

Abstract
As applications in mass spectrometry continue to expand into the field of structural biology, there have been an increasing number of studies on noncovalent biological assemblies. Ensuring that protein complexes maintain native-like conformations and architectures during the transition from solution to the gas phase is a key aim. Probing composition and arrangement of subunits of multi-charged complexes via tandem mass spectrometry (MS/MS) may lead to protein unfolding and the redistribution of charges on the constituent subunits, leading to asymmetric charge partitioning and ejection of a high-charged monomer. Additionally, the overall dissociation efficiency of many ion activation methods is often suppressed for low charge states, hindering the effectiveness of MS/MS for complexes that have low charge density. Ultraviolet photodissociation (UVPD) of proteins using 193-nm photons is a high-energy alternative to collisional activation and demonstrates little to no charge state dependence. Here the symmetry of charge partitioning upon UVPD is evaluated for an array of multimeric protein complexes as a function of initial charge state. The results demonstrate that high laser energies (3 mJ) for UVPD induces more symmetric charge partitioning and ejection of low-charged, presumably compact monomers than higher-energy collisional dissociation (HCD).
Graphical Abstract
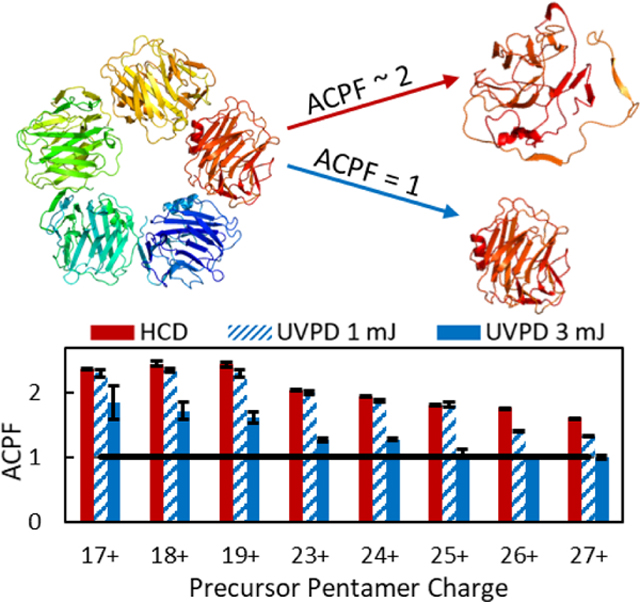
Introduction
Since the original integration of electrospray ionization (ESI) with mass spectrometry (MS) by Fenn et al.1 and extensive development for biological applications over the past 30 years, MS has expanded into the arena of structural biology, making possible the analysis of transmembrane proteins,2,3 multimeric complexes,4–7 and even virus capsids up to 9 MDa.8,9 Such protein assemblies have been traditionally difficult to characterize owing to the molecular size constraints or rigorous sample preparation required of more established methods such as nuclear magnetic resonance spectroscopy and X-ray crystallography.10–12 With only ~14% of proteins functioning as monomers, understanding the networks of noncovalent interactions that create the protein architectures and stabilize protein assemblies is crucial to providing insight into molecular processes.13 To that end, ESI-MS along with various activation methods have been employed to probe the stability,5,14–18 assembly and unfolding,15,17,19–21 topology,15–17,22 and primary sequence18,21–24 of multimeric proteins.
