

Abstract
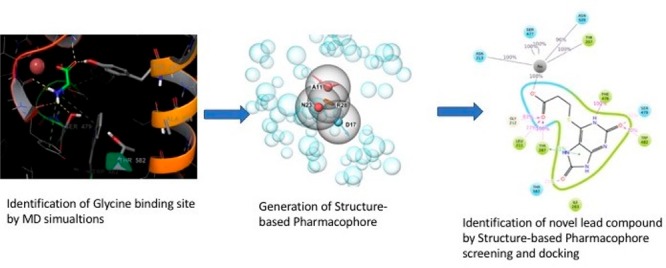
In recent years, mammalian Glycine transporter 2 (GlyT2) has emerged as a promising target for the development of compounds against chronic pain states. In our current work, we discovered a new set of promising hits that inhibit the glycine transporter at nano- and micromolar activity and have excellent selectivity over GlyT1 (as shown by in vitro studies) using a newly designed virtual screening (VS) protocol that combines a structure-based pharmacophore and docking screens with a success rate of 75%. Furthermore, the free energy perturbation calculations and molecular dynamics (MD) studies revealed the GlyT2 amino acid residues critical for the binding and selectivity of both Glycine and our Hit1 compound. The FEP+ results well-matched with the available literature mutational data proving the quality of the generated GlyT2 structure. On the basis of these results, we propose that our hit compounds may lead to new chronic pain agents to address unmet and challenging clinical needs.
Keywords: GlyT2, virtual screening, FEP+ molecular dynamics, docking, pharmacophore, Glycine Transporter 2
In the mammalian CNS, glycine and GABA function as inhibitory neurotransmitters by binding to postsynaptic chloride receptors, which results in chloride influx and hyperpolarization. These glycinergic neurons participate in inhibition of pain signal transmission by releasing glycine onto pain fibers, and dysfunction of these neurons leads to augmented excitation and transmission of neuropathic pain signals in the dorsal horn.1,2 The glycine transporter 2 (GlyT2) is responsible for glycine reuptake from the extracellular space back into the glycinergic neuron to terminate neurotransmission; therefore, pharmacological GlyT2 inhibition would allow prolonged glycine binding and activation of glyine receptor (GlyR) onto pain fibers, reducing their ability to transmit pain signals to a higher center in the brain.
Chronic pain is a difficult-to-treat medical condition, and new pharmacological targets are awaiting discovery. The most widely used analgesic class for pain includes NSAIDs (COX inhibitors, aspirin, ibuprofen, paracetamol, among many others). However, they are ineffective for chronic neuropathic pain, and long-term use leads to gastric bleeding or perforation and myocardial infarction.3−8 The second choice of analgesics are opioids, which are effective yet highly problematic due to their addictive properties and high mortality rate by overdose.9−11 Finally, gabapentin and pregabalin are widely used to treat neuropathic pain, but growing concerns have been raised because of their misuse by drug abusers.12,13 All of these concerns collectively represent a therapeutic challenge that requires discovery of additional pharmacological targets and drug development efforts. Among the most recent targets to intervene in chronic pain, GlyT2 (SLC6A5) has emerged as a promising candidate.3−6,14 Therefore, insights into GlyT2 structure–function relationships and active screening of GlyT2 blockers are currently under development. Currently there are no FDA-approved drugs targeting the GlyT2 transporter. VVZ-149 is a dual GlyT2 and 5HT2A inhibitor, with a GlyT2 activity of 0.86 μM, and is in clinical trials (2b stage); unfortunately, it shows low selectivity.15 Several other drug candidates did not advance to clinical trials because either they had a poor ADMET profile (ALX1393) or were irreversible binders (Org-25543), which produced serious toxic effects by decreasing GlyT2 expression.16 Several drug candidates for schizophrenia, which inhibit GlyT1 homologues, failed for similar reasons.17 This has given researchers the impression that GlyT1/2 are not relevant targets. However, the correct balance of a number of factors may provide optimal drug candidates, which may replace current antichronic pain medicines. A good blood–brain barrier (BBB) ratio along with an optimal ADMET profile, reversible GlyT2 inhibition, and selectivity are the key factors for new modulators. For instance, a recently developed second-generation inhibitor GT-0198 showed excellent preclinical data18 but exhibited some inhibition on other transporters.
