

Abstract
BACKGROUND
Hereditary spastic paraplegias (HSPs) refer to a group of heterogeneous neurodegenerative diseases characterized by lower limbs spasticity and weakness. So far, over 72 genes have been found to cause HSP (SPG1-SPG72). Among autosomal dominant HSP patients, spastic paraplegia 4 (SPG4/SPAST) gene is the most common pathogenic gene, and atlastin-1 (ATL1) is the second most common one. Here we reported a novel ATL1 mutation in a Chinese spastic paraplegia 3A (SPG3A) family, which expands the clinical and genetic spectrum of ATL1 mutations.
CASE SUMMARY
A 9-year-old boy with progressive spastic paraplegia accompanied by right hearing loss and mental retardation for five years was admitted to our hospital. Past history was unremarkable. The family history was positive, and his grandfather and mother had similar symptoms. Neurological examinations revealed hypermyotonia in his lower limbs, hyperreflexia in knee reflex, bilateral positive Babinski signs and scissors gait. The results of blood routine test, liver function test, blood glucose test, ceruloplasmin test and vitamin test were all normal. The serum lactic acid level was significantly increased. The testing for brainstem auditory evoked potential demonstrated that the right side hearing was impaired while the left was normal. Magnetic resonance imaging showed mild atrophy of the spinal cord. The gene panel test revealed that the proband carried an ATL1 c.752A>G p.Gln251Arg (p.Q251R) mutation, and Sanger sequencing confirmed the existence of family co-segregation.
CONCLUSION
We reported a novel ATL1 Q251R mutation and a novel clinical phenotype of hearing loss in a Chinese SPG3A family.
Keywords: Hereditary spastic paraplegia, SPG3A, Atlastin-1 (ATL1) gene, Hearing loss, Case report
Core tip: Hereditary spastic paraplegias are a group of genetically and clinically heterogeneous neurodegenerative diseases characterized by lower limbs spasticity and weakness. Here we reported a novel ATL1 Q251R mutation predicted to be pathogenic and a novel clinical phenotype of hearing loss in a Chinese SPG3A family, which expands the clinical and genetic spectrum of ATL1 mutations.
INTRODUCTION
Hereditary spastic paraplegias (HSPs), also called spastic paraplegias (SPGs), are a group of genetically and clinically heterogeneous neurodegenerative diseases characterized by lower limbs spasticity and weakness. HSP can be classified into pure and complicated HSP based on symptoms. In pure HSP, the patient simply develops spasticity and weakness in lower limbs, while in complicated HSP, the patient presents with lower limbs spasticity accompanied by other symptoms, such as seizure and ataxia[1]. Over 72 genes have been identified to cause HSP and named by the order of discovery (SPG1-SPG72). HSP can be inherited in autosomal dominant, autosomal recessive or X-linked forms[2].
Among autosomal dominant HSP (AD-HSP) patients, spastic paraplegia 4 (SPG4/SPAST) is the most common pathogenic gene while the second most common one is atlastin-1 (ATL1)[3,4]. The patients presenting with walking disturbances sometimes initially visit orthopaedic outpatient clinic for treatment. It is crucial to distinguish HSP from other orthopaedic diseases. Drugs, stretching and physiotherapy can reduce spasticity of HSP patients. In some severe HSP cases, orthopaedic surgery is also needed for improving contracture in the lower limbs[5]. Here we reported a novel ATL1 Q251R mutation in a Chinese family with spastic paraplegia 3A (SPG3A), with a novel phenotype of hearing loss.
CASE PRESENTATION
Chief complaints
A 9-year-old male student was admitted to our hospital orthopaedic outpatient clinic because of progressive spastic paraplegia accompanied by right hearing loss and mental retardation for five years.
History of present illness
Five years ago, the patient began to have difficulty in walking and climbing stairs progressively accompanied by right hearing loss and mental retardation.
History of past illness
His medical history was not remarkable.
Personal and family history
His family history was positive for spastic paraplegia (Figure 1). His grandfather (subject I:1 Figure 1) developed unsteady walking at 3 years old, while his mother presented the same symptoms (subject II:2 Figure 1) at 8 years old. His mother had no other symptoms, while his grandfather had mental retardation (Table 1).
Figure 1.
