

Abstract
The p38αMAPK is a serine/threonine protein kinase and a key node in the intracellular signaling networks that transduce and amplify stress signals into physiological changes. A preponderance of preclinical data and clinical observations established p38αMAPK as a brain drug discovery target involved in neuroinflammatory responses and synaptic dysfunction in multiple degenerative and neuropsychiatric brain disorders. We summarize the discovery of highly selective, brain-penetrant, small molecule p38αMAPK inhibitors that are efficacious in diverse animal models of neurologic disorders. A crystallography and pharmacoinformatic approach to fragment expansion enabled the discovery of an efficacious hit. The addition of secondary pharmacology screens to refinement delivered lead compounds with improved selectivity, appropriate pharmacodynamics, and efficacy. Safety considerations and additional secondary pharmacology screens drove optimization that delivered the drug candidate MW01-18-150SRM (MW150), currently in early stage clinical trials.
Introduction
A theme in pathophysiology progression among diverse central nervous system (CNS) diseases is the involvement of synaptic dysfunction and neuroinflammation as part of pathophysiology progression. In some cases, a neuroinflammation–synaptic dysfunction axis is considered a common disease progression mechanism. For example, innate immune cells, such as glia in the brain, communicate with neurons through an array of secreted and contact-dependent signals. The glia-neuron interactions influence axon homeostasis, plasticity, and synaptic transmission.1,2 As a result of these reciprocal interactions, pathological processes are destined to affect both innate immunity and synaptic communication, either locally or across physiological axes. The identification of risk-modifying genetic polymorphisms and pathology studies3−6 provide clinical evidence in support of the neuroimmune–synaptic dysfunction mechanism of disease progression. On the basis of p38αMAPK involvement in both neuroinflammation and synaptic dysfunction, it is a logical discovery target for alteration of disease or disease susceptibility when the neuroimmune and synaptic dysfunction pathophysiology mechanisms are involved.
Previous clinical trials with p38MAPK inhibitors (Table 1S in Supporting Information) used experimental therapeutics not optimized for CNS pharmacological exposure and employed mixed-kinase inhibitors, some of which exhibited dose limiting toxicity. Clearly, when targeting CNS disorders, there is a need for clinical therapeutic candidates that are selective p38αMAPK inhibitors with improved pharmacological properties and adequate brain exposure. CNS disorders offer an enhanced potential for p38αMAPK inhibitor drug efficacy due to pleiotropic pharmacology driven by multiple pharmacological actions from a single target engagement versus pleiotropic pharmacology due to multiple targets for a single drug. In attempts to address the unmet need, we developed a selective, brain penetrant p38αMAPK inhibitor, MW01-18-150SRM (Figure 1), that is a novel drug candidate currently in phase 1 clinical trials. In this review, we summarize the biological rationale and refinement approach.
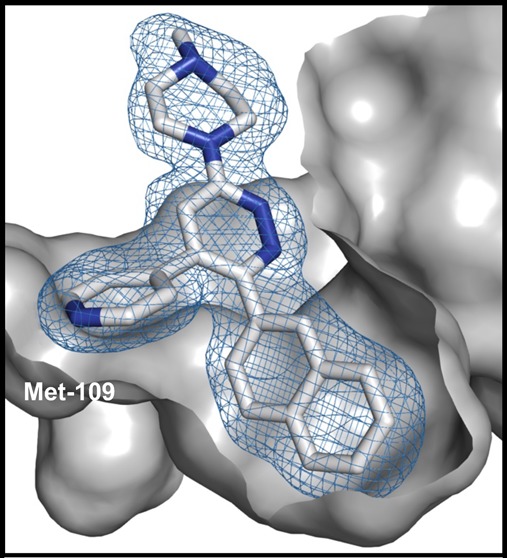
Figure 1.

Structure of the human p38αMAPK:11 complex (PDB code 4R3C). The protein is shown as a gray surface representation, and compound 11 is shown in a stick representation with the atoms colored white for carbon and blue for nitrogen. The mesh over compound 11 reflects the refined experimental electron density for compound 11. (A) Compound 11 binds within the active site cleft, located between the bilobed catalytic domain, and has interactions with the connecting hinge region and a proximal hydrophobic pocket. (B) The compound 11 pyridine ring nitrogen forms a hydrogen bond interaction with the amide backbone at amino acids Leu108 and Met109, while the vicinal 2-naphthyl substituent fills the proximal hydrophobic pocket that adds to kinome selectivity.
Overview
The biological rationale of our approach was a focus on pathophysiology progression mechanisms common to multiple CNS diseases. In this regard, the discovery process was somewhat disease agnostic because common pathophysiology progression mechanisms were targeted in medicinal chemistry refinement and secondary pharmacology screening. This is in contrast to focusing solely on single disease efficacy and primary target affinity that is a prevailing theme in drug discovery and preclinical development. However, deliverables from each stage of refinement (hit-to-lead and lead-to-candidate) were studied in discrete models of disease that exhibit the targeted pathophysiology progression processes.
A key aspect of medicinal chemistry refinement was the extensive use of secondary pharmacology assays during the SAR analyses, which embed a systems analysis dimension to the single target process. Secondary pharmacology outcomes at each stage restrict the chemical diversification process in order to minimize potential pharmacological liabilities and enhance CNS drug related properties.9−14,20,21 Therefore, the overall number of new compound syntheses is restricted as diversifications into chemical space associated at early stages with adverse pharmacology and toxicity are avoided.
High resolution crystallographic structures of enzyme:ligand complexes were used in the design and in the interpretation of deliverables from each phase of refinement, starting with fragment expansion based on crystallography of enzyme:ligand complexes.7,8 Chemical diversifications during refinement were restricted to those that retained key target recognition features of human p38αMAPK:small molecule complexes (PDB codes 4ZTH, 4EWQ, 4F9Y, 4F9W, and 4R3C; CCDC 1851508 and 1851509) and avoided introduction of potential pharmacological liabilities based on pharmacoinformatics or secondary pharmacology screen outcomes.13,14,20,21 As is typical of structure-assisted fragment expansion, the number of syntheses required to attain the campaign goal is lessened. Recognition features included retention of the pyridine ring nitrogen charge properties, as evidenced by potentiometric titration, and orientation relative to the vicinal aromatic substituent, as evidenced by the crystallographic structure of human p38αMAPK:small molecule complexes at each stage.
Primary data and outcomes at discrete stages of the preclinical development of MW150 are described in the literature.4,12,13,15−19 The SAR studies driven by structure and secondary pharmacology filtering improved kinome-wide selectivity, metabolic stability, bioavailability, and function. Dose-limiting toxicology and adverse pharmacology liabilities were identified and removed during early stage hit-to-lead refinement. The process established that p38αMAPK is not a neurotoxic target as previously hypothesized. However, the process did not allow a single off-target kinase or GPCR to be identified as a toxicology associated target. While progression from hit to lead compound removed crossover to a subset of kinase targets seen in earlier campaigns, it also resulted in removal of GPCR agonist and antagonist off-target activity.13 Lead compounds that were inhibitors of both p38αMAPK and p38βMAPK, yet had kinome selectivity and no GPCR agonist or antagonist activity, also did not reveal overt toxicity in secondary pharmacology screens.13,14 Therefore, there is inadequate evidence to support the perspective that p38αMAPK and p38βMAPK crossover activity is a significant contributor to dose-related adverse pharmacology observed in previous p38MAPK campaigns (Table 1S). Overall, an accumulating body of evidence from studies of genetically modified mice and comparative studies of compounds 9–11 raises the possibility that inhibitors engaging both p38αMAPK and p38βMAPK, but having kinome-wide selectivity for the p38MAPK family and lacking key GPCR agonist activity, might be adequate for a variety of pharmacological indications.
