner ID, Wingo CS. Hypokalemia−consequences, causes, and correction. J Am Soc Nephrol 8: 1179–1188, 1997. [DOI] [PubMed] [Google Scholar]
- 51.Wen D, Cornelius RJ, Sansom SC. Interacting influence of diuretics and diet on BK channel-regulated K homeostasis. Curr Opin Pharmacol 15: 28–32, 2014. doi: 10.1016/j.coph.2013.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Youn JH, McDonough AA. Recent advances in understanding integrative control of potassium homeostasis. Annu Rev Physiol 71: 381–401, 2009. doi: 10.1146/annurev.physiol.010908.163241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zaika O, Mamenko M, Berrout J, Boukelmoune N, O’Neil RG, Pochynyuk O. TRPV4 dysfunction promotes renal cystogenesis in autosomal recessive polycystic kidney disease. J Am Soc Nephrol 24: 604–616, 2013. doi: 10.1681/ASN.2012050442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhang DX, Mendoza SA, Bubolz AH, Mizuno A, Ge ZD, Li R, Warltier DC, Suzuki M, Gutterman DD. Transient receptor potential vanilloid type 4-deficient mice exhibit impaired endothelium-dependent relaxation induced by acetylcholine in vitro and in vivo. Hypertension 53: 532–538, 2009. doi: 10.1161/HYPERTENSIONAHA.108.127100. [DOI] [PMC free article] [PubMed] [Google Scholar]