

Abstract
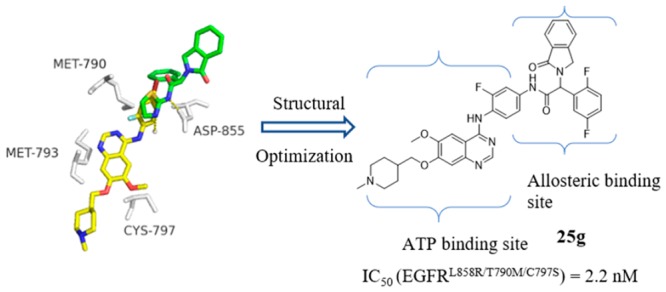
In this paper, we describe the discovery and optimization of a series of noncovalent reversible epidermal growth factor receptor inhibitors of EGFRL858R/T790M/C797S. One of the most promising compounds, 25g, inhibited the enzymatic activity of EGFRL858R/T790M/C797S with an IC50 value of 2.2 nM. Cell proliferation assays showed that 25g effectively and selectively inhibited the growth of EGFRL858R/T790M/C797S-dependent cells. This series of compounds, which occupy both the ATP binding site and the allosteric site of the EGFR kinase, may serve as a basis for the development of fourth-generation EGFR inhibitors for L858R/T790M/C797S mutants.
Keywords: EGFR, mutant, inhibitor, C797S
Introduction
Epidermal growth factor receptor (EGFR), a member of the HER family, is an essential transmembrane glycoprotein in cell signaling pathways that regulate cell proliferation, differentiation, and apoptosis.1 The overexpression of EGFR has been observed in many types of solid tumors.2−4 Nonsmall cell lung cancer (NSCLC) is one of the most malignant cancer types worldwide.5 With the drug development, various small molecular EGFR inhibitors (Figure S1, Supporting Information) have been developed as therapeutic agents for NSCLC.
The first generation of reversible EGFR inhibitors, gefitinib and erlotinib, delivered significant therapeutic effects for NSCLC patients with activating EGFR mutations.6−8 The L858R point mutation and exon 19 deletion are the most common activating mutations, with enhanced sensitivity to inhibitors.9 However, after 12 months of clinical treatment, the T790M mutation appeared in 50%–60% of drug-resistant patients.10 The presence of T790M increases the affinity of the receptor for ATP, thereby reducing the ability of EGFR inhibitors to effectively compete with ATP.11 Then, the second- and third-generation EGFR irreversible inhibitors were developed12 that had increasing cellular potency against T790M mutants, mainly by covalently binding to Cys797.13 However, because the aniline moiety of the second-generation EGFR inhibitors may not interact as effectively with the side chain of Met790, the T790M activity is lower against the activating EGFR mutations.14 The third-generation EGFR inhibitors selectively and irreversibly target EGFRT790M and other activated EGFR mutations. AZD9291 (4, osimertinib, Figure S1, Supporting Information),15,16 the only FDA approved third generation inhibitor,17 has good potency against the EGFRT790M mutant and minimal toxicities, with excellent selectivity for wild-type EGFR. Nevertheless, a clinical study showed that 20–30% of patients treated with AZD9291 developed the tertiary point mutation C797S,18 which prevents irreversible inhibitors from covalently binding to Cys797. Loss of the covalent interaction results in a marked decrease in inhibition, which then leads to the development of resistance.
Jia et al. reported on compound EAI001 (6, Figure S1, Supporting Information) as the first non-ATP competitive EGFRL858R/T790M/C797S inhibitor.19 After structural optimization, a more potent compound 1 (EAI045,Figure 1) was obtained. EAI045 binds to the allosteric site created by the outward displacement of the αC-helix of the EGFR kinase, located next to the ATP binding pocket.19 The discovery of this allosteric site laid the theoretical foundation for our research.
Figure 1.

Structures of compound 1 (EAI045) and compound 2.