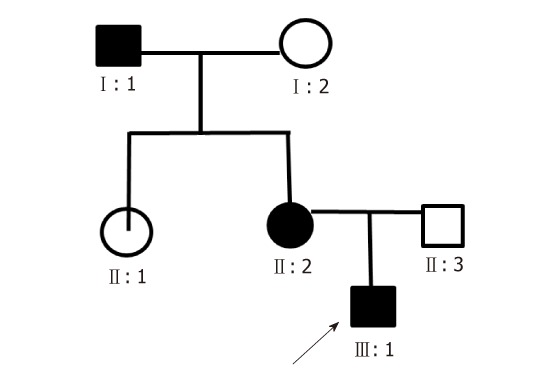
The pedigree of the SPG3A family. The patient is indicated with arrow (III:1) and the affected families are indicated by solid boxes (I:1 and II:2).
Table 1.
Clinical characteristics of the patient and affected family members
| Characteristics | I:1 | II:2 | III:1 |
| Gender | Male | Female | Male |
| Age at onset (yr) | 3 | 8 | 4 |
| Past history | Lumbar disc herniation | None | None |
| Clinical presentations | Walking disturbance, mental retardation | Walking disturbance | Walking disturbance, mental retardation, right hearing loss |
| Physical examination | |||
| Muscle strength | Normal | Normal | Lower limbs: grade 5- |
| Muscle tension | Lower limbs: increase | Lower limbs: increase | Lower limbs: increase |
| Sensory | Normal | Normal | Normal |
| Tendon reflex | Bilateral knee reflex1 | Bilateral knee reflex1 | Bilateral knee reflex2 |
| Babinski signs | Positive | Positive | Positive |
| Gait | Scissors gait | Scissors gait | Scissors gait |
| Auxiliary examination | |||
| MRI | Normal | Normal | Mild atrophy of the spinal cord |
| EMG/NCS | NA | Normal | Right tibial nerve’s F wave: Wide |
| BAEP | NA | NA | Right side hearing was impaired |
Active;
Hyperreflexia. MRI: Magnetic resonance imaging; EMG: Electromyography; NCS: Nerve conduction study; BAEP: Brainstem auditory evoked potential; NA: Not available.
Physical examination upon admission
Vital signs were in the normal ranges: Body temperature, 37.0 °C, respiratory rate, 21 breaths/min, pulse rate, 92 bpm and blood pressure, 98/60 mmHg. Neurological examinations revealed hypermyotonia in his lower limbs, hyperreflexia in knee reflex, and bilateral positive Babinski signs. He had scissors gait when walking. His lower limbs’ muscle strengths were grade 5-/5.
Laboratory examinations
The results of blood routine test, urine routine test, stool routine test, liver function test, renal function test, serum creatase, serum electrolyte, plasma ammonia, blood glucose, ceruloplasmin test and vitamin test were all within normal ranges. The serum lactic acid level was significantly raised to 4.36 mmol/L (normal range: 1.42-1.90 mmol/L). The gene panel included 72 known pathogenic genes associated with spastic paraplegia (Supplement Table 1). Genetic testing revealed that the proband carried an ATL1 c.752A>G p.Gln251Arg (p.Q251R) mutation, and Sanger sequencing confirmed the existence of family co-segregation (Figure 2).
Figure 2.
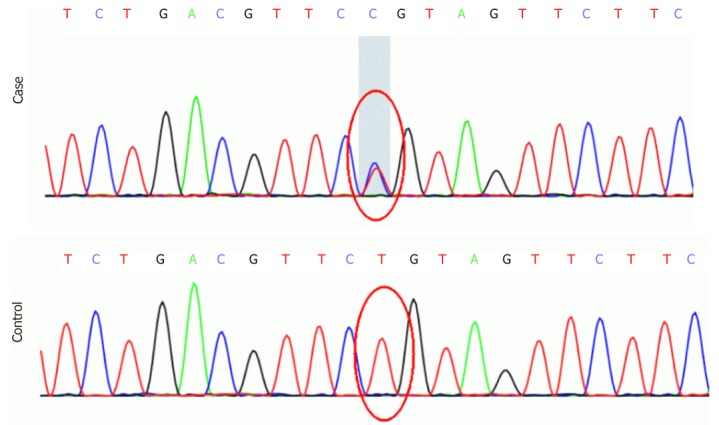
DNA sequencing identified a novel ATL1 c.752A>G, p.Q251R mutation (top: sequence of the patients; bottom: sequence of healthy individuals).
Imaging examinations
Magnetic resonance imaging (MRI) of the proband showed mild atrophy of the spinal cord (Figure 3), while the MRI results of his grandfather and mother were normal.
Figure 3.
