

Abstract
Toll-like receptors (TLR) are key components of the innate immune system that elicit inflammatory responses through the adaptor proteins myeloid differentiation protein 88 (MyD88) and Toll-interleukin receptor domain-containing adaptor protein-inducing interferon-β (TRIF). Previously, we demonstrated that TRIF mediates the signaling of angiotensin II (ANG II)- induced hypertension and cardiac hypertrophy. Since TRIF is activated selectively by TLR3 and TLR4, our goals in this study were to determine the roles of TLR3 and TLR4 in mediating ANG II-induced hypertension and cardiac hypertrophy, and associated changes in proinflammatory gene expression in heart and kidney. In wild-type (WT) mice, ANG II infusion (1,000 ng·kg−1·min−1 for 3 wk) increased systolic blood pressure and caused cardiac hypertrophy. In ANG II-infused TLR4-deficient mice (Tlr4del), hypertrophy was significantly attenuated despite a preserved or enhanced hypertensive response. In contrast, in TLR3-deficient mice (Tlr3−/−), both ANG II-induced hypertension and hypertrophy were abrogated. In WT mice, ANG II increased the expression of several proinflammatory genes in hearts and kidneys that were attenuated in both TLR4- and TLR3-deficient mice compared with WT. We conclude that ANG II activates both TLR4-TRIF and TLR3-TRIF pathways in a nonredundant manner whereby hypertension is dependent on activation of the TLR3-TRIF pathway and cardiac hypertrophy is dependent on both TLR3-TRIF and TLR4-TRIF pathways.
NEW & NOTEWORTHY Angiotensin II (ANG II)-induced hypertension is dependent on the endosomal Toll-like receptor 3 (TLR3)-Toll-interleukin receptor domain-containing adaptor protein-inducing interferon-β (TRIF) pathway of the innate immune system but not on cell membrane localized TLR4. However, ANG II-induced cardiac hypertrophy is regulated by both TLR4-TRIF and TLR3-TRIF pathways. Thus, ANG II-induced rise in systolic blood pressure is independent of TLR4-TRIF effect on cardiac hypertrophy. The TLR3-TRIF pathway may be a potential target of therapeutic intervention.
Keywords: angiotensin II, cardiac hypertrophy, hypertension, innate immune system, MyD88, TLR3, TLR4, Toll-like receptors, TRIF
INTRODUCTION
The immune system plays an important role in the onset and maintenance of hypertension (6, 8, 27, 51, 59). Several clinical studies and experimental models of hypertension have been shown to involve inflammation, proinflammatory cytokines, and specific immune cells of the adaptive and innate immune systems (11, 26, 31, 37, 39, 58, 62). Despite the demonstrated role of the immune system in hypertension, the process that initiates the immune response is not understood. Since the innate immune system primes and activates the adaptive immune system by direct molecular interactions and release of immune mediators (12, 24), components of innate immunity are likely involved as priming events in hypertension. The innate immune system is triggered by several pattern recognizing receptors including multiple Toll-like receptors (TLRs) that detect exogenous pathogen-associated molecular patterns (PAMPs) or endogenous damage-associated molecular patterns (DAMPs). TLRs initiate intracellular signaling cascades culminating in proinflammatory gene expression, including cytokines, via activation of the well-known NF-κB and interferon-regulatory factor transcription factors. These functions lead to dendritic cell (DC) maturation and may also be responsible for autoimmune responses (43). Based on pharmacological inhibition, several TLRs have been implicated in hypertension (14, 34, 50). These TLRs utilize myeloid differentiation protein 88 (MyD88) as an adaptor for intracellular signaling. However, another adaptor protein, Toll interleukin receptor-domain-containing adaptor-inducing interferon-β (TRIF), is utilized only by TLR3 and TLR4 signaling in a receptor-specific manner. Thus, TLR4 signaling initiates the activation of both MyD88 and TRIF-mediated signaling pathways, whereas TLR3 initiates only the TRIF-mediated pathway.
Using genetically null mice for MyD88 and TRIF genes, we have previously shown that MyD88, the canonical adaptor protein required for proinflammatory signaling by most TLRs, is not required for angiotensin II (ANG II)-induced hypertension in mice (52). Instead, in this instance, MyD88 signaling is anti-inflammatory and suppresses the ANG II pressor response. Moreover, we found that the other adaptor protein TRIF that induces an interferon pathway is essential for ANG II-induced hypertension. However, both TLR3 and TLR4 activate TRIF-dependent signaling. Since the two TLRs display disparate ligand specificity, triggering mechanisms for activation and cellular localization (23, 30), we investigated which of the TLRs plays the dominant role in ANG II-induced hypertension and cardiac hypertrophy. As TLR3 is an endosomal receptor, whereas TLR4 is localized on the cell membrane, the identification of the dominant TLR in the Ang II response would provide insight into the nature of the triggering ligand and the molecular mechanism responsible for ANG II-induced activation of the innate immune system.
In this study, we used mice deficient in TLR4 and TLR3 to determine the contribution of each of these innate immune receptors to the responses to ANG II. We found that TLR3 but not TLR4 is required for ANG II-induced hypertension. However, both TLR3- and TLR4-mediated TRIF-dependent pathways contribute to ANG II-induced cardiac hypertrophy, and hypertrophy in TLR4-deficient mice is suppressed despite an enhanced increase in blood pressure. Thus, TLR3 and TLR4 differentially induce ANG II hypertension and cardiac hypertrophy.
MATERIALS AND METHODS
Mouse strains.
All animal experimental protocols were performed in compliance with National Institutes of Health regulations and were approved by the University of Iowa Institutional Review Committee. Wild-type (WT; C57BL/10, Jackson Laboratory stock number 0665), Tlr4del (C57BL/10ScNJ, Jackson Laboratory stock number 003752), and Tlr3−/− (Jackson Laboratory stock number 009675) mice were obtained from Jackson Laboratories. The Tlr4del mice have a C57BL/10 background with a spontaneous deletion of the Tlr4 locus resulting in a lack of mRNA (44). Tlr3−/− mice have been bred to C57BL/6 background and are homozygous for targeted deletion of exon 1 of Tlr3 gene and do not produce functional gene product (1). Male mice of 10–16 wk age were used for the experiments as described below.
ANG II infusion pumps and blood pressure measurement.
ANG II infusion and tail-cuff pressure measurements were done as described previously (52). Briefly, micro-osmotic pumps (Alzet model 1004, 0.11 μl/h, 28 days), containing either saline or ANG II (1,000 ng·kg−1·min−1), were inserted subcutaneously in the interscapular space under anesthesia (2.5% isoflurane vapor). Infusions were maintained for 3 wk.
Tail-cuff pressure was recorded using Visitech-2000 (Visitech Systems). Mice were acclimatized in the tail-cuff apparatus at least twice in the week before the start of the experiments. Baseline tail-cuff pressure data were recorded before pump insertion. All tail-cuff recordings were performed three times a week before noon to avoid diurnal variations in the blood pressure. Thirty measurements of tail-cuff systolic blood pressure (SBP) in each session were obtained from each mouse. After discarding the initial 10 measurements of acclimation readings, the measurements were exported to Microsoft Excel for analyses. Measurements from each mouse in each session were culled into quartiles based on SBP level, and the two central quartiles were included in SBP analyses. The mean SBP and day of ANG II or saline infusion for individual mice were plotted using Prism 7.0 software for Mac (GraphPad Software, La Jolla, CA). After 3 wk of infusion with saline or ANG II, mice were euthanized by decapitation under anesthesia (3% isoflurane), and organs were obtained for further analyses. Hearts, kidneys, spleen, brain, and aorta were snap frozen in liquid nitrogen and stored at −80°C.
Histology.
Hearts were fixed in Zn2+-formalin and embedded in paraffin. Histological sections were stained with either hematoxylin and eosin or by Masson’s trichrome and visualized by light microscopy. Cardiac hypertrophy was assessed by measurement of total heart weight (HW)/body weight (BW) ratio. Left ventricular cardiomyocyte cross-sectional area was measured from four different cross sections of three mouse hearts from each group. The measurements were made by an investigator who was blinded to the identity of the samples. Digitized micrographs of cardiomyocytes were obtained, and cross-sectional area was determined using a stage-micrometer reference scale in the image processing software Fiji (48). From each heart, more than 250 cardiomyocyte cross-sectional areas were measured.
RNA isolation and quantitative real-time PCR.