For identification and characterization of new GlyT2 inhibitors in this study, we have undertaken an approach that combines state-of-the-art computational methodologies with biochemical assays to fully characterize the inhibitors. We paid special attention to the development of a high-quality GlyT2 structure that can be used for both hit identification and lead optimization, revealing ligands mechanism of action and selectivity. The identified new hit compounds should shed light on a novel family of molecules that, after proper optimization, can be tested in animal models of chronic pain.
To obtain a high-quality GlyT2 structure that can be suitable for identification of new hit molecules by virtual screening (VS), we performed a series of intensive MD simulations. On the basis of the initial homology model, which was constructed using as templates the structures of Drosophila dopamine (pdb id: 4xpt) and human serotonin (pdb id: 5i6x) transporters (see SI Methods), we executed five independent 500 ns-long MD simulations on both the apo (without ligand) and holo (with ligand) conformations. Execution of multiple simulations ensured improved sampling and detection of the main structural binding pocket conformations. We obtained the center of the most populated cluster of structures (>50% of the population) from all of the MD simulations by cluster analysis. Furthermore, this structure was energy-minimized for our in silico drug discovery study. We selected also an average structure for further examinations. Finally, 2 × 250 ns long simulations on the serotonin transporter (SLC6A4) structure, for which an X-ray structure is available, pdb id: 5i6x, were performed (control runs). This allowed us to assess the quality of the MD studies with respect to the membrane parameters, Na+/Cl– coordination, and overall stability. Several MD simulations via a combination of Amber14SB and lipid17 force fields were also executed, and the structures were in close agreement to those obtained by CHARMM36 FF.
These simulations indicated that the GlyT2 transporter has a similar structure to those of serotonin, dopamine, and leucine transporters. We identified the amino acid residues that interact with glycine (Figures 1 and S1). The average root–mean–square deviation (RMSD) value of the backbone atoms between GlyT2 and the serotonin transporter was approximately 3 Å, but this difference was mainly due to the rearrangement of the extracellular flexible loops; the RMSD of the TM helixes was 1.33 Å. Residues interacting with glycine did not undergo any significant changes after the simulations were equilibrated (150 ns), which was confirmed by the good convergence observed (data not shown). Glycine, chlorine, and the major two Na+ ions were very stable along the simulations. The glycine carbonyl group forms H-bonds with the first sodium, Gly212, and Tyr287, whereas the nitrogen undergoes H-bonding with Ala208, Ser477, and Ser479. The π–π stacking with Tyr207, Phe476, and Trp482 was well-pronounced. The position of the third Na+ ion was revealed. It was coordinated mainly by Glu248, Met276, Trp263, and Glu648 and rapidly (after 50 ns of simulation) occupied this pocket (Figures S2 and S3A,B).
Figure 1.
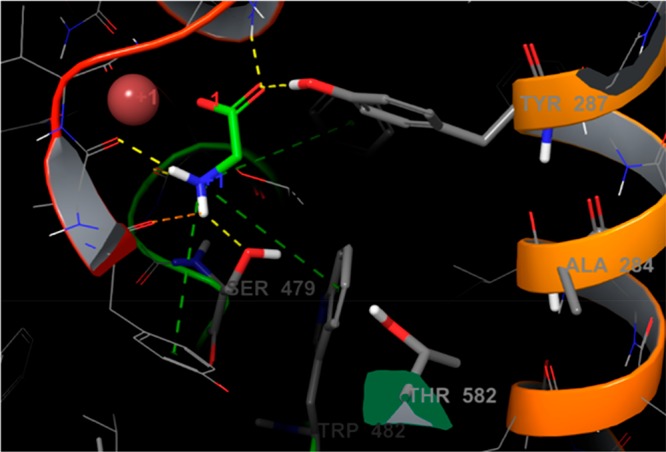
Glycine molecule in the binding site of GlyT2 as identified by our MD studies. The hydrogen bonds and π-cation are shown in yellow and green dotted lines.
Our hypothesis is that the third Na+ ion is required for an additional glycine binding site stabilization, and in particular it affects the conformation of residue Trp482, critical for GlyT2 function and the residues neighboring amino acids. To prove this, we ran 5 × 500 ns-long MD simulations on the GlyT1 transporter where the third Na+ was not present. The second sodium was less stable in GlyT1, and we even captured one of the likely unbinding paths in one of the MD runs (Figure S4). However, different paths are also possible.
We also studied the role of chloride ion on stability. Destabilization of this ion via the Tyr233Phe mutation completely cancels the glycine activity. The lack of interactions between the chlorine and the OH group of Tyr233 led to a rapid dissociation of the Cl– ion, which in turn destabilized the neighboring sodium and finally the glycine (Figure S5, Movie S1). At the end of these simulations, both the first sodium and glycine left the transporter, but the glycine moved out into extracellular space via its entry path, which confirmed that transport is not possible without chlorine.
