

Abstract
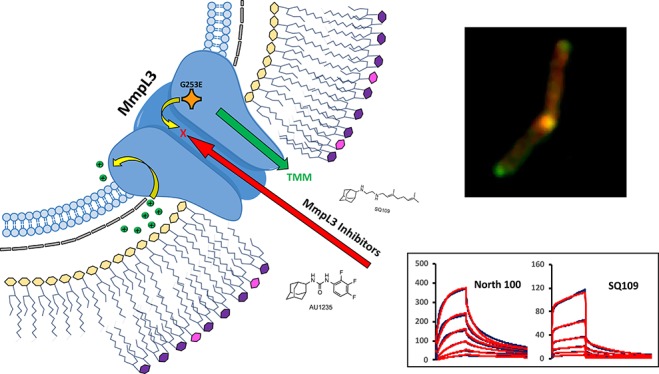
MmpL3, an essential transporter involved in the export of mycolic acids, is the proposed target of a number of antimycobacterial inhibitors under development. Whether MmpL3 serves as the direct target of these compounds, however, has been called into question after the discovery that some of them dissipated the proton motive force from which MmpL transporters derive their energy. Using a combination of in vitro and whole-cell-based approaches, we here provide evidence that five structurally distinct MmpL3 inhibitor series, three of which impact proton motive force in Mycobacterium tuberculosis, directly interact with MmpL3. Medium- to high-throughput assays based on these approaches were developed to facilitate the future screening and optimization of MmpL3 inhibitors. The promiscuity of MmpL3 as a drug target and the mechanisms through which missense mutations located in a transmembrane region of this transporter may confer cross-resistance to a variety of chemical scaffolds are discussed in light of the exquisite vulnerability of MmpL3, its apparent mechanisms of interaction with inhibitors, and evidence of conformational changes induced both by the inhibitors and one of the most commonly identified resistance mutations in MmpL3.
Keywords: Mycobacterium, tuberculosis, MmpL3, mycolic acids, drug development, proton motive force
The mycolic acid transporter MmpL3 has received much attention lately as the putative target of multiple novel series of compounds with activity against Mycobacterium tuberculosis (Mtb) or nontuberculous mycobacteria (NTM). MmpL3 is required for the translocation of mycolic acids in the form of trehalose monomycolates (TMM) from the cytoplasm to the periplasmic space where mycolic acids can then be used in assembly of the mycobacterial outer membrane.1−4 The steep decrease in Mtb viability that follows the chemical or genetic inhibition of MmpL3 in vitro, inside macrophages, and in acute and chronic mouse models of tuberculosis (TB) infection points to the exquisite vulnerability of this transporter.1−3,5−10 The potency that MmpL3 inhibitors display against multidrug resistant (MDR) strains of Mtb and their synergistic interactions with a number of anti-TB drugs and drug candidates11,12 further highlight the potential that MmpL3 inhibitors have to reduce the duration of TB and MDR-TB treatments. Accordingly, a number of MmpL3 inhibitors are currently under development; among them, SQ109,13 which has completed phase II efficacy studies in TB patients in Africa, and a number of indolecarboxamide- and tetrapyrazolopyrimidine-based inhibitors selected on the basis of their mycobactericidal activity, tolerability, favorable pharmacokinetic profiles and efficacy in acute and chronic murine models of TB and NTM infections.6−8,14−20 The lack of simple and relatively high-throughput assays to rapidly screen optimized analogues of these compounds currently represents an obstacle to their further development. The finding that some of these inhibitors have more than one target in Mtb (including other targets in the mycolic acid biosynthetic pathway)16,21 together with the observation that a subset of them may exert their inhibitory effect on MmpL3 by dissipating the proton motive force (PMF) from which MmpL transporters derive their energy21−24 has further raised questions as to their direct or indirect mechanism of inhibition of MmpL3. Recently, Xu and collaborators25 provided evidence of a direct interaction between MmpL3 and one of its inhibitors, known as BM212,26 by showing that the [14C]-labeled inhibitor bound to the purified MmpL3 protein from Mycobacterium smegmatis (Msmg). The same investigators developed a spheroplast-based flippase assay for MmpL3.25 Although informative in that this assay provided the first direct evidence of the buildup of TMM in the inner leaflet of the plasma membrane following MmpL3 inhibition, the rather cumbersome preparation of spheroplasts that this assay requires and the fact that it does not readily distinguish between direct and indirect mechanisms of inhibition of the transporter limit its usefulness for compound screening.
Because of the growing interest in MmpL3 as the putative target of multiple new small molecule inhibitors of Mtb and NTM and the importance of understanding the mechanism of action of these compounds to drive their optimization process, we here report on the development of in vitro and whole-cell-based assays enabling the identification of direct inhibitors of MmpL3 from Mtb and their use to validate the interaction of five of the most studied series of inhibitors to date with the transporter. Biolayer interferometry- and surface-plasmon-resonance-based assays point to some inhibitors inducing conformational changes in MmpL3. Limited proteolysis experiments further point to one of the most commonly identified resistance mutations in MmpL3 causing conformational changes in the protein, thereby providing a plausible mechanism through which missense mutations may confer cross-resistance to a broad variety of inhibitors. Finally, the disclosure of the crystal structure of MmpL3 alone and in complex with SQ109, an adamantyl urea and indolecarboxamide,27 while we were in the final stages of preparing this manuscript largely confirms our conclusion of a common inhibitor binding site located in the middle region of the transmembrane domain of MmpL34 and provides a strong structural rationale for the functionality of our assays.
Results
Cross-Resistance between MmpL3 Inhibitors
Six representative MmpL3 inhibitors were selected for the purpose of this study, including the adamantyl urea AU1235,1 the 1,2-diamine SQ109,2 the tetrahydropyrazolopyrimidine THPP1,8 the 1,5-diarylpyrrole BM212,26 and the indolecarboxamides NITD-304 and NITD-3496 (Figure 1A). The first four compounds have previously been reported to inhibit the transfer of mycolic acids to their cell envelope acceptors in Mtb or M. bovis BCG.1,2,8,16 That NITD-304 and NITD-349 displayed the same expected property of MmpL3 inhibitors was verified by metabolic labeling of Mtb H37Rv with [1,2-14C]acetate upon treatment with increasing concentrations of the two compounds (Figure S1).
Figure 1.

Chemical structures of the six MmpL3 inhibitors (A) and four inhibitor probes (B) used in this study..
A number of mutations in mmpL3 were reported to increase the resistance of Mtb to one or more of the compounds listed above. To rigorously compare the level of resistance conferred by these mutations to each of the six compounds and more precisely delineate the regions of MmpL3 associated with cross-resistance, 77 different variants of the Mtb mmpL3 gene (mmpL3tb), including variants harboring missense mutations previously reported to confer resistance to the inhibitors listed above, were expressed in the background of a Msmg mmpL3 deletion mutant (MsmgΔmmpL3), and the resulting recombinant strains were tested for MICs against all six compounds. Of all the MmpL3tb variants tested, 11 increased at least 4-fold the level of resistance of the recombinant strains to one or more compounds. These variants are listed in Table 1. With few exceptions that are described below, the variants conferred resistance to at least three out of the six compounds indicative of extensive cross-resistance between structurally unrelated chemotypes. The Q372R resistance mutation, in contrast, was found to be specific to the NITD compounds, whereas F644C specifically impacted susceptibility to SQ109 and THPP1, and T311I impacted the MICs of only AU1235 and THPP1. BM212 was the compound for which the fewest resistance mutations were found.
Table 1. Resistance Profile of Msmg mmpL3 Mutants Rescued with Mutated Variants of mmpL3tba.

MIC values are in μg/mL. Red boxes indicate an increase in MIC over MsmgΔmmpL3 expressing wild-type mmpL3tb of eightfold or more; green indicates a fourfold increase in MIC. No color indicates a maximum of twofold change in MIC, which is considered within the experimental margin of error. The MICs of North 131 and North 132 against Msmg expressing WT mmpL3tb are 32 and 64 μg/mL, respectively.