From ESI-MS spectra alone, determination of subunit stoichiometry of protein complexes is possible.4 Composition can be further confirmed by employing activation methods for disassembly of the multimeric protein complexes into subcomplexes and individual subunits. The most common activation method used for these types of applications has been collision-induced dissociation (CID), in which the protein experiences many low-energy collisions. Collisional activation of protein complexes is believed to be dominated by a mechanism in which typically a single subunit unfolds, causing Coulombically-favored charge redistribution; this process results in release of highly-charged, unfolded monomers from the “charged-stripped” (n-1)mers.14–16,19,20,25,26 Relative stabilities of protein complexes ranging from dimers to 24-mers have been determined by monitoring dissociation profiles into monomers and subcomplexes.14,15 Obtaining deeper structural information is limited by the asymmetric dissociation mechanism of unfolding of one subunit at a time with concomitant asymmetric charge re-distribution; however, “atypical” CID of multimers into more symmetric subcomplexes is possible depending on charge density and subunit interfacial strength.15,20 Robinson and coworkers have manipulated the charge states of protein complexes via strategic use of solution additives, thus allowing the detailed examination of the impact of charge state on the dissociation of protein complexes.14,15,19,20 Reducing the charge state mitigates extensive unfolding of the protein complexes (as observed from ion mobility data) and suppresses dissociation of the complexes, likely owing to the increased energy barrier of activation caused by decreased Coulombic repulsion.15,19,20 In the case of complexes with large cavities (e.g. ring-like structures), compaction is observed prior to unfolding.16,19 Increasing the charge state via supercharging agents enhanced access to atypical CID pathways in which ejected monomers had proportionally lower charge states and more dimers were released, akin to products formed in solution.15,19 This outcome was attributed to a decrease in the difference in energy required to unfold a protein compared to disassembly of the complex. Ion mobility also confirmed that monomers ejected with lower charge states retained compact, folded structures, although it is unclear if the monomers retained the native folding adopted in solution.16,19,20
In addition to the impact of charge state, the disassembly process in the gas phase was modulated by subunit flexibility, the presence of salt bridges, and the degree of interfacial areas of the protein complexes.15 In fact, salt bridge rearrangement (SaBRe) provides an alternative mechanism for the dissociation of protein complexes, during which intermolecular salt bridges (i.e. ones that span subunits) are cleaved heterolytically, resulting in oppositely charged ion pairs on an ejected monomer and the corresponding (n-1)mer.27 This behavior would explain the asymmetric dissociation observed during CID and its charge-dependency because the number of salt bridges is inversely related to the precursor charge. Supercharged precursors, for example, may contain fewer ion pairs and thus have a lower probability of asymmetric charge partitioning resulting from heterolytic salt bridge scission. This scenario supports the atypical dissociation of some supercharged protein complexes.15,19,27
Although the gas-phase unfolding mechanism was presumed to vary for different charge states based on variations in CID patterns, the collisional cross sections (CCS) of several protein complexes (such as tetrameric transthyretin (TTR) and pentameric serum amyloid P (SAP)) varied by less than 1% across a wide range of charge states (9+ to 15+ for TTR and 18+ to 30+ for SAP).19 The CCS measurements suggested similar initial conformations of the gas-phase complexes (regardless of the use of charging additives) that maintain native-like topologies in the gas phase with little evident for compaction or unfolding in the ESI mass spectra using gentle (low energy) tuning conditions.19
An alternative to slow-heating CID is the fast, high-energy method of surface-induced dissociation (SID).5,16,17,28–30 The picosecond timescale of SID prevents extensive unfolding that occurs on the microsecond to millisecond timescale. Therefore, symmetric dissociation into compact subunits and subcomplexes that retain a proportional amount of precursor charge and cross sections that correspond to solution structures are observed.5,16,17,28–31 Specifically, SID of several proteins from charge-reduced and supercharged conditions consistently resulted in symmetric dissociation of complexes into a full array of subcomplexes with minimal unfolding or expansion, providing insight into quaternary structure and topology.16 The SaBRe rationalization for the symmetric dissociation observed with SID suggests that the high energy deposition caused by a single, fast collision with a surface allows homolytic cleavage of salt bridges, resulting in symmetric charge partitioning.27 Regardless of mechanism, the dissociation patterns promoted by SID are less influenced by precursor mass and charge state, allowing a range of subcomplexes to be observed for increasingly large assemblies, such as the native 801 kDa GroEL 14-mer.5
Electron-based activation methods, such as electron transfer and electron capture dissociation (ETD and ECD, respectively), result in production of sequence ions that arise from backbone cleavages in the most surface exposed regions of proteins.21,23,24,32,33 Noncovalent interactions can be preserved during ETD and ECD, generating sequence ions that still retain a ligand, referred to as holo-fragments. Such holo-fragments are useful in localizing the active sites of proteins.21,23,24,32,33 Both the efficiency of electron-based dissociation and the production of holo-fragments increased with precursor charge state.21,23,24,32,33 In an outcome similar to that observed for collisional activation, asymmetric charge partitioning of ejected monomers was observed upon electron activation of protein complexes, and a means to quantify this phenomenon based on an asymmetric charge partitioning factor (ACPF, Eq. 1) was developed.24 In Eq. 1, MES and CES are the mass and weighted average charge of the ejected subunit, respectively, and MI and CI are the mass and charge of the intact precursor.
| Eq. 1 |
Although ETD and ECD have been applied successfully for dissociation of supercharged complexes in high charge states, fragmentation was limited for lower charge states generated from standard native or charge-reducing solutions.21,23,24,32,33 The limited fragmentation was attributed to the significant dependence of the energy deposition of electron activation on precursor charge that has been consistently reported for electron-based activation methods.