To quantify the residue contributions to glycine–GlyT2 interactions, we conducted FEP+ calculations. We identified 10 residues that are important for compound binding (Table 1). Some of them are already known to significantly impact glycine activity.19,20 For instance, these include Ser477Ala, Phe476Ala, Ser479Ala, and Trp482Ala, which completely abolish glycine transport.21 Our data match previously published qualitative and quantitative experimental studies. For instance, Ser479Ala decreased the EC50 value of glycine from 12 to 1070 μM, a difference of ΔΔG = 2.6 kcal/mol, for example, in a frame of the error of that predicted by the FEP+ value. These data indicate differences in the glycine binding affinity upon mutations in the range of 1.5–4.0 kcal/mol, transforming glycine binding into the millimolar range, for example, fully canceling it. Furthermore, the Tyr287Phe mutation weakens the glycine binding by 0.7 kcal/mol, due to the loss of one H-bond, which is in agreement with the commonly accepted strength of a typical hydrogen bond in a protein environment,22 suggesting that our FEP+ data and generated structure are precise. Thus, the preliminary FEP+ results provide a structural basis for the observed significance of the selected binding site residues to glycine transport. Most of these residues are conserved in GlyT1, dopamine, and serotonin transporters, yet some of them are unique to GlyT2, which provide us helpful data on selectivity.
Table 1. Changes in Glycine Binding Activity (ΔΔG; kcal/mol) upon 11 Mutations As Calculated by FEP+.
| mutation | predicted ΔΔG | predicted error |
|---|---|---|
| SER477ALA | 4.04 | 0.44 |
| SER479ALA | 3.37 | 0.43 |
| TRP482ALA | 2.62 | 0.45 |
| PHE476ALA | 1.58 | 0.43 |
| TYR207ALA | 1.43 | 0.46 |
| TYR287ALA | 0.97 | 0.44 |
| VAL209ALA | 0.89 | 0.41 |
| LEU211ALA | 0.69 | 0.42 |
| TYR287PHE | 0.69 | 0.42 |
| GLY212ALA | 0.44 | 0.79 |
| THR578ALA | –0.68 | 0.44 |
We developed a special protocol for our high-throughput virtual screening (HTVS) with the aim of discovering new selective GlyT2 inhibitors. It has been established that a combination of the pharmacophore and docking methods provides superior hit identification success compared to employing a single approach.23 Currently, there are few known specific GlyT2 inhibitors. Their mechanism of action is diverse, and these sets of ligands have low structural similarity. Thus, a ligand-based pharmacophore search was not suitable for our research needs. We used our newly developed in-house strategy that combines a recently refined structure-based pharmacophore approach and docking method in a frame of Schrodinger 2017–3 software (see SI Methods) [ref 4 in SI]. This type of pharmacophore model is based on the docking of several hundred fragments into the binding site, and the pharmacophore points are defined in accordance with their best position and binding score. These are the places within the binding site that were predicted to be essential for ligand binding.
We generated four pharmacophore points, requested four of four matching points, and screened over 3.5 million lead like compounds from the ZINC15 library (Figure 3).24 Point D17 (donor) represents the requested H-bond with Ser479, which is, as it was shown above, important for selectivity and is mutated to Gly in GlyT1. Furthermore, we subjected 35 000 best candidates to docking and visually inspected the top-scoring 100 ligands. The top 20 ligands were selected by visual inspection and redocked then by induced-fit docking (IFD). The IFD procedure was accomplished by metadynamics refining the binding modes as shown previously (Figure S6) [ref 6 in SI]. The combination IFD and metadynamics basically evaluates and compares the stability of IFD poses after several independent runs. Two different structurally similar groups of ligands have been found to be most promising GlyT2 inhibitors (Figure 2A,B). We selected four of them (two from each group) for biological testing (Figure 4).
Figure 3.