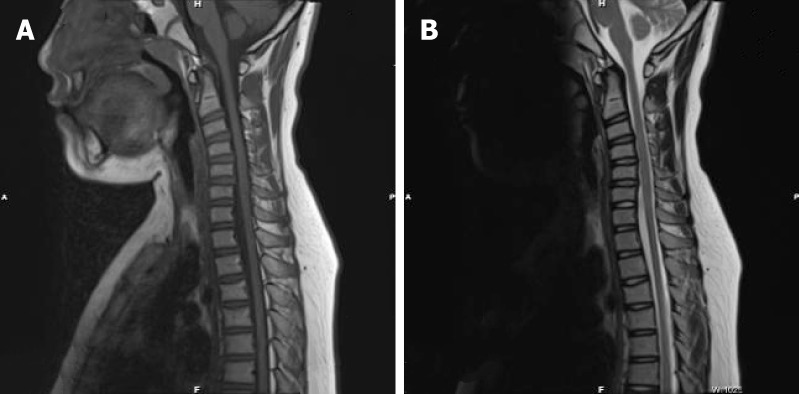
Magnetic resonance imaging showed the mild atrophy the spinal cord. A: T1 sagittal view B: T2 sagittal view.
FINAL DIAGNOSIS
A diagnosis of autosomal-dominant SPG3A was made based on previously published criteria[6].
TREATMENT
Mecobalamine 0.5 mg three times a day, coenzyme Q10 400 mg twice a day and baclofen 5 mg three times a day were administrated to the patient.
OUTCOME AND FOLLOW-UP
No adverse effects were observed. The patient’s symptoms deteriorated gradually in a follow-up visit after two months.
DISCUSSION
To date, 68 ATL1 pathogenic mutation types have been identified, most of which are missense mutations, followed by small insertions, small deletions and whole exon deletions. The mutation types were located in exon 12 (n = 29, 42.65%), exon 4 (n = 12, 17.65%), exon 8 (n = 8, 11.77%), exon 10 (n = 6, 8.82%), exon 7 (n = 4, 5.88%), exon 5 (n = 2, 2.94%), exon 11 (n = 2, 2.94%), exon 3 (n = 1, 1.47%), exon 6 (n = 1, 1.47%), exon 9 (n = 1, 1.47%), exon 13 (n = 1, 1.47%), and intron 1 (n = 1, 1.47%). The most common mutation genetic model is autosomal dominant (AD) inheritance (n = 57, 83.82%) while the sporadic is the second most common one (n = 7, 10.30%), and autosomal recessive (AR) inheritance is rare (n = 2, 2.94%) while two mutations’ types are not available (n = 2, 2.94%) (Table 2)[4,7-12]. Most ATL1 mutation carriers develop pure HSP[4,13,14], while a few of them present with complicated phenotypes, such as seizure, optic atrophy, mental retardation and ataxia[15]. In China, the most common phenotype of ATL1 mutation carriers is pure HSP while only one complicated phenotype was observed, namely muscular atrophy[16-21].
Table 2.
ATL1 pathogenic mutations in hereditary spastic paraplegia
| Exon | Nucleotide changes | Amino acid changes | Genetic model |
| 3 | G353A | R118Q | AR |
| 4 | T452C | F151S | AD |
| 4 | G458C | S153T | AD |
| 4 | C460G | Q154E | AD |
| 4 | C467T | T156I | AD |
| 4 | T470G | L157W | AD |
| 4 | T470C | L157S | AD |
| 4 | G473C | R158T | AD |
| 4 | G481C | A161P | AD |
| 4 | A484C | T162P | AD |
| 4 | T488C | V163A | AD |
| 4 | G493A | A165T | AD |
| 5 | C565G | H189D | AD |
| 5 | A572G | Q191R | AD |
| 6 | A587G | Y196C | AD |
| 7 | C649T | R217* | AR |
| 7 | G650A | R217Q | AD |
| 7 | C715T | R239C | AD |
| 7 | G716T | R239L | AD |
| 8 | A740C | H247P | AD |
| 8 | T749C | L250P | AD |
| 8 | C751A | Q251K | AD |
| 8 | G757A | V253I | AD |
| 8 | A773G | H258R | AD |
| 8 | C777A | S259Y | AD |
| 8 | C776T | S259Y | AD |
| 8 | T776G | S259F | AD |
| 9 | T944G | I315S | AD |
| 10 | C1006T | Y336H | AD |
| 10 | C1025A | P342Q | AD |
| 10 | C1030T | P344S | S |
| 10 | T1036G | S346A | AD |
| 10 | T1040C | M347T | AD |
| 10 | G1048T | A350S | AD |
| 11 | A1064T | N355I | S |
| 11 | C1065A | N355K | S |
| 12 | T1123C | C375R | AD |
| 12 | C1193A | S398Y | AD |
| 12 | C1193T | S398F | S |
| 12 | T1202C | L401P | S |
| 12 | A1220G | K407R | AD |
| 12 | A1222G | M408V | AD |
| 12 | T1223C | M408T | AD |
| 12 | A1222G | M408T | AD |
| 12 | G1226A | G409D | S |
| 12 | G1228A | G410R | AD |
| 12 | A1237C | F413V | AD |
| 12 | T1239C | F413L | AD |
| 12 | C1242G | S414R | AD |
| 12 | C1243T | R415W | AD |
| 12 | A1244G | R415Q | AD |
| 12 | C1246T | R416C | AD |
| 12 | G1247A | R416H | AD |
| 12 | T1308A | N436K | S |
| 12 | A1319C | N440T | AD |
| 12 | A1376G | Y459C | AD |
| 12 | G1406C | G469A | AD |
| 12 | G1445T | G482V | AD |
| 12 | C1483T | R495W | AD |
| 13 | G1556A | S519N | AD |
| 12 | 1306-1308delAAT | N436del | AD |
