

Abstract
Host ecological factors and external environmental factors are known to influence the structure of gut microbial communities, but few studies have examined the impacts of environmental changes on microbiotas in free‐ranging animals. Rapid land‐use change has the potential to shift gut microbial communities in wildlife through exposure to novel bacteria and/or by changing the availability or quality of local food resources. The consequences of such changes to host health and fitness remain unknown and may have important implications for pathogen spillover between humans and wildlife. To better understand the consequences of land‐use change on wildlife microbiotas, we analyzed long‐term dietary trends, gut microbiota composition, and innate immune function in common vampire bats (Desmodus rotundus) in two nearby sites in Belize that vary in landscape structure. We found that vampire bats living in a small forest fragment had more homogenous diets indicative of feeding on livestock and shifts in microbiota heterogeneity, but not overall composition, compared to those living in an intact forest reserve. We also found that irrespective of sampling site, vampire bats which consumed relatively more livestock showed shifts in some core bacteria compared with vampire bats which consumed relatively less livestock. The relative abundance of some core microbiota members was associated with innate immune function, suggesting that future research should consider the role of the host microbiota in immune defense and its relationship to zoonotic infection dynamics. We suggest that subsequent homogenization of diet and habitat loss through livestock rearing in the Neotropics may lead to disruption to the microbiota that could have downstream impacts on host immunity and cross‐species pathogen transmission.
Keywords: Desmodus rotundus, diet homogenization, land‐use change, livestock, microbiota, resource provisioning
1. INTRODUCTION
The animal gut microbiota plays an essential role in maintaining host health, including modulating effects of nutrition and immunity (Amato et al., 2014; Hanning & Diaz‐Sanchez, 2015; Khosravi & Mazmanian, 2013; O'Sullivan et al., 2013). However, the microbiota is not a static entity, and community composition can change rapidly in response to shifts in host diet (David et al., 2014; Turnbaugh, Ridaura, Faith, Rey, & Gordon, 2009). If such shifts lead to functional aberrations in community membership or composition—a pathological state called “dysbiosis”—nutritional fitness and host capacity to resist infection may be reduced (Khosravi & Mazmanian, 2013; Stecher, Maier, & Hardt, 2013; Williams et al., 2016). A primary mechanism by which gut microbial communities can influence infection is by altering the maintenance or development of the host immune system; bacteria associated with the gut epithelium can produce ligands that interact with toll‐like receptors in host cells, stimulating immune response cascades (Hooper, Littman, & Macpherson, 2012; Kau, Ahern, Griffin, Goodman, & Gordon, 2011; Macpherson & Harris, 2004; O'Sullivan et al., 2013; Thaiss, Zmora, Levy, & Elinav, 2016). For example, laboratory studies of mice illustrate that commensal microbiota contribute to the host's ability to mount proper immune responses (e.g., adaptive immunity) against viral infection (Ichinohe et al., 2011). Similarly, administration of probiotic bacteria to brown trout (Salmo trutta) increases the activity of the complement system, also suggesting links between the microbiota and innate immune function in animals (Balcázar et al., 2007). Compared to studies of laboratory animals and humans, interactions between endosymbiotic microbial communities and the host immune system are less well understood for wildlife (Evans, Buchanan, Griffith, Klasing, & Addison, 2017; Pedersen & Babayan, 2011), which are regularly exposed to pathogens and to environmental variation (McKenzie et al., 2017).
Environmental variation can be stark when humans rapidly alter the landscape for other uses. A growing body of research suggests that animal microbiotas may respond to land‐use change, particularly where such change leads to altered or deficient food resources. For example, black howler monkeys (Alouatta pigra) from fragmented forests display low dietary diversity that is associated with less diverse microbiota (Amato et al., 2013). The gut microbiotas of Udzungwa red colobus monkeys (Procolobus gordonorum) in fragmented forests were found to have significantly lower microbiota alpha diversity than conspecifics living in undisturbed forests and have reduced functional capacity to digest toxic xenobiotics naturally present in their diet (Barelli et al., 2015). These studies suggest that land‐use change and habitat loss may impact the fitness of animal microbiotas through changes in host diet. However, these patterns are not evident for all host species, nor across all spatial scales. One study of a community of African primates found that gut microbiotas were most strongly structured by host species identity and were largely resistant to perturbation even across fragmentation gradients (Mccord et al., 2014). Significant associations between microbial community structure and land‐use change have also been reported for some select taxa (e.g., amphibians; Becker, Longo, Haddad, & Zamudio, 2017; Reyes et al., 2017), but for most host taxa, there is little understanding whether land‐use change influences the gut microbiota.
Even where evidence exists that habitat quality impacts microbiota structure, how such ecological variation in diversity and structure relates to host immune defense in natural systems remains largely unquantified (Woodhams et al., 2014) in spite of relevance for the monitoring and control of reservoir hosts for zoonotic pathogens (Altizer et al., 2018). From a conservation perspective, understanding the potentially powerful but indirect effects of habitat destruction on animal microbiotas may also play a critical role in host conservation and management (Trevelline, Fontaine, Hartup, & Kohl, 2019; Wei et al., 2018; West et al., 2019). Under the “One Health” concept, which recognizes that the health of wildlife is interdependent with the health of humans and livestock (Zinsstag, Schelling, Waltner‐Toews, & Tanner, 2011), microbiotas may dually serve to monitor host health and track potential emerging zoonoses.
In this study, we investigated the interplay among diet, microbiota structure, and innate immune function in a free‐ranging bat species, the common vampire bat (Desmodus rotundus, hereafter “vampire bats”) (Figure 1). Bats as a group harbor more zoonotic viruses than other mammalian orders and have been implicated in the spillover of pathogens such as Hendra virus, Bartonella mayotimonensis, and Marburg virus (Amman et al., 2012; Olival et al., 2017; Plowright et al., 2015; Veikkolainen, Vesterinen, Lilley, & Pulliainen, 2014). Vampire bats, in particular, are widely distributed from northern Mexico to northern Argentina and, owing to their blood‐feeding diet, can transmit pathogens such as rabies virus. Rabies is a major threat to livestock and human health in Latin America for which vampire bats are the main reservoir host (Greenhall & Schmidt, 1988; Schneider et al., 2009). Feeding on vertebrate blood may also facilitate cross‐species transmission of other pathogens such as Bartonella, hemoplasmas, influenza, and trypanosomes (Becker, Bergner, et al., 2018a; Hoare, 1965; Tong et al., 2013; Volokhov et al., 2017). Agricultural intensification in Latin America (i.e., the conversion of forests into livestock pasture) provides vampire bats with an abundant and accessible source of mammalian prey that has been implicated in shaping bat immune phenotypes. However, exactly how land conversion influences the vampire bat gut microbiota, and if this helps explain observed immune profiles, remains unknown (Altizer et al., 2018; Becker, Bergner, et al., 2018a; Streicker & Allgeier, 2016).
Figure 1.

The common vampire bat, Desmodus rotundus. Photo credit: Brock and Sherri Fenton
We approached these questions by sampling blood, rectal microbiota, and hair from vampire bats at two adjacent sites in Belize that contrast in land use. We first characterized the vampire bat gut microbiota using 16S amplicon sequencing of rectal swab samples. We chose this sampling scheme because previous studies have shown that rectally collected stool communities are more diverse than the intestinal lumen and record a strong signature of diet, one our principle drivers of interest (Araújo‐Pérez et al., 2012; Ingala, Simmons, Wultsch, & Krampis, 2018). Next, we measured long‐term diet using stable isotopes of carbon (δ13C) and nitrogen (δ15N) from hair. Vampire bats feeding primarily on livestock can be differentiated from those feeding on wildlife using δ13C, as most grasses consumed by livestock (e.g., cattle) use the C4 photosynthetic pathway while most forest plants consumed by forest wildlife (e.g., peccary) use the C3 pathway (Streicker & Allgeier, 2016; Voigt & Kelm, 2006a). δ15N also provides inference into the trophic level of prey species, as consumer δ15N is enriched by 3‰−4‰ relative to its diet (Post, 2002). We next tested if bat sampling site and long‐term diet (derived from hair samples) predict gut microbial diversity and composition. Lastly, we assessed whether microbiota attributes were associated with a functional measure of innate immune defense in the bats using plasma. Because vampire bats with access to domestic animals feed primarily on these prey (Streicker & Allgeier, 2016), we predicted that bats residing in a more agriculturally intensified site would show evidence of a more homogenous diet and have a gut microbiota with decreased alpha diversity (Becker, Czirják, et al., 2018b; Streicker & Allgeier, 2016; Voigt & Kelm, 2006a). We next predicted that less diverse vampire bat microbiotas would be associated with weaker immune defenses as measure by a bacterial killing assay (Khosravi & Mazmanian, 2013).