To gain further insight into the apparent lack of specificity of a number of resistance mutations, we modified the nature of the mutations at positions L189, G253, S591, F644, and V684 of MmpL3tb so as to change the size, polarity, and/or charge of these cross-resistance-associated residues and analyzed how these changes affected the MICs of each inhibitor. Interestingly, while amino acid changes introduced at positions 189, 591, and 644 of MmpL3tb restored the transporter’s susceptibility to all inhibitor series, compound-specific susceptibility patterns were observed in response to some of the mutations introduced at positions 253 (compare G253R and G253W) and 684 (compare V684W and V684I) (Table 2). Of note is the importance of a negative charge at position 253 (G253E) for resistance to THPP1, SQ109, AU1235, and both indolecarboxamides, whereas a positive charge at position 189 (L189R) appears to be needed for resistance to the same inhibitors (Table 2). These results provide support to the hypothesis that direct and specific interactions between the inhibitors and the side chains of some MmpL3 amino acid residues are critical to their activity.
Table 2. Effect of Modifying the Nature of the Amino Acid Residue at Positions of MmpL3tb Associated with Cross-Resistance on MICsa.

Mutations in bold letters correspond to the original resistance mutations identified by whole genome sequencing of spontaneous resistant mutants. They are identical to the resistance mutations shown in Table 1 and are referred to as the parent mutants. Other mutations were generated for the purpose of this experiment and similarly expressed in the background of MsmgΔmmpL3 as described under Methods. Mutations that reverted the resistance phenotype of the parent mutants to at least three inhibitor series are indicated in red font. Mutations that reverted the resistance phenotype of the parent mutants to no more than one or two inhibitors are indicated in green font. MIC values are in μg/mL. CIP, ciprofloxacin (control drug).
Consistent with earlier findings, all cross-resistance mutations mapped to the central region of transmembrane segments of MmpL3 (Figure S2) that are thought to be important for proton translocation or proton-driven conformational changes in the transporter.4 Accordingly, the majority of these mutations were found to negatively impact the transport activity of MmpL3, as evidenced by the slower growth of the mutants on agar medium and reduced rates of mycolic acid transfer to the cell wall (Figure S3). That the resistance phenotype associated with these mutations did not result from a significant increase in the level of expression of mmpL3tb in the mutants was verified by qRT-PCR (Figure S4).
Collectively, our results support the hypothesis that MmpL3 inhibitors interact directly and with a relative specificity to a common transmembrane region of MmpL3tb that is critical to the activity of the transporter.
Synthesis of IC and Urea-Based Inhibitor Probes and Evidence of Colocalization with MmpL3tb in Intact Cells
The finding of what appeared to be a common inhibitor binding region in MmpL3tb next prompted us to design fluorescent inhibitor probes targeted to the same region of the transporter. Both adamantyl urea-based (North 132) and indolecarboxamide-based (North 100, North 114, North 131) probes harboring either tetramethylrhodamine (TAMRA) or BODIPY FL fluorophores (Figure 1B) were designed and synthesized as detailed in the Supplementary Methods. That all of the probes were on-target and thus displayed the expected inhibitory effect on the transfer of mycolic acids to trehalose dimycolate and arabinogalactan in intact MsmgΔmmpL3/pMVGH1-mmpL3tb cells was first verified by metabolic labeling with [1,2-14C]acetate (Figure S5). Importantly, the same MmpL3tb mutations found to confer resistance to multiple series of compounds also increased resistance to the inhibitor probes North 100 and/or North 114 (Table 1), indicating that these probes should be able to effectively compete with test inhibitors for the same binding region of MmpL3tb. North 131 and North 132 had too high MICs against MsmgΔmmpL3 expressing wild-type mmpL3tb to be tested against the mutants (Table 1).
The TAMRA-labeled North 100 and North 114 probes were incubated with the Msmg strain MsmgΔmmpL3/pMVGH1-mmpL3tb-GFP expressing MmpL3tb C-terminally fused to GFP. As reported previously,28 the MmpL3tb-GFP fusion protein concentrated at the old pole and septum of the cells with the greatest fluorescence intensity mapping to the septum (Figure 2). Fluorescence microscopic analyses of MsmgΔmmpL3/pMVGH1-mmpL3tb-GFP cells labeled with North 100 and North 114 clearly pointed to the colocalization of the probes with MmpL3tb-GFP (Figure 2 and Figure S6). In contrast, no colocalization was observed when a TAMRA-labeled probe unrelated to MmpL3 inhibitors (TAMRA-mannose) was used (Figure S6).
Figure 2.
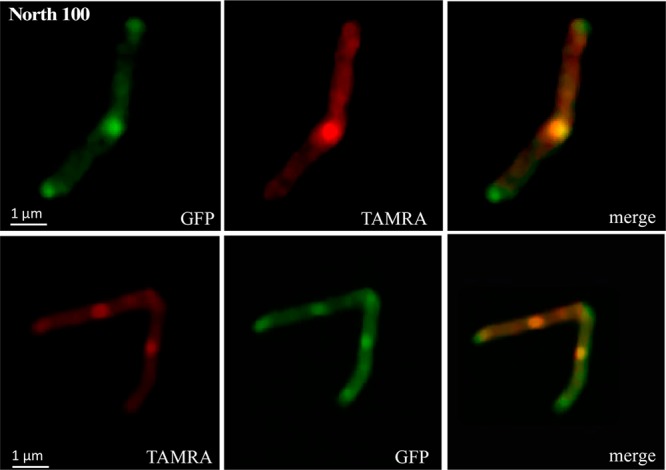
Colocalization of North 100 with MmpL3tb-GFP at the old poles and septa of an M. smegmatis mmpL3 deletion mutant expressing an mmpL3tb-gfp fusion.
Evidence of Inhibitor Probe Binding to MmpL3tb in Vitro and Development of a Competition Binding Assay
To directly assess the ability of the fluorescent probes to interact with purified MmpL3tb in vitro, the MmpL3tb and MmpL3tb-GFP proteins that we purified from the Msmg strains MsmgΔmmpL3/pMVGH1-mmpL3tb and MsmgΔmmpL3/pMVGH1-mmpL3tb-gfp, respectively, were bound to polystyrene particles as described in the Methods, and the particles were exposed to various concentrations of North 100, North 114, and North 132 for 15 min at room temperature. Flow cytometry analysis of the MmpL3tb- and MmpL3tb-GFP-coated particles revealed a concentration-dependent binding of all probes to the particles (Figure 3A; Figure S7). Negligible probe binding was observed in the absence of MmpL3tb or MmpL3tb-GFP on the particles (i.e., both untreated particles and particles treated with the mouse anti-His IgG antibody only) (data not shown), on particles harboring the GFP protein alone, or when the TAMRA-mannose probe was used (Figure 3A).
Figure 3.

Evidence of inhibitor probe binding to purified MmpL3tb-GFP and flow-cytometry-based competition binding assay using purified MmpL3tb-GFP. (A) Comparative analysis of the binding of indolecarboxamide probes to MmpL3tb-GFP bound to polystyrene particles. MmpL3tb-GFP-coated beads were incubated for 15 min at room temperature with different concentrations of North 100 and North 114. After three washes in PBS pH 7.0–5% glycerol, the TAMRA mean fluorescence intensity (MFI) of the beads was analyzed by flow cytometry. The histograms show the absence of binding of the TAMRA-mannose probe to MmpL3tb-GFP-coated beads, while North 100 and North 114 bind to particles coated with different concentrations of MmpL3tb-GFP. The graph shows the concentration-dependent binding of North 100 and North 114 to particles coated with MmpL3tb-GFP; no binding is observed on particles coated with GFP alone. The MFI reported are mean values ± SD of technical triplicates and are representative of at least two independent experiments. (B) Flow-cytometry-based competition binding assay performed on purified MmpL3-GFP. Cells were cotreated with 2 μM North 114 and increasing concentrations of the inhibitors as described in the Methods. The concentrations of inhibitors are indicated under the x-axis. Shown on the y-axis are the MFI of the polystyrene particles from each treatment group expressed relative to that of particles not treated with any inhibitor (relative fluorescence value [RFI] arbitrarily set to 1). MFIs were determined by analyzing 10 000 particles under each condition. The data reported are mean values ± SD of technical duplicates and are representative of at least three independent experiments. Asterisks denote statistically significant decreases in fluorescence intensity between no inhibitor control (blue bars) and beads cotreated with North 114 and various concentrations of the inhibitors pursuant to the Student’s t-test (P < 0.05). No significant displacement was seen with the negative control drugs isoniazid (INH) and rifampicin (RIF), whose mechanisms of action are independent of MmpL3tb (with the exception of INH at 2 μM but not 4 μM).