We found that compound 2 (2, Figure 1), a known EGFR inhibitor (vandetinib),20 exhibited modest potency (IC50 = 369.2 nM) against the EGFRL858R/T790M/C797S mutant. Both the reference21 and our molecular docking simulation (gscore = −8.2 kcal/mol) indicated that compound 2 could extend into the ATP binding pocket of EGFR (Figure 2A). Inspired by the binding model of EAI045, we attempted to modify 2 to occupy both the ATP binding site and the allosteric site of the EGFR kinase, with the aim of enhancing the binding affinity of the inhibitor to EGFRL858R/T790M/C797S, to effectively compete with ATP and thus overcome resistance. Then, 25a was developed (Figure 2B). Based on 25a, we designed and synthesized a series of novel, highly potent, and noncovalent reversible inhibitors of EGFRL858R/T790M/C797S and explored their structure–activity relationships. In this study, one of the most promising compounds is 25g (Figure 2B), which inhibited the enzymatic activity of EGFRL858R/T790M/C797S with an IC50 value of 2.2 nM. Cell proliferation assays showed that 25g effectively and selectively inhibited the growth of EGFRL858R/T790M/C797S-dependent cells. These findings may help overcome acquired resistance to third-generation EGFR inhibitors.
Figure 2.

(A) Overlaid model of the docked pose of compound 2 (yellow) and the bound conformation of the allosteric inhibitor EAI001 (cyan) in EGFRT790M/V948R (PDB: 5d41), key residues are shown as gray sticks; (B) Chemical structures of compounds 25a and 25g; (C) The 2D interactions diagram for the quinazoline scaffold to show strategies of structural modifications, with chemical structures of the representative compounds. n = 0, 1; Z = −CH3, −CH(CH3)2, −Ph.
Results and Discussion
The docking model of 2 with EGFRT790M/V948R shows that 2 binds at the ATP binding site of EGFR with its phenyl group occupying a position similar to that of the thiazole moiety of EAI001 (Figure 2A). EAI001 binds as a “Y shaped” configuration in the allosteric site.22 Modifying 2 to occupy both the ATP binding site and the allosteric site may be a promising way to increase the bioactivity against the EGFRL858R/T790M/C797S triple mutant.
To facilitate the occupation of the allosteric site of EGFR, different hydrophobic groups were introduced to the R1 position of 2 with an amide bond as the linker. The resultant compounds, 18a–18i (Figure 2C), had no inhibitory activity against EGFRL858R/T790M/C797S (Table S1, Supporting Information). Referring to the structure of EAI045, oxoisoindolin-2-phenylacetamide was introduced into the R1 position, synthesizing compound 25a. The kinase assay showed that 25a has a nanomolar level bioactivity (IC50 = 9.3 nM) against EGFRL858R/T790M/C797S. We surmised that the substituted group at the R1 position of 25a has a “Y-shaped” configuration,22 making it more likely to embed in the allosteric site. To further explore the structure–activity relationship and acquire compounds with higher potency, we selected compound 25a as the new lead compound.
After a docking simulation, we found that the interactions between 25a and EGFR include three parts (Figure 2C and Figure 3): (1) the quinazoline scaffold of 25a forms a hydrogen bond with residue Met793 in the hinge region; (2) the “Y-shaped” R1 group oxoisoindolin-2-phenylacetamide extends into the EGFR kinase allosteric site with hydrophobic interaction; and (3) the alkoxy side chain R2, R3 of the quinoline scaffold faces toward the solvent-exposed region.
Figure 3.
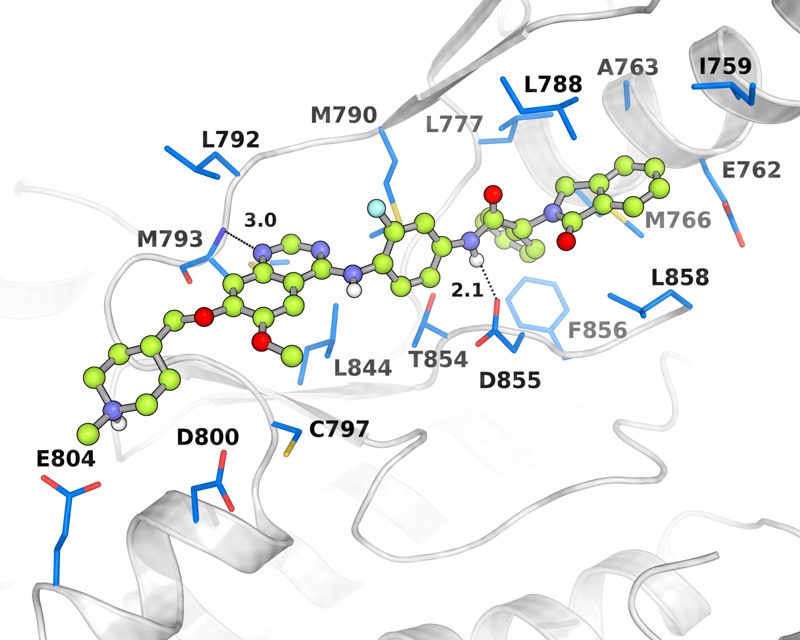
Docked pose of compound 25a. The EGFR protein (PDB: 5d41) is shown as a gray cartoon, and the key residues are shown as blue sticks. Key H-bonds are displayed as black dashes and measured by distances. The figure was generated using Pymol 1.3.