| 4 | Exon 4 del | 140-174del | NA |
| Intron 1 | c.35-3C>T | G13fsX16 | AD |
| 12 | 1462_1463insTG | T490Afs | NA |
| 12 | 1466-1467insTG | T490fsX508 | AD |
| 12 | 1474insG | A492fsX522 | AD |
| 12 | 1504-1505insG | E502fsX522 | AD |
| 12 | 1520insA | I507fsX522 | AD |
AR: Autosomal recessive; AD: Autosomal dominant; S: Sporadic; NA: Not available.
The impairments of the upper motor system can lead to spastic paraplegia, including cerebral palsy, brain injury, spinal cord infection, spinal cord tumor, and spinal cord injury[22-26]. Among them, the most common cause of spastic paraplegia in children is cerebral palsy, which can mimic HSP[27]. Consequently, it is important to identify HSP in orthopedic patients presenting with spastic paraplegia. Lumbosacral dorsal rhizotomy, botulinum toxin, and physiotherapy are effective ways to treat spasticity in children[28,29].
In the present study, we detected a novel ATL1 Q251R mutation, which is located in exon 8. ATL1 Q251R was considered as a novel mutation, as it is absent in the Human Gene Mutation Database (HGMD) (http://www.hgmd.cf.ac.uk/ac/index.php) and clinvar database (www.ncbi.nlm.nih.gov/clinvar/). Besides, no previous case has been reported with ATL1 Q251R by searching it in PubMed and Web of Science. Protein Variation Effect Analyzer (PROVEN), Mutation Taster and Mutation Assessor were utilized to predict the pathogenicity of ATL1 Q251R, and the results were described as deleterious, disease-causing and medium credible pathogenic, respectively. The amino-acid substitution replaced a neutrally charged glutamine for a positively charged arginine. Besides, ATL1 Q251K was also reported to be a disease-causing mutation in HSP[30]. Consequently, the above evidence suggests that ATL1 Q251R is likely to be a pathogenic mutation of HSP. Further functional studies are warranted to confirm its pathogenicity.
ATL1 was firstly identified and reported to be pathogenic in five HSP kindreds[31]. It encodes for atlastin-1 (ATL1) protein that belongs to the dynamin family of guanosine triphosphatases (GTPases). ATL1 protein has a vital role in homotypic endoplasmic reticulum fusion, which is likely to be the underlying mechanism in the pathogenesis of HSP[32].
In our SPG3A family, we found that the proband and affected family members exhibit different clinical manifestations despite having the same mutation. The proband developed progressive walking disturbance accompanied by hearing loss and mental retardation, while his mother exhibited pure HSP symptoms and his grandfather also had mental retardation but no hearing dysfunction. This clearly indicates that SPG3A is clinically heterogeneous. The intra-family variable penetrance may result from environmental modifiers as well as regulatory variants[33]. Furthermore, sex and mutation types are of great importance in modifying the penetrance in HSP[34]. In our SPG3A family, regulatory variants, gender differences and environmental factors may be the underlying contributors to different phenotypes.
Our group previously analyzed the clinical spectrum of HSP in China and found that most of cases were pure one, whereas a few showed complicated phenotypes like atrophy in extremities[35]. Only a few HSP patients develop deafness or hearing loss in the course of the disease, however, none of SPG3A patients with deafness or hearing loss has been reported[36]. The patient we presented here developed progressive walking disturbance accompanied by hearing loss. Therefore, we also presented a novel clinical phenotype in SPG3A, hearing loss.