Having established that our indolecarboxamide and adamantyl urea-based probes bound to purified MmpL3tb, we next determined whether a displacement of North 114 would occur upon addition of increasing concentrations of each of the six prototypical inhibitors to the reaction mixture. The results that are presented in Figure 3B revealed a clear concentration-dependent displacement of North 114 by NITD-304, NITD-349, AU1235, BM212, and THPP1. The displacement was not significant for SQ109. The residual binding of North 114 to MmpL3tb might be explained by the relatively high hydrophobicity of this probe, making it prone to accumulate in the hydrophobic environment of MmpL3tb. In summary, whether due to a competition between the test inhibitors and the fluorescent probe for the same binding site on MmpL3tb or to conformational changes in MmpL3tb induced by the test inhibitors27 reducing the affinity of the protein for the fluorescent probe, five out the six test compounds clearly proved capable of displacing North 114 in a concentration-dependent manner, indicative of their direct interaction with the mycolic acid transporter.
Competition Binding between MmpL3 Inhibitor and Inhibitor Probes in Intact Mycobacterial Cells
Since the metabolic labeling of probe-treated MsmgΔmmpL3/pMVGH1-mmpL3tb cells (Figure S5) and the colocalization of North 100 and North 114 with MmpL3tb-GFP in intact bacilli (Figure 2 and Figure S6) both indicated that the inhibitor probes reached their target in whole cells, we next adapted the flow-cytometry-based displacement assay described above to intact cells and assessed the potential of this relatively simple assay in the rapid screening of MmpL3tb inhibitors. Flow-cytometry-based competition binding assays between North 114 or North 100 and MmpL3 inhibitors were run using MsmgΔmmpL3/pMVGH1-mmpL3tb cells as a source of MmpL3tb target. Prior to performing these assays, we first verified by metabolic labeling with [1,2-14C]acetate that BM212, THPP1, NITD-304, NITD-349, and SQ109 actually targeted MmpL3tb in our Msmg recombinant system (Figure S8). That this was the case for AU1235 was previously established by Grzegorzewicz et al.1
The addition of increasing concentrations of the six prototypical compounds to MsmgΔmmpL3/pMVGH1-mmpL3tb cells previously labeled with either North 100 or North 114 revealed a concentration-dependent displacement of the probe by all six inhibitors that was not observed with the control drugs INH and RIF (Figure 4).
Figure 4.
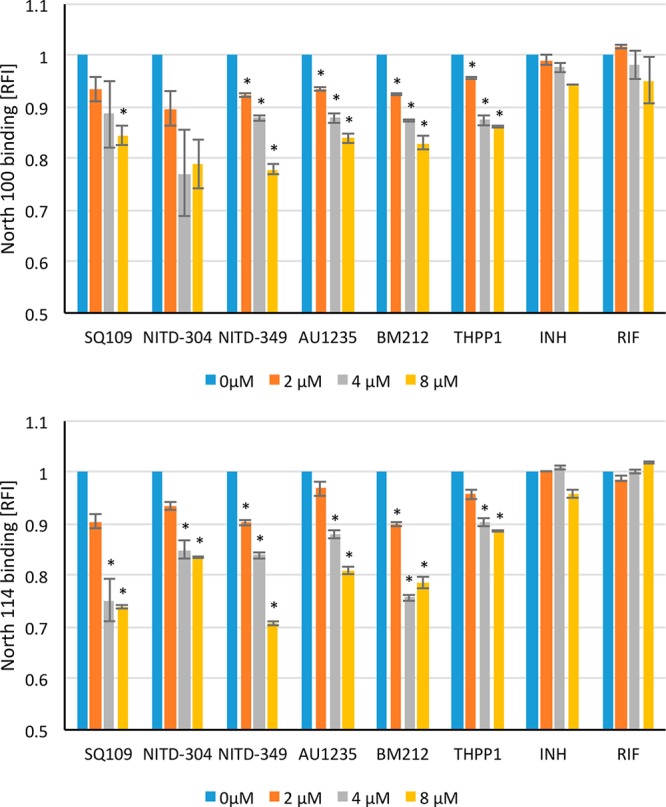
Flow-cytometry-based competition binding assay using intact Msmg cells expressing MmpL3tb. Flow-cytometry-based competition binding assay was performed in an Msmg mmpL3 deletion mutant expressing the wild-type mmpL3tb gene fused to gfp (MsmgΔmmpL3/pMVGH1-mmpL3tb). Cells were labeled with 4 μM North 100 (top graph) or North 114 (bottom graph) and subsequently treated with increasing concentrations of the inhibitors as described in the Methods. The concentrations of inhibitors are indicated under the x-axis. Shown on the y-axis are the MFI of the bacilli from each treatment group expressed relative to that of bacilli not treated with any inhibitor (relative fluorescence value [RFI] arbitrarily set to 1). MFIs were determined by analyzing 10 000 bacilli under each condition. The data reported are mean values ± SD of technical duplicates and are representative of at least three independent experiments. Asterisks denote statistically significant decreases in fluorescence intensity between no inhibitor controls (blue bars) and bacilli cotreated with North 100 or North 114 and various concentrations of the inhibitors pursuant to the Student’s t-test (P < 0.05).
Detection of Inhibitor Binding to MmpL3 Using Biolayer Interferometry and Surface Plasmon Resonance
As an alternative and more direct approach to monitor inhibitor binding to purified MmpL3tb, a biolayer interferometry (BLI) assay and a surface plasmon resonance (SPR) assay were further developed.
For the BLI assay, MmpL3tb purified from Msmg was immobilized onto the surface of sensor tips, and compound interaction with MmpL3tb was monitored by first dipping the sensors into microplate wells containing increasing concentrations of test compounds (association), followed by dipping into wells containing buffer alone (dissociation). The kinetics of compound binding and dissociation to MmpL3tb was monitored in real-time. Among the compounds tested, SQ109, North 4 and North 21 (the structural analogues of North 114 and North 100 devoid of TAMRA fluorophore, respectively) (Table 1) consistently bound to MmpL3tb with the strongest signals (Figure S9). A weak binding signal was also detected for AU1235. BM212 is poorly soluble and yielded only weak binding signals. No signal was detected for THPP1, NITD-304, or NITD-349. Finally, the fluorescence of North 100 and North 114 interferes with the BLI detection method; these probes could thus not be tested for binding using this method.
For the SPR assay, the purified MmpL3tb protein was immobilized onto the surface of CM5 chips, and increasing concentrations of inhibitors were injected over the surface. Unlike BLI, SPR is insensitive to fluorescence, and both the North 100 and North 114 probes were thus included in the SPR experiments. We found that all tested compounds bind to MmpL3tb specifically, albeit with signals of different strengths and different apparent affinities (Figure 5). Fitting the data into kinetic models yielded the on- and off-rates and the dissociation constants for each compound (Table S1). The best fit for all (with the exception of North 114) was found for the two-state reaction model that postulates a conformational change following inhibitor binding. Indeed, binding of small molecule inhibitors to large proteins such as immobilized MmpL3tb is unlikely to lead to large changes in mass. Hence, the strong SPR signals observed in the case of North 100, North 114, AU1235, SQ109, NITD-304, and THPP1 are most likely due to conformational changes in the transporter induced by the binding of these inhibitors, a hypothesis now supported by the high-resolution crystal structure of MmpL3 from Msmg in complex with SQ109 and an indolecarboxamide inhibitor.27 The dissociation constants (KD) describing the binding of the inhibitors to MmpL3tb range from the low millimolar to low micromolar concentrations with NITD-349, NITD-304, AU1235, and North 100 having the highest affinities to the transporter (Table S1). North 114 was the only compound presenting a submicromolar affinity for one of the MmpL3tb states and further studies are needed to understand the molecular mechanism of these interactions.
Figure 5.

Direct interactions of inhibitors with MmpL3tb as measured by surface plasmon resonance. The MmpL3tb protein was immobilized at the density ∼12 000 response units (RU), and compounds were injected in HEPES-TX buffer supplemented with 5% DMSO at concentrations 1.625, 3.75, 7.5, 15, 30, and 60 μM for BM212 and 12.5, 25, 50, 100, 200, and 400 μM for the rest of the compounds. The SPR sensorgrams (black lines) were fitted into different kinetic models, and the best fits are shown as red lines.