Fragment to Hit to Lead Compound
An arylpyridazine fragment (compound 1) was chosen as the starting fragment based on pharmacological precedents (see ref (13) for discussion). Briefly, the pyridazine ring can be exploited as a biologically friendly fragment for kinase inhibitor development through at least two distinct mechanisms. First, it has the potential to serve a ligand recognition role via its ring nitrogens engaged in H-bonding with the hinge region. Second, the pyridazine ring can serve a nonhinge binding role when it is a chemotype in the context of a more basic substituent that fulfills the ligand recognition role, such as the pyridine ring nitrogen of compound 1. The nonhinge binding role provides a framework for pyridazine ring diversifications that sample alternative chemical space as long as the relative charge and orientation of the pyridine ring nitrogen are retained. The hydrogen bonding interaction with the hinge region of the kinase is an interaction also used by ATP. However, the vicinal aromatic group occupancy of the proximal hydrophobic pocket provides potential for selectivity as the pocket is not occupied by ATP and is rare among known kinase structures. The proximal hydrophobic pocket also offers opportunity to increase apparent affinity through increased space filling by lipophilic substituents. The structure of the p38αMAPK:compound 1 complex (PDB code 4ZTH) reveals adequate space for diversification at R3 without disturbing these two key target interactions. The amine, piperazinylpyrimidine, chosen for diversification of compound 1 to generate hit compound 3 was selected based on prior use of this pharmacophore. Especially relevant was its presence in MW01-6-189WH, a clinically safe CNS aminoarylpyridazine therapeutic candidate (NCT02942771, NCT01357421) that contains a pyridine substituent but is not a protein kinase inhibitor (Tables 2S and 3S and compound 2 in Table 5S).
The p38αMAPK inhibitor hit, compound 3, retains the two key target recognition features (Figure 2; PDB code 4EWQ) and is within the multiproperty molecular confines identified by in vivo data for orally bioavailable CNS drugs.20 Independent confirmation of the functional importance of the interaction between the phenyl substituent and the hydrophobic pocket is provided by the failure of compounds to inhibit the point mutant p38αMAPK (T106M).12−14The methionine side chain in the enzymatically active p38αMAPK (T106M) does not allow phenyl group access to the pocket. The hit compound 3 was found to be comparatively selective in terms of kinome crossover (Figure 3 and Table 4S). It exhibited brain exposure and in vivo function in attenuating neuroinflammation, synaptic dysfunction, and cognition-related behavioral deficits.12 However, it has the same liability of prior p38MAPK inhibitors in that it was a multikinase inhibitor (Figure 3).12,13 Further, an initial dose range finding screen revealed a dose limiting therapeutic index (including neurotoxicity) observed at doses >12-fold above efficacy doses.13
Figure 2.

Hit design via fragment expansion. Diversification of the inactive fragment compound 1 by amination yields the active hit compound 3. The crystal structure of the p38αMAPK:1 (PDB code 4ZTH) identifies key molecular recognition features that are retained in the p38αMAPK:3 (PDB code 4EWQ). The key features include the position and net charge of the pyridine ring nitrogen that allows H-bonding with the peptide backbone of the kinase hinge region and the occupancy of the localized hydrophobic pocket by the vicinal phenyl substituent. The representation and orientation of structures shown are as in Figure 1B.
Figure 3.

Kinome target selectivity of hit compound 3 vs off-target kinases of widely used p38MAPK inhibitors. Red circles denote inhibition at specific kinases with IC50 < 1 μM. (A) Kinase inhibition profile for the widely used p38αMAPK inhibitors VX-745, BIRB-796, and SB203580.24−26 Crossover kinases with IC50 < 1 μM for VX-745 include ABL1, ABL2, p38β, PDGFRβ, SRC; for BIRB-796 include BLK, CDK5, CDK8, DDR1, DDR2, EPHA3, EPHA7, EPHA8, EPHB2, p38β p38γ, FLT1, FRK, NTRK1, JNK1, JNK2, JNK3, KIT, MAP4K4, MRCKβ, PTK2β, RET, SLK, STK10, TIE1, TIE2, TNIK, TRKB, TRKC, ZAK; and for SB203580 include BRAF, CIT, CK1δ, CK1ε, DMPK, GAK, JNK2, JNK3, NLK, p38β, RIPK2, STK36, TNIK. (B) Kinase targets (IC50 < 1 μM) for compound 3 (Table 4S). Crossover kinases with IC50 < 1 μM for compound 3 include CK1α, CK1δ, CK1ε, PKD3, RIPK2, and TNIK.
The next step to explore improvement in safety, improvement in comparative kinase inhibition activity, and retention of the H-bond interaction with the hinge with vicinyl aromatic occupancy of the proximal pocket was simplification of the amine substituent of compound 3 (Table 1). Simplification was required so that subsequent SAR could explore expansion of the aromatic at R6 while remaining within the property constraints that pharmacoinformatics identified among CNS drug profiles.20 The crystallographic structures revealed exploitable chemical space around R3 that should retain the key recognition features. The substitution of the piperazinylpyrimidine substituent with methylpiperazine yielded compound 4 (Table 1). The use of secondary pharmacology screens revealed that simplification of the amine at R3 also removed13 the dose limiting toxicity found in dose range finding studies (increasing maximal tolerated dose in mice from 60 mg/kg for compound 3 to >200 mg/kg for compound 4). While a range of simplified R3 amines that retained the IC50 were identified, secondary pharmacology screens revealed that compound 4 did not solve the perplexing issue13 that widely used p38αMAPK inhibitors can be good inhibitors of CK1δ. CK1 is a challenging off-target kinase13 with potential toxicology associations and pharmacodynamics that can overlap those of multikinase p38αMAPK inhibitors. The preference of p38MAPK inhibitors for CK1δ and crossover to other kinome targets also places severe limits on the interpretation of efficacy studies. A logical next step provided by compound 4 was to explore variation of the amine substituent and screen for selectivity for p38MAPKs vs CK1δ (Table 1).13 However, this strategy, exemplified by compounds 5–8, generated compounds that retained or improved CK1δ inhibitory activity (Table 1). Nevertheless, these more simplified structures with retention of p38αMAPK inhibitor activity allowed pursuit of vicinal aromatic substituents while staying within the overall multiproperty limits of the approach (MW < 400, log P < 4, PSA ∼ 40).
Table 1. Hit Compound 3 to Lead Compound 9a.

Footnote: *Primary screen assay linearity 0.01 to 10 μM. Values are the mean ± SD. Values outside linearity are reported as >10 μM.
The crystallographic structures of the human p38αMAPK:fragment and p38αMAPK:hit compound complexes (PDB codes 4ZTH and 4EWQ; Figure 2) revealed significant unoccupied space within the pocket. Inspection of available structures in the PDB for other kinases also did not reveal a similar pocket in close proximity to the established kinase hinge binding region. The hypothesis that better occupancy of the proximal pocket via aryl substituent diversification could generate additional enhancement of kinase selectivity was explored using CK1δ as an internal control (Table 1), resulting in the delivery of compound 9.(13) The kinase activity screen and the crystal structure of the human p38αMAPK:9 complex (Figure 5; PDB code 4F9Y) demonstrated13 the feasibility of this approach.
Figure 5.

Comparison of the p38αMAPK:9 and p38αMAPK:10 complexes. Shown are the representation and the orientation of (A) compound 9 and (B) compound 10 in the kinase active site. Perspective is the same as for compound 11 in Figure 1B. The hydrogen bond interaction of the pyridine ring nitrogen with the amide backbone of the kinase is retained in all structures, and the vicinal naphthyl groups of compounds 9–11 occupy the proximal hydrophobic pocket.
Subjecting compound 9 to kinome-wide, hierarchal, concentration dependent inhibitor screens documented (Figure 4A) its improved kinome-wide selectivity (within drug target class).13 Secondary pharmacology screening for functional GPCR agonist and antagonist activity demonstrated13 that there was no crossover to this other major CNS drug target class, a concern when molecular recognition is altered by increased lipophilicity. The absence of certain GPCR agonist activities, such as those associated with cardiotoxicity,10 also reduced campaign risk for later stage drug development. Additional in vitro and in vivo pharmacological screens of the lead compound 9 confirmed drug target engagement and pharmacodynamic biomarkers.16−18 Compound 9 was subjected to a final check for function and shown in distinct animal models to have in vivo efficacy.13,17
Figure 4.

Kinome selectivity of lead compound 9 and candidate compounds 10 and 11. (A) Progression from compound 3 to compound 9 improved kinome selectivity to p38αMAPK, p38βMAPK, and atypical p38MAPK (NLK). In parallel with kinome selectivity improvement, crossover activity in GPCR agonist and antagonist cell based screens was removed as was in vivo dose limiting toxicity. (B) Refinement to compounds 10 and 11 biased inhibition toward p38αMAPK with retention of overall kinome selectivity, absence of GPCR agonist or antagonist activity, and absence of dose limiting toxicity. Red circles reflect inhibition at specific kinases with IC50 < 1 μM.13,14
At this stage of the campaign, compound 9 delivered a new and more selective p38MAPK inhibitor with in vivo function, had an IC50 for CK1δ well above the canonical 1 μM mark, and retained activity below 1 μM for p38αMAPK and p38βMAPK.