Total RNA from mouse heart and kidneys was isolated using mirVana RNA isolation kit (Ambion) or RNeasy RNA Isolation Kit (Qiagen). A 2-μg aliquot of RNA sample was used to synthesize cDNA in 50-μl reactions using Oligo(dT) as primers and SuperScript III Reverse Transcriptase (Invitrogen). Quantitative (q) real-time PCR was performed on qPCR cycler (ABI) using SYBR green-based PCR reactions, as described (52). Quantifications were done using ΔΔCT method where Gapdh was used as a reference gene for normalization of RNA expression. Results are presented as relative RNA expression normalized to the expression in saline-infused mice. The primers used in this study have been previously described and are listed in Table 1.
Table 1.
Primers used for real-time PCR
| Primer | Sequence |
|---|---|
| Tlr3-forward | 5′-CCAGAAGAATCTAATCAAATTAGATTTGTC-3′ |
| Tlr3-reverse | 5′-TTTTGCTAAGAGCAGTTCTTGGAG-3′ |
| Tlr4-forward | 5′-GGCAACTTGGACCTGAGGAG-3′ |
| Tlr4-reverse | 5′-CATGGGCTCTCGGTCCATAG-3′ |
| AT1R-forward | 5′-GCTGGCATTTTGTCTGGATAAC-3′ |
| AT1R -reverse | 5′-GCTTTTCTGGGTTGAGTTGG-3′ |
| Mmp9-forward | 5′-GAAGGCAAACCCTGTGTGTT-3′ |
| Mmp9-reverse | 5′-AGAGTACTGCTTGCCCAGGA-3′ |
| Nox4-forward | 5′-CTCTACTGGATGACTGGAAACC-3′ |
| Nox4-reverse | 5′-AGTCAGGTCTGTTTTCTTGCC-3′ |
| Nppa-forward | 5′-GTCTTGGCCTTTTGGCTTC-3′ |
| Nppa-reverse | 5′-TTCCTCAGTCTGCTCACTC-3′ |
| Tnf-forward | 5′-TGCCTATGTCTCAGCCTCTTC-3′ |
| Tnf-reverse | 5′-GAGGCCATTTGGGAACTTCT-3′ |
| Tgfb2-forward | 5′-GCTAATGTTGTTGCCCTCCT-3′ |
| Tgfb2-reverse | 5′-GCAGCAATTATCCTGCACATT-3′ |
| Gapdh-forward | 5′-CATTTCCTGGTATGACAATGAATACG-3′ |
| Gapdh-reverse | 5′-TCCAGGGTTTCTTACTCCTTGGA-3′ |
AT1R, angiotensin II receptor type 1; Mmp9, matrix metalloprotease 9; Nppa, atrial natriuretic peptide; Nox4, NADPH-oxidase 4; Tgfb2, transforming growth factor beta 2; Tlr3, Toll-like receptor 3; Tlr4, Toll-like receptor 4; Tnf, tumor necrosis factor-alpha.
Statistical analyses.
Statistical analyses were performed using an unpaired t-test or one-way ANOVA in Prism software (GraphPad Software) as shown in respective figures. Results are presented as means ± SE. Post hoc analyses on multiple group data were done using Tukey’s multiple comparisons test. A P value of ≤ 0.05 was considered statistically significant. Comparisons of ANG II pressor responses between the three groups were made on measurements obtained during the first 9 days of infusion (1st phase) and the last 10 days (2nd phase). Differences between SBP obtained during ANG II and saline infusions on corresponding days were aggregated and compared between the three groups by one-way ANOVA and Tukey’s post hoc test. A separate comparison of differences in SBP between saline- and ANG II-infused mice was done by unpaired t-tests.
RESULTS
TLR3 pathway but not TLR4 is required for ANG II-induced hypertension.
We infused ANG II (1,000 ng·kg−1·min−1) or saline subcutaneously in WT, Tlr4del, and Tlr3−/− mice and measured the SBP three times a week for 3 wk. Prior to the infusions, the baseline SBP in the saline- and ANG II-infused groups was not significantly different within each genotype. The baseline SBP of Tlr4del and Tlr3−/− mice was not different from that of WT, although SBP of Tlr3−/− mice at the baseline was statistically slightly lower than Tlr4del. The SBP during saline infusions over the 3-wk period was not significantly different between the three groups (Fig. 1A). The responses to ANG II, however, differed in the three groups. In WT mice, the SBP during the first 9 days of infusion of ANG II (1st phase) was not significantly higher than in the saline infusion group. The increase in SBP was pronounced and significantly higher than saline after first 9 days of infusion (2nd phase; Fig. 1, A and B). In Tlr4del, there was a rapid and significant increase in SBP with ANG II infusion compared with saline infusion that was evident during the first 9 days (1st phase) and was sustained thereafter (Fig. 1, A and B). In Tlr3−/−, there was no significant increase in SBP with ANG II compared with saline throughout the infusion period (Fig. 1, A and B).
Fig. 1.

A: measurement of systolic blood pressure (SBP) during saline (shaded circles) or angiotensin II (ANG II) (closed circles, 1,000 ng·kg−1·min−1) infusions for 3 wk in wild-type mice (WT, n = 6, 8), mice with TLR4 deletion (Tlr4del, n = 9, 8), and TLR3 knockout (Tlr3−/−, n = 6, 6) mice. Values represent means ± SE. Dotted horizontal line corresponds to the peak SBP in WT and is displayed for comparison with SBP of Tlr4del and Tlr3−/− mice. Statistically significant increases in SBP by ANG II in WT and Tlr4del are marked by * (P ≤ 0.05 for values compared with the saline infusion on corresponding days, using unpaired t-test). Increase in SBP in ANG II-infused Tlr3−/− was not different from that seen with saline throughout the 3-wk infusion period. B: comparison of the effect of ANG II on SBP (ΔSBP) in WT, Tlr4del, and Tlr3−/− during the first 9 days (1st phase) and last 10 days (2nd phase) of infusion. These results were analyzed by one-way ANOVA and Tukey’s multiple comparison test (*statistically significant changes, P ≤ 0.05; n.s., not significant). Compared with the WT, response to ANG II infusion in Tlr4del was significantly greater during the 1st phase but not during the 2nd phase, whereas response in Tlr3−/− was suppressed during both 1st and 2nd phases of ANG II infusion. C: peak SBP in ANG II-infused WT, Tlr4del, and Tlr3−/− mice. Peak SBP values in ANG II-infused WT and Tlr4del mice were significantly greater than saline-infused mice but were not different between saline- and ANG II-infused Tlr3−/− (*significant difference from saline-infused values, unpaired t-test, P ≤ 0.05; n.s., not significant).
Direct comparison of ANG II-induced increases in SBP between genotypes showed that in Tlr4del, the initial increase in mean SBP (1st phase) was significantly larger than seen in the WT (WT ΔSBP 4.35 ± 2.76 mmHg, Tlr4del ΔSBP 19.43 ± 3.56 mmHg; P = 0.01) or in Tlr3−/− (ΔSBP 2.9 ± 3.03 mmHg; P = 0.01, 1st phase, Fig. 1B). In the 2nd phase (from 10 days of ANG II infusion to end of experiment), ANG II-induced increase in SBP was significantly different between WT versus Tlr3−/− (WT ΔSBP 21.62 ± 2.65 mmHg vs. Tlr3−/− ΔSBP 8.93 ± 1.74 mmHg; P = 0.01) and Tlr4del versus Tlr3−/− (Tlr4del ΔSBP 21.87 ± 3.35 mmHg vs. Tlr3−/− ΔSBP 8.93 ± 1.74 mmHg; P = 0.009) but not between the WT and Tlr4del (P = 0.99).
However, the SBP in ANG II-infused Tlr4del was sustained, reaching a peak value of 146.0 ± 13.1 mmHg on day 18, which was higher than the corresponding peak SBP in WT of 139.3 ± 5.3 mmHg on day 18, but that difference was not statistically significant (P = 0.84, Fig. 1C). In contrast, in Tlr3−/−, there was an increase in SBP with ANG II infusion, and the values attained were significantly lower than in WT or Tlr4del (Fig. 1, A and B). Importantly, in Tlr3−/−, the increase in SBP with ANG II infusion was not significantly different than the corresponding increase with infusion of saline in the Tlr3−/−.
The pulse rate of three genotypes was similar at the end of infusions of saline, averaging 607 ± 27, 666 ± 18, and 625 ± 27 in WT, Tlr4del, and Tlr3−/−, respectively. The corresponding values in the three groups at the end of ANG II infusion were 594 ± 20, 675 ± 15, and 663 ± 20 beats/min.
TLR4- and TLR3-mediated pathways contribute to ANG II-induced cardiac hypertrophy.