2. MATERIALS AND METHODS
2.1. Capture and sampling of vampire bats
Between April 20 and 25, 2015, we sampled 36 vampire bats from two adjacent areas in the Orange Walk District of Belize: Lamanai Archaeological Reserve (LAR) and Ka'Kabish (KK). LAR is a protected area bordered by the New River Lagoon, forest, and agricultural habitat, whereas KK is a site of remnant forest surrounded by clear‐cut agricultural fields (Herrera, Duncan, Clare, Fenton, & Simmons, 2018). The forest at LAR consists largely of closed‐canopy semi‐deciduous forest interspersed with areas of old secondary growth located within several kilometers of cattle pastures. While the forest at LAR is of higher quality than at KK, its proximity to cattle pastures still falls within the home range area for D. rotundus (Trajano, 1996). At KK, the forest is ecologically similar but more degraded, with smaller trees, a lower canopy, and more secondary vegetation. The area sampled at KK was limited entirely to forest regrown on Maya structures and plazas. Sampling sites were 8 km apart and consisted of roosts in a hollow tree and a colonial cistern (LAR) and looter's tunnels in an unexcavated Maya temple complex (KK) (Figure 2). Bats were captured with mist nets at roost entrances or along nearby flight paths from 19:00 until 22:00; a harp trap was also set from 18:00 to 05:00. Upon capture, bats were placed in individual cloth bags and issued a uniquely coded wing band (3.5 mm; Porzana Inc.).
Figure 2.

Detailed map of study sites in northern Belize. The shaded inset shows the location of Orange Walk District. Colored borders show the bounds of Lamanai Archaeological Reserve (LAR) and Ka'Kabish (KK). Prior to 2015, these sites were connected by contiguous forest (green), but now are disconnected by agricultural matrix (white and tan). Satellite imagery was derived from Google Maps, retrieved March 2019
For analyses of the vampire bat gut microbiota, we collected rectal samples from 30 bats (n LAR = 14, n KK = 16) using sterile miniature rayon swabs (1.98 mm; Puritan). Rectal samples were flash‐frozen in liquid nitrogen for optimal bacterial DNA preservation (Hale, Tan, Knight, & Amato, 2015). For analyses of vampire bat diet, we trimmed <5 mg hair from the interscapular region of each individual for stable isotope analysis. To quantify innate immune defense, we obtained blood samples by lancing the propatagial vein with sterile 23‐gauge needles and collecting 20–50 μl of blood in heparinized capillary tubes. Plasma was isolated by centrifuging blood in serum separator tubes and was subsequently frozen at −20°C until transfer to −80°C storage at the University of Georgia. Following sampling, all bats were released at the sites where they were captured. We were unable to collect all sample types (hair, blood, rectal swabs) for all bats, and sample sizes vary (swabs = 30 individuals, hair = 29, swabs and hair = 23, blood = 20).
All field procedures followed guidelines of the American Society of Mammalogists (Sikes et al., 2016) and were approved by the University of Georgia Animal Care and Use Committee (AUP A2014 04‐016‐Y3‐A5). Fieldwork was approved by the Belize Forestry Department under permit CD/60/3/15(21).
2.2. Microbiota diversity and community composition
Host and microbial DNA was extracted using Macherey–Nagel Nucleospin Soil kit according to the manufacturers’ protocol (Macherey‐Nagel, Inc.). PCR‐based library formation targeting the 16S rRNA gene variable region 4 (V4) was performed using forward primer 515F (5′ AATGATACGGCGACCACCGAGATCTACAC NNNNNNNN TATGGTAATTGTGTGCCAGCMGCCGCGGTAA 3′, where “N” indicates the nucleotides of the barcode sequence) and reverse primer 806R (5′ CAAGCAGAAGACGGCATACGAGAT NNNNNNNNNNNN AGTCAGTCAGCCGGACTACHVGGGTWTCTAAT 3′) with Illumina adaptor sequences on the 5′ end (Apprill, Mcnally, Parsons, & Weber, 2015; Caporaso et al., 2011). Prepared libraries were sequenced on the Illumina MiSeq platform using 2 × 350 bp v3 chemistry.
We recovered an average raw read depth of 40,945 sequences per sample (SE mean = 3,160). Raw reads were processed using QIIME2 v. 2019.1 (Bolyen et al., 2018). We used the DADA2 QIIME2 plugin to denoise and quality filter reads, call amplicon sequence variants (ASVs), and generate a feature table of ASV counts and host metadata (Callahan et al., 2016). After quality filtering with DADA2, the average read depth across samples was 28,415 (SE mean = 2,345). We assigned bacterial taxonomy to the ASV feature table using the Naive Bayesian Q2 feature classifier as implemented in QIIME2, comparing against a SILVA reference database trained on the 515F/806R region of the 16S gene (Bokulich et al., 2018; Karst et al., 2016). Next, we frequency‐filtered potential contaminants from the ASV feature table using the initial sample DNA concentrations and R package decontam (v.1.2.1) (Figure A1A1; Davis, Proctor, Holmes, Relman, & Callahan, 2018). To determine the core set of ASVs characterizing the vampire bat microbiome, we used the core_members function from the microbiome R package (http://microbiome.github.io), setting the prevalence threshold at 50% (Caporaso et al., 2010). Prior to statistical analysis of microbiota, we produced rarefaction curves in QIIME2 in order to choose an appropriate minimum rarefying depth (n = 10,000).
2.3. Quantifying vampire bat diet
We used previously published data on stable isotopes of carbon (δ13C) and nitrogen (δ15N) from hair samples (n total = 29; n LAR = 13, n KK = 16). to infer long‐term feeding patterns of each individual bat (Becker, Czirják, et al., 2018b). δ13C and δ15N were quantified from dried fur with a Thermo Delta V isotope ratio mass spectrometer at the University of Georgia Center for Applied Isotope Studies.
2.4. Measuring functional immune defense
Innate immune function data for this study were published previously (Becker, Czirják, et al., 2018b); briefly, we assessed a functional measure of the innate immune defense in individual vampire bats by quantifying the ex vivo bacterial killing ability (BKA) of plasma samples against Escherichia coli ATCC 8739 (Tieleman, Williams, Ricklefs, & Klasing, 2005). In bat plasma, this pathogen is cleared mainly through complement proteins (Moore et al., 2011). Using the methods described in Becker, Chumchal, et al. (2017), we used the microplate reader method with 1:8 dilutions of plasma to phosphate‐buffered saline run in duplicate and challenged with a 104 bacteria/ml solution (E power Microorganisms #0483E7; Microbiologics Inc). BKA is thus expressed at the percentage of E. coli cleared by the sample relative to the positive control.
2.5. Statistical analyses
We first used linear regressions to assess if δ13C and δ15N varied per site. We also derived the standard ellipse area corrected for small sample size (SEAc) for δ13C and δ15N as a proxy for the diversity of feeding strategies and thus dietary homogenization per site (Jackson, Inger, Parnell, & Bearhop, 2011). We used a permutational multivariate analysis of variance (PERMANOVA) to assess differences in isotopic position (matrix of δ13C and δ15N) according to site.
We next assessed whether site and long‐term bat diet (δ13C and δ15N) predicted microbiota diversity using univariate and multivariate tests. For alpha diversity, we fit linear regression models with log‐transformed Shannon diversity as the dependent variable and separately included site (n = 30) and both δ13C and δ15N (n = 23) as predictors; site and diet were not included in the same model owing to the different sample size and to avoid overfitting. To test for differences in microbial beta diversity according to site and diet, we performed a PERMANOVA on both weighted and unweighted UniFrac distances using the vegan (v. 2.5‐4) package (Lozupone, Hamady, & Knight, 2006; Oksansen et al., 2017). Mantel tests were performed to test for associations between δ13C/δ15N distance matrices and both weighted and unweighted Unifrac matrices. We visualized differences in microbial membership and composition according to sampling site using principal coordinates analysis (PCoA) using package phyloseq (v. 1.26.1) (Gower, 1966; McMurdie & Holmes, 2013).
We used DESeq2 (v. 1.22.2) (Love, Huber, & Anders, 2014; McMurdie & Holmes, 2014) to test the effects of diet (as inferred by δ13C and δ15N) on microbiota composition on an ASV‐by‐ASV basis. DESeq2 models raw OTU counts using a negative binomial distribution that accounts for differences in library sizes and determines significance of explanatory variables using the Wald test (Love et al., 2014). Using this model, we tested for ASV enrichment as a function of binned δ13C values. Following previous studies of vampire bats, we considered values within the interval of −13 to −7 to be representative of more‐livestock diets, with values outside this interval corresponding to less‐livestock diets (Voigt & Kelm, 2006a). We considered ASVs to be significantly enriched if the Benjamini–Hochberg adjusted p value was <0.05.
Finally, we assessed the relationship between BKA and (a) the relative abundance of core ASVs identified above and (b) alpha diversity to test how the community composition and diversity of the gut microbiota correlate with innate immune defense. We transformed proportional BKA to be bound within the 0 and 1 interval and used the logit‐transformed values as the dependent variable in a series of robust linear mixed effects models with the robustlmm package (Koller, 2016). Robust estimation methods help minimize the effect of potential outliers, which is particularly helpful when sample sizes are small. We modeled separate univariate relationships between microbial data (we used a square‐root transformation for the relative abundance of each core ASV; Zuur, Ieno, & Elphick, 2009) and BKA and included assay plate as a random effect. As rlmer() does not report p values, we report the slope and 95% confidence interval for each predictor (e.g., each core relative abundance and alpha diversity), interpreting no overlap with zero as statistically significant.