Furthermore, both neurological defects and orthopaedic diseases can result in movement abnormalities[5]. In fact, orthopaedic surgeons are usually the first doctors who are visited by patients with walking disturbances or gait abnormalities, including HSP patients presenting with progressive spasmodic paraplegia. For example, a Caucasian girl was misdiagnosed with cerebral palsy and a final correct diagnosis of SPG3A was made by genetic testing[12]. Consequently, careful medical history inquiry and physical examination are extremely important for diagnosis. In some cases, no definite diagnosis can be established by an orthopaedic surgeon alone. The evaluation of a neurologist or multidisciplinary team including a neurologist is essential for correct diagnosis. Besides, the treatments of HSP also involve appropriate orthopaedic therapies, such as surgery in severe HSP patients[37].
CONCLUSION
In conclusion, we reported a novel ATL1 Q251R mutation which is likely to be pathogenic and a clinically novel phenotype of hearing loss in a Chinese SPG3A family, which expands the clinical and genetic spectrum of ATL1 mutations. SPG3A was clinically heterogeneous even with the same pathogenic mutation. In addition, this report emphasizes the importance of distinguishing HSP patients from other patients in orthopaedic outpatient clinic.
ACKNOWLEDGEMENTS
The authors are grateful to all subjects for participation in our study.
Footnotes
Informed consent statement: Informed written consent was obtained from the patient for publication of this report.
Conflict-of-interest statement: The authors declare that they have no conflict of interest.
CARE Checklist (2016) statement: The manuscript was prepared and revised according to the CARE Checklist (2016).
Manuscript source: Unsolicited manuscript
Peer-review started: February 11, 2019
First decision: March 9, 2019
Article in press: April 9, 2019
Specialty type: Medicine, research and experimental
Country of origin: China
Peer-review report classification
Grade A (Excellent): 0
Grade B (Very good): B, B, B
Grade C (Good): C
Grade D (Fair): 0
Grade E (Poor): 0
P-Reviewer: Demonacos C, Kiselev AV, Radenovic L, Rodrigues-Lisoni FC S-Editor: Ji FF L-Editor: A E-Editor: Xing YX
Contributor Information
Xue-Wen Xiao, Department of Neurology, Xiangya Hospital, Central South University, Changsha 410008, Hunan Province, China.
Juan Du, Department of Neurology, Xiangya Hospital, Central South University, Changsha 410008, Hunan Province, China; National Clinical Research Center for Geriatric Disorders, Xiangya Hospital, Central South University, Changsha 410008, Hunan Province, China.
Bin Jiao, Department of Neurology, Xiangya Hospital, Central South University, Changsha 410008, Hunan Province, China; National Clinical Research Center for Geriatric Disorders, Xiangya Hospital, Central South University, Changsha 410008, Hunan Province, China; Key Laboratory of Hunan Province in Neurodegenerative Disorders, Central South University, Changsha 410008, Hunan Province, China.
Xin-Xin Liao, Department of Geriatrics Neurology, Xiangya Hospital, Central South University, Changsha 410008, Hunan Province, China.
Lu Zhou, Department of Neurology, Xiangya Hospital, Central South University, Changsha 410008, Hunan Province, China.
Xi-Xi Liu, Department of Neurology, Xiangya Hospital, Central South University, Changsha 410008, Hunan Province, China.
Zhen-Hua Yuan, Department of Neurology, Xiangya Hospital, Central South University, Changsha 410008, Hunan Province, China.
Li-Na Guo, Department of Neurology, Xiangya Hospital, Central South University, Changsha 410008, Hunan Province, China.
Xin Wang, Department of Neurology, Xiangya Hospital, Central South University, Changsha 410008, Hunan Province, China.
Lu Shen, Department of Neurology, Xiangya Hospital, Central South University, Changsha 410008, Hunan Province, China; National Clinical Research Center for Geriatric Disorders, Xiangya Hospital, Central South University, Changsha 410008, Hunan Province, China; Key Laboratory of Hunan Province in Neurodegenerative Disorders, Central South University, Changsha 410008, Hunan Province, China; Key Laboratory of Organ Injury, Aging and Regenerative Medicine of Hunan Province, Changsha 410008, Hunan Province, China.
Zhang-Yuan Lin, National Clinical Research Center for Geriatric Disorders, Xiangya Hospital, Central South University, Changsha 410008, Hunan Province, China. linzhangyuan2505@sina.com; Department of Orthopedics, Xiangya Hospital, Central South University, Changsha 410008, Hunan Province, China.