We measured HW/BW ratio in mice at the end of 3-wk infusion of saline or ANG II. In the WT, ANG II infusion resulted in significantly greater HW/BW ratio compared with saline infusion (38% increase, ANG II 6.73 ± 0.42 mg/g vs. saline 4.88 ± 0.27 mg/g, P = 0.005, Fig. 2A). In Tlr4del mice, ANG II caused a lesser increase in HW/BW ratio than in WT when compared with the saline-infused controls (15% increase, ANG II 5.38 ± 0.19 mg/g vs. saline 4.61 ± 0.17 mg/g, P = 0.012). In Tlr3−/− mice, the HW/BW with ANG II infusion was not significantly different from that with saline infusions (ANG II 4.99 ± 0.15 mg/g vs. saline 4.70 ± 0.13 mg/g, P = 0.17).
Fig. 2.

A: cardiac hypertrophy measured as heart weight/ body weight (HW/BW) ratios after 3 wk of either saline or angiotensin II (ANG II) infusion in wild-type (WT; n = 6 for saline and n = 8 for ANG II), Tlr4del (n = 9 for saline and n = 6 for ANG II), and Tlr3−/−mice (n = 6 in each group). HW/BW ratio was significantly greater in ANG II-infused vs. saline-infused WT mice (38% increase), but the increase in ANG II-infused vs. saline-infused Tlr4del mice was significantly less than in WT (15% increase, P = 0.004). There was no significant difference in the HW/BW ratio between saline- and ANG II-infused Tlr3−/− mice at the end of the infusion period with ANOVA and post hoc Tukey’s multiple comparison test for ANG II-infused HW/BW ratios. *P < 0.05. B: graph representing cross-sectional areas of left ventricular cardiomyocytes from WT (saline 197.3 ± 1.9 µm2, n = 417 cells; ANG II 269.7 ± 13.5 µm2, n = 537 cells), Tlr4del (saline 186.0 ± 6.4 µm2, n = 260 cells; ANG II 178.6 ± 10.4 µm2, n = 287), and Tlr3−/− (saline 202.8 ± 12.3 µm2, n = 225; ANG II 207.5 ± 19.3 µm2, n = 311). Cross sections obtained from three mice in each group at the end of 3-wk infusions of saline or ANG II were stained with hematoxylin-eosin. Compared with saline infusion, ANG II infusion caused significant increases in cross-sectional area of cardiac myocytes in WT hearts but not in Tlr4del or Tlr3−/− hearts. (*P < 0.05 for unpaired t-tests; n = 3 mice each group). C: thrombi were seen histologically in hearts from ANG II-infused WT mice (four hearts examined) but not in the Tlr4del or Tlr3−/− hearts (three hearts of each strain examined). Thrombi were associated with inflammation that is marked by arrows showing infiltration of leukocytes (purple nuclei) extending into adjacent cardiac walls. Hematoxylin-eosin stains, magnification ×100. n.s., not significant; RV, right ventricle.
We further assessed the effect of TLR4 and TLR3 deficiency on cardiac hypertrophy by measuring the size of cardiomyocytes. The cross-sectional area of cardiac myocytes with ANG II versus saline infusions increased significantly in WT (ANG II 269.7 ± 13.5 vs. saline 197.3 ± 1.9 µm2, P = 0.006) but did not change in Tlr4del mice (ANG II 178.6 ± 10.4 vs. saline 186.0 ± 6.5 µm2, P = 0.57) or in Tlr3−/− mice (ANG II 207.5 ± 19.3 vs. saline 202.8 ± 12.3 µm2, P = 0.84; Fig. 2B). These results indicate that ANG II-induced cardiac myocyte hypertrophy and inflammation seen in WT are abrogated in Tlr3−/− mice as well as in Tlr4del mice despite a significant pressor response in the latter.
In addition, examination of histological slides of ANG II-infused hearts revealed the presence of thrombi with infiltration of inflammatory leukocytes with basophilic nuclei that extended into the adjacent myocardium in two of four WT hearts examined. These were absent from hearts of three Tlr4del and three Tlr3−/− mice (Fig. 2C).
Cardiac and renal proinflammatory gene expression with ANG II are dependent on TLR3 and TLR4.
Since TLRs are a major class of receptors in the innate immune system that play a prominent role in inflammatory signaling and since hypertension has a major immunological component, we measured the mRNA expression of a set of proinflammatory genes in the hearts and kidneys of WT, Tlr4del, and Tlr3−/− mice infused with either saline or ANG II for 3 wk (Figs. 3 and 4). These selected genes represent inflammatory cytokines, oxidative stress genes, and tissue remodeling genes that are known to be induced during hypertension and by ANG II infusion (7, 9, 15, 19, 36, 38, 53, 60, 66). Compared with saline-infused WT mice, the hearts from ANG II-infused WT mice had increased expression of atrial natriuretic peptide (Nppa, 3.2-fold), tumor necrosis factor-alpha (Tnf, 1.4-fold), NADPH-oxidase 4 (Nox4, threefold), matrix metalloprotease 9 (Mmp9, 1.7-fold), interleukin-6 (Il6, 7.6-fold), and interleukin-beta 1 (Il1b, 1.8-fold). These hearts from ANG II-infused WT mice also had significantly increased cardiomyocyte cross-sectional area. In contrast, the increase in expression of most of these genes in Tlr4del and Tlr3−/− mice, which did not show increased myocyte cross-sectional area or hypertrophy with ANG II infusion, was either attenuated or abrogated, with the exception of Il1b expression that had a greater increase in ANG II-infused Tlr4del hearts (1.8-fold in WT vs. 2.4-fold in Tlr4del, Fig. 3). In Tlr3−/− mice, the attenuation and abrogation of increases in gene expression were even more pronounced than in Tlr4del (Nppa, Nox4, and Il6). Moreover, cardiac Il1b expression was significantly repressed (2.2-fold down) in ANG II-infused Tlr3−/− mice. Clearly, the ANG II-mediated expression of cardiac proinflammatory genes and the associated cardiac hypertrophy are dependent on TLR3 and to a large extent also on TLR4.
Fig. 3.

Real-time PCR comparisons of changes in inflammation-related cardiac gene expression induced by angiotensin II (ANG II) infusion in wild-type (WT), Tlr4del, and Tlr3−/−mice. RNA expression was measured by ΔΔCt method using Gapdh RNA as a reference. Values displayed represent ANG II-induced changes in expression relative to values in saline-infused mice. Increases with ANG II seen in all cardiac genes in WT were lesser in Tlr4del except for Il1b expression that was greater. All gene expressions were more uniformly attenuated in Tlr3−/− and particularly Il1b that was actually decreased. *Significant changes (P ≤ 0.05; n.s., not significant) by unpaired t-tests (n = 3 samples in each group). Il1b, interleukin-beta 1 (P–R); Il6, interleukin-6 (M–O); Mmp9, matrix metalloprotease 9 (J–L); Nppa, atrial natriuretic peptide (A–C); Nox4, NADPH-oxidase 4 (D–F); Tnf, tumor necrosis factor-alpha (G–I).
Fig. 4.

A: real-time PCR comparison of angiotensin II (ANG II)-induced proinflammatory gene expression in kidneys of wild-type (WT), Tlr4del, and Tlr3−/− mice after 3-wk infusion of ANG II. Relative change in RNA expression was determined by ΔΔCt method using Gapdh RNA expression as a reference. Values of ANG II-induced RNA expression relative to their expression in kidneys from saline-infused mice were compared by t-test (n ≥ 3 samples in each group, *P < 0.05 by t-tests; n.s., not significant). Significant increases with ANG II infusion were seen in the expression of Il6 in WT and of both Il6 and Tgfb2 in Tlr4del. In contrast, there were decreases in Nox4 and Tgfb2 expression in Tlr3−/−. These changes would coincide with the loss of pressor response in Tlr3−/−. B: immunoblot of NOX4 and GAPDH proteins in kidneys of saline- or ANG II-infused WT, Tlr4del, and Tlr3−/− mice. Equal amounts of total protein (30 µg per lane) were resolved on a bis-tris gel and immunodetected using an anti-NOX4 or anti-GAPDH antibody and enhanced chemiluminescence. Lanes 1 and 2 (WT saline), lanes 3 and 4 (WT ANG II), lanes 5 and 6 (Tlr4del saline), lanes 7 and 8 (Tlr4del ANG II), lanes 9 and 10 (Tlr3−/− saline), and lanes 11 and 12 (Tlr3−/− ANG II) show samples from individual mice. Band intensities were measured within the linear dynamic range of signals by Gel Imager (Bio-Rad). A representative image of an X-ray film exposure is shown here. NOX4 band intensities were quantified and normalized to the intensities of GAPDH band in the corresponding samples. C: graph showing comparison of NOX4 protein between the kidneys of saline- and ANG II-infused WT, Tlr4del, and Tlr3−/− mice. Mean values of the ratios of NOX4 to GAPDH band intensities from kidneys of saline- or ANG II-infused mice are presented. Changes are compared from saline-infused samples of the same genotype. Increase in NOX4 in samples of ANG II-infused WT mice was significantly greater than in ANG II-infused Tlr4del or Tlr3−/− (unpaired t-test). There was no significant difference between NOX4 protein from ANG II-infused Tlr4del and Tlr3−/−. *Statistically significant difference (P ≤ 0.05; n.s., not significant). Il1b, interleukin-beta 1; Il6, interleukin-6; Nox4, NADPH-oxidase 4; Tgfb2, transforming growth factor beta 2.