Conformational Changes in MmpL3 upon Inhibitor Binding
Assuming that the inhibition of MmpL3 by some of the compounds results from conformational changes in the transporter upon inhibitor binding, it is possible that mutations conferring resistance to these inhibitors themselves alter the conformation of MmpL3 to protect its activity. Such mutations may either prevent the inhibitor from binding to MmpL3 or restore TMM export activity despite inhibitor-induced conformational changes. The G253E variant of MmpL3tb confers resistance to all prototypic inhibitors with the exception of BM212 (Table 1). We thus purified this MmpL3tb variant and compared the interactions of inhibitors with WT MmpL3tb versus MmpL3tb(G253E) by BLI. Surprisingly, while no binding to MmpL3tb(G253E) was detected for SQ109 and North 21, the binding of North 4 to MmpL3tb was unaltered by the G253E mutation (Figure S9). No differences in BLI signals were found for the other tested compounds. Thus, the G253E substitution specifically affects the interactions of some inhibitors with MmpL3tb.
To determine whether the binding of inhibitors and the G253E substitution affected the conformation of MmpL3tb, we next used a partial proteolysis approach. To this end, the purified parent and G253E MmpL3tb variants were treated with increasing concentrations of trypsin in the presence and absence of inhibitors. We found that the proteolytic patterns of the WT and G253E variants differ from one another. The tryptic profiles of WT MmpL3tb comprise four fragments with approximate molecular masses of 78 kDa (band 1), 69 kDa (band 2), 65 kDa (band 3), and 59 kDa (band 4) that are reproducibly present on the gels (Figure 6). The last three tryptic fragments are the most stable and accumulate at the highest concentration of trypsin (1 μg/mL). In contrast, in addition to the 69, 65, and 59 kDa fragments that are common with the WT, the proteolytic profiles of MmpL3tb(G253E) contain two unique bands with estimated molecular masses of 90 kDa (band 5) and 75 kDa (band 6). This result strongly suggests that the G253E substitution stabilizes a different conformation of MmpL3tb, which could be contributing to changes in inhibitor binding and resistance.
Figure 6.
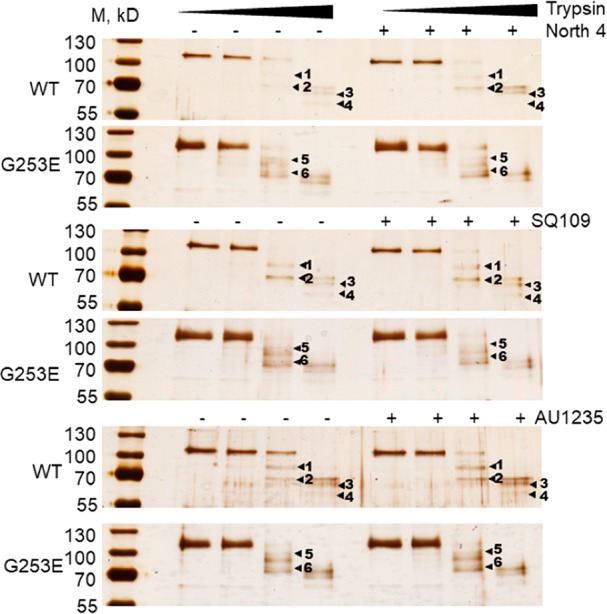
Proteolytic profiles of the wild-type MmpL3WT and its resistant variant MmpL3G253E in the presence and absence of inhibitors. Purified proteins (100 nM final concentrations) were incubated with increasing concentrations of trypsin (0.01, 0.1, and 1.0 μg/mL). Inhibitors were added where indicated to the final concentration of 200 μM prior to trypsin. The digest was carried out for 30 min at 37 °C, and the tryptic fragments were separated by 12% SDS-PAGE followed by silver nitrate staining. The dominant bands are indicated by arrowheads.
The addition of a molar excess of inhibitors changed the relative amounts of tryptic fragments but not the proteolytic profiles of the MmpL3tb WT and G253E variants (Figure 6 and Figure S10). This result is consistent with transient changes in MmpL3tb upon inhibitor binding and is in agreement with the fast on- and off-kinetics of their interactions with the protein (Figure 5 and Figure S9).
Effect of MmpL3 Inhibition versus Inhibitors on the PMF of Mtb
The deleterious impact of MmpL3 inhibition on the PMF and energy production of mycobacterial cells has been suggested by two independent studies. First was the report by Degiacomi et al.10 which showed that genetically silencing mmpL3 in Mtb leads to the repression of a number of genes involved in energy production, including atpB, which encodes a component of the ATP synthase, and nuoB, nuoD, and nuoH, which encode subunits of the NADH dehydrogenase. Second was the observation that Msmg mutants resistant to a novel class of MmpL3 inhibitors and whose growth properties were consistent with reduced MmpL3 activity displayed a membrane potential significantly higher than that of their WT parent strain.29 Because treating mycobacteria with some MmpL3 inhibitors has been reported to alter the transmembrane potential (ΔΨ), the transmembrane electrochemical proton gradient (ΔpH), or both components of the PMF,21−24 these observations called into question whether the inhibitors themselves were responsible for PMF dissipation (i.e., independent of their binding to MmpL3) as suggested earlier,21 or whether their impact on PMF was secondary to the inhibition of MmpL3.
To differentiate between these two hypotheses, we first measured the effect of genetically silencing mmpL3 on each of the two components of the PMF of Mtb. To this end, an mmpL3 conditional knock-down previously generated in our laboratory (MmpL3-DUC)3 was used to measure the impact of mmpL3 silencing on the ΔΨ and ΔpH of Mtb by labeling with 3,3′-diethyloxacarbocyanine iodide [DiOC2(3)] and 5-chloromethyl-fluorescein diacetate (CMFDA), respectively. While the progressive silencing of mmpL3 with increasing concentrations of anhydro-tetracycline (ATc) did not significantly impact the intracellular pH of the cells, a clear ATc concentration-dependent increase in ΔΨ (up to 2-fold above WT levels) was noted, which stabilized at ATc concentrations at and above 1 ng/mL (Figure 7). Thus, the decrease in proton uptake associated with MmpL3-mediated substrate export that follows mmpL3 silencing leads to a significant increase in ΔΨ in Mtb.
Figure 7.
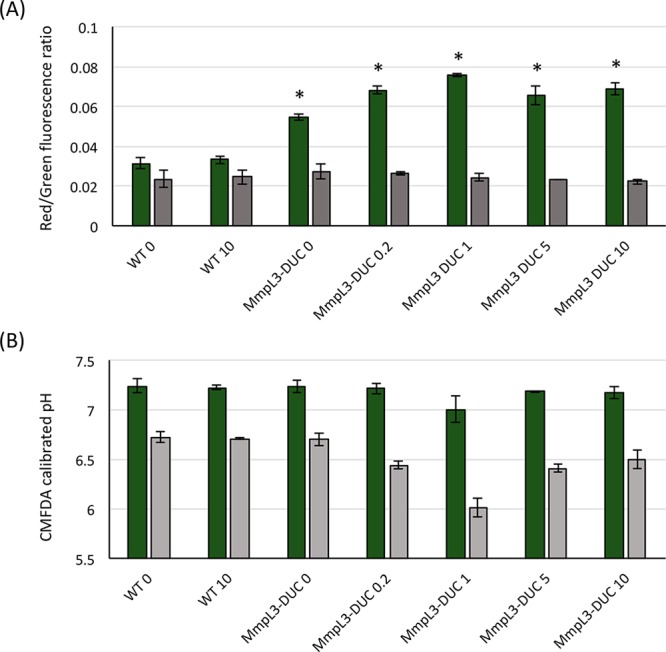
Effect of silencing mmpL3 expression on the membrane potential (ΔΨ) and electrochemical proton gradient (ΔpH) of Mtb. The ΔΨ (A) and inner bacterial pH (B) of wild-type (WT) Mtb and mmpL3 knock-down (MmpL3-DUC) cells grown in the presence of 0, 0.2, 1, 5, or 10 ng/mL anhydro-tetracycline were determined. The expression of mmpL3 was previously shown to be inhibited in an anhydro-tetracycline concentration-dependent manner in the MmpL3-DUC knock-down strain.3 The values represent the averages and standard deviations of measurements performed on three independent bacterial suspensions after exposure to CCCP (4× MIC; 25 μM) (gray bars) or the DMSO solvent (1%) solvent alone (green bars) for 30 min at 37 °C. Results are representative of two independent tests. Asterisks denote statistically significant differences between WT and DUC mutant cells pursuant to the Student’s t-test (P < 0.05).