References
- 1.Kara E, Tucci A, Manzoni C, Lynch DS, Elpidorou M, Bettencourt C, Chelban V, Manole A, Hamed SA, Haridy NA, Federoff M, Preza E, Hughes D, Pittman A, Jaunmuktane Z, Brandner S, Xiromerisiou G, Wiethoff S, Schottlaender L, Proukakis C, Morris H, Warner T, Bhatia KP, Korlipara LV, Singleton AB, Hardy J, Wood NW, Lewis PA, Houlden H. Genetic and phenotypic characterization of complex hereditary spastic paraplegia. Brain. 2016;139:1904–1918. doi: 10.1093/brain/aww111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gan-Or Z, Bouslam N, Birouk N, Lissouba A, Chambers DB, Vérièpe J, Androschuk A, Laurent SB, Rochefort D, Spiegelman D, Dionne-Laporte A, Szuto A, Liao M, Figlewicz DA, Bouhouche A, Benomar A, Yahyaoui M, Ouazzani R, Yoon G, Dupré N, Suchowersky O, Bolduc FV, Parker JA, Dion PA, Drapeau P, Rouleau GA, Ouled Amar Bencheikh B. Mutations in CAPN1 Cause Autosomal-Recessive Hereditary Spastic Paraplegia. Am J Hum Genet. 2016;98:1038–1046. doi: 10.1016/j.ajhg.2016.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Vandebona H, Kerr NP, Sue CM. Mutation Analysis of Spastin (SPG4) in Patients with Hereditary Spastic Paraplegia. J Clin Neurosci. 2009;16:476. [Google Scholar]
- 4.Zhao GH, Liu XM. Clinical features and genotype-phenotype correlation analysis in patients with ATL1 mutations: A literature reanalysis. Transl Neurodegener. 2017;6:9. doi: 10.1186/s40035-017-0079-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Houlden H, Charlton P, Singh D. Neurology and orthopaedics. J Neurol Neurosurg Psychiatry. 2007;78:224–232. doi: 10.1136/jnnp.2006.092072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gasser T, Finsterer J, Baets J, Van Broeckhoven C, Di Donato S, Fontaine B, De Jonghe P, Lossos A, Lynch T, Mariotti C, Schöls L, Spinazzola A, Szolnoki Z, Tabrizi SJ, Tallaksen CM, Zeviani M, Burgunder JM, Harbo HF EFNS. EFNS guidelines on the molecular diagnosis of ataxias and spastic paraplegias. Eur J Neurol. 2010;17:179–188. doi: 10.1111/j.1468-1331.2009.02873.x. [DOI] [PubMed] [Google Scholar]
- 7.Dong EL, Wang C, Wu S, Lu YQ, Lin XH, Su HZ, Zhao M, He J, Ma LX, Wang N, Chen WJ, Lin X. Clinical spectrum and genetic landscape for hereditary spastic paraplegias in China. Mol Neurodegener. 2018;13:36. doi: 10.1186/s13024-018-0269-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mészárosová AU, Grečmalová D, Brázdilová M, Dvořáčková N, Kalina Z, Čermáková M, Vávrová D, Smetanová I, Staněk D, Seeman P. Disease-Causing Variants in the ATL1 Gene Are a Rare Cause of Hereditary Spastic Paraplegia among Czech Patients. Ann Hum Genet. 2017;81:249–257. doi: 10.1111/ahg.12206. [DOI] [PubMed] [Google Scholar]
- 9.Willkomm L, Heredia R, Hoffmann K, Wang H, Voit T, Hoffman EP, Cirak S. Homozygous mutation in Atlastin GTPase 1 causes recessive hereditary spastic paraplegia. J Hum Genet. 2016;61:571–573. doi: 10.1038/jhg.2016.6. [DOI] [PubMed] [Google Scholar]
- 10.Lu C, Li LX, Dong HL, Wei Q, Liu ZJ, Ni W, Gitler AD, Wu ZY. Targeted next-generation sequencing improves diagnosis of hereditary spastic paraplegia in Chinese patients. J Mol Med (Berl) 2018;96:701–712. doi: 10.1007/s00109-018-1655-4. [DOI] [PubMed] [Google Scholar]
- 11.Duz MB, Dasdemir S, Kalayci Yigin A, Akalin MA, Seven M. Three novel mutations in 20 patients with hereditary spastic paraparesis. Neurol Sci. 2018;39:1551–1557. doi: 10.1007/s10072-018-3454-7. [DOI] [PubMed] [Google Scholar]
- 12.Andersen EW, Leventer RJ, Reddihough DS, Davis MR, Ryan MM. Cerebral palsy is not a diagnosis: A case report of a novel atlastin-1 mutation. J Paediatr Child Health. 2016;52:669–671. doi: 10.1111/jpc.13200. [DOI] [PubMed] [Google Scholar]