We also measured mRNA expression of Il6, Il1b, Nox4, and transforming growth factor beta 2 (Tgfb2) in kidneys from these mice (Fig. 4A). In the WT mice, only the expression of Il6 mRNA was significantly increased by ANG II compared with saline (1.6-fold). In Tlr4del mice that exhibited an exaggerated pressor response to ANG II, there were significantly greater increases in renal expression of Il6 (2.8-fold) and Tgfb2 (1.2-fold). In Tlr3−/−, the absence of any pressor response to ANG II (Fig. 1A) was associated with a significant reduction in renal expression of both Nox4 and Tgfb2 compared with the saline responses (Fig. 4).
Although immunoblotting attempts to measure protein expression of cytokines to complement mRNA expressions were not successful because of the very limited sensitivity of antibodies, an immunoblot analysis of NOX4 protein in kidney lysates of saline- and ANG II-infused mice was possible. The results confirmed an increase in protein expression in WT mice with ANG II that was not seen in Tlr4del or Tlr3−/− mice (Fig. 4, B and C). Thus, the changes in gene expression induced by ANG II in heart and kidney parallel the effect of ANG II on cardiac hypertrophy and hypertension, respectively.
Cardiac and renal expression of TLR3, TLR4, and ANG II type 1a receptor.
We investigated the baseline mRNA expression of Tlr3, Tlr4, and ANG II type 1a receptor (Agtr1a) in tissues known to be involved in ANG II-induced hypertension and cardiac hypertrophy. In WT mice, the expressions of Tlr3, Tlr4, and Agtr1a in spleen, heart, kidney, aorta, and brain of WT mice were variable. Compared with spleen, the expression of Tlr3 and Tlr4 in these tissues was significantly less than 50%. Yet these levels were sufficient and essential to mediate the responses to ANG II. In contrast, the expression of Agtr1a in heart and kidney was almost double the splenic expression and significantly higher than in aorta and brain (Fig. 5).
Fig. 5.

Expression of mRNA of Tlr3, Tlr4, and Agtr1a in organs of wild-type (WT) mice relative to the spleen. Relative expression in heart, kidney, aorta, and brain were compared with expression in spleen by ΔΔCt method using Gapdh RNA expression as reference (n = 3 in each group). RNA expression in different tissues was analyzed by one-way ANOVA and Tukey’s multiple comparisons test. *Significant differences from spleen (P ≤ 0.05). Agtr1a, angiotensin II receptor type 1a; Tlr3, Toll-like receptor 3; Tlr4, Toll-like receptor 4.
Moreover, compared with the WT, the expression of Agtr1a mRNA in the heart of Tlr4del was the same, but the expression in the hearts of Tlr3−/− was markedly enhanced by 10-fold (Fig. 6). Despite this increase, the ANG II-mediated cardiac hypertrophy was abrogated in both Tlr4del and Tlr3−/−.
Fig. 6.
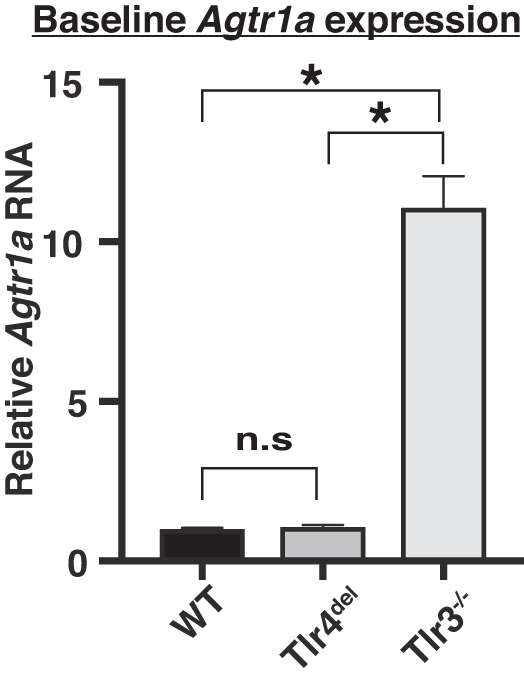
Comparison of baseline expression of Agtr1a in the hearts of wild-type (WT), Tlr4del, and Tlr3−/− mice. Expression of Agtr1a in the hearts of WT and Tlr4del was similar but markedly increased in the hearts of Tlr3−/− mice (n = 3 in each group, *P ≤ 0.05, one-way ANOVA and Tukey’s post hoc test; n.s., not significant). Agtr1a, angiotensin II receptor type 1a.
ANG II infusion for 3 wk did not increase the expression of either Tlr3 or Tlr4 in the hearts and kidneys of WT mice except for an increase in cardiac Tlr4 (1.4-fold increase, Fig. 7). Tlr4 expression was not increased by ANG II infusion in the hearts or kidneys of Tlr3−/− mice (Fig. 7). Tlr3 expression was also unchanged in ANG II-infused heart and kidneys of Tlr4del mice.
Fig. 7.
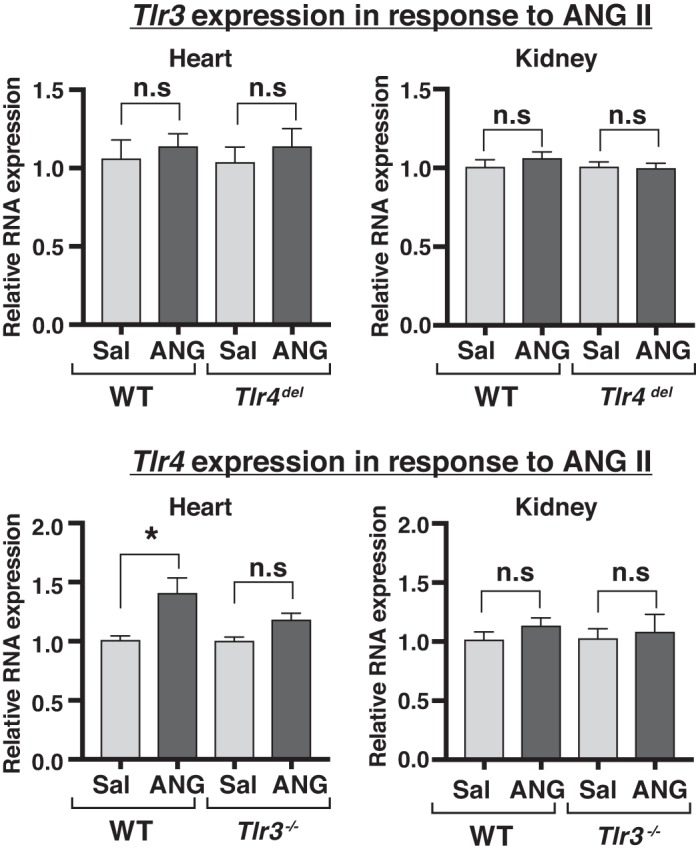
Effect of angiotensin II (ANG II) infusion on cardiac and renal expression of Tlr3 and Tlr4. ANG II infusion for 3 wk did not alter the expression of Tlr3 or Tlr4 in hearts or kidneys of Tlr4del or Tlr3−/− mice, respectively. Only hearts from ANG II-infused wild-type (WT) mice showed significant increase in Tlr4 expression. (n = 3 each group, *P < 0.05). n.s., not significant; sal, saline; Tlr3, Toll-like receptor 3; Tlr4, Toll-like receptor 4.
Thus, Tlr3, Tlr4, and Agtr1a are coexpressed in the tissues tested, and the failure of ANG II to increase blood pressure or cardiac hypertrophy in Tlr4del or Tlr3−/− mice was not due to the intrinsic variability in expression of Tlr3 and Tlr4 or the reduced expression of Agtr1a.