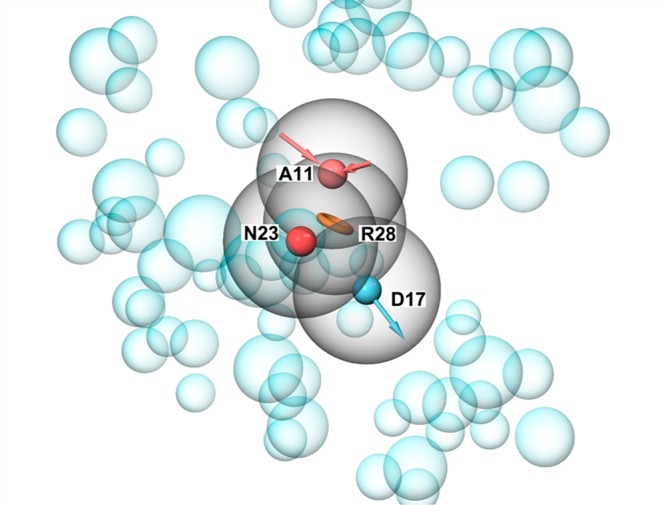
Structural based pharmacophore model based on the GlyT2MD generated structure. Four pharmacophores (acceptor (A11), negative charge (N23), aromatic ring (R28), and donor (D17)) were used.
Figure 2.
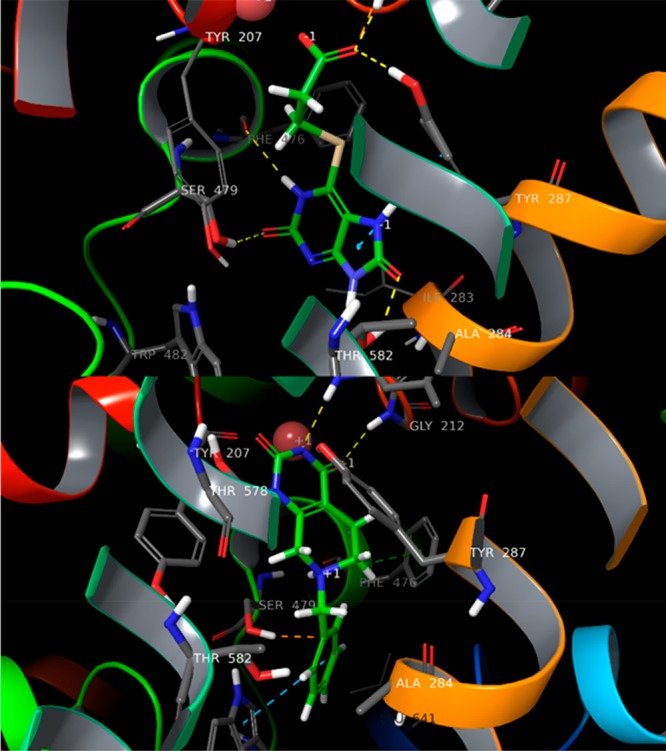
(A) Hit1 and (B) Hit2 in the binding site of GlyT2, identified by HTVS and MD studies.
Figure 4.
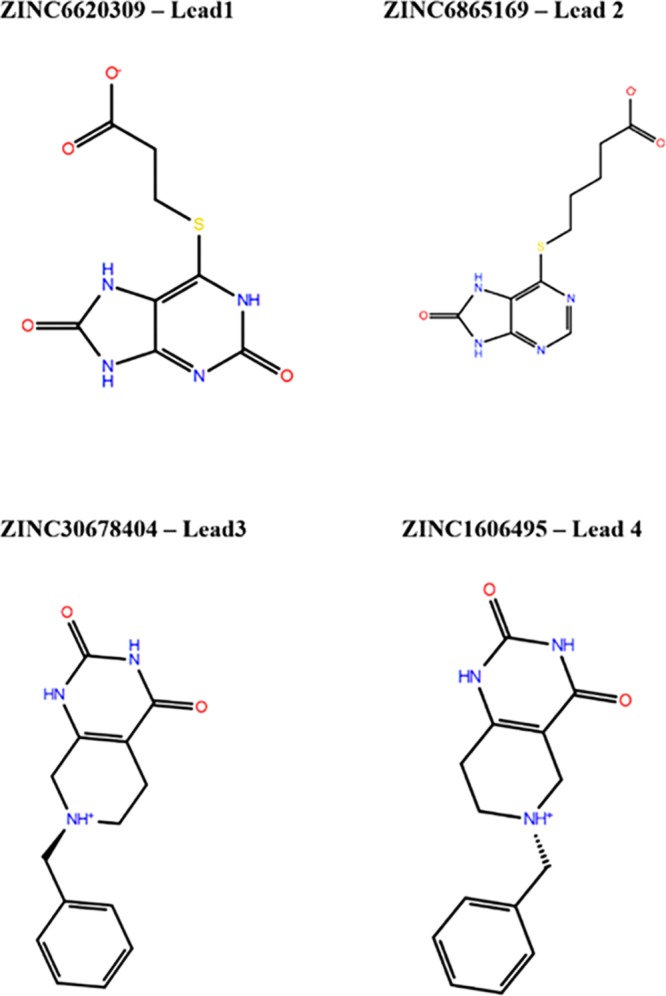
Identified by VS hits, which were confirmed by in vitro studies.