We then optimized 25a mainly from three aspects: (1) the allosteric region; (2) the hinge region; and (3) the solvent-exposed region.
In the allosteric region, “the Y-shaped” group oxoisoindolin-2-phenylacetamide was introduced at the R1 position (Figure 2C). Compound 25b was first synthesized, and the “Y-shaped” group was attached to the ortho position (5′-position) of anilino-quinazoline. Compound 25b displayed an IC50 value of 37.1 nM against EGFRL858R/T790M/C797S, a 4-fold decrease compared to that of 25a. Compounds 25c and its isomer 25d exhibited IC50 values of 7.9 nM and 19.2 nM, respectively, against EGFRL858R/T790M/C797S. This result indicates that the S-enantiomer is preferred over R, but both are acceptable. Then, a fluorine atom was introduced to different positions of the phenyl to acquire 25e–25g. Kinase assay results showed that 25g was the most potent, increasing the inhibitory activity by over 4-fold compared with 25a (IC50 = 2.2 nM). The introduction of the two fluorine atoms plays a crucial role in strengthening the binding affinity. Replacing the phenyl group of 25a within a cyclohexane group led to compound 25h. Compound 25h displayed less potent inhibitory activity, with an IC50 value of 179.6 nM against EGFRL858R/T790M/C797S, a significant decrease in activity compared with 25a, suggesting that the π–π stacking interaction between the phenyl of the “Y-shaped” group and residue Phe856 of the hydrophobic allosteric cavity plays an important role in maintaining the bioactivities of this series against EGFRL858R/T790M/C797S (Figure 3).
In the hinge region, only one hydrogen-bond interaction can form between the quinazoline scaffold of 25a and Met793 (Figure 2C). To enhance the binding strength, we proposed that another hydrogen bond might be formed between the compound and Met793 by introducing a substituent at the R position containing a hydrogen bond donor such as −NH2; thus, 25j was synthesized. Kinase assay results showed that 25j displayed no inhibitory activity against either T790M/L858R or L858R/T790M/C797S mutant, indicating that “-NH2” was not tolerated at the R position.
In the solvent-exposed region, to investigate the effect of the hydrophilic tail on bioactivity against EGFRL858R/T790M/C797S, we performed structural derivatization using piperidine, morpholine, and alkyl chains at R2 or R3 to synthesize compounds 25i and 25k–o. The kinase inhibitory activities of these compounds were lower than that of 25a, which has an IC50 value of 9.3 nM against EGFRL858R/T790M/C797S (Table S2, Supporting Information).
After a series of optimizations, compound 25g, with the best kinase inhibitory activity against EGFRL858R/T790M/C797S, was obtained (IC50 = 2.2 nM). Like 25a, docking simulations suggested that 25g also occupies both the ATP binding site and the allosteric site (Figure 4), exhibiting similar intermolecular interactions.
Figure 4.