DISCUSSION
The immune system plays a dominant role in the initiation and maintenance of hypertension. We have previously shown that the TRIF adaptor protein, which mediates responses to both TLR3 and TLR4, is required for ANG II-induced hypertension, whereas the canonical proinflammatory MyD88 adaptor protein, which mediates TLR4 but not TLR3 signaling, is paradoxically antihypertensive in mice (52). Here, we report that 1) the TLR3-TRIF pathway is responsible for ANG II-induced hypertension, 2) deletion of TLR4 enhances the early pressor response to ANG II, likely through the lack of the MyD88-mediated suppressive influence, which we had reported earlier, and 3) both TLR3 and TLR4 contribute to cardiac hypertrophy. The increases in SBP and in cardiac hypertrophy are independently regulated in ANG II hypertension. This is most evident in Tlr4del mice, in which the pressor response to ANG II is enhanced whereas the myocyte cross-sectional area is unchanged. We define clear and different roles of the two TLRs and their adaptor proteins in ANG II hypertension.
Differential roles of TLR3 and TLR4 in TRIF-mediated ANG II-induced hypertension.
ANG II infusion in the WT mice led to hypertension, whereas ANG II infusion in Tlr3−/− mice did not raise blood pressure significantly more than the saline infusion. On the other hand, Tlr4del mice showed exaggerated early increases in SBP with ANG II infusion. These results suggest that the TLR3-mediated TRIF pathway, but not the TLR4-TRIF pathway, is necessary for sustained ANG II-induced hypertension.
Although the baseline SBP of the Tlr3−/− mice was lower than the Tlr4del, this difference was not maintained over the period of 3-wk infusion of saline during which the arterial pressure levels were not statistically different and could not possibly account for the enhanced pressor response to ANG II in Tlr4del and its absence in Tlr3−/−.
Moreover, the early enhanced hypertensive response during the initial phase of ANG II infusion in the Tlr4del mice compared with the WT mice also suggests that TLR4 activation partly restrains the hypertensive response to ANG II through activation of MyD88 as mentioned above. This finding is consistent with our previous results (52).
Proinflammatory gene expression.
In the heart, hypertrophic signaling often involves overexpression of fetal and inflammatory genes (53, 63). We observed the formation of thrombi in ANG II-infused WT hearts that were not seen in Tlr4del or Tlr3−/− mice (Fig. 2C). Aggregates of dense cellular leukocytic infiltrates were located along the periphery of each thrombus and at sites of thrombus attachment, extended into the cardiac wall. The presence and localization of cellular infiltrates are consistent with the expected inflammatory response to a thrombus. ANG II-infused WT mice also have increased expression of Nppa, a prognostic marker for outcome of inflammatory responses in cardiovascular diseases, including experimental and human hypertension (2, 7, 10). Expression of TNF-α also has been shown to increase in numerous studies during inflammatory processes including ANG II-induced hypertension (13, 20, 36, 56, 64). NOX4 activity has a distinct role in basal and ANG II-stimulated superoxide and hydrogen peroxide production (15) and hypertension (5, 38, 54). MMP9 plays a key role in tissue remodeling, and its expression increases with ANG II infusion (46, 57, 60). Similarly, increased expression of IL-6 is related to cardiac hypertrophy, and ANG II-induced hypertension and cardiac hypertrophy are attenuated in IL-6 knockout mice (9, 32). The expression of these proinflammatory genes, which are known to sustain a cardiac inflammatory hypertrophic response (10, 19, 47, 49, 53, 55, 66), was consistently attenuated in both Tlr4del and Tlr3−/− mice that did not show cardiac hypertrophy. In addition, increased Tlr4 expression in the hearts of ANG II-infused WT but not in Tlr3−/− mice (Fig. 7) suggests a possible role of Tlr3 in ANG II-mediated Tlr4 gene regulation. We also observed that despite a marked elevation of Agtr1a mRNA expression in Tlr3−/− hearts (Fig. 6), ANG II-induced cardiac hypertrophy was attenuated. These results suggest that TLR3 might be operative downstream from angiotensin II receptor type 1 (AT1R), and its deficiency disrupts the cascade of molecular events leading to hypertension and cardiac hypertrophy.
Renal Nox4 generates reactive oxygen species with ANG II infusion, which is blocked by AT1-receptor blockers (54). Thus, a significant reduction in renal Nox4 expression observed in ANG II-infused Tlr3−/− mice that had no increase in blood pressure upon ANG II infusion would be consistent with these reports.
Similarly, Tgfb expression, responsible for glomerular fibrosis (4, 16, 17), was induced, along with Il6, by ANG II in the kidneys of Tlr4del mice, which also had enhanced pressor response. However, Tgfb2 expression was significantly reduced in kidneys of ANG II-infused Tlr3−/− mice.
We observed increased Il1b expression in ANG II-infused Tlr3−/− kidneys. Although deficiency of IL-1 receptor that binds to both IL-1α and IL-1β results in lower blood pressure in ANG II-infused mice (65), the role of IL-1β in hypertension is not well defined (29). Lower IL-1β levels are associated with impaired renal function (45). Interestingly, IL-1β receptors signal through MyD88, the ubiquitous adaptor protein, which, as we have recently reported, restrains ANG II-induced hypertension (52).
Together, these results suggest that the inflammatory signaling from ANG II activation of TLR3 involves cardiac and renal gene expression to promote hypertrophy and hypertension.
Differential response to TRIF activation by TLR3 and TLR4.
Our results also reveal nonredundant signaling by different TLRs (TLR3 and TLR4) through the same (TRIF) adaptor protein. How does the same adaptor protein, TRIF, propagate different signaling outcomes when engaged to either TLR3 or TLR4? TLR3 and TLR4 interact with TRIF adaptor differently (41). Whereas TRIF directly associates with the TLR3 C-terminal, its association with TLR4 is mediated by another adaptor protein, TRIF-related adaptor molecule (TRAM) (61). These differences in molecular organization could result in differential effects as summarized in Fig. 8. One such effect is observed during maturation of DCs, in which TRIF signaling by TLR3 requires expression of interferon-α (IFN-α), whereas TLR4-TRIF signaling is sufficient without the requirement of IFN-α for DC maturation (23). Moreover, in autoimmune diabetes, the MyD88- and TRIF-mediated pathways also have different effects, and the TRIF-dependent responses antagonize pathogenic MyD88 signaling in that disease state (3).
Fig. 8.

Schematic of proposed model of differential effects of angiotensin II (ANG II) on hypertension and cardiac hypertrophy. TLR3 mediated TRIF activation is required for ANG II hypertension, whereas TLR-TRIF is not. This is likely due to intrinsic differences between TLR4-TRIF and TLR3-TRIF interactions. The former requires TRAM as adaptor protein to activate TRIF, whereas TLR3 directly interacts with TRIF. Moreover, TLR4 activates an additional adaptor protein, MyD88, which in contrast to TRIF, exerts a restraint on ANG II hypertension. In contrast, ANG II-induced cardiac hypertrophy is dependent on the TRIF-mediated pathway by both TLR4 and TLR3. DAMP, damage-associated molecular patterns; MyD88, myeloid differentiation protein 88; TLR, Toll-like receptor; TRAM, TRIF-related adaptor molecule; TRIF, Toll-interleukin receptor domain-containing adaptor protein-inducing interferon-β.
Shared role of TLR3- and TLR4-mediated TRIF signaling in ANG II-induced cardiac hypertrophy.
Unlike the ANG II-induced hypertension, ANG II-induced cardiomyocyte hypertrophy was suppressed in Tlr4del mice despite an enhanced pressor response and was also abrogated in Tlr3−/− as the pressor response was suppressed. These results highlight two aspects of cardiac hypertrophy: 1) a component of cardiac hypertrophy is likely independent of the mechanical afterload of the increased blood pressure in ANG II hypertension, and 2) ANG II has differential effects on hypertension and cardiac hypertrophy through activation of both TLR3 and TLR4. A similar effect of increased blood pressure in the absence of cardiac hypertrophy in TLR4-deficient mice has been reported by Matsuda et al. (33). The mechanism of AT1R-mediated induction of TLR signaling pathways is not known. However, it is likely to arise from coexpression of the AT1R, TLR3, and TLR4 in the same tissue, which might determine their respective contribution in the pathology of the organ. We found that both TLR3 and TLR4, and AT1R, are coexpressed in the heart and kidney. Thus, both TLR3 and TLR4 pathways can be engaged in these tissues depending on the ligand generated by ANG II action.
Interaction between AT1R and TLR.