3. RESULTS
3.1. Dietary differences between sites
Within our sample of 29 vampire bats for which stable isotope data were available, individuals foraging within LAR (intact) and KK (fragmented) varied by δ13C (β LAR = −1.53, t = −2.29, p = 0.03) but only marginally by δ15N (β LAR = −0.86, t = −1.89, p = 0.07). However, KK bats showed narrower isotopic niche width (SEAc = 1.21) compared to LAR bats (SEAc = 14.41; Figure 3). Accordingly, the PERMANOVA showed that site predicted bat isotopic position (F 1,27 = 4.7, R 2 = 0.15, p = 0.01).
Figure 3.

Site variability in stable isotopes of carbon (δ13C) and nitrogen (δ15N) from vampire bats sampled in Belize (n = 29). Points show individual diet data from all bats sampled in 2015, while lines show the standard ellipse corrected for small sample size (SEAc). Red points = LAR (n = 13), blue points = Ka'Kabish (n = 16)
3.2. Core microbiota summary
Contamination filtering identified a total of seven putative contaminants which were filtered from the feature table (Figure A1A1), and the resulting cleaned feature table was used for the core microbiota summary and all statistical analyses. The overall structure of the 30 vampire bat microbiotas revealed that the dominant bacterial families were the Peptostreptococcaceae (Phylum Firmicutes), Enterobacteriaceae (Phylum Proteobacteria), Helicobacteraceae (Phylum Epsilonbacteraeota), Staphylococcaceae (Phylum Firmicutes), Mycoplasmataceae (Phylum Tenericutes), Halomonadaceae (Phylum Proteobacteria), and Bacillaceae (Phylum Firmicutes) (Figure 4; Table 1). The core microbiome, defined as those ASVs represented in >50% of the 30 samples (rarefied to 10,000 reads per sample), contained 12 ASVs (Table 1). Two ASVs, an unidentified member of the family Peptostreptococcaceae and a member of the genus Helicobacter, were detected in all samples (Table 1). The average relative abundance of the Peptostreptococcaceae core ASV was 45.0%, whereas the average relative abundance of Helicobacter across samples was 4.5%.
Figure 4.

Composition of vampire bat microbiotas (n = 30), showing relative abundance of top 25 bacteria at the genus level. Each stacked bar represents an individual bat. The dendrogram illustrates the similarity between the samples based on Bray–Curtis distance using the mean linkage method. The left bar illustrates relative abundance of bacterial genera across all samples
Table 1.
Amplicon sequence variants (ASVs) defining the core microbiota of Belizean vampire bats. To be considered part of the “core” microbiota, ASVs had to be present in >50% of the samples
| ASV ID | Phylum | Family | Genus | Prevalence | Prevalence (%) | Avg. abundance (%) |
|---|---|---|---|---|---|---|
| fcd55911156149fdda9de94a733cc51c | Epsilonbacteraeota | Helicobacteraceae | Helicobacter | 25 | 83.3 | 1.2 |
| 470e48a9a6ed554db28a235a56d894d2 | Epsilonbacteraeota | Helicobacteraceae | Helicobacter | 30 | 100 | 4.5 |
| c17deeb81d27df533b8c5d09f80f672f | Proteobacteria | Enterobacteriaceae | Edwardsiella | 21 | 70 | 13.9 |
| 50d98760f933669facdf7e0f5f4c7d2b | Proteobacteria | Halomonadaceae | Halomonas | 26 | 86.7 | 0.2 |
| 952668c4024daf247606aa20c6f6d96e | Firmicutes | Bacillaceae | Aeribacillus | 20 | 66.7 | 0.08 |
| aa9d7191529c931ebd368615d0531c8a | Firmicutes | Staphylococcaceae | Staphylococcus | 9 | 30 | 1.8 |
| 15d888a8d8e6c158974710b615dcbcdf | Firmicutes | Staphylococcaceae | Staphylococcus | 21 | 70 | 3.5 |
| 56b584fbd736b3d1f678e06b8a059663 | Tenericutes | Mycoplasmataceae | Mycoplasma | 16 | 53.3 | 0.42 |
| da16e0be25b62f12a3d9750211d42bd4 | Tenericutes | Mycoplasmataceae | Mycoplasma | 16 | 53.3 | 0.09 |
| e261c20b5c702a2249153b9f5d1eac42 | Firmicutes | Peptostreptococcaceae | Unclassified | 16 | 53.3 | 0.53 |
| c733ab7b076b07b518f42063ebc3351a | Firmicutes | Peptostreptococcaceae | Unclassified | 30 | 100 | 45 |
| 993f939073eee4a78f5e61cc57784442 | Firmicutes | Peptostreptococcaceae | Unclassified | 16 | 53.3 | 0.32 |
3.3. Effects of site and diet on microbiome communities
Alpha diversity was not significantly different between sites (F 1,28 = 1.6, R 2 = 0.06, p = 0.21), although LAR had weakly higher richness than KK (β = 0.15, t = 1.28). Diet variation did not predict alpha diversity through either δ13C (β = 0.05, t = 0.99, p = 0.33) or δ15N (β = 0.01, t = 0.22, p = 0.83). We found no statistically significant differences in bat gut microbial beta diversity between sites, as inferred by PERMANOVA on both unweighted (Figure 5; F 1,28 = 1.13, R 2 = 0.039, p = 0.27) and weighted (Figure 5; F 1,28 = 1.01, R 2 = 0.035, p = 0.34) Unifrac distances. However, multivariate dispersion around the centroid for unweighted Unifrac distances was significantly different between sites, indicating higher variability in community membership in KK compared with LAR (Figure 5; F 1,28 = 5.0344, N perm = 999, p = 0.04). Similarly, beta diversity of bat gut microbiotas did not differ as a function of δ13C (Mantel test, unweighted Unifrac r = −0.27, p = 0. 99; weighted Unifrac r = 0.05, p = 0.29) or δ15N (Mantel test, unweighted Unifrac, r = −0.29, p = 0.99; weighted Unifrac r = 0.10, p = 0.20).
Figure 5.

Principal coordinates analysis (PCoA) plots of vampire bat microbiotas sampled in Belize (2015) according to sampling site. Points represent (a) unweighted and (b) weighted UniFrac distances for individual microbiotas, while lines indicate 95% confidence ellipses around group centroid
To assess whether individual ASVs vary in response to diet, we performed an enrichment test using the DESeq2 function. Because δ13C, but not δ15N, differed significantly between KK and LAR vampire bats, we performed enrichment analysis on δ13C values only. Analysis of binned δ13C values revealed that several core taxa belonging to the genera Edwardsiella, Streptococcus, and Staphylococcus were enriched in the high‐livestock group compared with the low‐livestock group (Figure 6, Table 2). Vampire bats from both sites fell across both δ13C bins (Figure B1B1), suggesting a decoupling of livestock consumption and sampling site. One ASV of the genus Staphylococcus was depleted in the high‐livestock group compared with the low‐livestock group (Figure 6).
Figure 6.

Significantly enriched ASVs stratified by low‐ or high‐livestock δ13C values. Points correspond to log2fold enrichment values, where values >0 reflect enrichment of the taxon in the high‐livestock bin compared to low‐livestock bin. Values <0 reflect enrichment of the taxon in the low livestock bin compared to the high livestock bin
Table 2.
Differentially enriched amplicon sequence variants (ASVs) binned by low‐ or high‐livestock δ13C values
| ASV ID | baseMean | log2FoldChange | padj | Phylum | Family | Genus |
|---|---|---|---|---|---|---|
| b4b643e846091df086bc9d98161b0f26 | 551.468504 | 25.86 | 5.96E‐09 | Proteobacteria | Enterobacteriaceae | Edwardsiella |
| acc3df6ad875dc570a8c718ec2e89391 | 193.62763 | 24.41 | 4.40E‐08 | Proteobacteria | Enterobacteriaceae | Edwardsiella |
| 42ad2e83d3859624e59a514ba1cd042d | 7.55500713 | 19.90 | 1.69E‐05 | Firmicutes | Staphylococcaceae | Staphylococcus |
| a2051ef322b2c87b3306d7a2d8754065 | 78.719487 | −28.46 | 2.66E‐11 | Firmicutes | Staphylococcaceae | Staphylococcus |
| 945bb343f8dc3dd656b9ca15c3c0f09d | 185.036165 | 23.88 | 7.69E‐08 | Firmicutes | Staphylococcaceae | Staphylococcus |
| 42398c699c71f476da7c10146dd41246 | 7.80057541 | 19.95 | 1.69E‐05 | Firmicutes | Streptococcaceae | Streptococcus |
Positive log2fold change coefficients indicate that the ASV is more abundant in the high‐livestock group compared to low‐livestock group. Negative values indicate depletion of that ASV in the high‐livestock group compared to the low‐livestock group.
3.4. Microbiota composition and innate immunity
The relative abundance of core ASVs was significantly associated with bat immune defense (i.e., BKA, n total = 20; n LAR = 8, n KK = 12), with the direction and strength of this relationship differing among individual core ASVs (Figure 7). After accounting for outliers and assay plate, the relative abundances of Edwardsiella and Mycoplasma were both positively associated with BKA (Edwardsiella: β = 1.76, 95% CI = 0.88–2.65; Mycoplasma: β = 4.58, 95% CI = 1.7–7.46), while the relative abundance of Staphylococcus was negatively associated with BKA (β = −1.65, 95% CI = −3.29 to −0.02). The relative abundances of Helicobacter, Halomonas, and Aeribacillus showed no relationship with BKA (Figure 7). Our data suggested a trend for BKA to be negatively associated with alpha diversity, but confidence intervals overlapped with zero (β = −1.18, 95% CI = −2.68 to 0.33) .