- 13.Alvarez V, Sánchez-Ferrero E, Beetz C, Díaz M, Alonso B, Corao AI, Gámez J, Esteban J, Gonzalo JF, Pascual-Pascual SI, López de Munain A, Moris G, Ribacoba R, Márquez C, Rosell J, Marín R, García-Barcina MJ, Del Castillo E, Benito C, Coto E Group for the Study of the Genetics of Spastic Paraplegia. Mutational spectrum of the SPG4 (SPAST) and SPG3A (ATL1) genes in Spanish patients with hereditary spastic paraplegia. BMC Neurol. 2010;10:89. doi: 10.1186/1471-2377-10-89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Elert-Dobkowska E, Stepniak I, Krysa W, Rajkiewicz M, Rakowicz M, Sobanska A, Rudzinska M, Wasielewska A, Pilch J, Kubalska J, Lipczynska-Lojkowska W, Kulczycki J, Kurdziel K, Sikorska A, Beetz C, Zaremba J, Sulek A. Molecular spectrum of the SPAST, ATL1 and REEP1 gene mutations associated with the most common hereditary spastic paraplegias in a group of Polish patients. J Neurol Sci. 2015;359:35–39. doi: 10.1016/j.jns.2015.10.030. [DOI] [PubMed] [Google Scholar]
- 15.Orlacchio A, Montieri P, Babalini C, Gaudiello F, Bernardi G, Kawarai T. Late-onset hereditary spastic paraplegia with thin corpus callosum caused by a new SPG3A mutation. J Neurol. 2011;258:1361–1363. doi: 10.1007/s00415-011-5934-z. [DOI] [PubMed] [Google Scholar]
- 16.Chen SQ, Zhou Y, Li XY, La B, Huang S, Huang WJ, Zhou CL, Maxwell PH, Wang YM. Severe hereditary spastic paraplegia caused by a de novo SPG3A mutation. Sci China. 2005;51:1854–1856. [Google Scholar]
- 17.Li XH, Song C, Chen SQ, Zhou Y, Guo H, Zhou CL, Yang ZY, Liang YX, Wang YM. A SPG3A mutation with a novel foot phenotype of hereditary spastic paraplegia in a Chinese Han family. Chin Med J (Engl) 2007;120:834–837. [PubMed] [Google Scholar]
- 18.Ming L. [SPG3A-hereditary spastin paraplegia with genetic anticipation and incomplete penetrance] Zhonghua Yi Xue Yi Chuan Xue Za Zhi. 2007;24:15–18. [PubMed] [Google Scholar]
- 19.Chan KY, Ching CK, Mak CM, Lam CW, Chan AY. Hereditary spastic paraplegia: identification of an SPG3A gene mutation in a Chinese family. Hong Kong Med J. 2009;15:304–307. [PubMed] [Google Scholar]
- 20.Lu X, Cen Z, Xie F, Ouyang Z, Zhang B, Zhao G, Luo W. Genetic analysis of SPG4 and SPG3A genes in a cohort of Chinese patients with hereditary spastic paraplegia. J Neurol Sci. 2014;347:368–371. doi: 10.1016/j.jns.2014.10.017. [DOI] [PubMed] [Google Scholar]
- 21.Shin JW, Jung KH, Lee ST, Moon J, Seong MW, Park SS, Lee SK, Chu K. Novel mutation in the ATL1 with autosomal dominant hereditary spastic paraplegia presented as dysautonomia. Auton Neurosci. 2014;185:141–143. doi: 10.1016/j.autneu.2014.06.001. [DOI] [PubMed] [Google Scholar]
- 22.Matsuura E, Yoshimura A, Nozuma S, Higuchi I, Kubota R, Takashima H. Clinical presentation of axial myopathy in two siblings with HTLV-1 associated myelopathy/tropical spastic paraparesis (HAM/TSP) BMC Neurol. 2015;15:18. doi: 10.1186/s12883-015-0275-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lee JY, Kim SN, Lee IS, Jung H, Lee KS, Koh SE. Effects of Extracorporeal Shock Wave Therapy on Spasticity in Patients after Brain Injury: A Meta-analysis. J Phys Ther Sci. 2014;26:1641–1647. doi: 10.1589/jpts.26.1641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chang CH, Chen YY, Yeh KK, Chen CL. Gross motor function change after multilevel soft tissue release in children with cerebral palsy. Biomed J. 2017;40:163–168. doi: 10.1016/j.bj.2016.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yan X, Lan J, Liu Y, Miao J. Efficacy and Safety of Botulinum Toxin Type A in Spasticity Caused by Spinal Cord Injury: A Randomized, Controlled Trial. Med Sci Monit. 