Next, the same labeling approach with the fluorescent dyes DiOC2 and CMFDA was used to investigate the effect of all six MmpL3 inhibitors on the PMF of the same Mtb H37Rv mc26206 wild-type parent strain as above (rather than that of Msmg as was done previously).21 Consistent with earlier findings in Msmg,21 SQ109 dissipated ΔΨ in a concentration-dependent manner, while BM212 showed an effect on ΔΨ only at the highest concentration tested (8× MIC), and other MmpL3 inhibitors failed to show any effect at all at concentrations up to 20× their MIC value (Figure 8A). BM212 at 8× MIC and SQ109 at 4× and 20× MIC further collapsed ΔpH, whereas other MmpL3 inhibitors showed no such effect in this assay (Figure 8B). The effect of BM212 and SQ109 on ΔpH further reflected in a succinate-driven proton translocation assay with the fluorescent substrate ACMA22 using Mtb H37Rv mc26206 inverted membrane vesicles (IMVs). Interestingly, AU1235 also collapsed ΔpH in this assay when used at fourfold MIC concentration (Figure S11A). Reasons why the effect of AU1235 on ΔpH was detected in the IMV assay but not in the assay using intact Mtb bacilli may be due to the inefficient or slow penetration of this compound inside cells and to the relatively short time of exposure of intact bacilli to the inhibitor (30 min). Speaking for the broad-spectrum uncoupling activity of SQ109, this inhibitor similarly dissipated the pH gradient of IMVs prepared from E. coli which are naturally devoid of MmpL3. BM212, AU1235, and the three other MmpL3 inhibitors, in contrast, showed no such activity (Figure S11B).
Figure 8.

Effect of SQ109, BM212, THPP1, AU1235, NITD-304, and NITD-349 on the membrane potential (ΔΨ) and electrochemical proton gradient (ΔpH) of intact Mtb bacilli. (A) Effect of inhibitors on ΔΨ (A) and ΔpH (B) of intact Mtb cells. The ΔΨ and inner bacterial pH of Mtb cells treated with DMSO and water (solvent controls; brown bars), control antibiotics (isoniazid, rifampicin, and ciprofloxacin), PMF dissipaters (valinomycin, nigericin, and CCCP), or the MmpL3 inhibitors SQ109, THPP1, AU1235, NITD-304, and NITD-349 at 4× and 20× their MIC value were determined. For solubility reasons, BM212 and nigericin were tested at 2× and 8× their MIC value (MIC values: NITD-304, 0.02 μM; NITD-349, 0.05 μM; SQ109, 2.36 μM; AU1235, 0.48 μM; BM212, 3.76 μM; THPP1, 13.44 μM; INH, 0.58 μM; RIF, 0.19 μM; nigericin, 4.31 μM; CCCP, 6.25 μM). Green bars are for the 2× and 4× MIC treatments; gray bars are for the 8× or 20× MIC treatments. Results are representative of three independent tests. The values represent the averages and standard deviations of measurements performed on three independent bacterial suspensions after exposure to the test compounds or solvents for 30 min at 37 °C. Asterisks denote statistically significant differences between water or DMSO controls and inhibitor-treated cells pursuant to the Student’s t-test (P < 0.05).
Because the impact of BM212, AU1235, and SQ109 on PMF dissipation is (i) not shared by all classes of MmpL3 inhibitors and (ii) does not phenocopy the effect of silencing mmpL3 in Mtb, we thus conclude from these experiments that the observed effects of a subset of MmpL3 inhibitors on ΔΨ and ΔpH is not consecutive to the suppression of MmpL3 activity but rather a consequence of secondary effects of these compounds on the bacilli.
Discussion
Multiple independent lines of evidence, including direct detection of fluorescent and unlabeled inhibitor binding to purified MmpL3tb, colocalization of inhibitor probes and MmpL3tb in intact bacilli, comparative proteolysis of MmpL3 WT in the presence and absence of inhibitors, and in vitro and whole-cell-based competition binding assays, point to the direct binding of all analyzed structurally different series of compounds to the transporter. BLI, SPR, and limited proteolysis of the MmpL3tb protein further point to important conformational changes in the transporter as a result of the binding of most compounds. Collectively, our results are in agreement with the recent analysis of cocrystal structures of MmpL3 in complex with SQ109, AU1235, and an indolecarboxamide.27 The observation by Zhang and collaborators27 that three series of inhibitors all bind to the same site within the proton translocation channel of the transporter further helps understand how some single missense mutations may confer cross-resistance to multiple compounds and provides a structural rationale for the functionality of our probe displacement assays.
The existence of both direct and indirect mechanisms of inhibition of MmpL3 (the latter involving the dissipation of PMF in treated cells) has been proposed as a possible explanation for the promiscuity of MmpL3 as a drug target. The results of our study now clearly favor a direct mechanism of inhibition of MmpL3 by all compound series analyzed to date, whether or not they display additional effects on PMF that potentiates their activity. The typical lipophilicity associated with MmpL3 inhibitors,12 which likely favors their partitioning in the phospholipid bilayer where they interact with MmpL3,4,27 is most probably a key driver of their efficacy. Given the extreme vulnerability of MmpL3,3 it is to be expected that compounds with even weak binding affinity to the transporter may still show potency, thereby explaining the bias of phenotypic screens toward hits targeting MmpL3.30 A correlate of this is that MmpL3 inhibition may easily mask potential secondary effects or targets of selected hits. Both the facts that SQ109, BM212, and some THPP compounds show activity against nonreplicating persistent Mtb bacilli21 and that SQ109, BM212, and AU1235 dissipate one or both components of the PMF in Mtb (unlike most other MmpL3 inhibitors) are clear indications that these inhibitors target more than one aspect of the physiology of mycobacteria. The interaction between THPP1 and MmpL3 revealed by our studies further helps explain why this compound, which was reported to target another protein required for mycolic acid synthesis known as EchA6,16,31 initially leads to the buildup of TMM in treated cells despite inhibiting mycolate synthesis and why spontaneous THPP1-resistant mutants consistently display mutations in MmpL3 rather than EchA6.8,16,32
The specificity of a subset of resistance mutations for some compound series contrasts with the finding of a number of mutations conferring broad-spectrum resistance to chemically diverse inhibitors (Table 1). In light of the conformational change induced by the G253E mutation (Figure 6), it is tempting to speculate that some of these broad-spectrum resistance mutations may function to either restore TMM export to levels sufficient to sustain growth or to simultaneously prevent multiple series of compounds from accessing their binding site on the transporter. Support for both hypotheses was in fact obtained from our BLI experiments wherein the MmpL3tb G253E variant lost the ability to bind some inhibitors (North 21; SQ109) yet retained the ability to bind others (North 4) (Figure S9) despite having become resistant to all of them (Table 1).
An intriguing observation resulting from the analysis of PMF in a Mtb mmpL3 conditional knock-down was the finding that mmpL3 silencing leads to a dramatic increase in ΔΨ (Figure 7). This finding is in line with the results of earlier transcriptomics studies that pointed to important perturbations in the membrane energetics of Mtb cells upon mmpL3 silencing.10 It is suggestive of the important impact of MmpL3 transport activity on the PMF of intact cells as a whole, and tentatively speaks for the amount of energy required by mycobacteria to carry out the essential physiological process of exporting mycolic acids during active growth.
Having now established that SQ109, THPP1, BM212, adamantyl ureas, and indolecarboxamides interact with MmpL3, future studies may focus on optimizing some of these series into next-generation inhibitors with improved potency by structure-based drug design and establishing through the development of ad hoc biochemical assays, whether these inhibitors abolish proton translocation in the transporter as suggested by recent structural studies27 or whether they act through different mechanisms.
Methods
Bacterial Strains and Growth Conditions
The avirulent auxotrophic Mtb H37Rv strain mc26206 (ΔpanCDΔleuCD) was grown at 37 °C in Middlebrook 7H9-OADC-0.05% tyloxapol supplemented with 0.2% casamino acids, 48 μg/mL pantothenate and 50 μg/mL l-leucine or on similarly supplemented Middlebrook 7H11-OADC agar medium. Msmg mc2155 was grown in Middlebrook 7H9 broth (Difco) with 10% albumin-dextrose-catalase (ADC) supplement and 0.05% Tween 80, Luria–Bertani (LB) medium (10 g/L Bacto-tryptone, 5 g/L yeast extract and 5 g/L NaCl) (Difco) and on 7H11-ADC or LB agar at 37 °C. Kanamycin (Kan; 25 μg/mL), streptomycin (Str, 20 μg/mL), and hygromycin (Hyg; 50 μg/mL) were added as needed.