Do TLRs and AT1R interact? ANG II and TLR ligands can reciprocally enhance downstream effects. We have reported that ANG II can significantly increase the release of cytokines by spontaneously hypertensive rat splenocytes, but not Wistar-Kyoto rat splenocytes, upon activation of specific TLR ligands (22). TLR4 activation by pertussis toxin has been shown to enhance the AT1R -mediated response in cardiac fibroblasts by increasing AT1R density on the cell surface (40). In a study by Sunagawa and colleagues (42), intracerebroventricular infusion of exogenous ANG II, which induces an increase in blood pressure, did not increase TLR4 expression but increased the expression of its downstream adaptor MyD88. In another report, ANG II-induced hypertension and cardiac hypertrophy were reduced by intracerebroventricular delivery of TLR4 inhibitor peptide (13). Our results show that cardiac Tlr4 expression was increased in ANG II-infused WT mice but not in Tlr3−/− mice, suggesting that TLR3 might influence the expression of TLR4 in this model. Although in myocardial infarction or aortic banding-induced cardiac hypertrophy the roles of the universal TLR adaptor protein MyD88 (53) and TLR4 are well established (25), a direct interaction between AT1R and TLR is yet to be demonstrated. Additionally, a role of endosomal TLR9 signaling in hypertension-associated vascular dysfunction has been described (35). Thus, it is possible that endosomal TLR3 also plays a similar vascular role in ANG II hypertension. In this regard, one may question whether direct activation of TLR3 with its exogenous ligand is sufficient to induce the hypertensive response. In our hands, mere intraperitoneal injection of a double-stranded RNA analog (poly-IC), a known ligand for TLR3, was not sufficient to increase blood pressure in WT mice (M. V. Singh and F. M. Abboud, unpublished observation). This suggests that although TLR3 is required for ANG II hypertension and cardiac hypertrophy, TLR3 activation by itself may not be sufficient to induce hypertension; additional molecular events initiated by ANG II and the activation of AT1R may also be required. As the endogenous agonists of TLR3 are less characterized, further studies will be required to define the TLR ligands generated by ANG II that lead to multi-organ damage.
Limitations of tail-cuff plethysmography.
It is important to note that our studies with tail-cuff plethysmography are inherently limited by lack of beat-by-beat pressure measurements, diurnal changes in blood pressure, and animal activity levels for which radiotelemetry is required. However, valid physiological and pathological experiments requiring long-term measurements of blood pressure have been performed using both methods. Indeed, several studies have shown a good correspondence between tail-cuff plethysmography and radiotelemetry in the chronic measurements of arterial pressure (18, 21, 28). We do not believe that the tail-cuff method interfered with our recording of increased pressure during the first 9 days in ANG II-infused WT because in ANG II-infused Tlr4del mice there was significant increase in the tail-cuff pressure during the same period, and this increase was sustained thereafter. Also, in our previous study the WT mouse strain did not increase tail-cuff pressure during the first week of similar dose of ANG II infusion, whereas MyD88-deficient mice had an enhanced pressor response during the first week (52).
In summary, we have shown that ANG II-induced hypertension and cardiac hypertrophy involve differential activation of two Toll-like receptors, TLR4 and TLR3. TLR3 activates only the TRIF-mediated pathway, which is the dominant determinant of ANG II hypertension and hypertrophy. TLR4, on the other hand, activates both MyD88- and TRIF-mediated pathways. The latter contributes to hypertrophy, whereas MyD88 restrains the proinflammatory hypertensive effect of ANG II.
GRANTS
This study was supported by National Heart, Lung, Blood, Institute Program Project Grant HL-14388 (to F. M. Abboud), Veterans Affairs Merit Review Award 1BX001414 (to M. W. Chapleau), and American Heart Association Innovative Research Grant 16IRG27260323 (to M. V. Singh).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
M.V.S. conceived and designed research; M.V.S. and M.Z.C. performed experiments; M.V.S., S.N., D.K.M., and F.M.A. analyzed data; M.V.S., D.K.M., M.W.C., and F.M.A. interpreted results of experiments; M.V.S., S.N., and D.K.M. prepared figures; M.V.S. and F.M.A. drafted manuscript; M.V.S., M.W.C., and F.M.A. edited and revised manuscript; M.V.S., M.Z.C., S.N., D.K.M., M.W.C., and F.M.A. approved final version of manuscript.
REFERENCES
- 1.Alexopoulou L, Holt AC, Medzhitov R, Flavell RA. Recognition of double-stranded RNA and activation of NF-kappaB by Toll-like receptor 3. Nature 413: 732–738, 2001. doi: 10.1038/35099560. [DOI] [PubMed] [Google Scholar]
- 2.An MR, Chung YJ, Kang DG, Nam SC, Lee J. Augmented expression of cardiac atrial natriuretic peptide system in hypertensive rats. J Korean Med Sci 14: 497–501, 1999. doi: 10.3346/jkms.1999.14.5.497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Androulidaki A, Wachsmuth L, Polykratis A, Pasparakis M. Differential role of MyD88 and TRIF signaling in myeloid cells in the pathogenesis of autoimmune diabetes. PLoS One 13: e0194048, 2018. doi: 10.1371/journal.pone.0194048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Border WA, Noble NA. Interactions of transforming growth factor-beta and angiotensin II in renal fibrosis. Hypertension 31: 181–188, 1998. doi: 10.1161/01.HYP.31.1.181. [DOI] [PubMed] [Google Scholar]
- 5.Bouabout G, Ayme-Dietrich E, Jacob H, Champy MF, Birling MC, Pavlovic G, Madeira L, Fertak LE, Petit-Demoulière B, Sorg T, Herault Y, Mudgett J, Monassier L. Nox4 genetic inhibition in experimental hypertension and metabolic syndrome. Arch Cardiovasc Dis 111: 41–52, 2018. doi: 10.1016/j.acvd.2017.03.011. [DOI] [PubMed] [Google Scholar]
- 6.Caillon A, Schiffrin EL. Role of inflammation and immunity in hypertension: recent epidemiological, laboratory, and clinical evidence. Curr Hypertens Rep 18: 21, 2016. doi: 10.1007/s11906-016-0628-7. [DOI] [PubMed] [Google Scholar]
- 7.Cantin M, Garcia R, Thibault G, Kuchel O, Gutkowska J, Larochelle P, Hamet P, Schiffrin EL, Genest J. Atrial natriuretic factor (ANF) in experimental and human hypertension. Clin Invest Med 10: 561–567, 1987. [PubMed] [Google Scholar]
- 8.Case AJ, Zimmerman MC. Sympathetic-mediated activation versus suppression of the immune system: consequences for hypertension. J Physiol 594: 527–536, 2016. doi: 10.1113/JP271516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen F, Chen D, Zhang Y, Jin L, Zhang H, Wan M, Pan T, Wang X, Su Y, Xu Y, Ye J. Interleukin-6 deficiency attenuates angiotensin II-induced cardiac pathogenesis with increased myocyte hypertrophy. Biochem Biophys Res Commun 494: 534–541, 2017. doi: 10.1016/j.bbrc.2017.10.119. [DOI] [PubMed] [Google Scholar]
- 10.Chen W, Spitzl A, Mathes D, Nikolaev VO, Werner F, Weirather J, Špiranec K, Röck K, Fischer JW, Kämmerer U, Stegner D, Baba HA, Hofmann U, Frantz S, Kuhn M. Endothelial actions of ANP enhance myocardial inflammatory infiltration in the early phase after acute infarction. Circ Res 119: 237–248, 2016. doi: 10.1161/CIRCRESAHA.115.307196. [DOI] [PubMed] [Google Scholar]
- 11.Cornelius DC, Hogg JP, Scott J, Wallace K, Herse F, Moseley J, Wallukat G, Dechend R, LaMarca B. Administration of interleukin-17 soluble receptor C suppresses TH17 cells, oxidative stress, and hypertension in response to placental ischemia during pregnancy. Hypertension 62: 1068–1073, 2013. doi: 10.1161/HYPERTENSIONAHA.113.01514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Courtney AH, Lo WL, Weiss A. TCR signaling: mechanisms of initiation and propagation. Trends Biochem Sci 43: 108–123, 2018. doi: 10.1016/j.tibs.2017.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dange RB, Agarwal D, Masson GS, Vila J, Wilson B, Nair A, Francis J. Central blockade of TLR4 improves cardiac function and attenuates myocardial inflammation in angiotensin II-induced hypertension. Cardiovasc Res 103: 17–27, 2014. doi: 10.1093/cvr/cvu067. [DOI] [PubMed] [Google Scholar]