Figure 7.

Robust LMMs testing the relationship between innate immunity and relative abundance of core gut microbiome taxa. Innate immunity was measured as % Escherichia coli killed during the assay by vampire bat blood serum. Hashed lines represent relationships where the 95% confidence interval overlaps with 0. Boxplot depicts Shannon diversity by site
4. DISCUSSION
Common vampire bats (D. rotundus) are one of three species of vampire bats found in the Neotropics and are the most specialized for feeding on mammalian prey (Goodwin & Greenhall, 1961; Greenhall, Joermann, Schmidt, & Seidel, 1983; Greenhall & Schmidt, 1988; Greenhall, Schmidt, & Lopez‐Forment, 1969; Turner, 1975). As such, vampire bats are uniquely poised to experience diet‐induced shifts in physiology and microbiota structure as a result of livestock rearing in the Neotropics. We demonstrate here that land‐use change even at relatively fine spatial scales (c. 8 km) is associated with differences in vampire bat diets that may influence the abundance of core microbiome members. We found significant differences in diet breadth between vampire bats captured at the fragmented‐forest site (KK) versus the contiguous forest site (LAR) (Figure 3). Vampire bats from KK had δ13C signatures more indicative of consistent feeding on livestock (Becker, Chumchal, et al., 2017; Voigt & Kelm, 2006b) and narrower dietary breadth, suggesting greater dietary homogenization at KK than LAR, where diets were far more variable. This pattern of diet homogenization at KK is generally consistent across sampling years (2014–2017; D. J. Becker, unpublished data), suggesting that these data are representative of the long‐term trends in vampire diets at the two sites. Studies of stable isotopes and bloodmeal analysis suggest that vampire bats likely prefer cattle because cattle populations are continuously present and exist in higher density than native prey mammals (Becker, Czirják, et al., 2018b; Bobrowiec, Lemes, & Gribel, 2015; Voigt & Kelm, 2006b). Our study supports this diet preference and suggests that bats in protected, mature forests have more variable diets than those in isolated forest fragments adjacent to cattle pastures, even though some individuals from LAR had δ13C signatures which fell into the high‐livestock group (Figure B1B1). A possible explanation for higher diet variability at LAR is that bats in larger forest tracts must forage over wider areas because their prey is more widely dispersed across the landscape. However, more detailed studies of vampire bat movement using global positioning systems (GPS) radiotracking are needed to confirm this hypothesis (Fenton et al., 1992).
We report here a core vampire bat microbiota composed mostly of members of the phyla Firmicutes, Proteobacteria, Tenericutes, and Epsilonbacteraeota (Figure 3, Table 1). This composition is consistent with previous reports of vampire bat microbiotas (Carrillo‐Araujo et al., 2015; Ingala et al., 2018; Phillips et al., 2012). Vampire bats have gut microbiotas that are compositionally distinct from those of other bats, most likely as a result of selective pressures to optimize bacterial metabolism of bloodmeals (Zepeda Mendoza et al., 2018). Indeed, vampire bats share several key hemolytic bacterial symbionts, such as Aeromonas spp., with other blood‐feeding animals such as leeches (Muller, Pinus, & Schmidt, 1980), suggesting that hematophagous animal microbiome composition is closely associated with this specialized diet.
Given that diet is known to be a strong driver of microbiota community composition and function (Muegge et al., 2012; Phillips et al., 2017) and that some differences exist in vampire bat dietary breadth between KK and the LAR, we tested for differences in the microbiotas of vampire bats between these contrasting sites. We found no significant differences in either alpha or beta diversity between sites (Figure 4). Previous studies in primates have found that degraded habitats are associated with lower microbiota diversity at similar spatial scales (Amato et al., 2013; Barelli et al., 2015), yet we failed to detect a difference here. It is possible that vampire bats in this region of Belize are not philopatric to particular roosts, such that some of our captures at KK could have been previously foraging or living in LAR, and vice versa. In this scenario, we would expect any differences in microbiota structure or membership between sites to be confounded. More detailed studies on vampire bat roost fidelity in Belize are needed, as there appears to be variability in roost philopatry across the range of D. rotundus. For example, a study of Argentine vampire bats found that only about 15% of individuals were recaptured at the roost where they were initially captured (Delpietro, Russo, Carter, Lord, & Delpietro, 2017), while another study found no movement among Peruvian vampire bats in roosts located just 2.2 km apart (Streicker et al., 2012). Though we found no difference in beta diversity between sites, we did note increased heterogeneity of KK gut microbiotas compared with LAR, despite the fact that the bats at KK have more homogenous diets (Figure 3). Previous studies have reported similar patterns, where perturbed animal gut communities are characterized by increased dispersion around a “healthy” centroid rather than deterministic community shifts (Zaneveld, McMinds, & Thurber, 2017). We speculate that a possible explanation for this observation is that local livestock is treated with antibiotics which may be consumed by the vampire bats during feeding, triggering increased microbial turnover and influencing the heterogeneity in community structure we report here. More detailed data collection on local antibiotic use in livestock could be incorporated to test this hypothesis in the future.
Because we found differences in dietary breadth (Figure 3) but not microbiota structure (Figure 5) between KK and LAR, we also tested for correlations between diet and microbiota structure irrespective of sampling site; however, we also did not detect significant correlations between either weighted or unweighted Unifrac and δ13C distance matrices. We did not test for correlations between Unifrac matrices and δ15N, because we found no significant differences in this diet metric (which approximates trophic level) among our samples. Previous studies have demonstrated a very narrow range of δ15N in vampire bats from non‐coastal habitats (Streicker & Allgeier, 2016), and that wildlife and domestic prey do not vary appreciably in this isotope in this region of Belize (Becker, Longo, et al., 2017). Isotopic ratios measure long‐term dietary trends (Tieszen, Boutton, Tesdahl, & Slade, 1983) recorded on the time scale it takes the animal to generate the tissue (in our case, hair), whereas the gut microbial community can experience turnover within hours or days following a dietary shift (David et al., 2014). In light of this evidence, it is possible that stable isotopes of hair do not capture dietary changes on a time scale appropriate for analyzing links with microbial turnover in the gut. Future studies should consider using short‐term diet monitoring techniques, such as DNA metabarcoding of fecal samples (Clare et al., 2014), to better assess how microbiota composition changes in response to diet shifts.
Though overall community composition and membership were not related to long‐term diet, differential enrichment analysis showed that some individual core ASVs were enriched in the higher‐livestock group of bats compared to those with lower‐livestock diets (Figure 6). Importantly, both high and low δ13C values were distributed across vampire bats from the two sampling sites, indicating that bats in the contiguous forest site (LAR) are also leaving the reserve to forage on cattle (Figure S2). The reserve (LAR) is within several kilometers of cattle pastures (Figure 2), which falls within the reported home range size of 2–3 km2 for D. rotundus (Streicker et al., 2012; Trajano, 1996). Our results suggest that the interplay between changes in long‐term diet and shifts in microbiota are perhaps more complex than previously thought, while overall community structure may resist perturbation across sites, shifts in the relative abundance of select taxa such as those reported here could still have functional implications for host. Future studies could incorporate shotgun metagenomic techniques to more directly measure the functions associated with microbial taxa changing in response to diet.
Because vampire bats may be important reservoirs of zoonotic pathogens (Condori‐Condori, Streicker, Cabezas‐Sanchez, & Velasco‐Villa, 2013; Mayen, 2003; Volokhov et al., 2017; Zetun, Hoffmann, Silva, Souza, & Langoni, 2009) and the microbiota is known to modulate immunity (Khosravi & Mazmanian, 2013; Thaiss et al., 2016), we tested for associations between innate immune defense and microbiota structure. We found significant relationships between the proportional abundance of some core microbiota with the ability of host plasma to kill E. coli (Figure 7). Future studies that incorporate additional measures of bat immune phenotypes, especially those quantifying different arms of the immune system or expression of various immune genes (e.g., cytokines), could shed further light on links between the microbiota and infection dynamics (Demas, Zysling, Beechler, Muehlenbein, & French, 2011; Fassbinder‐Orth, 2014).
A limitation of amplicon‐based studies is that 16S regions often lack the sensitivity to distinguish among bacterial species and/or strains, which may have different metabolic functions despite sharing identical 16S sequences (Antony‐Babu et al., 2017). Future studies using shotgun metagenomic techniques could be used to analyze how the microbiota changes on a gene‐by‐gene basis in response to habitat quality, which may give more detailed insight to how these changes in vampire bats impact metagenome functions related to immunity. For example, a hologenomic study of vampire bats found that their microbiotas were characterized by an enrichment of potentially protective bacterial genes originating from Amycolatopsis mediterranei, which is known to produce antiviral compounds (Zepeda Mendoza et al., 2018). Expansion of our study (a) across a quantitative fragmentation gradient and (b) using shotgun metagenomic techniques could provide detailed insights into the functional consequences of habitat fragmentation on vampire bat microbiotas. In particular, the inclusion of a truly pristine forest site would better contextualize our isotopic data by providing a site where cattle are infrequently available or altogether absent.