From Lead Compound to Candidate
The crystal structure of the p38αMAPK:9 complex (PDB code 4F9Y; Figure 5) forecast as a reasonable next step continued exploration of better pocket occupancy to further enhance kinome selectivity and metabolic stability. A logical extension was testing a 2-naphthyl substituent at R6. As summarized in Table 2, compound 10 yielded an improved compound that also provided some separation of p38αMAPK inhibitor activity from that for p38βMAPK while not allowing CK1δ crossover activity to move below the 1 μM cutoff. The crystal structure of the p38αMAPK:10 complex confirmed a more intimate filling of the proximal pocket (Figure 5; PDB code 4F9W).
Table 2. Lead Compound 9 to Candidate Compound 11a.

Footnote: *Activity screen assay linearity from 0.01 to 10 μM. Values are mean ± SD. Values outside linearity, >10 μM. Human liver microsome assay, linear to 60 min.
Secondary pharmacology screens such as liver microsome assays can add drug-related considerations, versus ligand focus, and can alert one to first-pass metabolism related liabilities during refinement. As indicated in Table 2, this secondary pharmacology screen embedded in the SAR efforts revealed that compound 10 had less than ideal liver microsomal stability. The methylpiperazine once again provided an excellent starting point for the R3 substituent as it doubled the liver microsome stability time in the context of the 2-naphthylpyridazine scaffold as evidenced by comparison of compound 10 to compound 11 (Table 2). Additional probing of improvement in metabolic stability potential involved the synthesis and testing of fluorine analogs of the 2-naphthyl substituent. However, there was not a straightforward trend. For example, compounds 12–15 offered no clear advantage over compound 11 in terms of liver microsome stability. Surprisingly, compound 14 lost the metabolic stability seen in compound 11. While there was a trend toward greater reduction in p38βMAPK and CK1δ inhibitory activity with other analogs, it sometimes came at the cost of worsening of p38αMAPK inhibitory activity. The results also suggested that the biosteric effect of fluorine might be more in line with localized lipophilic or electronegative effects of fluorine dependent on intermolecular interactions.23 Consistent with that hypothesis, introduction of an indoyl analog (compound 16) provided a differential effect on kinase crossover with p38βMAPK vs CK1δ. Additional exploration of changes at the R3 position in the context of 2-naphthyl at the R6 position (compounds 17–20) did not yield advantages in kinase selectivity or liver microsome stability compared to compound 11. Therefore, compound 11 was chosen for further characterization. While not pursued further as part of this study, the trend suggests that future optimization based on strategic introduction of more electronegative atoms in the context of next generation analogs of compound 11 is worth pursuit for additional separation of kinase crossover to p38βMAPK or CK1δ and potential increase in selective affinity for p38αMAPK.
The more advanced secondary pharmacology screens documented the selectivity of compounds 10 and 11 within the kinome drug target class.13,14 Secondary pharmacology screening for functional GPCR agonist and antagonist activity demonstrated that there was no crossover of compounds 10 and 11 to this major CNS drug target class.13,14 Secondary pharmacology screens showed compound 11 had the potential for improved metabolic stability and exposure.
Candidate Compound 11
Compound 11, produced by the GMP scheme shown in Figure 6, is a crystalline drug substance. It has good oral bioavailability, selectivity, metabolic stability, safety, and exposure in plasma and brain tissue (key attributes summarized in Tables 3 and 4; Figure 7). Tables 7S and 8S provide a summary of additional key chemical and physical data for compound 11 that contributed to its selection14,15 as a development candidate.
Figure 6.

Production scheme for compound 11. The regulatory starting material (RSM) was prepared as previously described.14 The intermediate compound was prepared by chlorination of the arylpyridazinone by POCl3 in acetonitrile (ACN).13,14,22 The product substance (API) was generated by treatment with 1-methylpiperazine in ethanol. The summary chemical and physical properties, including crystal structure (CCDC deposit numbers 1851508 and 1851509), are in Tables 8S and 9S.
Table 3. Pharmacokinetic Parameters of Compound 11 in Rata.
| dose (route) | male | female |
|---|---|---|
| 2 mg/kg (iv) | T1/2 (h): 2.07 ± 0.313 | T1/2 (h): 3.73 ± 0.288 |
| CL (L h–1 kg–1): 1.46 ± 0.236 | CL (L h–1 kg–1): 0.677 ± 0.099 | |
| Vss (L/kg): 3.57 ± 0.424 | Vss (L/kg): 3.02 ± 0.372 | |
| 2 mg/kg (po) | T1/2 (h): 3.13 ± 2.25 | T1/2 (h): 4.71 ± 0.543 |
| Cmax (ng/mL): 222 ± 69.6 | Cmax (ng/mL): 363 ± 116 | |
| AUC∞ (h·ng/mL): 739 ± 84.8 | AUC∞ (h·ng/mL): 2035 ± 193 | |
| oral bioavailability | 53.0% ± 6.09 | 67.8% ± 6.43 |
| 6 mg/kg (po) | T1/2 (h): 2.87 ± 0.504 | T1/2 (h): 5.07 ± 0.244 |
| Cmax (ng/mL): 392 ± 216 | Cmax (ng/mL): 646 ± 98.7 | |
| AUC∞ (h·ng/mL): 2601 ± 965 | AUC∞ (h·ng/mL): 6632 ± 949 | |
| 10 mg/kg (iv) | T1/2 (h): 2.38 ± 0.210 | T1/2 (h): 3.73 ± 0.676 |
| CL (L h–1 kg–1): 1.71 ± 0.0512 | CL (L h–1 kg–1): 0.816 ± 0.206 | |
| Vss (L/kg): 4.63 ± 0.365 | Vss (L/kg): 3.76 ± 1.19 | |
| 10 mg/kg (po) | T1/2 (h): 3.47 ± 1.26 | T1/2 (h): 4.45 ± 0.383 |
| Cmax (ng/mL): 649 ± 180 | Cmax (ng/mL): 1203 ± 72.3 | |
| AUC∞ (h·ng/mL): 3841 ± 152 | AUC∞ (h·ng/mL): 10615 ± 738 | |
| oral bioavailability | 55.1% ± 2.18 | 70.7% ± 4.92 |
Values are the mean ± SD.
Table 4. Summary of Compound 11 Secondary Pharmacology Screen Outcomes.
| screen | results |
|---|---|
| Kinome-wide off-target (305 kinases) | Negative except for p38αMAPK (see ref (14)) |
| Functional GPCR agonist and antagonist (166 GPCRs) | Negative (see ref (14)) |
| Panlabs and others (44 targets) | Negative (see Experimental Section) |
| Enzymes: MAO A/B, acetyl cholinesterase, COX-1, COX-2, PDE3A, PDE4D2 | |
| Receptors: Adenosine A2A, adrenergic (α1A, α2A, β1, β2), androgen, cannabinoid (CB1, CB2), GABAA, dopamine (D1, D2S), cholecystokinin CCK1, glucocorticoid, histamine (H1, H2), muscarinic (M1, M2, M3), NMDA, α4β2 nicotinic acetylcholine, opiate (OP1, OP2, OP3), serotonin (5-HT1A, 5- HT1B, 5-HT2A, 5-HT2B, 5-HT3), vasopression V1A | |
| Channels: L-type calcium channel, potassium channel ([KA], hERG), sodium channel | |
| Transporters: Dopamine, norepinephrine, Serotonin | |
| Liver microsome stability | Human, T1/2 > 60 min; rat, T1/2 > 40 min (see ref (14)) |
| Hepatocyte stability | Human, T1/2 > 120 min; dog, T1/2 > 120 min (see ref (14)) |
| CYP inhibition: (1A2, 2B6, 2D6, 2C8, 2C9, 2C19, 3A4) | Negative (see ref (14)) |
| CYP substrate: (1A2, 2B6, 2D6, 2C8, 2C9, 2C19, 3A4) | Negative (see ref (14)) |
| CYP induction | Negative (see Experimental Section) |
| Permeability, pump substrate status: | |
| P-gp/Caco-2 | Not substrate/highly permeable (see ref (14)) |
| BCRP/MDCK | Not substrate/highly permeable (see ref (14)) |
| Dose range finding: 14-day daily oral with TK (rat) | NOAELa = 125 mg/kg; HEDa = 20.0 mg/kg (see ref (14)) |
| Aged AD KI mouse, oral, single administration: behavior and pathology | NOAELa ≥ 250 mg/kg; HEDa = 20.0 mg/kg (see ref (14)) |
NOAEL= no observable adverse effect level; HED = human equivalent dose.