- 14.Dange RB, Agarwal D, Teruyama R, Francis J. Toll-like receptor 4 inhibition within the paraventricular nucleus attenuates blood pressure and inflammatory response in a genetic model of hypertension. J Neuroinflammation 12: 31, 2015. doi: 10.1186/s12974-015-0242-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dikalov SI, Dikalova AE, Bikineyeva AT, Schmidt HH, Harrison DG, Griendling KK. Distinct roles of Nox1 and Nox4 in basal and angiotensin II-stimulated superoxide and hydrogen peroxide production. Free Radic Biol Med 45: 1340–1351, 2008. doi: 10.1016/j.freeradbiomed.2008.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fakhouri F, Placier S, Ardaillou R, Dussaule JC, Chatziantoniou C. Angiotensin II activates collagen type I gene in the renal cortex and aorta of transgenic mice through interaction with endothelin and TGF-beta. J Am Soc Nephrol 12: 2701–2710, 2001. [DOI] [PubMed] [Google Scholar]
- 17.Feng W, Chumley P, Hua P, Rezonzew G, Jaimes D, Duckworth MW, Xing D, Jaimes EA. Role of the transcription factor erythroblastosis virus E26 oncogen homolog-1 (ETS-1) as mediator of the renal proinflammatory and profibrotic effects of angiotensin II. Hypertension 60: 1226–1233, 2012. doi: 10.1161/HYPERTENSIONAHA.112.197871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fraser TB, Turner SW, Mangos GJ, Ludbrook J, Whitworth JA. Comparison of telemetric and tail-cuff blood pressure monitoring in adrenocorticotrophic hormone-treated rats. Clin Exp Pharmacol Physiol 28: 831–835, 2001. doi: 10.1046/j.1440-1681.2001.03531.x. [DOI] [PubMed] [Google Scholar]
- 19.Goruppi S, Patten RD, Force T, Kyriakis JM. Helix-loop-helix protein p8, a transcriptional regulator required for cardiomyocyte hypertrophy and cardiac fibroblast matrix metalloprotease induction. Mol Cell Biol 27: 993–1006, 2007. doi: 10.1128/MCB.00996-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Guzik TJ, Hoch NE, Brown KA, McCann LA, Rahman A, Dikalov S, Goronzy J, Weyand C, Harrison DG. Role of the T cell in the genesis of angiotensin II induced hypertension and vascular dysfunction. J Exp Med 204: 2449–2460, 2007. doi: 10.1084/jem.20070657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Haggerty CM, Mattingly AC, Gong MC, Su W, Daugherty A, Fornwalt BK. Telemetric blood pressure assessment in angiotensin ii-infused ApoE-/- mice: 28 day natural history and comparison to tail-cuff measurements. PLoS One 10: e0130723, 2015. doi: 10.1371/journal.pone.0130723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Harwani SC, Chapleau MW, Legge KL, Ballas ZK, Abboud FM. Neurohormonal modulation of the innate immune system is proinflammatory in the prehypertensive spontaneously hypertensive rat, a genetic model of essential hypertension. Circ Res 111: 1190–1197, 2012. doi: 10.1161/CIRCRESAHA.112.277475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hu W, Jain A, Gao Y, Dozmorov IM, Mandraju R, Wakeland EK, Pasare C. Differential outcome of TRIF-mediated signaling in TLR4 and TLR3 induced DC maturation. Proc Natl Acad Sci USA 112: 13994–13999, 2015. doi: 10.1073/pnas.1510760112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jain A, Pasare C. Innate control of adaptive immunity: beyond the three-signal paradigm. J Immunol 198: 3791–3800, 2017. doi: 10.4049/jimmunol.1602000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jiang DS, Zhang XF, Gao L, Zong J, Zhou H, Liu Y, Zhang Y, Bian ZY, Zhu LH, Fan GC, Zhang XD, Li H. Signal regulatory protein-α protects against cardiac hypertrophy via the disruption of toll-like receptor 4 signaling. Hypertension 63: 96–104, 2014. doi: 10.1161/HYPERTENSIONAHA.113.01506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ju X, Ijaz T, Sun H, Ray S, Lejeune W, Lee C, Recinos A III, Guo D-CC, Milewicz DM, Tilton RG, Brasier AR. Interleukin-6-signal transducer and activator of transcription-3 signaling mediates aortic dissections induced by angiotensin II via the T-helper lymphocyte 17-interleukin 17 axis in C57BL/6 mice. Arterioscler Thromb Vasc Biol 33: 1612–1621, 2013. doi: 10.1161/ATVBAHA.112.301049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Justin Rucker A, Crowley SD. The role of macrophages in hypertension and its complications. Pflugers Arch 469: 419–430, 2017. doi: 10.1007/s00424-017-1950-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Krege JH, Hodgin JB, Hagaman JR, Smithies O. A noninvasive computerized tail-cuff system for measuring blood pressure in mice. Hypertension 25: 1111–1115, 1995. doi: 10.1161/01.HYP.25.5.1111. [DOI] [PubMed] [Google Scholar]
- 29.Krishnan SM, Sobey CG, Latz E, Mansell A, Drummond GR. IL-1β and IL-18: inflammatory markers or mediators of hypertension? Br J Pharmacol 171: 5589–5602, 2014. doi: 10.1111/bph.12876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kurdi M, Booz GW. New take on the role of angiotensin II in cardiac hypertrophy and fibrosis. Hypertension 57: 1034–1038, 2011. doi: 10.1161/HYPERTENSIONAHA.111.172700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Laresgoiti-Servitje E. A leading role for the immune system in the pathophysiology of preeclampsia. J Leukoc Biol 94: 247–257, 2013. doi: 10.1189/jlb.1112603. [DOI] [PubMed] [Google Scholar]
- 32.Lee DL, Sturgis LC, Labazi H, Osborne JB Jr, Fleming C, Pollock JS, Manhiani M, Imig JD, Brands MW. Angiotensin II hypertension is attenuated in interleukin-6 knockout mice. Am J Physiol Heart Circ Physiol 290: H935–H940, 2006. doi: 10.1152/ajpheart.00708.2005. [DOI] [PubMed] [Google Scholar]
- 33.Matsuda S, Umemoto S, Yoshimura K, Itoh S, Murata T, Fukai T, Matsuzaki M. Angiotensin II activates MCP-1 and induces cardiac hypertrophy and dysfunction via Toll-like receptor 4. J Atheroscler Thromb 22: 833–844, 2015. doi: 10.5551/jat.27292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.McCarthy CG, Wenceslau CF, Goulopoulou S, Baban B, Matsumoto T, Webb RC. Chloroquine suppresses the development of hypertension in spontaneously hypertensive rats. Am J Hypertens 30: 173–181, 2017. doi: 10.1093/ajh/hpw113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.McCarthy CG, Wenceslau CF, Goulopoulou S, Ogbi S, Baban B, Sullivan JC, Matsumoto T, Webb RC. Circulating mitochondrial DNA and Toll-like receptor 9 are associated with vascular dysfunction in spontaneously hypertensive rats. Cardiovasc Res 107: 119–130, 2015. doi: 10.1093/cvr/cvv137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mehaffey E, Majid DS. Tumor necrosis factor-α, kidney function, and hypertension. Am J Physiol Renal Physiol 313: F1005–F1008, 2017. doi: 10.1152/ajprenal.00535.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Meng X, Yang J, Dong M, Zhang K, Tu E, Gao Q, Chen W, Zhang C, Zhang Y. Regulatory T cells in cardiovascular diseases. Nat Rev Cardiol 13: 167–179, 2016. doi: 10.1038/nrcardio.2015.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Montezano AC, Touyz RM. Reactive oxygen species, vascular Noxs, and hypertension: focus on translational and clinical research. Antioxid Redox Signal 20: 164–182, 2014. doi: 10.1089/ars.2013.5302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Muñoz-Durango N, Barake MF, Letelier NA, Campino C, Fardella CE, Kalergis AM. Immune system alterations by aldosterone during hypertension: from clinical observations to genomic and non-genomic mechanisms leading to vascular damage. Curr Mol Med 13: 1035–1046, 2013. doi: 10.2174/1566524011313060015. [DOI] [PubMed] [Google Scholar]