To validate the use of our in vitro assay for screening purposes, we initially carried out inhibition studies with the selective GlyT1 inhibitor ALX5407 and GlyT2 inhibitor ALX1393 (Figure S8).25,26 The inhibitor ALX1393 has been the most extensively studied in different models of pain in rodents, shows 40-fold selectivity for GlyT2 over GlyT1, and inhibits the human GlyT2 at nanomolar concentrations. To investigate the response of the selected compounds, we used porcine aorta epithelial cells stable expressing the human GlyT2. In these cells, the transporter traffics and accumulates at the plasma membrane where shows a Km of 123 μM and Vmax of 8.2 nmol/min/mg (Figure S7). In control experiments shown in Figure 5A, we measured the concentration dependence of ALX1393 and observed a rapid inhibition of transport, with an average IC50 = 25.9 nM, a value that is in an agreement with literature data.26 This observation confirms that our model system expresses a functional human transporter, which transports glycine with the expected specificity, and the activity can be inhibited by the traditional inhibitor ALX1393. As expected, incubation with the GlyT1 inhibitor ALX5407 showed a slow rate of inhibition with a calculated IC50 = 1.8 μM (data not shown).
Figure 5.

Inhibition curves of GlyT2 activity by ALX1393, ZINC6620309, and ZINC6865169. We incubated PAE cells expressing GlyT2 with varying concentrations of inhibitors in the uptake mix and measured the activity for 10 min. We obtained sigmoidal curves and calculated IC50 values via Sigma Plot.
Furthermore, we tested four selected compounds identified by in silico screening (Figure 4). Interestingly, a small lead molecule 1, termed Hit1/Lead1 (Figures 2A, 4, and 5; ZINC6620309), consistently inhibited the transport at high nanomolar concentrations and showed an IC50 = 0.48 μM. The structural analog, Hit2, which consists of two more carbon atoms after the propionic acid segment, and modification of the aromatic ring, was also active but featured slightly decreased (0.52 μM) inhibition; that is, it was still a good inhibitor. This is presumably due to the longer chain that cannot be accommodated well in the ligand binding domain (LBD) or lack of significance for the binding interactions with Ser479 (Table 2). The second class of ligands was also active. Hit3 showed an IC50 = 33.2 μM. This is because that simple compound provides only interactions close to the first Na+ binding pocket (Figure 2B). Finally, Hit4 was not potent due to the position of its long chain, providing less access to the key GlyT2 residues.
Table 2. Inhibition of Glycine Uptake by Zinc Compounds in Cells Expressing Human GlyT2 and Mouse GlyT1, Respectivelya.
| inhibitor | GlyT2 IC50 (μM) | GlyT1 IC50 (μM) |
|---|---|---|
| ALX1393 | 0.026 ± 0.003 | N/A |
| ZINC6620309 (Hit1) | 0.478 ± 0.028 | >200 |
| ZINC6865169 (Hit2) | 0.518 ± 0.066 | >200 |
| ZINC30678404 (Hit3) | 33.2 ± 3.28 | >200 |
IC50 value was determined by varying the concentration of inhibitor from 0 to 10 or 100, up to 200 μM for GlyT1, at pH 7.5. Values are the average of three to five determinations with an average standard error of less than 12%.
In the process of developing inhibitors for glycine transporter 2, one vital concern that must be addressed is the possibility of unspecific activity of inhibitors in both glycine transporters. To address this concern, we tested the molecules in a mammalian cell line expressing the GlyT1a. As control we used ALX5407, which is a very well-known inhibitor of GlyT1 in vitro. ALX5407 showed a significant inhibition of around 80% in GlyT1 at a very low concentration (IC50:14.16 nM, Figure S9). All these results suggest that GlyT1 is a functional glycine transporter that can be targeted for pharmacological studies. Furthermore, we tested and validated the specificity of our hit molecules in GlyT1. The results showed that Hit1–3 did not have any detectable activity in GlyT1, even at high micromolar concentrations (>200 μM, see Table 2) (Figures S10–S12).