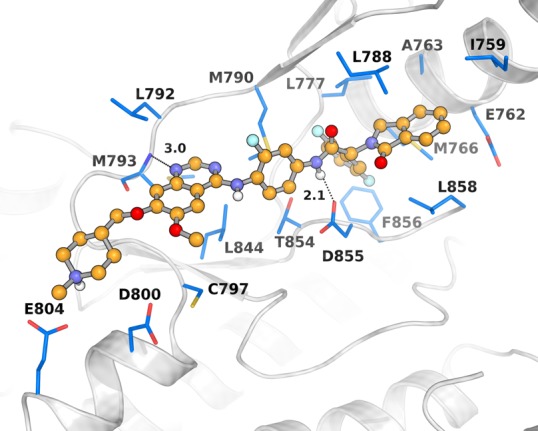
Docked pose of compound 25g. The EGFR protein (PDB: 5d41) is shown as a gray cartoon, and the key residues are shown as blue sticks. Key H-bonds are displayed as black dashes and measured by distance. The figure was generated using Pymol 1.3.
The synthetic procedures of all compounds are shown in the Supporting Information (Schemes S1–S3).
The kinase profile of 25g across 37 kinases is shown in Figure 5. Treatment with 25g at 100 nM demonstrated more than 90% inhibition of EGFRL858R/T790M/C797S as well as the EGFR family of kinases (ErbB2 and ErbB4) and BLK, and it exhibited moderate potency against Src, Abl, PDGFs, Txk, AXL, BTK, and VEGFR2 with inhibition rates from 50% to 75%, However, 25g showed less than 50% inhibition of 25 other kinases (VEGFR1, 3, EPH-A2, -B2, IGF1R, FGFR1-4, JAK1, 3, Trk-A, -B, -C, ALK, IKK, RET, C-kit, and so on), indicating that 25g showed a favorable selectivity.
Figure 5.

Kinase profile of 25g cross 37 kinases. *EGFRLTC: abbreviation for EGFRL858R/T790M/C797S.
Then, we tested the antiproliferative activities of 25g against a panel of cell lines with different EGFR status. As shown in Table 1, 25g exhibited better antiproliferative effects against BaF3-EGFRL858R/T790M/C797S cell lines than AZD9291 (IC50: 0.64 μM vs 3.93 μM) and EAI045 (IC50 > 10 μM, Table S5, Supporting Information). Meanwhile, 25g showed moderate antiproliferative effects against the parental BaF3 cells (IC50 = 7.68 μM), H1975 (IC50 = 3.03 μM) cells, and A431 cells (IC50 = 1.24 μM). Therefore, 25g was selected as the EGFRL858R/T790M/C797S inhibitor for further study of cell signaling pathways.
Table 1. In Vitro EGFR Antiproliferative Activitya.
| Antiproliferative
IC50 (μM) |
|||
|---|---|---|---|
| Cells | 25g | AZD9291 | Brigatinib |
| BaF3 | 7.68 ± 0.73 | 5.11 ± 0.49 | 7.31 ± 0.98 |
| BaF3-EGFRL858R/T790M/C797S | 0.64 ± 0.14 | 3.93 ± 0.38 | 0.42 ± 0.09 |
| H1975 | 3.03 ± 0.49 | 0.03 ± 0.01 | NDb |
| A431 | 1.24 ± 0.16 | 1.44 ± 0.03 | NDb |
Antiproliferative activity was examined by the Resazurin assay or the SRB assay. Date are averages of at least three independent determinations and reported as the means ± SD (standard deviations).
Not determined.
The inhibitory activity of the representative compound 25g against the phosphorylation of EGFR was further confirmed by Western blot analysis. As shown in Figure 6, compound 25g dose-dependently inhibited the phosphorylation of EGFR in BaF3-EGFRL858R/T790M/C797S cells, while AZD9291 was unable to inhibit EGFR phosphorylation. Compound 25g showed significant inhibitory effects against p-EGFR at a concentration as low as 0.1 μM and almost completely inhibited the phosphorylation of EGFR at a concentration of 1 μM, which was comparable to the potency of the fourth-generation allosteric inhibitor EAI045.
Figure 6.

Bioactivity of 25g on the phosphorylation of EGFR in BaF3-EGFRL858R/T790M/C797S cells.
In addition, we also evaluated the potency of 25g against EGFR19Del/T790M/C797S, another triple mutant protein that contains an exon 19 deletion activating mutation. The results showed that 25g exhibited weak activity against the EGFR19Del/T790M/C797S kinase, with an IC50 value of 331.3 nM (Table S3) and weak antiproliferative activity against BaF3-EGFR19D/T790M/C797S cells, with an IC50 value of 3.54 μM (Table S4). Accordingly, 25g is more potent against EGFRL858R/T790M/C797S than against EGFR19D/T790M/C797S.