Expression of Mutated Variants of MmpL3tb
Seventy-seven mutated variants of MmpL3tb were tested for susceptibility to MmpL3 inhibitors. They include the C-terminally truncated and point-mutated MmpL3tb variants generated as part of functional studies reported earlier4 (total of 59 variants). Other mutants (L189A, L189R, G253E, G253R, G253W, S288T, T311I, Q372R, L567P, S591I, S591W, S591G, F644C, F644Y, V684A, V684G, V684I, and V684W) were generated using a standard two-step PCR overlap method or PCR-amplified from the genomic DNA of AU1235-, NITD-304-, and NITD-349-Mtb spontaneous resistant mutants generated in-house. All variants were cloned in the mycobacterial expression mycobacterial plasmid, pMVGH1, under control of the hsp60 promoter and expressed in the background of a Msmg mmpL3 deletion mutant (MsmgΔmmpL3) as described previously.1,4MsmgΔmmpL3/pMVGH1-mmpL3tb-gfp was similarly generated by expressing in MsmgΔmmpL3 the mmpL3 gene from Mtb fused to gfp at its 3′-end.
Drug-Susceptibility Testing
The susceptibility of MsmgΔmmpL3 expressing different variants of MmpL3tb to inhibitors and control drugs was determined in 96-well microtiter plates at 37 °C in 7H9-ADC-0.05% Tween 80 medium using the resazurin blue test.33
Whole Cell Radiolabeling Experiments
Radiolabeling of Msmg and Mtb (0.5 μCi/ml; specific activity, 54.3 Ci/mol, PerkinElmer) was performed for 3 h (Msmg) or 24 h (Mtb) at 37 °C with shaking. MmpL3 inhibitors and inhibitor probes were added at the same time as [1,2-14C]acetic acid to the bacterial cultures.
Lipid and Mycolic Acid Analyses
Total lipid extraction from bacterial cells and preparation of fatty acid and mycolic acid methyl esters from extractable lipids and delipidated cells followed earlier procedures.1 [1,2-14C]acetic acid-derived lipids and fatty acid/mycolic acid methyl esters were separated by TLC on aluminum-backed silica gel 60-precoated plates F254 (E. Merck) and revealed by PhosphorImaging.
Membrane Potential and Electrochemical Proton Gradient Measurements in Intact Mtb Cells
The effects of inhibitors on the transmembrane potential (ΔΨ) and transmembrane electrochemical proton gradient (ΔpH) of intact Mtb H37Rv mc26206 cells were determined by fluorescence quenching of the membrane potential-sensitive probe 3-3′diethyloxacarbocyanine iodide [DiOC2(3)] (ThermoFisher) and the pH-sensitive probe 5-chloromethyl-fluorescein diacetate (CMFDA) (ThermoFisher), respectively, essentially as described.24 For ΔΨ measurements, Mtb cells were labeled with 60 μM DiOC2 in PBS supplemented with 50 mM KCl, pH 7.2, for 4 h at 37 °C. For ΔpH measurements, Mtb cells were labeled with 10 μM CMFDA in PBS, pH 7.0, for 4 h at 37 °C. Changes in fluorescence due to the disruption of ΔΨ or ΔpH upon inhibitor treatment were monitored for 30 min with a fluorescence spectrophotometer (Victor X5, PerkinElmer). DiOC2(3) was excited at 485 nm, and emission was measured at 615 and 535 nm. The ratio of fluorescence intensity at 615 vs 535 nm is relative to the strength of ΔΨ. The excitation wavelengths for the ΔpH assay were 440 and 490 nm, and fluorescence emission was measured at 520 nm. The pH of the bacterial cytoplasm was measured by calculating the ratio of fluorescence intensity excited at 490 vs 440 nm.
Microscopy
MsmgΔmmpL3/pMVGH1-mmpL3tb-gfp cultures grown to exponential phase and treated for 4 h with 2 μM of North 100 and North 114 at 37 °C were collected, washed twice in phosphate-buffered saline containing 0.05% Tween 80, and fixed in freshly prepared 2% paraformaldehyde for 30 min at room temperature. Approximately 106 cells were next transferred to a glass slide by Cytospin, mounted with Fluoro-Gel (Electron Microscopy Science) and visualized using a KEYENCE BZ-X700 fluorescence microscope. Alternatively, cells were visualized using an Olympus FV-1000 confocal microscope. Multiple independent experiments were performed, and images from one representative experiment are shown.
Binding of Purified MmpL3tb to Flow Beads and Competition Binding Assays with Inhibitor Probe(s)
Purified MmpL3tb-GFP and MmpL3tb (see Supplementary Methods) were coated onto goat antimouse IgG (H&L)-coated polystyrene particles (Spherotech) previously treated with a mouse anti-His IgG antibody (Sigma). To assess fluorescent probe binding and for competition binding assays, MmpL3tb- and MmpL3tb-GFP-coated beads were incubated for 15 min at room temperature with different concentrations of the probes in the presence or absence of test inhibitors. After three washes in PBS pH 7.0–5% glycerol, the TAMRA and BODIPY FL mean fluorescence intensities of the beads was analyzed by flow cytometry on a Cytek Aurora Spectral cytometer. Flow Cytometry Standard (FCS) file data were analyzed using Flowjo software (Treestar Inc., Ashland, OR).
Competition Binding Assays Using Intact Msmg Cells
Competition binding assays in intact MsmgΔmmpL3/pMVGH1-mmpL3tb bacilli were conducted by treating the cells with 4 μM of probe North 114 or North 100 for 1 h at 37 °C, prior to washing the cells twice with 7H9-ADC-0.05% Tween 80 and resuspending them with different concentrations of the test compounds for another hour at 37 °C. Treated cells washed with 7H9-ADC-0.05% Tween 80 and fixed with 2% paraformaldehyde were finally resuspended in PBS-0.05% Tween 80 and subjected to flow cytometry analysis as described above.
Surface Plasmon Resonance
For the amino-coupling of MmpL3tb, CM5 chip surfaces were activated with 0.05 M N-hydroxysuccinimide and 0.2 M N-ethyl-N-(3-diethylaminopropyl)carbodiimide (BIAcore). MmpL3tb was injected over surfaces immediately after activation. After immobilization, the excess of reactive groups was blocked by injecting 0.5 M ethanolamine HCl (pH 8.0). The immobilization and subsequent binding experiments were conducted in running buffer containing 25 mM HEPES-KOH (pH 7.0), 150 mM NaCl, 0.2% Triton X-100 (HEPES-TX). The CM5 chip contains four chambers, one of which contained the immobilized MmpL3tb (ligand), whereas the second (control surface) was activated and processed in the same way but protein was omitted during the immobilization step. For kinetic modeling, we considered only the simplest models that would be compatible with one or two distinct events during both inhibitor binding and dissociation. These four models are (i) simple 1:1 binding model; (ii) heterogeneous ligand (HL), in which different protein populations on chip surface have different kinetic properties; (iii) two-state reaction or ligand-induced conformational change, wherein conformational change occurs on the same time scale as ligand binding; and (iv) bivalent analyte, where multiple analytes bind independently at nonidentical sites. Distinguishing between these models is possible if the data are fit globally; that is, by fitting all sensorgrams obtained at various protein concentrations using the same set of parameters.34
Limited Proteolysis
Proteolysis of MmpL3tb was carried as follows. The purified protein was diluted in assay buffer (150 mM NaCl, 20 mM HEPES-KOH pH 8.0, 0.2% Triton X-100) to a final concentration of 100 nM. Compounds were added to the protein sample to a final concentration of 200 μM (solubility permitting) in DMSO (5% final concentration) and allowed to incubate for 5 min at room temperature prior to digestion with 0.01, 0.1, and 1.0 μg/mL trypsin for 30 min at 37 °C. Protein samples were analyzed by SDS-PAGE and visualized by silver staining.
Acknowledgments
This work was supported by a grant from the National Institutes of Health/National Institute of Allergy and Infectious Diseases (AI116525) (to M.J., H.I.Z. and E.J.N.), a grant from the Bill and Melinda Gates Foundation (OPP1181207) (to M.J. and H.I.Z.), and a sponsored research contract with the Global Alliance for TB Drug Development (to M.J.). The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH. We are grateful to the Global Alliance for TB Drug Development for the provision of NITD-304 and NITD-349, to Dr. Remuinan-Blanco (GSK Tres Cantos Open Lab Foundation) for the provision of THPP1, and to Dr. Borlee (Colorado State University) for his help with confocal imaging.
Glossary
Abbreviations
- ATc
anhydro-tetracycline
- BLI
biolayer interferometry
- IMV
inverted membrane vesicles
- Mtb
Mycobacterium tuberculosis
- Msmg
Mycobacterium smegmatis
- PMF
proton motive force
- SPR
Surface Plasmon Resonance
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsinfecdis.9b00048.