Figure 7.

Exposure of compound 11 in plasma and brain after oral administration. Oral gavage administration results in comparable profiles for plasma (A) and brain (B). Oral doses (mg/kg): 1.25 (circles), 2.5 (squares), 5 (triangles), 10 (diamonds). Sampling time points (h): 0.25, 0.5, 1, 3, 6, 8, 12. Drug concentrations in biological matrices were determined by LC–MS/MS as described in Experimental Section.
Efficacy and Pharmacodynamics of Compound 11
Table 5 summarizes the previously described4,14 efficacy and pharmacodynamics of compound 11 in amyloid-based Alzheimer’s disease (AD) pathophysiology progression models14,15 and in autism spectrum disorder (ASD) models.4
Table 5. Compound 11 Efficacy and Pharmacodynamics.
| property | experimental system | end point |
|---|---|---|
| Efficacy | Autism spectrum disorder (ASD) model: SERT Ala56 mouse (ip injection daily for 7 d) | MW150 repeat administration mitigates SERT Ala56-mediated 5-HT receptor hypersensitivities and altered social interaction; reverses SERT Ala56-mediated reductions in intestinal motility |
| APP/PS1 transgenic (oral gavage daily for 1–2 mos) | Improved performance in contextual fear memory and radial arm water maze (RAWM) with no effect on sensory, motor, and motivational mechanisms; no effects on open field tasks; dose-dependent efficacy at 0.5–2.5 mM | |
| APPNlh/Nlh/PS1P264L/P264L knock-in (KI) (ip injection daily for 14 d) | Improved performance in RAWM | |
| Tauopathy model: infusion of synthetic 4R/2N human tau into dorsal hippocampus (single ip injection of MW150) | Attenuates defects in associative memory (contextual fear learning paradigm); attenuates defects in short-term spatial memory (RAWM); no effect on sensory, motor, and motivational mechanisms; no effects on open field tasks | |
| Tauopathy model: rTg4510 (human tau, P301L mutation) (oral gavage daily for 45 d) | Attenuates defects in associative memory (contextual fear learning paradigm); attenuates defects in short-term spatial memory (RAWM) | |
| Pharmacodynamic end points | Autism spectrum disorder (ASD) model: SERT Ala56 mouse | MW150 repeat administration normalizes 5-HT clearance in SERT Ala56 mice; no effects on SERT protein levels; no effects on brain 5-HT levels or turnover |
| Anisomycin-treated CHO cell line | Decrease in anisomycin-induced 5-HT uptake | |
| LPS treated BV2 microglia cell line | Endogenous kinase inhibition measuring phosphorylated substrate pMK2: IC50 = 332 nM. Inhibition of IL-1β overproduction: IC50 = 936 nM | |
| BV2 microglia cell line | No effect on cell proliferation, migration, or phagocytosis | |
| APPNlh/Nlh/PS1P264L/P264L KI | Cortex IL-1β and TNFα levels decreased in mice showing improved performance in RAWM | |
| APP/PS1 transgenic | No effect on Aβ plaque burden | |
| APPNlh/Nlh/PS1P264L/P264L KI | No effect on Aβ plaque levels or volume; no effect on levels of PBS-soluble or formic acid-soluble Aβ40 or Aβ42 | |
| p38αMAPKT106M KI | Cortex IL-1β and TNFα levels did not change with MW150 treatment of drug resistant mice |
In two distinct transgenic and knock-in amyloid based models there is efficacy as measured by molecular end points and quantitative behavioral assays.14 Mechanism based pharmacodynamic end points are also demonstrated. Importantly, the efficacy and pharmacodynamics are evident in the absence of detectable effects on the amyloid pathway or plaques.15
The potential of p38αMAPK as a therapeutic target in neuropsychiatric disorders is provided by outcomes of compound 11 testing in genetics-based animal models of ASD susceptibility.4 For example, a specific type of ASD involving alterations in p38αMAPK mediated serotonin transporter (SERT) activity was reversed by compound 11 treatment of adult mice. The phenotypes generated by compound 11 treatment mimic the behavioral and biochemical dysregulation of ASD in patients with genetic alterations in SERT.4 For example, compound 11 showed efficacy as measured by end points such as altered social interactions and gastrointestinal disturbances due to the SERT point mutation. Further, clinically translatable pharmacodynamic end points were observed in treated animals, such as normalization of disease associated serotonemia.
To explore the broader potential of compound 11 in non-amyloid dementia models, we tested efficacy in two distinct tauopathy animal models. The oligomeric tau infusion model induces reproducible deficits in spatial and associative memory performance, which are attenuated by treatment with compound 11 (Figure 8). The reversible transgenic (rTg4510) mouse model overexpresses a human, mutant tau form (P301L) known to lead to dementia, with animals developing age-related cognitive impairment, neurofibrillary tangles, and neuronal loss. Repeat administration of compound 11 treatment normalized cognitive and behavioral deficits (Figure 9A,B) and mitigated brain tissue degeneration (Figure 9C,D). The improved outcomes in cognitive function with two different tauopathy models are consistent with the potential utility of compound 11 for additional neurodegenerative therapeutic indications such as Lewy body dementia, age-related tauopathy, and progressive supranuclear palsy.
Figure 8.

Compound 11 treatment rescues tau-induced memory impairment. The effects of compound 11 administration on hippocampus-dependent reference memory in the radial arm water maze (RAWM) and contextual fear memory in the contextual fear conditioning (CFC) assay were tested. (A) Human tau oligomer infusion (white triangles) into mice brains creates short-term memory impairment as shown by the significantly greater numbers of errors in the RAWM test compared to control, vehicle-infused mice (black circles) (two-way ANOVA, p < 0.01). Compound 11 administration (light gray diamonds) to the tau-infused animals (5 mg/kg ip, 20 min prior to start of testing) attenuates memory impairment (p < 0.05). Compound 11 administration in control, vehicle-infused mice (dark gray squares), had no effect. (B) Fear conditioning was assessed by exposing mice to a mild shock and then reassessed 24 h later to gauge “freezing” behavior. Tau-infused mice (white bar) experience reduced rates of freezing relative to control mice (“vehicle”; black bar) or control mice administered compound 11 (dark gray bar). Compound 11 administration in tau-infused mice (light gray bar) significantly increases freezing rates, demonstrating protection against tau-related memory impairment (p < 0.05).
Figure 9.

Compound 11 treatment rescues cognitive deficits and cortex size in tau transgenic animal model. (A) Compound 11 treatment of rTg4510 mice (white triangles) results in a reduction in errors in the RAWM cognitive function assay compared to vehicle-treated rTg4510 animals (gray squares). Outcomes for compound 11 treated mice are comparable to disease state control mice (black circles; doxy-treated rTg4510 mice). (B) Contextual fear conditioning shows that compound 11 treatment (white bar) rescues the freezing deficit seen in rTg4510 mice treated with vehicle (gray bar), reaching levels similar to the control doxy-treated rTg4510 animals (black bar). (C) Vehicle-treated rTg4510 mice (gray bar) show reduced cerebral cortex size compared to control doxy-treated mice (black bar), and compound 11 treatment of rTg4510 mice (white bar) prevents the cortical tissue loss (t test: p < 0.05 vs Tg4510 vehicle). (D) Reductions in hippocampal size also manifest in the rTg4510 mice (gray bar) compared to the doxy mice (black bar). Compound 11 treatment of the rTg4510 mice (white bar) shows a trend toward rescuing the size deficit but does not reach statistical significance (p = 0.104 comparing compound 11-treated rTg4510 mice vs vehicle treated rTg4510 mice).