We calculated some drug-likeness properties and tested the aqueous solubility of 25g (Table S7 and Table S8, Supporting Information). The results showed that 25g has poor solubility. Preliminary studies of in vivo pharmacokinetic (PK) properties of compound 25g in rats following intravenous (IV) and oral (PO) administration (Table S6, Supporting Information) showed that 25g has a high clearance rate (CL = 960.8 mL/h/kg) and poor oral bioavailability (F = 0.55%).
Conclusion
Based on a report of the EGFR allosteric site19 and an iterative process of molecular docking, synthesis, and biological testing, we developed a series of potent and noncovalent reversible EGFR inhibitors that occupy both the ATP binding site and the allosteric site. The structure–activity relationships of this series of inhibitors were summarized against EGFRL858R/T790M/C797S. One of the most promising compounds, 25g inhibited the enzymatic activity of EGFRL858R/T790M/C797S with an IC50 value of 2.2 nM. Western blot results showed that 25g inhibits the phosphorylation of EGFRL858R/T790M/C797S and downstream signal transduction at the cellular level. Cell proliferation assays confirmed that 25g effectively and selectively inhibited the growth of EGFRL858R/T790M/C797S-dependent cells. With the aim of further structural optimization to improve PK properties in the future, this series of compounds might serve as a good basis for the development of fourth-generation EGFR inhibitors of L858R/T790M/C797S mutants.
Glossary
Abbreviations
- EGFR
epidermal growth factor receptor
- NSCLC
nonsmall cell lung cancer
- PK
pharmacokinetic
- IV
intravenous
- PO
oral.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.8b00564.
Author Contributions
§ Q.L., T.Z., and S.L. contributed equally to this work. All authors have given approval to the final version of the manuscript.
This work was supported by the National Key Research and Development Program [2016YFA0502304 to H.L.]; the National Natural Science Foundation of China (grant 81825020 to H.L., 81803437 to S.L.); the National Science & Technology Major Project “Key New Drug Creation and Manufacturing Program”, China (Number: 2018ZX09711002); and Fundamental Research Funds for the Central Universities, Special Program for Applied Research on Super Computation of the NSFC-Guangdong Joint Fund (the second phase) under Grant No.U1501501. S.L. is sponsored by the Shanghai Sailing Program (No. 18YF1405100). H.L. is sponsored by the National Program for Special Support of Eminent Professionals and the National Program for Support of Top-notch Young Professionals.
The authors declare no competing financial interest.
Supplementary Material
References
- Sharma S. V.; Bell D. W.; Settleman J.; et al. Epidermal growth factor receptor mutations in lung cancer. Nat. Rev. Cancer 2007, 7, 169–181. 10.1038/nrc2088. [DOI] [PubMed] [Google Scholar]
- Shia J.; Klimstra D. S.; Li A. R.; Qin J.; Saltz L.; Teruya-Feldstein J.; Akram M.; Chung K. Y.; Yao D.; Paty P. B.; Gerald W.; Chen B. Epidermal growth factor receptor expression and gene amplification in colorectal carcinoma: an immunohistochemical and chromogenic in situ hybridization study. Mod. Pathol. 2005, 18, 1348–1350. 10.1038/modpathol.3800417. [DOI] [PubMed] [Google Scholar]
- Wang X.; Zhang S.; MacLennan G. T.; Eble J. N.; Lopez-Beltran A.; Yang X. J.; Pan C.-X.; Zhou H.; Montironi R.; Cheng L. Epidermal growth factor receptor protein expression and gene amplification in small cell carcinoma of the urinary bladder. Clin. Cancer Res. 2007, 13 (3), 953–957. 10.1158/1078-0432.CCR-06-2167. [DOI] [PubMed] [Google Scholar]