2018;24:8160–8171. doi: 10.12659/MSM.911296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Celiktas M, Asik MO, Gezercan Y, Gulsen M. Pigmented villonodular synovitis of the thoracic vertebra presenting with progressive spastic paraparesis. Case Rep Orthop. 2013;2013:870324. doi: 10.1155/2013/870324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lee RW, Poretti A, Cohen JS, Levey E, Gwynn H, Johnston MV, Hoon AH, Fatemi A. A diagnostic approach for cerebral palsy in the genomic era. Neuromolecular Med. 2014;16:821–844. doi: 10.1007/s12017-014-8331-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Health Quality Ontario. Lumbosacral Dorsal Rhizotomy for Spastic Cerebral Palsy: A Health Technology Assessment. Ont Health Technol Assess Ser. 2017;17:1–186. [PMC free article] [PubMed] [Google Scholar]
- 29.Pavone V, Testa G, Restivo DA, Cannavò L, Condorelli G, Portinaro NM, Sessa G. Botulinum Toxin Treatment for Limb Spasticity in Childhood Cerebral Palsy. Front Pharmacol. 2016;7:29. doi: 10.3389/fphar.2016.00029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dürr A, Camuzat A, Colin E, Tallaksen C, Hannequin D, Coutinho P, Fontaine B, Rossi A, Gil R, Rousselle C, Ruberg M, Stevanin G, Brice A. Atlastin1 mutations are frequent in young-onset autosomal dominant spastic paraplegia. Arch Neurol. 2004;61:1867–1872. doi: 10.1001/archneur.61.12.1867. [DOI] [PubMed] [Google Scholar]
- 31.Zhao X, Alvarado D, Rainier S, Lemons R, Hedera P, Weber CH, Tukel T, Apak M, Heiman-Patterson T, Ming L, Bui M, Fink JK. Mutations in a newly identified GTPase gene cause autosomal dominant hereditary spastic paraplegia. Nat Genet. 2001;29:326–331. doi: 10.1038/ng758. [DOI] [PubMed] [Google Scholar]
- 32.Bian X, Klemm RW, Liu TY, Zhang M, Sun S, Sui X, Liu X, Rapoport TA, Hu J. Structures of the atlastin GTPase provide insight into homotypic fusion of endoplasmic reticulum membranes. Proc Natl Acad Sci U S A. 2011;108:3976–3981. doi: 10.1073/pnas.1101643108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Castel SE, Cervera A, Mohammadi P, Aguet F, Reverter F, Wolman A, Guigo R, Iossifov I, Vasileva A, Lappalainen T. Modified penetrance of coding variants by cis-regulatory variation contributes to disease risk. Nat Genet. 2018;50:1327–1334. doi: 10.1038/s41588-018-0192-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Parodi L, Fenu S, Barbier M, Banneau G, Duyckaerts C, Tezenas du Montcel S, Monin ML, Ait Said S, Guegan J, Tallaksen CME, Sablonniere B, Brice A, Stevanin G, Depienne C, Durr A SPATAX network. Spastic paraplegia due to SPAST mutations is modified by the underlying mutation and sex. Brain. 2018;141:3331–3342. doi: 10.1093/brain/awy285. [DOI] [PubMed] [Google Scholar]
- 35.Luo Y, Chen C, Zhan Z, Wang Y, Du J, Hu Z, Liao X, Zhao G, Wang J, Yan X, Jiang H, Pan Q, Xia K, Tang B, Shen L. Mutation and clinical characteristics of autosomal-dominant hereditary spastic paraplegias in China. Neurodegener Dis. 2014;14:176–183. doi: 10.1159/000365513. [DOI] [PubMed] [Google Scholar]
- 36.Tesson C, Koht J, Stevanin G. Delving into the complexity of hereditary spastic paraplegias: how unexpected phenotypes and inheritance modes are revolutionizing their nosology. Hum Genet. 2015;134:511–538. doi: 10.1007/s00439-015-1536-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cottalorda J, Violas P, Seringe R French Society of Pediatric Orthopaedics. Neuro-orthopaedic evaluation of children and adolescents: a simplified algorithm. Orthop Traumatol Surg Res. 2012;98:S146–S153. doi: 10.1016/j.otsr.2012.04.015. [DOI] [PubMed] [Google Scholar]