Detailed experimental procedures, supplementary figures, and additional references (PDF)
Author Present Address
□ A.N.P: Sigma-Aldrich Corp., Milwaukee, WI 53209, United States.
Author Present Address
○ W.T.: Department of Laboratory Medicine, Shengjing Hospital of China Medical University, Liaoning, People’s Republic of China.
Author Contributions
W.L. and C.M.S. contributed equally to the work. W.L., C.M.S., Z.D., A.N.P., E.J.N., H.I.Z. and M.J. designed research. W.L., C.M.S., W.T., A.N.P., P.B., Z.D., and M.G.J. performed research. W.L., C.M.S., Z.D., A.N.P., P.B., M.G.J., E.J.N., H.I.Z., and M.J. analyzed data. C.M.S., E.J.N., H.I.Z., and M.J. wrote the main manuscript text. W.L., M.J., C.M.S., and E.J.N. prepared the figures. All authors reviewed the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Grzegorzewicz A. E.; Pham H.; Gundi V. A. K. B.; Scherman M. S.; North E. J.; Hess T.; Jones V.; Gruppo V.; Born S. E. M.; Korduláková J.; Chavadi S. S.; Morisseau C.; Lenaerts A. J.; Lee R. E.; McNeil M. R.; Jackson M. (2012) Inhibition of mycolic acid transport across the Mycobacterium tuberculosis plasma membrane. Nat. Chem. Biol. 8, 334–341. 10.1038/nchembio.794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tahlan K.; Wilson R.; Kastrinsky D. B.; Arora K.; Nair V.; Fischer E.; Barnes S. W.; Walker J. R.; Alland D.; Barry C. E. 3rd; Boshoff H. I. (2012) SQ109 targets MmpL3, a membrane transporter of trehalose monomycolate involved in mycolic acid donation to the cell wall core of Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 56, 1797–1809. 10.1128/AAC.05708-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W.; Obregon-Henao A.; Wallach J. B.; North E. J.; Lee R. E.; Gonzalez-Juarrero M.; Schnappinger D.; Jackson M. (2016) Therapeutic Potential of the Mycobacterium tuberculosis Mycolic Acid Transporter, MmpL3. Antimicrob. Agents Chemother. 60, 5198–5207. 10.1128/AAC.00826-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belardinelli J. M.; Yazidi A.; Yang L.; Fabre L.; Li W.; Jacques B.; Angala S. K.; Rouiller I.; Zgurskaya H. I.; Sygusch J.; Jackson M. (2016) Structure-Function Profile of MmpL3, the Essential Mycolic Acid Transporter from Mycobacterium tuberculosis. ACS Infect. Dis. 2, 702–713. 10.1021/acsinfecdis.6b00095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanley S. A.; Grant S. S.; Kawate T.; Iwase N.; Shimizu M.; Wivagg C.; Silvis M.; Kazyanskaya E.; Aquadro J.; Golas A.; Fitzgerald M.; Dai H.; Zhang L.; Hung D. T. (2012) Identification of novel inhibitors of M. tuberculosis growth using whole cell based high-throughput screening. ACS Chem. Biol. 7, 1377–1384. 10.1021/cb300151m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao S. P.; Lakshminarayana S. B.; Kondreddi R. R.; Herve M.; Camacho L. R.; Bifani P.; Kalapala S. K.; Jiricek J.; Ma N. L.; Tan B. H.; Ng S. H.; Nanjundappa M.; Ravindran S.; Seah P. G.; Thayalan P.; Lim S. H.; Lee B. H.; Goh A.; Barnes W. S.; Chen Z.; Gagaring K.; Chatterjee A. K.; Pethe K.; Kuhen K.; Walker J.; Feng G.; Babu S.; Zhang L.; Blasco F.; Beer D.; Weaver M.; Dartois V.; Glynne R.; Dick T.; Smith P. W.; Diagana T. T.; Manjunatha U. H. (2013) Indolcarboxamide is a preclinical candidate for treating multidrug-resistant tuberculosis. Sci. Transl. Med. 5, 214ra168. 10.1126/scitranslmed.3007355. [DOI] [PubMed] [Google Scholar]
- Lun S.; Guo H.; Onajole O. K.; Pieroni M.; Gunosewoyo H.; Chen G.; Tipparaju S. K.; Ammerman N. C.; Kozikowski A. P.; Bishai W. R. (2013) Indoleamides are active against drug-resistant Mycobacterium tuberculosis. Nat. Commun. 4, 2907. 10.1038/ncomms3907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Remuinan M. J.; Perez-Herran E.; Rullas J.; Alemparte C.; Martinez-Hoyos M.; Dow D. J.; Afari J.; Mehta N.; Esquivias J.; Jimenez E.; Ortega-Muro F.; Fraile-Gabaldon M. T.; Spivey V. L.; Loman N. J.; Pallen M. J.; Constantinidou C.; Minick D. J.; Cacho M.; Rebollo-Lopez M. J.; Gonzalez C.; Sousa V.; Angulo-Barturen I.; Mendoza-Losana A.; Barros D.; Besra G. S.; Ballell L.; Cammack N. (2013) Tetrahydropyrazolo[1,5-a]Pyrimidine-3-Carboxamide and N-Benzyl-6’,7’-Dihydrospiro[Piperidine-4,4’-Thieno[3,2-c]Pyran] Analogues with Bactericidal Efficacy against Mycobacterium tuberculosis Targeting MmpL3. PLoS One 8, e60933 10.1371/journal.pone.0060933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tantry S. J.; Degiacomi G.; Sharma S.; Jena L. K.; Narayan A.; Guptha S.; Shanbhag G.; Menasinakai S.; Mallya M.; Awasthy D.; Balakrishnan G.; Kaur P.; Bhattacharjee D.; Narayan C.; Reddy J.; Naveen Kumar C. N.; Shandil R.; Boldrin F.; Ventura M.; Manganelli R.; Hartkoorn R. C.; Cole S. T.; Panda M.; Markad S. D.; Ramachandran V.; Ghorpade S. R.; Dinesh N. (2015) Whole cell screen based identification of spiropiperidines with potent antitubercular properties. Bioorg. Med. Chem. Lett. 25, 3234–3245. 10.1016/j.bmcl.2015.05.087. [DOI] [PubMed] [Google Scholar]
- Degiacomi G.; Benjak A.; Madacki J.; Boldrin F.; Provvedi R.; Palu G.; Korduláková J.; Cole S. T.; Manganelli R. (2017) Essentiality of mmpL3 and impact of its silencing on Mycobacterium tuberculosis gene expression. Sci. Rep. 7, 43495. 10.1038/srep43495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W., Sanchez-Hidalgo A., Jones V., de Moura V. C., North E. J., and Jackson M. (2017) Synergistic Interactions of MmpL3 Inhibitors with Antitubercular Compounds In Vitro. Antimicrob. Agents Chemother. 61 10.1128/AAC.02399-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W.; Yazidi A.; Pandya A. N.; Hegde P.; Tong W.; Calado Nogueira de Moura V.; North E. J.; Sygusch J.; Jackson M. (2018) MmpL3 as a Target for the Treatment of Drug-Resistant Nontuberculous Mycobacterial Infections. Front. Microbiol. 9, 1547. 10.3389/fmicb.2018.01547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sacksteder K. A.; Protopopova M.; Barry C. E. 3rd; Andries K.; Nacy C. A. (2012) Discovery and development of SQ109: a new antitubercular drug with a novel mechanism of action. Future Microbiol. 7, 823–837. 10.2217/fmb.12.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokokawa F.; Wang G.; Chan W. L.; Ang S. H.; Wong J.; Ma I.; Rao S. P.; Manjunatha U.; Lakshminarayana S. B.; Herve M.; Kounde C.; Tan B. H.; Thayalan P.; Ng S. H.; Nanjundappa M.; Ravindran S.; Gee P.; Tan M.; Wei L.; Goh A.; Chen P. Y.; Lee K. S.; Zhong C.; Wagner T.; Dix I.; Chatterjee A. K.; Pethe K.; Kuhen K.; Glynne R.; Smith P.; Bifani P.; Jiricek J. (2013) Discovery of tetrahydropyrazolopyrimidine carboxamide derivatives as potent and orally active antitubercular agents. ACS Med. Chem. Lett. 4, 451–455. 10.1021/ml400071a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stec J.; Onajole O. K.; Lun S.; Guo H.; Merenbloom B.; Vistoli G.; Bishai W. R.; Kozikowski A. P. (2016) Indole-2-carboxamide-based MmpL3 Inhibitors Show Exceptional Antitubercular Activity in an Animal Model of Tuberculosis Infection. J. Med. Chem. 59, 6232–6247. 10.1021/acs.jmedchem.6b00415. [DOI] [PubMed] [Google Scholar]