In summary, we demonstrate here that changes in land use in Belize have measurable associations with both dietary breadth and gut microbiota variability, but not composition, in common vampire bats. We found bacterial genera respond to diet, but not site, suggesting that diet is related to but decoupled from sampling site in our dataset. Our results join a growing body of evidence suggesting that the microbiota can respond to environmental change. Even though we did not detect dysbiosis in these animals, we recognize that dysbiosis of the microbiota may be monitored as an early warning sign about potential downstream effects on host ecology and health (Bahrndorff, Alemu, Alemneh, & Lund Nielsen, 2016; Cheng et al., 2015; Lemieux‐Labonté, Simard, Willis, & Lapointe, 2017; Thomason, Mullen, Belden, May, & Hawley, 2017). We further showed that the relative abundances of some core microbiota members are related to innate immune function. While future immunological work could expand upon these findings and their implication for infection states, this pattern suggests that shifts in this subset of the microbiota could have especially profound impacts on host susceptibility to infections and on fitness. As agricultural land conversion continues in Belize (Cherrington et al., 2010), humans and their domestic animals will come into more frequent contact with wildlife, including vampire bats. It will be essential to continue monitoring the diets, habitat use, and microbiotas of vampire bats to test hypotheses about the impacts of these variables on zoonotic disease cycles, especially of rabies but also of other bacterial pathogens. Future studies integrating microbiota analysis, detailed dietary analysis, GPS radiotracking, and other tools can leverage the power of these techniques to expand on the foundational work we present here.
5. CONFLICT OF INTEREST
None declared.
AUTHOR CONTRIBUTIONS
MRI analyzed and interpreted the data and wrote the manuscript. DJB collected field data, analyzed stable isotope and BKA data, secured funding, and helped write the manuscript. JBH collected rectal swabs, performed 16S amplicon sequencing, analyzed microbiota data, and helped write the manuscript. KK participated in discussions on metagenomics and analysis of 16S amplicon sequencing. NBS secured permits for field collection of samples, coordinated the field expedition, and helped write the manuscript.
Supporting information
ACKNOWLEDGMENTS
We thank M. Brock Fenton, John Hermanson, Alexandra Bentz, Neil Duncan, numerous members of the Lamanai bat crew, and the staff of the Lamanai Field Research Center for assistance with sampling logistics, bat capture, and research permits. We also thank Tom Maddox and the University of Center for Applied Isotope Studies for stable isotope analyses, Sonia Altizer for providing laboratory space, and Katie Smith for laboratory assistance with the bacterial killing assay. MRI was supported by a Graduate Research Fellowship from the Richard Gilder Graduate School at the American Museum of Natural History. DJB was funded by an NSF Graduate Research Fellowship, ARCS Foundation Award, Sigma Xi, Odum School of Ecology, UGA Graduate School, the Explorer's Club, and UGA Biomedical and Health Sciences Institute. JBH and KK were supported by the Danish Natural Science Research Council and the Carlsberg Foundation. NBS was supported by the Department of Mammalogy at the American Museum of Natural History. The authors thank three anonymous reviewers whose comments greatly improved the quality of the manuscript.
APPENDIX A.
CONTAMINATION ELIMINATION METHODS AND RESULTS
We eliminated potential contaminants using the decontam R package v. 1.2.1. Because we did not sequence a negative extraction control, we used decontam's frequency‐filtering technique, which assumes a negative linear relationship between initial DNA concentration and frequency of the potential contaminant (Davis et al., 2018). If a contaminant model is a better fit to the data than a null model (where frequency is independent of DNA concentration), the amplicon sequence variant (ASV) is identified as a contaminant. The identified contaminants were removed from the phyloseq ASV feature table, and the cleaned feature table was used for all downstream analyses. The contaminants are shown in Figure A1A1.
Figure A1.

Contaminating ASVs identified by decontam frequency filtering. X‐axis shows the initial DNA concentration of microbiome samples as measured by fluorescent quantitation. The Y‐axis shows the frequency of the sequence variant. All ASVs shown above fit the contaminant model with p < 0.05
APPENDIX B.
DESEq2 ENRICHMENT RESULTS BY INDIVIDUAL AMPLICON SEQUENCE VARIANT
In the DESeq2 enrichment analysis, vampire bat samples from Ka'Kabish (KK) and Lamanai Archaeological Reserve (LAR) fell across both low‐ and high‐livestock bins, indicating that bats from these sites vary in the amount of livestock included in their diets. Of the six amplicon sequence variants (ASVs) found to be differentially enriched as a function of livestock consumption (Figure 6), nearly all had a mixture of individuals from KK and LAR (Figure B1B1).
Figure B1.
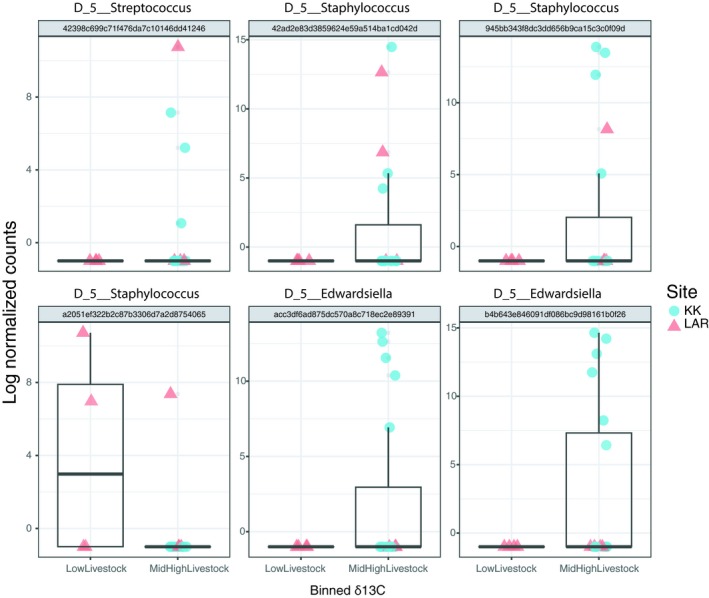
Log normalized counts of ASVs identified as differentially enriched between the low and high livestock δ13C bins (p adj < 0.05). Points correspond to vampire bat individuals. X‐axis depicts whether that sample was included in the high‐livestock interval or the low‐livestock interval. Boxplots depict the median count bounded by lower and upper quartiles. The whiskers represent the standard error of the median values
Ingala MR, Becker DJ, Bak Holm J, Kristiansen K, Simmons NB. Habitat fragmentation is associated with dietary shifts and microbiota variability in common vampire bats. Ecol Evol. 2019;9:6508–6523. 10.1002/ece3.5228
Data Availability Statement: Raw, demultiplexed 16S sequence reads are archived publicly on the NCBI Short Read Archive (SRA) under BioProject #PRJNA506223. Sample metadata, ASV phylogeny, and taxonomically annotated ASV feature table are available on FigShare: https://figshare.com/projects/Habitat_Fragmentation_is_Associated_with_Dietary_and_Microbiome_Variability_in_Common_Vampire_Bats/57041.
DATA AVAILABILITY STATEMENT
Raw, demultiplexed 16S sequence reads are archived publicly on the NCBI Short Read Archive (SRA) under BioProject #PRJNA506223. Sample metadata, ASV phylogeny, and taxonomically annotated ASV feature table are available on FigShare: https://figshare.com/projects/Habitat_Fragmentation_is_Associated_with_Dietary_and_Microbiome_Variability_in_Common_Vampire_Bats/57041.