Conclusions
Compound 11 (MW150) is a novel, CNS-penetrant, pharmacologically selective p38αMAPK inhibitor clinical candidate for neurological disorders. The discovery approach involved the integrated use of high resolution crystallography of p38αMAPK:ligand complexes, pharmacoinformatics-driven novel compound design, and outcomes from secondary pharmacology screens as progressive Go or No Go criteria. Disease-relevant efficacy screens of best-in-class lead compounds and the drug candidate provided preclinical proof-of-concept for attenuation of disease progression and the biological rationale for clinical development. The overall approach provided a focused route to high value lead compounds. The small number of novel compounds needed to arrive at a viable clinical candidate is characteristic of fragment-based approaches, especially when the process is driven by safety and pharmacodynamic considerations inherent in secondary pharmacology outcomes. Treatment with compounds 9–11 or cell-selective genetic knock-down of p38αMAPK documents that inhibition of p38αMAPK can mitigate detrimental proinflammatory cytokine overproduction and neuronal/synaptic damage in a variety of animal models,4,13−17,19,30,31 outcomes consistent with clinical observations and hypotheses. Overall, compound 11’s selectivity, comparative lack of prevailing off-target liabilities, and derisked pharmacology, including safety potential, make it attractive for potential use as a monotherapy or as a component of a therapeutic combination regimen for complex CNS disorders.27−29
Experimental Section
General
All NMR spectra were recorded on Bruker Avance III 500 MHz system equipped with a DCH CryoProbe and Mercury 300 MHz FT-NMR spectrometer. Data are reported in the following order: chemical shifts δ values in ppm downfield with the deuterated solvents are reported in parts per million (ppm) using tetramethylsilane (TMS) as an internal standard (signal = 0 ppm) (s = singlet, d = doublet, t = triplet, br = broad, m = multiplate), integration, coupling constant (Hz). LC/MS data were determined with a Waters Alliance 2695 HPLC/MS (Waters Symmetry C18, 4.6 mm × 75 mm, 3.5 μm) with a 2996 diode array detector from 210–400 nm; the solvent system is 5–95% ACN in water (0.1% TFA) over 9 min using a linear gradient, and retention times are in minutes. Accurate-mass/high-resolution mass spectrometry (HRMS) analysis was performed using an Agilent 6210A LC-TOF mass spectrometer in positive ion mode (ambient temperature). Purity of all final compounds was determined by HPLC (Dionex System, Sunnyvale, CA), with UVD170U ultraviolet detector and P680 pump. The column was a Phenomenex (Torrance, CA) Luna C18, 5 μm particle size (250 mm × 2.0 mm), supported by Phenomenex Security Guard cartridge kit C18 (4.0 mm × 2.0 mm) at a flow rate of 0.2 mL/min and using a mobile phase composed of 0.1% (v/v) formic acid (Fluka) in water as solvent A and 80% acetonitrile, 0.08% formic acid/water as solvent B (gradient from 0% to 100% B, 15 min; isocratic 22 min). Compounds were analyzed at ∼0.50 μg level, and peak quantification was performed based upon relative area% absorption at 260 nm. Purity of all compounds was determined to be >95% by HPLC. Unless otherwise noted, all synthesis was carried out in reagent grade solvents and was used as purchased without further purification unless otherwise stated. Compound 11 crystal structure was determined as previously described .34−36
Compounds listed in Tables 1 and 2 were synthesized using synthetic Schemes 1 and 2 below. Details of preparation and characterization for key compounds in Tables 1 and 2 are found in prior literature.12−14,22 Analytical data for additional compounds are summarized in Table 6S.
Scheme 1. Synthesis of Compound 3–8, 11, and 17–20.

Reagents and conditions: (a) 1-butanol/ethanol, reflux, 15–48 h at 110–130 °C.
Scheme 2. Synthesis of Compounds 9–16.

Reagents and conditions: (a) 1-butanol, R3-amiine, 120 °C; (b) R6-boronic acid, DME/water, Na2CO3, tetrakis(triphenylphosphine)palladium(0), 110 °C .
In Scheme 1, starting materials (previously described13,14 or commercially available) were taken in 1-butanol and reacted with an approximate (3–10) mole excess of the respective amine, the reaction mixture was cooled to ambient temperature, deionized water added, and the mixture subjected to repeat extraction with dichloromethane. The organic layers were taken to drying with anhydrous sodium sulfate and concentration in vacuo. The final products were purified by silica gel column chromatography using volatile solvents for elution and final processing.
In Scheme 2, amination was done using 3,6-dichloro-4-(pyridin-4-yl)pyridazine and workup, organic phase extraction, and purification were done as in Scheme 1 in combination with Suzuki reaction using boronic acid and tetrakis(triphenylphosphine)palladium(0) to yield compounds 9–16.
3-Phenyl-4-(pyridin-4-yl)-6-(4-(pyrimidin-2-yl)piperazin-1-yl)pyridazine (MW069a) (3)
Compound 3 was prepared following Scheme 1 above as previously described12 using discovery chemistry and starting with 6-chloro-3-phenyl-4-(pyridin-4-yl)pyridazineas previously described13 using a 5-fold mole excess of 2-(piperazin-1-yl)pyrimidine in the Scheme 1 protocol to give compound 3 as pale yellowish powder in 62% yield (gravimetric). Purity of >98% was determined by HPLC. 1H NMR (500 MHz, CDCl3) δ: 8.58–8.57 (m, 2H), 8.36–8.33 (dd, J = 1.4, 4.75 Hz, 2H), 7.35–7.25 (m, 5H), 7.15–7.13 (dd, J = 1.65, 4.35 Hz, 2H), 6.88 (s, 1H), 6.57–6.35 (dd, J = 1.4, 4.75 Hz, 1H), 4.05–4.02 (dd, J = 3.5, 5.1 Hz, 4H), 3.89–3.87 (dd, J = 3.15, 5.4 Hz, 4H). ESI-MS m/z: 396.3 [M + 1]+. HRMS calculated for C23H21N7 395.18584, found 395.18542.
N,N-Dimethyl-6-(naphthalen-1-yl)-5-(pyridin-4-yl)pyridazin-3-amine (MW181) (9)
Compound 9 was prepared following Scheme 2 above. Amination was done using 3,6-dichloro-4-(pyridin-4-yl)pyridazine using a 6-fold mole of 40% dimethylamine in combination with Suzuki reaction using 1-naphthylboronic acid and tetrakis(triphenylphosphine)palladium(0) (1:0.09) as described previously13 to give compound 9 as a light yellow crystals in 56% yield (gravimetric). Purity of >98% was determined by HPLC. 1H NMR (500 MHz, chloroform-d) δ: 8.34 (dd, J = 1.65, 4.6 Hz, 2H), 7.82 (dd, J = 1.3, 8.7 Hz, 2H), 7.68 (dd, J = 1.3, 8.8 Hz, 1H), 7. 43–7.32 (m, 3H), 7.28 (dd, J = 1.25, 7.0 Hz, 1H), 6.99–6.97 (m, 2H), 6.84 (s, 1H), 3.32 (s, 6H). HRMS calculated for C21H18N4 326.15315, found 326.1526.
6-(4-Methylpiperazin-1-yl)-3-(naphthalen-2-yl)-4-(pyridin-4-yl)pyridazine (MW150) (11)
Compound 11 was prepared following both Scheme 1 and Scheme 2. Following the same method as above for compound 9, amination was done using 3,6-dichloro-4-(pyridin-4-yl)pyridazine as in Scheme 2 using a 5-fold mole of 1-methylpiperazine in combination with Suzuki reaction using 2-naphthylboronic acid and tetrakis(triphenylphosphine)palladium(0) (1:0.09) as described previously14 to give compound 11 as a pale yellow crystals in 68% yield (gravimetric). Purity of >97% was determined by HPLC.
Large scale production of compound 11 was made as in Scheme 1 starting with 6-chloro-3-(naphthalen-2-yl)-4-(pyridin-4-yl)pyridazine as previously described14 using a 5 fold mole excess of 1-methylpiperazine in the protocol to give compound 11 as a pale yellow solid in 92% yield (gravimetric). Purity of >97% was determined by HPLC. 1H NMR (500 MHz, CDCl3) δ: 8.55–8.53 (dd, J = 1.7, 4.35 Hz, 2H), 7.94 (d, J = 1.9 Hz, 1H), 7.80–7.78 (dd, J = 1.85, 7.45 Hz, 1H), 7.73–7.69 (m, 2H), 7.49–7.43 (m, 2H), 7.36–7.34 (m, 1H), 7.15–7.13 (dd, J = 1.7, 4.25 Hz, 2H), 6.87 (s, 1H), 3.84 (t, J = 4.85, 5.1 Hz, 4H), 2.63 (t, J = 4.85, 4.95 Hz, 4H), 2.40 (s, 3H); HRMS calculated for C24H23N5 381.19535, found 381.19603.