In conclusion, these data collectively demonstrate that our structure is of high quality, allowing us to discover a new set of compounds with good inhibitory properties and selectivity that can be further optimized. Moreover, they also show that our VS approach leads to a high hit rate of 75%, although a homology/MD derived structure was used. We suggest that matching all key structural pharmacophores (4/4 herein) is a good strategy and an explanation of obtained results. On the basis of the aforementioned data, we chose Hit1 as our primary candidate for further lead optimization and studied this ligand in more detail.
To provide a structural basis of the observed activity, we procured relaxed Hits–GlyT2 complex structures by executing 2 × 250 ns MD simulations via Amber16. The carbonyl group of Hit1 interacts with the first sodium, Leu211, Gly212, and Tyr287. On the basis of this structure, it was evident that Tyr287, Ser479, and the backbone oxygen of Thr578 make stable H-bonding with the ligand and stabilize its aromatic component, in a conformation that provides π–π staking with Phe476 and Trp482 (Figure 2A). We also observed a stable H-bond between Hit1 and the Phe476 backbone oxygen. Thr578 coordinated the second sodium but also could flip its −OH group to form an H-bond with Tyr287, thus accelerating the Na+ unbinding dissociating and demonstrating that Hit1, like ALX1393, is presumably a reversible inhibitor. Both Ser479 and Thr582 are dynamic in the presence of Hit1. Thr582 can form H-bonds with the ligand, the backbone of Thr578, and Tyr207, whereas Ser479 can do so with Hit1 and Trp482. These variations are due to the significant solvation of the LBD, which was formed by the ligand’s multiple polar groups. Furthermore, we confirmed these results during our FEP+ guided lead optimization by execution of different sets of Hit1 derivatives (unpublished data). The percentage of H-bond formation of Hit1 with Thr582 and Ser479 varied between 25% and 65% for both of these residues (Figure 6A,B).
Figure 6.

Identified Hit1 binding mode and Hit1–GlyT2 interactions, during the FEP+ calculations, represented by (A) 2D view and (B) presence of GlyT2 interactions with GlyT2 residues. Purple and green lines show H-bonds and π–π staking, respectively, whereas green, red, and blue bar charts indicate H-bond, ionic, and hydrophobic interactions, respectively.
Similarly, the Hit2 pyrimidine group interacts with residues close to the first sodium but provides limited connection with the Na+ ion itself. The phenyl ring facilitates π–π staking with Phe476 and Trp482, but only hydrophobic interactions with Ser479 and Thr582 were observed (Figure 2B). Instead, these interactions were more pronounced with Ile283, a residue which is conserved between GlyT2 and GlyT1. We obtained similar results for Hit3, yet we observed one additional H-bond with Phe476 and staking with Tyr287 (Figure S13).
Despite the high sequence similarity between the GlyT2 and GlyT1 binding sites, the experimental data for our discovered Hit1 compound indicate that it is a highly selective GlyT2 inhibitor. The difference between the binding sites of these subclasses of transporters is only five residues. Thus, Hit1 is an ideal compound for revealing the selectivity. In GlyT1, the Ser479 and Thr582 residues are mutated to Gly and Leu, respectively, whereas Phe476 is mutated to tyrosine. Thus, it is clear that these mutations would have a great impact on Hit1 selectivity.
To quantify the contribution of all five mutated residues in GlyT1, and in particular Ser479Gly and Thr582Leu, we executed FEP+ ligand selectivity calculations. We employed our recently improved FEP+ sampling protocol,27,28 which has been shown to significantly improve the FEP+ results. Moreover, an additional equilibration in the pre-REST stage of FEP+ on that system should be performed due to the structural rearrangements introduced by the mutations in the aforementioned GlyT2 structure. Indeed, the Ser479Gly mutation negatively impacted the selectivity and contributed ΔΔG = +0.98 kcal/mol, whereas the Thr582Leu had the most significant contribution, ΔΔG = +2.29 kcal/mol (Table 3). This is in excellent agreement with the recent mutations studies for both ALX1393 and glycine.29
Table 3. Changes in Hit1 Binding Affinity (ΔΔG; kcal/mol) upon Five Mutations Constituting GlyT2/1 Differences As Calculated by FEP+.