REFERENCES
- Altizer, S. , Becker, D. J. , Epstein, J. H. , Forbes, K. M. , Gillespie, T. R. , Hall, R. J. , … Streicker, D. G. (2018). Food for contagion: Synthesis and future directions for studying host–parasite responses to resource shifts in anthropogenic environments. Philosophical Transactions of the Royal Society B: Biological Sciences, 373, 20170102 10.1098/rstb.2017.0102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amato, K. R. , Leigh, S. R. , Kent, A. , Mackie, R. I. , Yeoman, C. J. , Stumpf, R. M. , … Garber, P. A. (2014). The gut microbiota appears to compensate for seasonal diet variation in the wild black howler monkey (Alouatta pigra). Microbial Ecology, 69, 434–443. 10.1007/s00248-014-0554-7 [DOI] [PubMed] [Google Scholar]
- Amato, K. R. , Yeoman, C. J. , Kent, A. , Righini, N. , Carbonero, F. , Estrada, A. , … Leigh, S. R. (2013). Habitat degradation impacts black howler monkey (Alouatta pigra) gastrointestinal microbiomes. ISME Journal, 716, 1344–1353. 10.1038/ismej.2013.16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amman, B. R. , Carroll, S. A. , Reed, Z. D. , Sealy, T. K. , Balinandi, S. , Swanepoel, R. , … Towner, J. S. (2012). Seasonal Pulses of Marburg Virus Circulation in Juvenile Rousettus aegyptiacus Bats Coincide with Periods of Increased Risk of Human Infection. PLoS Pathogens, 8, e1002877 10.1371/journal.ppat.1002877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antony‐Babu, S. , Stien, D. , Eparvier, V. , Parrot, D. , Tomasi, S. , & Suzuki, M. T. (2017). Multiple Streptomyces species with distinct secondary metabolomes have identical 16S rRNA gene sequences. Scientific Reports, 7, 11089 10.1038/s41598-017-11363-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apprill, A. , Mcnally, S. , Parsons, R. , & Weber, L. (2015). Minor revision to V4 region SSU rRNA 806R gene primer greatly increases detection of SAR11 bacterioplankton. Aquatic Microbial Ecology, 75, 129–137. 10.3354/ame01753 [DOI] [Google Scholar]
- Araújo‐Pérez, F. , McCoy, A. N. , Okechukwu, C. , Carroll, I. M. , Smith, K. M. , Jeremiah, K. , … Keku, T. O. (2012). Differences in microbial signatures between rectal mucosal biopsies and rectal swabs. Gut Microbes, 3, 530–535. 10.4161/gmic.22157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bahrndorff, S. , Alemu, T. , Alemneh, T. , & Lund Nielsen, J. (2016). The microbiome of animals: Implications for conservation biology. International Journal of Genomics, 2016, 6508–7. 10.1155/2016/5304028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balcázar, J. L. , De Blas, I. , Ruiz‐Zarzuela, I. , Vendrell, D. , Calvo, A. C. , Márquez, I. , … Muzquiz, J. L. (2007). Changes in intestinal microbiota and humoral immune response following probiotic administration in brown trout (Salmo trutta). British Journal of Nutrition, 97, 522–527. [DOI] [PubMed] [Google Scholar]
- Barelli, C. , Albanese, D. , Donati, C. , Pindo, M. , Dallago, C. , Rovero, F. , … De Filippo, C. (2015). Habitat fragmentation is associated to gut microbiota diversity of an endangered primate: Implications for conservation. Scientific Reports, 5, 14862 10.1038/srep14862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker, C. G. , Longo, A. V. , Haddad, C. F. B. , & Zamudio, K. R. (2017). Land cover and forest connectivity alter the interactions among host, pathogen and skin microbiome. Proceedings of the Royal Society B: Biological Sciences, 284(1861), 20170582 10.1098/rspb.2017.0582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker, D. J. , Bergner, L. M. , Bentz, A. B. , Orton, R. J. , Altizer, S. , & Streicker, D. G. (2018a). Genetic diversity, infection prevalence, and possible transmission routes of Bartonella spp. in vampire bats. PLoS Neglected Tropical Diseases, 12(9), e0006786 10.1371/journal.pntd.0006786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker, D. J. , Chumchal, M. M. , Bentz, A. B. , Platt, S. G. , Czirják, G. Á. , Rainwater, T. R. , … Streicker, D. G. (2017). Predictors and immunological correlates of sublethal mercury exposure in vampire bats. Royal Society Open Science, 4, 170073 10.1098/rsos.170073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker, D. J. , Czirják, G. Á. , Volokhov, D. V. , Bentz, A. B. , Carrera, J. E. , Camus, M. S. , … Streicker, D. G. (2018b). Livestock abundance predicts vampire bat demography, immune profiles and bacterial infection risk. Philosophical Transactions of the Royal Society B: Biological Sciences, 373, 20170089 10.1098/rstb.2017.0089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bobrowiec, P. E. D. , Lemes, M. R. , & Gribel, R. (2015). Prey preference of the common vampire bat (Desmodus rotundus. Chiroptera) using molecular analysis. Journal of Mammalogy, 96, 54–63. 10.1093/jmammal/gyu002 [DOI] [Google Scholar]
- Bokulich, N. A. , Kaehler, B. D. , Rideout, J. R. , Dillon, M. , Bolyen, E. , Knight, R. , … Gregory Caporaso, J. (2018). Optimizing taxonomic classification of marker‐gene amplicon sequences with QIIME 2 ’ s q2‐feature‐classifier plugin. Microbiome, 6, 6508–17. 10.1186/s40168-018-0470-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolyen, E. , Rideout, J. R. , Dillon, M. R. , Bokulich, N. A. , Abnet, C. , Ghalith, G. A. , … Bai, Y. (2018). QIIME 2: Reproducible, interactive, scalable, and extensible microbiome data science. PeerJ Preprints, e27295v1 10.7287/peerj.preprints.27295v1. [DOI] [Google Scholar]
- Callahan, B. J. , McMurdie, P. J. , Rosen, M. J. , Han, A. W. , Johnson, A. J. A. , & Holmes, S. P. (2016). DADA2: High resolution sample inference from Illumina amplicon data. Nature Methods, 13, 48–56. 10.1016/j.semcancer.2015.04.010.Targeting [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caporaso, J. G. , Kuczynski, J. , Stombaugh, J. , Bittinger, K. , Bushman, F. D. , Costello, E. K. , … Knight, R. (2010). QIIME allows analysis of high‐throughput community sequencing data. Nature Methods, 7, 335–336. 10.1038/nmeth.f.303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caporaso, J. G. , Lauber, C. L. , Walters, W. A. , Berg‐Lyons, D. , Lozupone, C. A. , Turnbaugh, P. J. , … Knight, R. (2011). Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. Proceedings of the National Academy of Sciences, 108(Suppl), 4516–4522. 10.1073/pnas.1000080107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrillo‐Araujo, M. , TaÅŸ, N. , Alcántara‐Hernández, R. J. , Gaona, O. , Schondube, J. E. , MedellÃn, R. A. , … Falcón, L. I. (2015). Phyllostomid bat microbiome composition is associated to host phylogeny and feeding strategies. Frontiers in Microbiology, 6, 6508–9. 10.3389/fmicb.2015.00447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng, Y. , Fox, S. , Pemberton, D. , Hogg, C. , Papenfuss, A. T. , & Belov, K. (2015). The Tasmanian devil microbiome‐implications for conservation and management. Microbiome, 3, 76 10.1186/s40168-015-0143-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherrington, E. A. , Ek, E. , Cho, P. , Howell, B. F. , Hernández, B. E. , Andersen, E. R. , … Irwin, D. E. (2010). SERVIR: Forest Cover & Deforestation in Belize 1980-2010. Technical report.
- Clare, E. L. , Goerlitz, H. R. , Drapeau, V. A. , Holderied, M. W. , Adams, A. M. , Nagel, J. , … Fenton, M. B. (2014). Trophic niche flexibility in Glossophaga soricina: How a nectar seeker sneaks an insect snack. Functional Ecology, 28, 632–641. 10.1111/1365-2435.12192 [DOI] [Google Scholar]
- Condori‐Condori, R. E. , Streicker, D. G. , Cabezas‐Sanchez, C. , & Velasco‐Villa, A. (2013). Enzootic and Epizootic Rabies Associated with Vampire Bats, Peru. Emerging Infectious Diseases, 19(9), 1463–1469. 10.3201/eid1909.130083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- David, L. A. , Maurice, C. F. , Carmody, R. N. , Gootenberg, D. B. , Button, J. E. , Wolfe, B. E. , … Turnbaugh, P. J. (2014). Diet rapidly and reproducibly alters the human gut microbiome. Nature, 505, 559–563. 10.1038/nature12820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis, N. M. , Proctor, D. , Holmes, S. P. , Relman, D. A. , & Callahan, B. J. (2018). Simple statistical identification and removal of contaminant sequences in marker‐gene and metagenomics data. Microbiome, 6, 221499 10.1101/221499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delpietro, H. A. , Russo, R. G. , Carter, G. G. , Lord, R. D. , & Delpietro, G. L. (2017). Reproductive seasonality, sex ratio and philopatry in Argentina's common vampire bats. Royal Society Open Science, 4, 160959 10.1098/rsos.160959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demas, G. E. , Zysling, D. A. , Beechler, B. R. , Muehlenbein, M. P. , & French, S. S. (2011). Beyond phytohaemagglutinin: Assessing vertebrate immune function across ecological contexts. Journal of Animal Ecology, 80(4), 710–730. 10.1111/j.1365-2656.2011.01813.x [DOI] [PubMed] [Google Scholar]
- Evans, J. K. , Buchanan, K. L. , Griffith, S. C. , Klasing, K. C. , & Addison, B. (2017). Ecoimmunology and microbial ecology: Contributions to avian behavior, physiology, and life history. Hormones and Behavior, 88, 112–121. 10.1016/j.yhbeh.2016.12.003 [DOI] [PubMed] [Google Scholar]