Pharmacology Screens. Kinome-Wide Inhibitor Screening
The hierarchal screening was done as previously described.13,14 Briefly, the screen was done using Eurofins kinase profiler test system that includes 299–412 mammalian kinases representative of all major kinome branches as well as isoforms of individual families. The list of protein and lipid kinases and their NCBI Entrez accession numbers are provided in Table 3S. The initial profiling was performed at inhibitor concentrations of 10 or 20 μM based on previous activity analysis. Preliminary hits were validated as true or false positive hits by a concentration-dependent determination of an IC50 value using a standardized assay protocol. An apparent Ki value is determined for any kinase for which the inhibitor exhibits IC50 < 1 μM.
Large Scale Functional Screen for GPCR Agonist and Antagonist Activity
As described previously,13,14 a hierarchical approach was used to test compounds 9–11 (10–12.5 μM) for any off-target activity with the largest known family of small molecule drug targets, G-protein-coupled receptors (GPCRs). The Eurofins GPCR profiler test system employs a cell-based functional screen and a real-time calcium flux FLIPR assay on a panel of 166 GPCRs to detect both antagonists and agonists.
PanLab Screens
Compound 11 was tested at 10 μM against a panel of 44 safety targets in a commercial screen (www.eurofinsdiscoveryservices.com) that includes both enzymatic and radioligand binding assays. Several targets in the panel are associated with in vivo adverse drug reactions. Biochemical assay results were presented as the percent inhibition of specific binding or activity. Reference standards were run as an integral part of each assay to ensure the validity of the results obtained.
Liver Microsome Stability
Human and rat liver microsome stability screens were done as previously described14 using a standard microsome buffer containing test compounds at 1 μM. The reactions were terminated at time points of 15, 30, 45, and 60 min, samples were processed, and analysis was done by LC–MS/MS to monitor parent compound. The peak area response ratio to internal standard at each time point was compared to the response ratio at time 0 to determine the percent remaining. The assay was linear over 60 min with r2 = 0.969–1.00. The half-life was calculated based on t1/2 = 0.693/k, where k is the elimination rate constant based on the results of nonlinear fitting. The intrinsic clearance (CLint) was calculated based on CLint = k/D, where k is the elimination rate constant and D is protein concentration.
Hepatocyte Stability
Compound 11 was tested for stability in mixed gender human cryopreserved hepatocytes and male beagle dog cryopreserved hepatocytes using a validated protocol at CiTox Lab (Hungary). Compound 11 (1 μM) was added into the hepatocyte suspension (1.5 × 106 cells/mL), and reaction mix aliquots were withdrawn at 0, 15, 30, 60, and 120 min. Reactions were terminated and protein was precipitated by adding three volumes of ice-cold acetonitrile (ACN) containing internal standard. After centrifugation, an aliquot of the supernatant was analyzed by LC–MS/MS. The peak area response ratio (PARR) to internal standard was compared to the PARR at time 0 to determine the percent remaining at each time point. Half-life and clearance values were calculated using GraphPad software, fitting to a single phase exponential decay equation. Positive controls were performed in parallel to confirm the activity of the hepatocytes.
CYP Inhibitor Screen
Compounds were screened as potential inhibitors of CYPs 1A2, 2B6, 2D6, 2C8, 2C9, 2C19, and 3A414 at a final concentration of 10 μM over 10–30 min at 37 °C in the presence of a CYP-specific probe substrate (at concentration approximating its Km value) and, after reaction termination, metabolites monitored by LC–MS/MS. Positive controls were performed in parallel using a known inhibitor of the individual CYP isoform. Percent inhibition was calculated as the percent of control metabolite formed in the reaction mix with test compound relative to percent of metabolite formed in reaction without the test compound.
where PARR is the peak area response ratio of analyte to internal standard.
CYP Substrate Status
Compounds at a final concentration of 0.1 μM were screened as potential substrates for the CYPs 1A2, 2B6, 2D6, 2C8, 2C9, 2C19, and 3A4.14T1/2 was estimated from the slope of the linear portion of the curve for percentage of remaining parent compound versus time assuming first order kinetics. Substrate stability, expressed as percent of the parent compound remaining, was calculated by comparing the peak area of the compound at the time point relative to that at time 0. A reference compound specific for each CYP was run in parallel. The half-life (T1/2) was estimated from the slope of the initial linear range of the logarithmic curve of compound remaining (%) vs time assuming the first-order kinetics.
CYP Induction
Primary hepatocyte cultures from rats, dogs, and humans were incubated with compound 11 at six concentrations ranging from 0.5 to 50 μM (N = 3) for 48 h, followed by extraction of total RNA and PCR-driven synthesis of cDNA using probes for rat CYP1A2, CYP2B1, and CYP3A1; dog CYP1A2, CYP2B11, and CYP3A12; and human CYP1A2, CYP2B6, and CYP3A4. The induction potential of compound 11 was assessed by comparing mRNA expression in cells treated with 0.1% DMSO (control), positive control, and compound 11. The potential for drug–drug interactions was assessed by comparing the expression of mRNA coding for the three human cytochromes with expected free Cmax concentration in human subjects at pharmacologically relevant doses.
Permeability and Efflux Pump Substrate Status
Cell permeability and efflux pump substrate status tests were done at a final compound concentration of 5 μM in the Caco-2/P-gp and MDCK/BCRP screening systems as previously described.14 Permeation and concentration of inhibitor on both apical side (A → B) and basolateral side (B → A) were determined by LC–MS/MS. The apparent permeability, Papp (10–6 cm s–1), was calculated as follows: Papp = (dCr/dt) × Vr/(A × CA). Substrate status was determined by measuring the effect of a specific pump inhibitor (1 μM or 10 μM) on the bidirectional flux. The compound was considered a nonsubstrate if the efflux ratio of Papp(A→B)/Papp(B→A) is <2.0.
Dose Range Finding Screen with Toxicokinetics
Compound 11 was administered orally once a day for 14 days to male and female Sprague Dawley rats. Blood samples were collected up to 24 h after dosing on days 1 and 14, and concentrations were determined by LC–MS/MS. Pharmacokinetic parameters were calculated using WinNonlin. NOAEL for compound 11 was the highest dose tested (125 mg/kg).
Aged Wild Type and AD-Model Mouse Toxicity Screen
Compound 11 was administered at 250 mg/kg, 100× efficacy dose, to 11-month-old APP/PS1 knock-in (KI) mice and wild-type (WT) control animals. Observed signs for toxicity included palpebral closure, abnormal lacrimation (tears), excessive defecation or urination, gait disturbances, and tremors or seizures. Animals were euthanized for liver, brain, and serum harvest at 48 h after administration. No drug-related abnormalities were observed in the liver tissue in either WT or APP/PS1 KI mice.
Oral Bioavailability Screen
Compound 11 was administered by oral gavage to male and female Sprague Dawley rats at 2, 6, and 10 mg/kg and by iv administration at 2 and 10 mg/kg. Blood samples were collected up to 24 h postdose and plasma concentrations determined by LC–MS/MS. Pharmacokinetic parameters were calculated using Phoenix WinNonlin.
Effects of Compound 11 in Tauopathy Animal Models
All protocols involving mice were approved by the Institutional Animal Care and Use Committee (IACUC) at Columbia University, New York, NY. C57BL/6J and rTg4510 mice and their littermates were obtained from breeding colonies at Columbia University. Both male and female mice were used, and equal gender distribution was used when possible. All animal experiments and data analyses were done by investigators who were blinded to the experimental groups.
Cognitive Behavior Testing
Mice were tested in the 2-day radial arm water maze (RAWM) test for reference memory impairment as previously described.13 As controls for these experiments, visible platform testing was conducted to exclude that visual, motor, or motivation deficits affect the mouse performance. Mice were also tested in a contextual fear conditioning (CFC) test as previously described.13 Controls for these experiments included a threshold assessment test to check sensory perception of electric shock, and the open-field test to evaluate exploratory behavior. For more details of these behavior tests, see Fiorito et al.32
Oligomeric Tau Infusion Model
Preparation of recombinant tau and tau oligomers was done as previously described.33 Oligomeric tau (500 nM) or vehicle was infused via bilateral cannulas into the dorsal mouse hippocampi of wild-type (WT) C57Bl/6 mice. Infusion solutions were diluted in a final volume of 1 μL and infused over 1 min bilaterally (one injection, 20 min prior to testing for fear conditioning, and 20 min prior to the first and sixth sessions for RAWM). MW150 (5 mg/kg) or saline vehicle was administered by intraperitoneal (ip) injection 20 min prior to testing for fear conditioning or prior to the first and sixth sessions of RAWM.