| mutations | predicted ΔΔG | predicted error |
|---|---|---|
| ALA284GLY | 0.35 | 0.09 |
| MET579GLN | –0.61 | 0.18 |
| PHE476TYR | –0.51 | 0.23 |
| 0SER479GLY | 0.98 | 0.19 |
| THR582LEU | 2.29 | 0.30 |
It has been shown that the Ser479 to Gly mutation decreased glycine affinity form 13 to 72 μM, which corresponds to 0.95 kcal/mol (ΔG = 0.59 ln Kd), for example, the same value calculated as per our FEP+ simulations. Further, a significant decrease in the inhibition for the same mutation was also observed in ALX1393, whereas Thr582Leu completely abolished the glycine transport.29 This is an indication that in the glycine binding site all of these ligands facilitate similar interactions, which is reasonable result considering their similar structure at this part of the GlyT2 LBD: a carbonyl group and donor placed close to the first sodium and Ser479, respectively. However, for the remainder of the LBD, it seems that ALX1393 and Hit1 exhibit different interactions. Additional support comes from the data for Hit2 (Figure S14). Essentially, it provides ligand–protein interactions similar to those of Hit1, but the contact with Ser479 is not so significant. As a result, the selectivity was mainly due to the Thr582 H-bond. Notably, our FEP+ calculations showed that the Ala284Gly mutation had a small negative effect for the Hit1 activity, whereas Phe476Tyr had a positive effect. In contrast, the last mutation abolishes the Glycine transport and reduces ALX1393 binding yet does not cancel it.29 Thus, Hit1 interactions in this part of the LBD are different than those of ALX1393. These results provide a structural basis to the experimentally observed Hit1 selectivity in GlyT2. The calculated ADMET properties of Hit1 and Hit2 are shown in the corresponding SI.
In summary, our study demonstrated that the combination of structural based pharmacophore and docking techniques can be helpful procedure for identification of novel biologically active and more selective compounds. This combination enabled us to identify new selective GlyT2 inhibitors. Finally, the FEP+ approach provides a detailed insight into the structural basis of the selectivity and ideas for the further lead optimization.
Acknowledgments
We thank Ricardo Avila and Michael Scott Long for reading and editing the paper.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.9b00003.
Author Contributions
S.S., F.F., and M.M.-A. conceived the project. S.S. supervised the computational part of the project, and M.M.-A. supervised the biology part of the project. F.F. performed all the computational work. E.P. performed the GlyT2 inhibition assays and A.B.L performed the selectivity studies on GlyT1. F.F., S.S., and M.M.-A. contributed to the analysis of results and wrote the manuscript. All authors reviewed and edited the manuscript.
This work is supported by Dr. Suman Sirimulla’s startup fund from UTEP School of Pharmacy.
The authors declare no competing financial interest.
Supplementary Material
References
- Zeilhofer H. U.; Wildner H.; Yévenes G. E. Physiol. Rev. 2012, 92, 193–235. 10.1152/physrev.00043.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Hehn C. A.; Baron R.; Woolf C. J. Neuron 2012, 73, 638–52. 10.1016/j.neuron.2012.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandenberg R. J.; Ryan R. M.; Carland J. E.; Imlach W. L.; Christie M. J. Trends Pharmacol. Sci. 2014, 35, 423–30. 10.1016/j.tips.2014.05.006. [DOI] [PubMed] [Google Scholar]
- Wiles A. L.; Pearlman R.-J.; Rosvall M.; Aubrey K. R.; Vandenberg R. J. J. Neurochem. 2006, 99, 781–786. 10.1111/j.1471-4159.2006.04107.x. [DOI] [PubMed] [Google Scholar]
- Isaac M.; Slassi A.; Silva K. D.; Arora J.; MacLean N.; Hung B.; McCallum K. Bioorg. Med. Chem. Lett. 2001, 11, 1371–1373. 10.1016/S0960-894X(01)00253-0. [DOI] [PubMed] [Google Scholar]