- Cox J. A.; Abrahams K. A.; Alemparte C.; Ghidelli-Disse S.; Rullas J.; Angulo-Barturen I.; Singh A.; Gurcha S. S.; Nataraj V.; Bethell S.; Remuinan M. J.; Encinas L.; Jervis P. J.; Cammack N. C.; Bhatt A.; Kruse U.; Bantscheff M.; Futterer K.; Barros D.; Ballell L.; Drewes G.; Besra G. S. (2016) THPP target assignment reveals EchA6 as an essential fatty acid shuttle in mycobacteria. Nat. Microbiol 1, 15006. 10.1038/nmicrobiol.2015.6. [DOI] [PubMed] [Google Scholar]
- Franz N. D.; Belardinelli J. M.; Kaminski M. A.; Dunn L. C.; Calado Nogueira de Moura V.; Blaha M. A.; Truong D. D.; Li W.; Jackson M.; North E. J. (2017) Design, synthesis and evaluation of indole-2-carboxamides with pan anti-mycobacterial activity. Bioorg. Med. Chem. 25, 3746–3755. 10.1016/j.bmc.2017.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozikowski A. P.; Onajole O. K.; Stec J.; Dupont C.; Viljoen A.; Richard M.; Chaira T.; Lun S.; Bishai W.; Raj V. S.; Ordway D.; Kremer L. (2017) Targeting Mycolic Acid Transport by Indole-2-carboxamides for the Treatment of Mycobacterium abscessus Infections. J. Med. Chem. 60, 5876–5888. 10.1021/acs.jmedchem.7b00582. [DOI] [PubMed] [Google Scholar]
- De Groote M. A.; Jackson M.; Gonzalez-Juarrero M.; Li W.; Young C. L.; Wong C.; Graham J.; Day J.; Hoang T.; Jarvis T. C.; Ribble W.; Sun X.; Ochsner U. A. (2018) Optimization and Lead Selection of Benzothiazole Amide Analogs Toward a Novel Antimycobacterial Agent. Front. Microbiol. 9, 2231. 10.3389/fmicb.2018.02231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandya A. N., Prathipati P. K., Hegde P., Li W., Graham K. F., Mandal S., Drescher K. M., Destache C. J., Ordway D., Jackson M., and North E. J. (2019) Indole-2-Carboxamides Are Active against Mycobacterium abscessus in a Mouse Model of Acute Infection. Antimicrob. Agents Chemother. 63 10.1128/AAC.02245-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W.; Upadhyay A.; Fontes F. L.; North E. J.; Wang Y.; Crans D. C.; Grzegorzewicz A. E.; Jones V.; Franzblau S. G.; Lee R. E.; Crick D. C.; Jackson M. (2014) Novel insights into the mechanism of inhibition of MmpL3, a target of multiple pharmacophores in Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 58, 6413–6423. 10.1128/AAC.03229-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li K.; Schurig-Briccio L. A.; Feng X.; Upadhyay A.; Pujari V.; Lechartier B.; Fontes F. L.; Yang H.; Rao G.; Zhu W.; Gulati A.; No J. H.; Cintra G.; Bogue S.; Liu Y. L.; Molohon K.; Orlean P.; Mitchell D. A.; Freitas-Junior L.; Ren F.; Sun H.; Jiang T.; Li Y.; Guo R. T.; Cole S. T.; Gennis R. B.; Crick D. C.; Oldfield E. (2014) Multitarget drug discovery for tuberculosis and other infectious diseases. J. Med. Chem. 57, 3126–3139. 10.1021/jm500131s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng X.; Zhu W.; Schurig-Briccio L. A.; Lindert S.; Shoen C.; Hitchings R.; Li J.; Wang Y.; Baig N.; Zhou T.; Kim B. K.; Crick D. C.; Cynamon M.; McCammon J. A.; Gennis R. B.; Oldfield E. (2015) Antiinfectives targeting enzymes and the proton motive force. Proc. Natl. Acad. Sci. U. S. A. 112, E7073–7082. 10.1073/pnas.1521988112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foss M. H.; Pou S.; Davidson P. M.; Dunaj J. L.; Winter R. W.; Pou S.; Licon M. H.; Doh J. K.; Li Y.; Kelly J. X.; Dodean R. A.; Koop D. R.; Riscoe M. K.; Purdy G. E. (2016) Diphenylether-Modified 1,2-Diamines with Improved Drug Properties for Development against Mycobacterium tuberculosis. ACS Infect. Dis. 2, 500–508. 10.1021/acsinfecdis.6b00052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Z.; Meshcheryakov V. A.; Poce G.; Chng S. S. (2017) MmpL3 is the flippase for mycolic acids in mycobacteria. Proc. Natl. Acad. Sci. U. S. A. 114, 7993–7998. 10.1073/pnas.1700062114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- La Rosa V.; Poce G.; Canseco J. O.; Buroni S.; Pasca M. R.; Biava M.; Raju R. M.; Porretta G. C.; Alfonso S.; Battilocchio C.; Javid B.; Sorrentino F.; Ioerger T. R.; Sacchettini J. C.; Manetti F.; Botta M.; De Logu A.; Rubin E. J.; De Rossi E. (2012) MmpL3 Is the Cellular Target of the Antitubercular Pyrrole Derivative BM212. Antimicrob. Agents Chemother. 56, 324–331. 10.1128/AAC.05270-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B.; Li J.; Yang X.; Wu L.; Zhang J.; Yang Y.; Zhao Y.; Zhang L.; Yang X.; Yang X.; Cheng X.; Liu Z.; Jiang B.; Jiang H.; Guddat L. W.; Yang H.; Rao Z. (2019) Crystal Structures of Membrane Transporter MmpL3, an Anti-TB Drug Target. Cell 176, 636–648 e613. 10.1016/j.cell.2019.01.003. [DOI] [PubMed] [Google Scholar]
- Carel C.; Nukdee K.; Cantaloube S.; Bonne M.; Diagne C. T.; Laval F.; Daffé M.; Zerbib D. (2014) Mycobacterium tuberculosis proteins involved in mycolic acid synthesis and transport localize dynamically to the old growing pole and septum. PLoS One 9, e97148. 10.1371/journal.pone.0097148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNeil M. B.; Dennison D.; Parish T. (2017) Mutations in MmpL3 alter membrane potential, hydrophobicity and antibiotic susceptibility in Mycobacterium smegmatis. Microbiology 163, 1065–1070. 10.1099/mic.0.000498. [DOI] [PubMed] [Google Scholar]
- Goldman R. C. (2013) Why are membrane targets discovered by phenotypic screens and genome sequencing in Mycobacterium tuberculosis?. Tuberculosis 93, 569–588. 10.1016/j.tube.2013.09.003. [DOI] [PubMed] [Google Scholar]
- Moynihan P. J.; Besra G. S. (2017) Colworth prize lecture 2016: exploiting new biological targets from a whole-cell phenotypic screening campaign for TB drug discovery. Microbiology 163, 1385–1388. 10.1099/mic.0.000522. [DOI] [PubMed] [Google Scholar]
- Ioerger T. R.; O’Malley T.; Liao R.; Guinn K. M.; Hickey M. J.; Mohaideen N.; Murphy K. C.; Boshoff H. I.; Mizrahi V.; Rubin E. J.; Sassetti C. M.; Barry C. E.; Sherman D. R.; Parish T.; Sacchettini J. C. (2013) Identification of New Drug Targets and Resistance Mechanisms in Mycobacterium tuberculosis. PLoS One 8, e75245. 10.1371/journal.pone.0075245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin A.; Camacho M.; Portaels F.; Palomino J.-C. (2003) Resazurin microtiter assay plate testing of Mycobacterium tuberculosis susceptibilities to second-line drugs: rapid, simple, and inexpensive method. Antimicrob. Agents Chemother. 47, 3616–3619. 10.1128/AAC.47.11.3616-3619.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tikhonova E. B.; Dastidar V.; Rybenkov V. V.; Zgurskaya H. I. (2009) Kinetic control of TolC recruitment by multidrug efflux complexes. Proc. Natl. Acad. Sci. U. S. A. 106, 16416–16421. 10.1073/pnas.0906601106. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.