Transgenic rTg4510 Model
Adult (3 month old) rTg4510 mice or rTg4510 mice that had been administered doxycycline to turn off tau expression (control mice) were treated with vehicle or MW150 (2.5 mg/kg, orally, every other day) for 45 days. Mice were then subjected to behavior testing in RAWM and CFC.
Measurement of Brain Tissue Size
Following behavioral studies, mice were perfused PBS. Brains were removed, split sagittally into two hemispheres along the midline, and immersion-fixed in 4% paraformaldehyde (PFA). Brain size was determined in blind using separate delineation of the regions of interest, the cerebral cortex and the hippocampus.
Acknowledgments
This research was supported in part by NIH Awards U01AG043415, R01NS056051, R01AG031311, and R01NS093920 as well as ADDF Award 261108. We thank Dr. Christos D. Malliakas for his assistance with crystallographic structure and physical property studies of compound 11 and James Schavocky for his assistance and help with manuscript preparation.
Glossary
Abbreviations Used
- ACN
acetonitrile
- API
active pharmaceutical ingredient
- ASD
autism spectrum disorder
- CK1δ
casein kinase 1δ
- GMP
good manufacturing practice
- GPCR
G-protein-coupled receptor
- ICH
International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use
- KI
knock-in
- MW150
MW01-18-150SRM
- PARR
peak area response ratio
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jmedchem.9b00058.
Tables 1S–8S listing clinical trials of previous p38MAPK inhibitors, SMILES for compounds mentioned in this article, accession numbers and summary data for protein kinases and GPCR tested as part of secondary pharmacology, X-ray crystal structure and additional characterizations of the GMP drug substance (API) (PDF)
Accession Codes
PDB codes for structures of p38αMAPK:compound complexes are the following: 4ZTH (p38αMAPK:1); 4EWQ (p38αMAPK:3); 4F9Y (p38αMAPK:9); 4F9W (p38αMAPK:10); 4R3C (p38αMAPK:11 (MW150)); 4FA2 (p38αMAPK:SB239063).
The authors declare no competing financial interest.
Notes
CCDC deposit numbers for compound 11 (MW150) are 1851508 and 1851509.
Supplementary Material
References
- Chung W. S.; Welsh C. A.; Barres B. A.; Stevens B. Do Glia Drive Synaptic and Cognitive Impairment in Disease?. Nat. Neurosci. 2015, 18, 1539–1545. 10.1038/nn.4142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudy C. C.; Hunsberger H. C.; Weitzner D. S.; Reed M. N. The Role of the Tripartite Glutamatergic Synapse in the Pathophysiology of Alzheimer’s Disease. Aging Dis. 2015, 6, 131–148. 10.14336/AD.2014.0423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huentelman M. J.; Piras I. S.; Siniard A. L.; De Both M. D.; Richholt R. F.; Balak C. D.; Jamshidi P.; Bigio E. H.; Weintraub S.; Loyer E. T.; Mesulam M. M.; Geula C.; Rogalski E. J. Associations of MAP2K3 Gene Variants with Superior Memory in SuperAgers. Front. Aging Neurosci. 2018, 10, 155. 10.3389/fnagi.2018.00155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robson M. J.; Quinlan M. A.; Margolis K. G.; Gajewski-Kurdziel P. A.; Veenstra-VanderWeele J.; Gershon M. D.; Watterson D. M.; Blakely R. D. p38α MAPK Signaling Drives Pharmacologically Reversible Brain and Gastrointestinal Phenotypes in the SERT Ala56 Mouse. Proc. Natl. Acad. Sci. U. S. A. 2018, 115 (43), E10245–E10254. 10.1073/pnas.1809137115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun A.; Liu M.; Nguyen X. V.; Bing G. p38 MAP Kinase is Activated at Early Stages in Alzheimer’s Disease Brain. Exp. Neurol. 2003, 183, 394–405. 10.1016/S0014-4886(03)00180-8. [DOI] [PubMed] [Google Scholar]
- Sutcliffe J. S.; Delahanty R. J.; Prasad H. C.; McCauley J. L.; Han Q.; Jiang L.; Li C.; Folstein S. E.; Blakely R. D. Allelic Heterogeneity at the Serotonin Transporter Locus (SLC6A4) Confers Susceptibility to Autism and Rigid-Compulsive Behaviors. Am. J. Hum. Genet. 2005, 77, 265–279. 10.1086/432648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gill A. L.; Frederickson M.; Cleasby A.; Woodhead S. J.; Carr M. G.; Woodhead A. J.; Walker M. T.; Congreve M. S.; Devine L. A.; Tisi D.; O’Reilly M.; Seavers L. C. A.; Davis D. J.; Curry J.; Anthony R.; Padova A.; Murray C. W.; Carr R. A.; Jhoti H. Identification of Novel p38h MAP Kinase Inhibitors Using Fragment-Based Lead Generation. J. Med. Chem. 2005, 48, 414–426. 10.1021/jm049575n. [DOI] [PubMed] [Google Scholar]
- Hartshorn M. J.; Murray C. W.; Cleasby A.; Frederickson M.; Tickle I. J.; Jhoti H. Fragment-Based Lead Discovery Using X-ray Crystallography. J. Med. Chem. 2005, 48, 403–413. 10.1021/jm0495778. [DOI] [PubMed] [Google Scholar]
- Papoian T.; Chiu H.-J.; Elayan I.; Jagadeesh G.; Khan I.; Laniyonu A. A.; Li C. X.; Saulnier M.; Simpson N.; Yang B. Secondary Pharmacology Data to Assess Potential Off-Target Activity of New Drugs: A Regulatory Perspective. Nat. Rev. Drug Discovery 2015, 14, 294–296. 10.1038/nrd3845-c1. [DOI] [PubMed] [Google Scholar]
- Papoian T.; Jagadeesh G.; Saulnier M.; Simpson N.; Ravindran A.; Yang B.; Laniyonu A. A.; Khan I.; Szarfman A. Regulatory Forum Review: Utility of In Vitro Secondary Pharmacology Data to Assess Risk of Drug-induced Valvular Heart Disease in Humans: Regulatory Considerations. Toxicol. Pathol. 2017, 45, 381–388. 10.1177/0192623317690609. [DOI] [PubMed] [Google Scholar]
- Bowes J.; Brown A. J.; Hamon J.; Jarolimek W.; Sridhar A.; Waldron G.; Whitebread S. Reducing Safety-Related Drug Attrition: The Use of In VitroPharmacological Profiling. Nat. Rev. Drug Discovery 2012, 11, 909–922. 10.1038/nrd3845. [DOI] [PubMed] [Google Scholar]
- Munoz L.; Ranaivo H. R.; Roy S. M.; Hu W.; Craft J. M.; McNamara L. K.; Chico L. W.; Van Eldik L. J.; Watterson D. M. A Novel p38 MAPK Inhibitor Suppresses Brain Proinflammatory Cytokine Up-Regulation and Attenuates Synaptic Dysfunction and Behavioral Deficits in an Alzheimer’s Disease Mouse Model. J. Neuroinflammation 2007, 4, 21. 10.1186/1742-2094-4-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watterson D. M.; Grum-Tokars V. L.; Roy S. M.; Schavocky J. P.; Bradaric B. D.; Bachstetter A. D.; Xing B.; Dimayuga E.; Saeed F.; Zhang H.; Staniszewski A.; Pelletier J. C.; Minasov G.; Anderson W. F.; Arancio O.; Van Eldik L. J. Development of Novel In Vivo Chemical Probes to Address CNS Protein Kinase Involvement in Synaptic Dysfunction. PLoS One 2013, 8, e66226 10.1371/journal.pone.0066226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy S. M.; Grum-Tokars V. L.; Schavocky J. P.; Saeed F.; Staniszewski A.; Teich A. F.; Arancio O.; Bachstetter A. D.; Webster S. J.; Van Eldik L. J.; Minasov G.; Anderson W. F.; Pelletier J. C.; Watterson D. M. Targeting Human Central Nervous System Protein Kinases: An Isoform Selective p38αMAPK Inhibitor that Attenuates Disease Progression in Alzheimer’s Disease Mouse Models. ACS Chem. Neurosci. 2015, 6, 666–680. 