- Perez E. A. The role of adjuvant monoclonal antibody therapy for breast cancer: rationale and new studies. Curr. Oncol. Rep. 2001, 3 (6), 516–522. 10.1007/s11912-001-0073-9. [DOI] [PubMed] [Google Scholar]
- Torre L. A.; Bray F.; Siegel R. L.; Ferlay J.; Lortet-Tieulent J.; Jemal A. Global cancer statistics. Ca-Cancer J. Clin. 2015, 65 (2), 87–108. 10.3322/caac.21262. [DOI] [PubMed] [Google Scholar]
- Cohen M. H.; Williams G. A.; Sridhara R.; Chen G.; McGuinn W. D.; Morse D.; Abraham S.; Rahman A.; Liang C.; Lostritto R.; Baird A.; Pazdur R. United States Food and Drug Administration Drug Approval summary: Gefitinib (ZD1839; Iressa) tablets. Clin. Cancer Res. 2004, 10 (4), 1212–1218. 10.1158/1078-0432.CCR-03-0564. [DOI] [PubMed] [Google Scholar]
- Xu Y.; Liu H.; Chen J.; Zhou Q. Acquired resistance of lung adenocarcinoma to EGFR-tyrosine kinase inhibitors gefitinib and erlotinib. Cancer Biol. Ther. 2010, 9 (8), 572–582. 10.4161/cbt.9.8.11881. [DOI] [PubMed] [Google Scholar]
- Tiseo M.; Bartolotti M.; Gelsomino F.; Bordi P. Emerging role of gefitinib in the treatment of non-small-cell lung cancer (NSCLC). Drug Des., Dev. Ther. 2010, 81–98. 10.2147/DDDT.S6594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gazdar A. F. Activating and resistance mutations of EGFR in non-small-cell lung cancer: role in clinical response to EGFR tyrosine kinase inhibitors. Oncogene 2009, 28, S24–S31. 10.1038/onc.2009.198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu H. A.; Arcila M. E.; Rekhtman N.; Sima C. S.; Zakowski M. F.; Pao W.; Kris M. G.; Miller V. A.; Ladanyi M.; Riely G. J. Analysis of tumor specimens at the time of acquired resistance to EGFR-TKI therapy in 155 patients with EGFR-mutant lung cancers. Clin. Cancer Res. 2013, 19 (8), 2240–2247. 10.1158/1078-0432.CCR-12-2246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yun C.-H.; Mengwasser K. E.; Toms A. V.; Woo M. S.; Greulich H.; Wong K.-K.; Meyerson M.; Eck M. J. The T790M mutation in EGFR kinase causes drug resistance by increasing the affinity for ATP. Proc. Natl. Acad. Sci. U. S. A. 2008, 105 (6), 2070–2075. 10.1073/pnas.0709662105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou W.; Ercan D.; Chen L.; Yun C.-H.; Li D.; Capelletti M.; Cortot A. B.; Chirieac L.; Iacob R. E.; Padera R.; Engen J. R.; Wong K.-K.; Eck M. J.; Gray N. S.; Jänne P. A. Novel mutant-selective EGFR kinase inhibitors against EGFR T790M. Nature 2009, 462 (7276), 1070–1074. 10.1038/nature08622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh M.; Jadhav H. Targeting non-small cell lung cancer with small-molecule EGFR tyrosine kinase inhibitors. Drug Discovery Today 2018, 23 (3), 745–753. 10.1016/j.drudis.2017.10.004. [DOI] [PubMed] [Google Scholar]
- Zhang L.; Yang Y. Y.; Zhou H. J.; Zheng Q. M.; Li Y. H.; Zheng S. S.; Zhao S. Y.; Chen D.; Fan C. W. Structure-activity study of quinazoline derivatives leading to the discovery of potent EGFR-T790M inhibitors. Eur. J. Med. Chem. 2015, 102, 445–463. 10.1016/j.ejmech.2015.08.026. [DOI] [PubMed] [Google Scholar]