- Fassbinder‐Orth, C. A. (2014). Methods for quantifying gene expression in ecoimmunology: From qPCR to RNA‐Seq. Integrative and Comparative Biology, 54(3), 396–406. 10.1093/icb/icu023 [DOI] [PubMed] [Google Scholar]
- Fenton, M. B. , Acharya, L. , Audet, D. , Hickey, M. B. C. , Merriman, C. , Obrist, M. K. , … Adkins, B. (1992). Phyllostomid Bats (Chiroptera: Phyllostomidae) as Indicators of Habitat Disruption in the Neotropics. Biotropica, 24, 440 10.2307/2388615 [DOI] [Google Scholar]
- Goodwin, G. G. , & Greenhall, A. M. (1961). A review of the bats of Trinidad and Tobago: Descriptions, rabies infection, and ecology. New York, NY: American Museum of Natural History. [Google Scholar]
- Gower, J. C. (1966). Some distance properties of latent root and vector methods used in multivariate analysis. Biometrika, 53(3/4), 325 10.2307/2333639 [DOI] [Google Scholar]
- Greenhall, A. M. , Joermann, G. , Schmidt, U. , & Seidel, M. R. (1983). Desmodus rotundus. Mammalian Species, 202, 6508–6. 10.2307/3503895 [DOI] [Google Scholar]
- Greenhall, A. M. , & Schmidt, U. (1988). Natural hist of vampire bats. Boca Raton, FL: CRC Press. [Google Scholar]
- Greenhall, A. M. , Schmidt, U. , & Lopez‐Forment, W. (1969). Field observations on the mode of attack of the vampire bat (Desmodus rotundus) in Mexico. Anales Del Instituto De Biologia UNAM Serie Zoologia, 245–252. [Google Scholar]
- Hale, V. L. , Tan, C. L. , Knight, R. , & Amato, K. R. (2015). Effect of preservation method on spider monkey (Ateles geoffroyi) fecal microbiota over 8 weeks. Journal of Microbiol Methods, 113, 16–26. 10.1016/j.mimet.2015.03.021 [DOI] [PubMed] [Google Scholar]
- Hanning, I. , & Diaz‐Sanchez, S. (2015). The functionality of the gastrointestinal microbiome in non‐human animals. Microbiome, 3, 51 10.1186/s40168-015-0113-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrera, J. P. , Duncan, N. , Clare, E. , Fenton, M. B. , & Simmons, N. (2018). Disassembly of fragmented bat communities in Orange Walk District, Belize. Acta Chiropterologica, 20, 147–159. 10.3161/15081109ACC2018.20.1.011 [DOI] [Google Scholar]
- Hoare, C. A. (1965). Vampire bats as vectors and hosts of equine and bovine trypanosomes. Acta Tropica, 22, 204. [PubMed] [Google Scholar]
- Hooper, L. V. , Littman, D. R. , & Macpherson, A. J. (2012). Interactions between the microbiota and the immune system. Science, 336(6086), 1268–1273. 10.1126/science.1223490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ichinohe, T. , Pang, I. K. , Kumamoto, Y. , Peaper, D. R. , Ho, J. H. , Murray, T. S. , & Iwasaki, A. (2011). Microbiota regulates immune defense against respiratory tract influenza A virus infection. Proceedings of the National Academy of Sciences, 108, 5354–5359. 10.1073/pnas.1019378108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingala, M. R. , Simmons, N. B. , Wultsch, C. , & Krampis, K. (2018). Comparing microbiome sampling methods in a wild mammal : Fecal and intestinal samples record different signals of host ecology, evolution. Frontiers in Microbiology, 9, 6508–13. 10.3389/fmicb.2018.00803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson, A. L. , Inger, R. , Parnell, A. C. , & Bearhop, S. (2011). Comparing isotopic niche widths among and within communities: SIBER – Stable Isotope Bayesian Ellipses in R. Journal of Animal Ecology, 80(3), 595–602. 10.1111/j.1365-2656.2011.01806.x [DOI] [PubMed] [Google Scholar]
- Karst, S. M. , Dueholm, M. S. , McIlroy, S. J. , Kirkegaard, R. H. , Nielsen, P. H. , & Albertsen, M. (2016). Thousands of primer‐free, high‐quality, full‐length SSU rRNA sequences from all domains of life. Biorxiv, 10, 070771 10.1101/070771 [DOI] [Google Scholar]
- Kau, A. L. , Ahern, P. P. , Griffin, N. W. , Goodman, A. L. , & Gordon, J. I. (2011). Human nutrition, the gut microbiome and the immune system. Nature, 474, 327–336. 10.1038/nature10213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khosravi, A. , & Mazmanian, S. K. (2013). Disruption of the gut microbiome as a risk factor for microbial infections. Current Opinion in Microbiology, 16, 221–227. 10.1016/j.mib.2013.03.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koller, M. (2016). robustlmm: An R package for robust estimation of linear mixed‐effects models. Journal of Statistical Software, 75(6), 6508–24. 10.18637/jss.v075.i06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemieux‐Labonté, V. , Simard, A. , Willis, C. K. R. , & Lapointe, F.‐J. (2017). Enrichment of beneficial bacteria in the skin microbiota of bats persisting with white‐nose syndrome. Microbiome, 5, 6508–14. 10.1186/s40168-017-0334-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love, M. I. , Huber, W. , & Anders, S. (2014). Moderated estimation of fold change and dispersion for RNA‐seq data with DESeq2. Genome Biology, 15, 550 10.1186/s13059-014-0550-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lozupone, C. , Hamady, M. , & Knight, R. (2006). UniFrac‐an online tool for comparing microbial community diversity in a phylogenetic context. BMC Bioinformatics, 7, 371 10.1186/1471-2105-7-371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macpherson, A. J. , & Harris, N. L. (2004). Interactions between commensal intestinal bacteria and the immune system. Nature Reviews Immunology, 4, 478–485. 10.1038/nri1373 [DOI] [PubMed] [Google Scholar]
- Mayen, F. (2003). Haematophagous bats in Brazil, their role in rabies transmission, impact on public health, livestock industry and alternatives to an indiscriminate reduction of bat population. Journal of Veterinary Medicine Series B, 50(10), 469–472. 10.1046/j.1439-0450.2003.00713.x [DOI] [PubMed] [Google Scholar]
- McCord, A. I. , Chapman, C. A. , Weny, G. , Tumukunde, A. , Hyeroba, D. , Klotz, K. , … Goldberg, T. L. (2014). Fecal microbiomes of non‐human primates in Western Uganda reveal species‐specific communities largely resistant to habitat perturbation. American Journal of Primatology, 76(4), 347–354. 10.1002/ajp.22238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenzie, V. J. , Song, S. J. , Delsuc, F. , Prest, T. L. , Oliverio, A. M. , Korpita, T. M. , … Knight, R. (2017). The effects of captivity on the mammalian gut microbiome. Integrative and Comparative Biology, 57, 690–704. 10.1093/icb/icx090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMurdie, P. J. , & Holmes, S. (2013). Phyloseq: An R package for reproducible interactive analysis and graphics of microbiome census data. PLoS ONE, 8, e61217 10.1371/journal.pone.0061217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMurdie, P. J. , & Holmes, S. (2014). Waste not, want not: Why rarefying microbiome data is inadmissible. PLoS Computational Biology, 10, e1003531 10.1371/journal.pcbi.1003531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore, M. S. , Reichard, J. D. , Murtha, T. D. , Zahedi, B. , Fallier, R. M. , & Kunz, T. H. (2011). Specific alterations in complement protein activity of little Brown Myotis (Myotis lucifugus) Hibernating in white‐nose syndrome affected sites. PLoS ONE, 6, e27430 10.1371/journal.pone.0027430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muegge, B. D. , Kuczynski, J. , Knights, D. , Clemente, J. C. , Fontana, L. , Henrissat, B. , … Gordon, J. I. (2012). Diet drives convergence in gut microbiome functions across mammalian phylogeny and within humans. Science, 332, 970–974. 10.1126/science.1198719.Diet [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller, H. E. , Pinus, M. , & Schmidt, U. (1980). Aeromonas hydrophila as a normal intestinal bacterium of the vampire bat (Desmodus rotundus). Zentralblatt für Veterinärmedizin Reihe B, 27, 419–424. [PubMed] [Google Scholar]
- O'Sullivan, A. , He, X. , McNiven, E. M. S. , Haggarty, N. W. , Lönnerdal, B. , & Slupsky, C. M. (2013). Early diet impacts infant rhesus gut microbiome, immunity, and metabolism. Journal of Proteome Research, 12(6), 2833–2845. 10.1021/pr400170 [DOI] [PubMed] [Google Scholar]
- Oksansen, J. , Blanchet, F. G. , Friendly, M. , Kindt, R. , Legendre, P. , & McGlinn, D. (2017). vegan: Community Ecology Package. v. 2.4‐5. Retrieved from https://cran.r-project.org/package=vegan
- Olival, K. J. , Hosseini, P. R. , Zambrana‐Torrelio, C. , Ross, N. , Bogich, T. L. , & Daszak, P. (2017). Host and viral traits predict zoonotic spillover from mammals. Nature, 546, 646–650. 10.1038/nature22975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedersen, A. B. , & Babayan, S. A. (2011). Wild immunology. Molecular Ecology, 20, 872–880. 10.1111/j.1365-294X.2010.04938.x [DOI] [PubMed] [Google Scholar]
- Phillips, C. D. , Hanson, J. , Wilkinson, J. E. , Koenig, L. , Rees, E. , Webala, P. , & Kingston, T. (2017). Microbiome structural and functional interactions across host dietary niche space. Integrative and Comparative Biology, 57, 743–755. 10.1093/icb/icx011 [DOI] [PubMed] [Google Scholar]