- 40.Nishida M, Suda R, Nagamatsu Y, Tanabe S, Onohara N, Nakaya M, Kanaho Y, Shibata T, Uchida K, Sumimoto H, Sato Y, Kurose H. Pertussis toxin up-regulates angiotensin type 1 receptors through Toll-like receptor 4-mediated Rac activation. J Biol Chem 285: 15268–15277, 2010. doi: 10.1074/jbc.M109.076232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.O’Neill LA, Bowie AG. The family of five: TIR-domain-containing adaptors in Toll-like receptor signalling. Nat Rev Immunol 7: 353–364, 2007. doi: 10.1038/nri2079. [DOI] [PubMed] [Google Scholar]
- 42.Ogawa K, Hirooka Y, Kishi T, Sunagawa K. Brain AT1 receptor activates the sympathetic nervous system through toll-like receptor 4 in mice with heart failure. J Cardiovasc Pharmacol 58: 543–549, 2011. doi: 10.1097/FJC.0b013e31822e6b40. [DOI] [PubMed] [Google Scholar]
- 43.Pasare C, Medzhitov R. Toll-like receptors: linking innate and adaptive immunity. Microbes Infect 6: 1382–1387, 2004. doi: 10.1016/j.micinf.2004.08.018. [DOI] [PubMed] [Google Scholar]
- 44.Petkov PM, Cassell MA, Sargent EE, Donnelly CJ, Robinson P, Crew V, Asquith S, Haar RV, Wiles MV. Development of a SNP genotyping panel for genetic monitoring of the laboratory mouse. Genomics 83: 902–911, 2004. doi: 10.1016/j.ygeno.2003.11.007. [DOI] [PubMed] [Google Scholar]
- 45.Pruijm M, Ponte B, Vollenweider P, Mooser V, Paccaud F, Waeber G, Marques-Vidal P, Burnier M, Bochud M. Not all inflammatory markers are linked to kidney function: results from a population-based study. Am J Nephrol 35: 288–294, 2012. doi: 10.1159/000335934. [DOI] [PubMed] [Google Scholar]
- 46.Pushpakumar S, Kundu S, Pryor T, Givvimani S, Lederer E, Tyagi SC, Sen U. Angiotensin-II induced hypertension and renovascular remodelling in tissue inhibitor of metalloproteinase 2 knockout mice. J Hypertens 31: 2270–2281, 2013. doi: 10.1097/HJH.0b013e3283649b33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Richards AM, Nicholls MG, Yandle TG, Frampton C, Espiner EA, Turner JG, Buttimore RC, Lainchbury JG, Elliott JM, Ikram H, Crozier IG, Smyth DW. Plasma N-terminal pro-brain natriuretic peptide and adrenomedullin: new neurohormonal predictors of left ventricular function and prognosis after myocardial infarction. Circulation 97: 1921–1929, 1998. doi: 10.1161/01.CIR.97.19.1921. [DOI] [PubMed] [Google Scholar]
- 48.Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, Tinevez JY, White DJ, Hartenstein V, Eliceiri K, Tomancak P, Cardona A. Fiji: an open-source platform for biological-image analysis. Nat Methods 9: 676–682, 2012. doi: 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sergeeva IA, Hooijkaas IB, Ruijter JM, van der Made I, de Groot NE, van de Werken HJ, Creemers EE, Christoffels VM. Identification of a regulatory domain controlling the Nppa-Nppb gene cluster during heart development and stress. Development 143: 2135–2146, 2016. doi: 10.1242/dev.132019. [DOI] [PubMed] [Google Scholar]
- 50.Singh MV, Abboud FM. Toll-like receptors and hypertension. Am J Physiol Regul Integr Comp Physiol 307: R501–R504, 2014. doi: 10.1152/ajpregu.00194.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Singh MV, Chapleau MW, Harwani SC, Abboud FM. The immune system and hypertension. Immunol Res 59: 243–253, 2014. doi: 10.1007/s12026-014-8548-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Singh MV, Cicha MZ, Meyerholz DK, Chapleau MW, Abboud FM. Dual activation of TRIF and MyD88 adaptor proteins by angiotensin II evokes opposing effects on pressure, cardiac hypertrophy, and inflammatory gene expression. Hypertension 66: 647–656, 2015. doi: 10.1161/HYPERTENSIONAHA.115.06011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Singh MV, Swaminathan PD, Luczak ED, Kutschke W, Weiss RM, Anderson ME. MyD88 mediated inflammatory signaling leads to CaMKII oxidation, cardiac hypertrophy and death after myocardial infarction. J Mol Cell Cardiol 52: 1135–1144, 2012. doi: 10.1016/j.yjmcc.2012.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sousa T, Oliveira S, Afonso J, Morato M, Patinha D, Fraga S, Carvalho F, Albino-Teixeira A. Role of H(2)O(2) in hypertension, renin-angiotensin system activation and renal medullary disfunction caused by angiotensin II. Br J Pharmacol 166: 2386–2401, 2012. doi: 10.1111/j.1476-5381.2012.01957.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Squire IB, O’Brien RJ, Demme B, Davies JE, Ng LL. N-terminal pro-atrial natriuretic peptide (N-ANP) and N-terminal pro-B-type natriuretic peptide (N-BNP) in the prediction of death and heart failure in unselected patients following acute myocardial infarction. Clin Sci (Lond) 107: 309–316, 2004. doi: 10.1042/CS20040087. [DOI] [PubMed] [Google Scholar]
- 56.Sriramula S, Haque M, Majid DS, Francis J. Involvement of tumor necrosis factor-alpha in angiotensin II-mediated effects on salt appetite, hypertension, and cardiac hypertrophy. Hypertension 51: 1345–1351, 2008. doi: 10.1161/HYPERTENSIONAHA.107.102152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Umesalma S, Houwen FK, Baumbach GL, Chan SL. Roles of caveolin-1 in angiotensin ii-induced hypertrophy and inward remodeling of cerebral pial arterioles. Hypertension 67: 623–629, 2016. doi: 10.1161/HYPERTENSIONAHA.115.06565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.von Vietinghoff S, Ley K. Interleukin 17 in vascular inflammation. Cytokine Growth Factor Rev 21: 463–469, 2010. doi: 10.1016/j.cytogfr.2010.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wenzel U, Turner JE, Krebs C, Kurts C, Harrison DG, Ehmke H. Immune Mechanisms in Arterial Hypertension. J Am Soc Nephrol 27: 677–686, 2016. doi: 10.1681/ASN.2015050562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yaghooti H, Firoozrai M, Fallah S, Khorramizadeh MR. Angiotensin II differentially induces matrix metalloproteinase-9 and tissue inhibitor of metalloproteinase-1 production and disturbs MMP/TIMP balance. Avicenna J Med Biotechnol 2: 79–85, 2010. [PMC free article] [PubMed] [Google Scholar]
- 61.Yamamoto M, Sato S, Hemmi H, Uematsu S, Hoshino K, Kaisho T, Takeuchi O, Takeda K, Akira S. TRAM is specifically involved in the Toll-like receptor 4-mediated MyD88-independent signaling pathway. Nat Immunol 4: 1144–1150, 2003. doi: 10.1038/ni986. [DOI] [PubMed] [Google Scholar]
- 62.Yao W, Sun Y, Wang X, Niu K. Elevated serum level of interleukin 17 in a population with prehypertension. J Clin Hypertens (Greenwich) 17: 770–774, 2015. doi: 10.1111/jch.12612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Yoshida T, Yamashita M, Horimai C, Hayashi M. Kruppel-like factor 4 protein regulates isoproterenol-induced cardiac hypertrophy by modulating myocardin expression and activity. J Biol Chem 289: 26107–26118, 2014. doi: 10.1074/jbc.M114.582809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zhang J, Patel MB, Griffiths R, Mao A, Song YS, Karlovich NS, Sparks MA, Jin H, Wu M, Lin EE, Crowley SD. Tumor necrosis factor-α produced in the kidney contributes to angiotensin II-dependent hypertension. Hypertension 64: 1275–1281, 2014. doi: 10.1161/HYPERTENSIONAHA.114.03863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zhang J, Rudemiller NP, Patel MB, Karlovich NS, Wu M, McDonough AA, Griffiths R, Sparks MA, Jeffs AD, Crowley SD. Interleukin-1 receptor activation potentiates salt reabsorption in angiotensin ii-induced hypertension via the NKCC2 co-transporter in the nephron. Cell Metab 23: 360–368, 2016. doi: 10.1016/j.cmet.2015.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zhao QD, Viswanadhapalli S, Williams P, Shi Q, Tan C, Yi X, Bhandari B, Abboud HE. NADPH oxidase 4 induces cardiac fibrosis and hypertrophy through activating Akt/mTOR and NFκB signaling pathways. Circulation 131: 643–655, 2015. doi: 10.1161/CIRCULATIONAHA.114.011079. [DOI] [PMC free article] [PubMed] [Google Scholar]