- Zeilhofer H. U.; Acuña M. A.; Gingras J.; Yévenes G. E. Cell. Mol. Life Sci. 2018, 75, 447–465. 10.1007/s00018-017-2622-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagi R.; Yashoda Devi B.; Rakesh N.; Reddy S. S.; Patil D. J. Oral Surgery, Oral Medicine, Oral Pathology and Oral Radiology 2015, 119, 264–271. 10.1016/j.oooo.2014.12.002. [DOI] [PubMed] [Google Scholar]
- Davies N. M.; Reynolds J. K.; Undeberg M. R.; Gates B. J.; Ohgami Y.; VegaVilla K. R. Expert Rev. Neurother. 2006, 6, 1643–1655. 10.1586/14737175.6.11.1643. [DOI] [PubMed] [Google Scholar]
- Dasgupta N.; Funk M. J.; Proescholdbell S.; Hirsch A.; Ribisl K. M.; Marshall S. Pain Med. 2016, 17, 85–98. [DOI] [PubMed] [Google Scholar]
- Bohnert A. S. B.; Valenstein M.; Bair M. J.; Ganoczy D.; McCarthy J. F.; Ilgen M. A.; Blow F. C. JAMA 2011, 305, 1315. 10.1001/jama.2011.370. [DOI] [PubMed] [Google Scholar]
- Delić M.; Kajdiž K.; Pregelj P. Psychiatria Danubina 2017, 29, 289–291. [PubMed] [Google Scholar]
- Bannister K.; Qu C.; Navratilova E.; Oyarzo J.; Xie J. Y.; King T.; Dickenson A. H.; Porreca F. Pain 2017, 158, 2386–2395. 10.1097/j.pain.0000000000001040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodman C. W.; Brett A. S. N. Engl. J. Med. 2017, 377, 411–414. 10.1056/NEJMp1704633. [DOI] [PubMed] [Google Scholar]
- Cioffi C. L. J. Med. Chem. 2018, 61, 2652–2679. 10.1021/acs.jmedchem.7b00956. [DOI] [PubMed] [Google Scholar]
- Novel benzamide derivative and use thereof. Patent WO 2013081400, 2012.
- Mingorance-Le Meur A.; Ghisdal P.; Mullier B.; De Ron P.; Downey P.; Van Der Perren C.; Declercq V.; Cornelis S.; Famelart M.; Van Asperen J.; Jnoff E.; Courade J. P. British journal of pharmacology 2013, 170, 1053–63. 10.1111/bph.12343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glycine transporter inhibitor. Patent WO 2015012400A1, 2014.
- Omori Y.; Nakajima M.; Nishimura K.; Takahashi E.; Arai T.; Akahira M.; Suzuki T.; Kainoh M. J. Pharmacol. Sci. 2015, 127, 377–381. 10.1016/j.jphs.2015.02.010. [DOI] [PubMed] [Google Scholar]
- Coothankandaswamy V.; Cao S.; Xu Y.; Prasad P. D.; Singh P. K.; Reynolds C. P.; Yang S.; Ogura J.; Ganapathy V.; Bhutia Y. D. British journal of pharmacology 2016, 173, 3292–3306. 10.1111/bph.13616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian N.; Scopelitti A. J.; Carland J. E.; Ryan R. M.; O’Mara M. L.; Vandenberg R. J. PLoS One 2016, 11, e0157583 10.1371/journal.pone.0157583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zafra F.; Ibañez I.; Gimenez C. Neuronal Signaling 2016, 1, NS20160009. 10.1042/NS20160009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheu S.-Y.; Yang D.-Y.; Selzle H. L.; Schlag E. W. Proc. Natl. Acad. Sci. U. S. A. 2003, 100, 12683–7. 10.1073/pnas.2133366100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peach M. L.; Nicklaus M. C. J. Cheminf. 2009, 1, 6. 10.1186/1758-2946-1-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sterling T.; Irwin J. J. J. Chem. Inf. Model. 2015, 55, 2324–2337. 10.1021/acs.jcim.5b00559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atkinson B. N.; Bell S. C.; De Vivo M.; Kowalski L. R.; Lechner S. M.; Ognyanov V. I.; Tham C.-S.; Tsai C.; Jia J.; Ashton D.; Klitenick M. A. Mol. Pharmacol. 2001, 60, 1414–1420. 10.1124/mol.60.6.1414. [DOI] [PubMed] [Google Scholar]
- Takahashi Y.; Hara K.; Haranishi Y.; Terada T.; Obara G.; Sata T. Pharmacol., Biochem. Behav. 2015, 130, 46–52. 10.1016/j.pbb.2015.01.001. [DOI] [PubMed] [Google Scholar]
- Fratev F.; Sirimulla S. ChemRxiv 2019, 10.26434/chemrxiv.6204167.v2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fratev F.; Steinbrecher T.; Jónsdóttir S. Ó. ACS Omega 2018, 3, 4357–4371. 10.1021/acsomega.8b00123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carland J. E.; Thomas M.; Mostyn S. N.; Subramanian N.; O’Mara M. L.; Ryan R. M.; Vandenberg R. J. ACS Chem. Neurosci. 2018, 9, 603–614. 10.1021/acschemneuro.7b00407. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.