- Phillips, C. D. , Phelan, G. , Dowd, S. E. , McDONOUGH, M. M. , Ferguson, A. W. , Delton hanson, J. , … Baker, R. J. (2012). Microbiome analysis among bats describes influences of host phylogeny, life history, physiology and geography. Molecular Ecology, 21, 2617–2627. 10.1111/j.1365-294X.2012.05568.x [DOI] [PubMed] [Google Scholar]
- Plowright, R. K. , Eby, P. , Hudson, P. J. , Smith, I. L. , Westcott, D. , Bryden, W. L. , … McCallum, H. (2015). Ecological dynamics of emerging bat virus spillover. Proceedings of the Royal Society B, 282, 20142124 10.1098/rspb.2014.2124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Post, D. M. (2002). Using stable isotopes to estimate trophic position: Models, methods, and assumptions. Ecology, 83, 703–718. 10.1890/0012-9658(2002)083[0703:USITET]2.0.CO;2 [DOI] [Google Scholar]
- Reyes, R. , Pena, J. A. , Burrowes, P. A. , Medina, D. , Belden, L. K. , & Hughey, M. C. (2017). Skin bacterial microbiome of a generalist Puerto Rican frog varies along elevation and land use gradients. PeerJ, 10.7717/peerj.3688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider, M. C. , Romijn, P. C. , Uieda, W. , Tamayo, H. , Silva, D. F. D. , Belotto, A. , … Leanes, L. F. (2009). Rabies transmitted by vampire bats to humans: An emerging zoonotic disease in Latin America? Revista Panamericana de Salud Pública, 25, 260–269. 10.1590/S1020-49892009000300010 [DOI] [PubMed] [Google Scholar]
- Sikes, R. S. , Bryan, J. A. II , Byman, D. , Danielson, B. J. , Eggleston, J. , Gannon, M. R. , … Willoughby, J. R. (2016). 2016 Guidelines of the American Society of Mammalogists for the use of wild mammals in research and education. Journal of Mammalogy, 97, 663–688. 10.1093/jmammal/gyw078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stecher, B. , Maier, L. , & Hardt, W.‐D. (2013). “Blooming” in the gut: How dysbiosis might contribute to pathogen evolution. Nature Reviews Microbiology, 11, 277–284. 10.1038/nrmicro2989 [DOI] [PubMed] [Google Scholar]
- Streicker, D. G. , & Allgeier, J. E. (2016). Foraging choices of vampire bats in diverse landscapes: Potential implications for land‐use change and disease transmission. Journal of Applied Ecology, 53, 1280–1288. 10.1111/1365-2664.12690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Streicker, D. G. , Recuenco, S. , Valderrama, W. , Gomez Benavides, J. , Vargas, I. , Pacheco, V. , … Altizer, S. (2012). Ecological and anthropogenic drivers of rabies exposure in vampire bats: Implications for transmission and control. Proceedings of the Royal Society B, 279, 3384–3392. 10.1098/rspb.2012.0538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thaiss, C. A. , Zmora, N. , Levy, M. , & Elinav, E. (2016). The microbiome and innate immunity. Nature, 535, 65–74. 10.1038/nature18847 [DOI] [PubMed] [Google Scholar]
- Thomason, C. A. , Mullen, N. , Belden, L. K. , May, M. , & Hawley, D. M. (2017). Resident microbiome disruption with antibiotics enhances virulence of a colonizing pathogen. Scientific Reports, 7(1), 16177 10.1038/s41598-017-16393-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tieleman, B. I. , Williams, J. B. , Ricklefs, R. E. , & Klasing, K. C. (2005). Constitutive innate immunity is a component of the pace‐of‐life syndrome in tropical birds. Proceedings of the Royal Society B: Biological Sciences, 272, 1715–1720. 10.1098/rspb.2005.3155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tieszen, L. L. , Boutton, T. W. , Tesdahl, K. G. , & Slade, N. A. (1983). Fractionation and turnover of stable carbon isotopes in animal tissues: Implications for δ13C analysis of diet. Oecologia, 57(1–2), 32–37. 10.1007/BF00379558 [DOI] [PubMed] [Google Scholar]
- Tong, S. , Zhu, X. , Li, Y. , Shi, M. , Zhang, J. , Bourgeois, M. , … Donis, R. O. (2013). New World Bats Harbor Diverse Influenza A Viruses. PLoS Pathogens, 9, e1003657 10.1371/journal.ppat.1003657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trajano, E. (1996). Movements of cave bats in southeastern Brazil, with emphasis on the population ecology of the common vampire bat, Desmodus rotundus (Chiroptera). Biotropica, 28(1), 121 10.2307/2388777 [DOI] [Google Scholar]
- Trevelline, B. K. , Fontaine, S. S. , Hartup, B. K. , & Kohl, K. D. (2019). Conservation biology needs a microbial renaissance : A call for the consideration of host‐associated microbiota in wildlife management practices. Proceedings of the Royal Society B, 286(1895), 20182448 10.1098/rspb.2018.2448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turnbaugh, P. J. , Ridaura, V. K. , Faith, J. J. , Rey, F. E. , & Gordon, J. I. (2009). The Effect of Diet on the Human Gut Microbiome: A Metagenomic Analysis in Humanized Gnotobiotic Mice. Science Translational Medicine, 1, 6508–19. 10.1126/scitranslmed.3000322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner, D. C. (1975). The vampire bat: A field study in behavior and ecology (6th ed.). Baltimore, MD: Johns Hopkins University Press. [Google Scholar]
- Veikkolainen, V. , Vesterinen, E. J. , Lilley, T. M. , & Pulliainen, A. T. (2014). Bats as reservoir hosts of human bacterial pathogen, Bartonella mayotimonensis . Emerging Infectious Diseases, 20, 960–967. 10.3201/eid2006.130956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voigt, C. C. , & Kelm, D. H. (2006a). Host preference of the common vampire bat (Desmodus rotundus; chiroptera) assessed by stable isotopes. Journal of Mammalogy, 87, 6508–6. 10.1644/05-MAMM-F-276R1.1 [DOI] [Google Scholar]
- Voigt, C. C. , & Kelm, D. H. (2006b). Host preference of the common vampire bat (Desmodus rotundus; chiroptera) assessed by stable isotopes. Journal of Mammalogy, 87(1), 6508–6. 10.1644/05-MAMM-F-276R1.1 [DOI] [Google Scholar]
- Volokhov, D. V. , Becker, D. J. , Bergner, L. M. , Camus, M. S. , Orton, R. J. , Chizhikov, V. E. , … Streicker, D. G. (2017). Novel hemotropic mycoplasmas are widespread and genetically diverse in vampire bats. Epidemiology and Infection, 145(15), 3154–3167. 10.1017/S095026881700231X [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei, F. , Wu, Q. , Hu, Y. , Huang, G. , Nie, Y. , & Yan, L. (2018). Conservation metagenomics: a new branch of conservation biology. Science China Life Sciences, 62(2), 168–178. 10.1007/s11427-018-9423-3 [DOI] [PubMed] [Google Scholar]
- West, A. G. , Waite, D. W. , Deines, P. , Bourne, D. G. , Digby, A. , McKenzie, V. J. , & Taylor, M. W. (2019). The microbiome in threatened species conservation. Biological Conservation, 229, 85–98. 10.1016/j.biocon.2018.11.016 [DOI] [Google Scholar]
- Williams, C. L. , Dill‐McFarland, K. A. , Vandewege, M. W. , Sparks, D. L. , Willard, S. T. , Kouba, A. J. , … Brown, A. E. (2016). Dietary shifts may trigger dysbiosis and mucous stools in giant pandas (Ailuropoda melanoleuca). Frontiers in Microbiology, 7, 661 10.3389/fmicb.2016.00661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodhams, D. C. , Brandt, H. , Baumgartner, S. , Kielgast, J. , Küpfer, E. , Tobler, U. , … McKenzie, V. (2014). Interacting symbionts and immunity in the Amphibian skin mucosome predict disease risk and probiotic effectiveness. PLoS ONE, 9, e96375 10.1371/journal.pone.0096375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaneveld, J. R. , McMinds, R. , & Thurber, R. V. (2017). An Anna Karenina Principle for microbiomes: Many stressors destabilize rather than predictably shift animal microbiomes. Nature Microbiology, 2 10.1038/nmicrobiol.2017.121 [DOI] [PubMed] [Google Scholar]
- Zepeda Mendoza, M. L. , Xiong, Z. , Escalera‐Zamudio, M. , Runge, A. K. , Thézé, J. , Streicker, D. , … Gilbert, M. P. T. (2018). Hologenomic adaptations underlying the evolution of sanguivory in the common vampire bat. Nature Ecology & Evolution, 2(4), 659–668. 10.1038/s41559-018-0476-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zetun, C. , Hoffmann, J. , Silva, R. , Souza, L. , & Langoni, H. (2009). Leptospira spp. and Toxoplasma gondii antibodies in vampire bats (Desmodus rotundus) in Botucatu region, SP, Brazil. Journal of Venomous Animals and Toxins including Tropical Diseases, 15(3), 546–552. 10.1590/S1678-91992009000300014 [DOI] [Google Scholar]
- Zinsstag, J. , Schelling, E. , Waltner‐Toews, D. , & Tanner, M. (2011). From “one medicine” to “one health” and systemic approaches to health and well‐being. Preventive Veterinary Medicine, 101(3‐4), 148–156. 10.1016/j.prevetmed.2010.07.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuur, A. F. , Ieno, E. N. , & Elphick, C. S. (2009). A protocol for data exploration to avoid common statistical problems. Methods in Ecology and Evolution, 1(1), 3–14. 10.1111/j.2041-210X.2009.00001.x [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Raw, demultiplexed 16S sequence reads are archived publicly on the NCBI Short Read Archive (SRA) under BioProject #PRJNA506223. Sample metadata, ASV phylogeny, and taxonomically annotated ASV feature table are available on FigShare: https://figshare.com/projects/Habitat_Fragmentation_is_Associated_with_Dietary_and_Microbiome_Variability_in_Common_Vampire_Bats/57041.