- Ward R. A.; Anderton M. J.; Ashton S.; Bethel P. A.; Box M.; Butterworth S.; Colclough N.; Chorley C. G.; Chuaqui C.; Cross D. A. E.; Dakin L. A.; Debreczeni J. É.; Eberlein C.; Finlay M. R. V.; Hill G. B.; Grist M.; Klinowska T. C. M.; Lane C.; Martin S.; Orme J. P.; Smith P.; Wang F.; Waring M. J. Structure- and reactivity-based development of covalent inhibitors of the activating and gatekeeper mutant forms of the epidermal growth factor receptor (EGFR). J. Med. Chem. 2013, 56, 7025–7048. 10.1021/jm400822z. [DOI] [PubMed] [Google Scholar]
- Finlay M. R.; Anderton M.; Ashton S.; Ballard P.; Bethel P. A.; Box M. R.; Bradbury R. H.; Brown S. J.; Butterworth S.; Campbell A.; Chorley C.; Colclough N.; Cross D.; Currie G.; Grist M.; Hassall L.; Hill G. B.; James G.; James M.; Kemmitt P.; Klinowska T.; Lamont G.; Lamont S. G.; Martin N.; McFarland H. L.; Mellor M. J.; Orme J. P.; Perkins D.; Perkins P.; Richmond G.; Smith P.; Ward R. A.; Waring M. J.; Whittaker D.; Wells S.; Wrigley G. Discovery of a potent and selective EGFR inhibitor (AZD9291) of both sensitizing and T790M resistancemutations that spares the wild type form of the receptor. J. Med. Chem. 2014, 57 (20), 8249–67. 10.1021/jm500973a. [DOI] [PubMed] [Google Scholar]
- Planchard D.; Brown K. H.; Kim D.-W.; Kim S.-W.; Ohe Y.; Felip E.; Leese P.; Cantarini M.; Vishwanathan K.; Jänne P. A.; Ranson M.; Dickinson P. A. Osimertinib Western and Asian clinical pharmacokinetics in patients and healthy volunteers: implications for formulation, dose, and dosing frequency in pivotal clinical studies. Cancer Chemother. Pharmacol. 2016, 77 (4), 767–776. 10.1007/s00280-016-2992-z. [DOI] [PubMed] [Google Scholar]
- Eberlein C. A.; Stetson D.; Markovets A. A.; Al-Kadhimi K. J.; Lai Z.; Fisher P. R.; Meador C. B.; Spitzler P.; Ichihara E.; Ross S. J.; Ahdesmaki M. J.; Ahmed A.; Ratcliffe L. E.; O’Brien E. L. C.; Barnes C. H.; Brown H.; Smith P. D.; Dry J. R.; Beran G.; Thress K. S.; Dougherty B.; Pao W.; Cross D. A. E. Acquired Resistance to the Mutant-Selective EGFR Inhibitor AZD9291 Is Associated with Increased Dependence on RAS Signaling in Preclinical Models. Cancer Res. 2015, 75 (12), 2489–2500. 10.1158/0008-5472.CAN-14-3167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia Y.; Yun C.-H.; Park E.; Ercan D.; Manuia M.; Juarez J.; Xu C.; Rhee K.; Chen T.; Zhang H.; Palakurthi S.; Jang J.; Lelais G.; DiDonato M.; Bursulaya B.; Michellys P.-Y.; Epple R.; Marsilje T. H.; McNeill M.; Lu W.; Harris J.; Bender S.; Wong K.-K.; Jänne P. A.; Eck M. J. Overcoming EGFR(T790M) and EGFR(C797S) resistance with mutant-selective allosteric inhibitors. Nature 2016, 534 (7605), 129–132. 10.1038/nature17960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morabito A.; Piccirillo M. C.; Falasconi F.; De Feo G.; Del Giudice A.; Bryce J.; Di Maio M.; De Maio E.; Normanno N.; Perrone F. Vandetanib (ZD6474), a dual inhibitor of vascular endothelial growth factor receptor (VEGFR) and epidermal growth factor receptor (EGFR) tyrosine kinases: current status and future directions. Oncologist 2009, 14 (4), 378–390. 10.1634/theoncologist.2008-0261. [DOI] [PubMed] [Google Scholar]
- Garofalo A.; Goossens L.; Lemoine A.; Farce A.; Arlot Y.; Depreux P. Quinazoline-urea, new protein kinase inhibitors in treatment of prostate cancer. J. Enzyme Inhib. Med. Chem. 2010, 25 (2), 158–171. 10.3109/14756360903169485. [DOI] [PubMed] [Google Scholar]
- Chen L.; Fu W.; Zheng L.; et al. Recent progress of small-molecule epidermal growth factor receptor (EGFR) inhibitors against C797S resistance in non-small-cell lung cancer. J. Med. Chem. 2018, 61, 4290–4300. 10.1021/acs.jmedchem.7b01310. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.