10.1021/acschemneuro.5b00002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Z.; Bachstetter A. D.; Späni C. B.; Roy S. M.; Watterson D. M.; Van Eldik L. J. Retention of Normal Glia Function by an Isoform-Selective Protein Kinase Inhibitor Drug Candidate that Modulates Cytokine Production and Cognitive Outcomes. J. Neuroinflammation 2017, 14, 75. 10.1186/s12974-017-0845-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachstetter A. D.; Watterson D. M.; Van Eldik L. J. Target Engagement Analysis and Link to Pharmacodynamic Endpoint for a Novel Class of CNS-Penetrant and Efficacious p38α MAPK Inhibitors. J. Neuroimmune Pharmcol. 2014, 9, 454–460. 10.1007/s11481-014-9543-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maphis N.; Jiang S.; Xu G.; Kokiko-Cochran O. N.; Roy S. M.; Van Eldik L. J.; Watterson D. M.; Lamb B. T.; Bhaskar K. Selective Suppression of the α Isoform of p38 MAPK Rescues Late-Stage Tau Pathology. Alzheimer’s Res. Ther. 2016, 8, 54. 10.1186/s13195-016-0221-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morfini G. A.; Bosco D. A.; Brown H.; Gatto R.; Kaminska A.; Song Y.; Molla L.; Baker L.; Marangoni M. N.; Berth S.; Tavassoli E.; Bagnato C.; Tiwari A.; Hayward L. J.; Pigino G. F.; Watterson D. M.; Huang C.-F.; Banker G.; Brown R. H. Jr.; Brady S. T. Inhibition of Fast Axonal Transport by Pathogenic SOD1 Involves Activation of p38 MAP Kinase. PLoS One 2013, 8, e65235 10.1371/journal.pone.0065235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutigliano G.; Stazi M.; Arancio O.; Watterson D. M.; Origlia N. An Isoform-Selective p38α Mitogen-Activated Protein Kinase Inhibitor Rescues Early Entorhinal Cortex Dysfunctions in a Mouse Model of Alzheimer’s Disease. Neurobiol. Aging 2018, 70, 86–91. 10.1016/j.neurobiolaging.2018.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chico L. K.; Van Eldik L. J.; Watterson D. M. Targeting Protein Kinases in Central Nervous System Disorders. Nat. Rev. Drug Discovery 2009, 8, 892–909. 10.1038/nrd2999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chico L. K.; Behanna H. A.; Hu W.; Zhong G.; Roy S. M.; Watterson D. M. Molecular Properties and CYP2D6 Substrates: Central Nervous System Therapeutics Case Study and Pattern Analysis of a Substrate Database. Drug Metab. Dispos. 2009, 37, 2204–2211. 10.1124/dmd.109.028134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu W.; RalayRanaivo H.; Roy S. M.; Behanna H. A.; Wing L. K.; Munoz L.; Guo L.; Van Eldik L. J.; Watterson D. M. Development of a Novel Therapeutic Suppressor of Brain Proinflammatory Cytokine Up-Regulation that Attenuates Synaptic Dysfunction and Behavioral Deficits. Bioorg. Med. Chem. Lett. 2007, 17, 414–418. 10.1016/j.bmcl.2006.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meanwell N. A. Fluorine and Fluorinated Motifs in the Design and Application of Bioisosteres for Drug Design. J. Med. Chem. 2018, 61, 5822–5880. 10.1021/acs.jmedchem.7b01788. [DOI] [PubMed] [Google Scholar]
- Alam J. J. Selective Brain-Targeted Antagonism of p38 MAPKp Reduces Hippocampal IL-1 Levels and Improves Morris Water Maze Performance in Aged Rats. J. Alzheimer's Dis. 2015, 48, 219–227. 10.3233/JAD-150277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabian M. A.; Biggs W. H. III; Treiber D. K.; Atteridge C. E.; Azimioara M. D.; Benedetti M. G.; Carter T. A.; Ciceri P.; Edeen P. T.; Floyd M.; Ford J. M.; Galvin M.; Gerlach J. L.; Grotzfeld R. M.; Herrgard S.; Insko D. E.; Insko M. A.; Lai A. G.; Lélias J. M.; Mehta S. A.; Milanov Z. V.; Velasco A. M.; Wodicka L. M.; Patel H. K.; Zarrinkar P. P.; Lockhart D. J. A Small Molecule–Kinase Interaction Map for Clinical Kinase Inhibitors. Nat. Biotechnol. 2005, 23, 329–336. 10.1038/nbt1068. [DOI] [PubMed] [Google Scholar]
- Karaman M. W.; Herrgard S.; Treiber D. K.; Gallant P.; Atteridge C. E.; Campbell B. T.; Chan K. W.; Ciceri P.; Davis M. I.; Edeen P. T.; Faraoni R.; Floyd M.; Hunt J. P.; Lockhart D. J.; Milanov Z. V.; Morrison M. J.; Pallares G.; Patel H. K.; Pritchard S.; Wodicka L. M.; Zarrinkar P. P. A Quantitative Analysis of Kinase Inhibitor Selectivity. Nat. Biotechnol. 2008, 26, 127–132. 10.1038/nbt1358. [DOI] [PubMed] [Google Scholar]
- Chrzaszcz M.; Venkatesan C.; Dragisic T.; Watterson D. M.; Wainwright M. S. MinozacTreatment Prevents Increased Seizure Susceptibility in a Mouse “two-hit” Model of Closed Skull Traumatic Brain Injury and ElectroconvulsiveShock-induced Seizures. J. Neurotrauma. 2010, 27, 1283–1295. 10.1089/neu.2009.1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graves L. M.; Duncan J. S.; Whittle M. C.; Johnson G. L. The Dynamic Nature of the Kinome. Biochem. J. 2013, 450, 1–8. 10.1042/BJ20121456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arthur J. S. C.; Ley S. C. Mitogen-Activated Protein Kinases in Innate Immunity. Nat. Rev. Immunol. 2013, 13, 679–692. 10.1038/nri3495. [DOI] [PubMed] [Google Scholar]
- Bachstetter A. D.; Rowe R. K.; Kaneko M.; Goulding D.; Lifshitz J.; Van Eldik L. J. The p383 MAPK Regulates Microglial Responsiveness to Diffuse Traumatic Brain Injury. J. Neurosci. 2013, 33, 6143–6153. 10.1523/JNEUROSCI.5399-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xing B.; Bachstetter A. D.; Van Eldik L. J. Microglia p38M MAPK is Critical for LPS-Induced Neuron Degeneration Through a Mechanism Involving TNF. Mol. Neurodegener. 2011, 6, 84. 10.1186/1750-1326-6-84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiorito J.; Saeed F.; Zhang H.; Staniszewski A.; Feng Y.; Francis Y. I.; Rao S.; Thakkar D. M.; Deng S.-X.; Landry D. W.; Arancio O. Synthesis of Quinoline Derivatives: Discovery of a Potent and Selective Phosphodiesterase 5 Inhibitor for the Treatment of Alzheimer’s Disease. Eur. J. Med. Chem. 2013, 60, 285–294. 10.1016/j.ejmech.2012.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fa M.; Puzzo D.; Piacentini R.; Staniszewski A.; Zhang H.; Baltrons M. A.; Li Puma D.D.; Chatterjee I.; Li J.; Saeed F.; Berman H. L.; Ripoli C.; Gulisano W.; Gonzalez J.; Tian H.; Costa J. A.; Lopez P.; Davidowitz E.; Yu W. H.; Haroutunian V.; Brown L. M.; Palmeri A.; Sigurdsson E. M.; Duff K. E.; Teich A. F.; Honig L. S.; Sierks M.; Moe J. G.; D’Adamio L.; Grassi C.; Kanaan N. M.; Fraser P. E.; Arancio O. Extracellular Tau Oligomers Produce an Immediate Impairment of LTP and Memory. Sci. Rep. 2016, 6, 19393. 10.1038/srep19393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolomanov O. V.; Bourhis L. J.; Gildea R. J.; Howard J. A. K.; Puschmann H. OLEX2: A Complete Structure Solution, Refinement and Analysis Program. J. Appl. Crystallogr. 2009, 42, 339–341. 10.1107/S0021889808042726. [DOI] [Google Scholar]
- Sheldrick G. M. A Short History of SHELX. Acta Crystallogr., Sect. A: Found. Crystallogr. 2008, A64, 112–122. 10.1107/S0108767307043930. [DOI] [PubMed] [Google Scholar]
- Sheldrick G. M. Crystal Structure Refinement with SHELXL. Acta Crystallogr., Sect. C: Struct. Chem. 2015, C71, 3–8. 10.1107/S2053229614024218. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.