The absorption of high-energy, 193-nm photons results in extensive fragmentation of proteins, including both denatured proteins in high charge states and native-like proteins in low charge states.34,35 Sequence coverage of ultraviolet photodissociation (UVPD) generally surpasses that obtained by other activation methods for proteins and protein complexes.34,35 UVPD causes disassembly of protein complexes with varying degrees of charge partitioning among the released sub-units as well as formation of an array of sequence ions, including holo-products, depending on laser power.18,22 Two previous studies of UVPD of protein complexes have shown that using relatively low laser pulse energies (≤1.5 mJ per pulse) resulted in asymmetric dissociation similar to that using CID.18,22 Using higher laser pulse energies (≥1.5 mJ per pulse) led to more symmetric dissociation of complexes with monomers retaining a size-proportional amount of the initial precursor charge.18,22 The efficiency of UVPD exhibits little or no dependence on charge state, making it well-suited for analysis of more complex, native-like protein assemblies that carry fewer charges than unfolded proteins generated using denaturing conditions. The impact of charge state on the fragmentation pathways of five protein complexes activated by absorption of 193-nm photons is examined in the present study.
Methods and Materials
Reagents:
Ammonium acetate, triethylammonium acetate (Sigma, St. Louis, MO), and m-nitrobenzyl alcohol (Aldrich) were dissolved in HPLC grade water (Millipore, Burlington, MA) to concentrations of 100 mM. Bovine Cu/Zn superoxide dismutase (SOD) was obtained from MP Biomedicals (Santa Ana, CA), streptavidin (SA) from ProteoChem (Hurricane, UT), human hemoglobin (Hb) from Sigma Aldrich (St. Louis, MO), transthyretin (TTR) from Lee BioSolutions (Maryland Heights, MO), and C-reactive protein (CRP) from EMD Millipore (Burlington, MA). Each protein was diluted to a 50 μM stock of the protein monomer in 100 mM ammonium acetate and passed through a size exclusion spin column (BioRad, Hercules, CA) for purification. Samples analyzed by nano-ESI were prepared as described previously by Wysocki and coworkers.16 Briefly, each protein was diluted to 4–10 μM monomer in three types of solutions: “standard” native solutions containing 100 mM ammonium acetate; charge-reducing solutions containing 20 mM triethylammonium acetate (TEAA) and 80 mM ammonium acetate; and supercharging solutions containing 20 mM m-nitrobenzyl alcohol (m-NBA) and 80 mm ammonium acetate. All protein solutions had a final pH of approximately 7.
Data Acquisition:
All data was collected on a prototype Thermo Scientific Q Exactive Plus UHMR™ mass spectrometer (Bremen, Germany) that was modified to allow UVPD with a 193 nm excimer laser (Coherent, Santa Clara, CA) in the HCD cell as previously described.36 Samples were loaded into gold-coated borosilicate capillaries fabricated in-house. An electrospray voltage between 0.9 – 1.2 kV was applied and optimized for each sample, and ion optics were tuned for efficient transfer of each complex. To minimize unfolding of complexes in the mass spectrometer, no in-source fragmentation or in-source trapping was utilized to desolvate or otherwise activate the ions. Trapping gas pressure was also optimized for signal of the intact protein complexes using nominal settings of 2 through 7, which typically correspond to ultra-high vacuum pressures between 1 and 7E-10 mbar. Ions were trapped in the HCD cell for 10 ms prior to Orbitrap detection. Each charge state was isolated and activated with energies varying between 200 and 2000 eV lab frame collision energy (equal to the product of the acceleration voltage and the precursor charge) for HCD. The collision energy that was used for this calculation was the value of the “Direct eV” setting, which dictates the potential difference between the C-trap exit lens and the HCD entrance lens. For UVPD experiments, a single unfocused laser pulse was used with an energy setting between 0.5 and 3.0 mJ. Analysis times in the Orbitrap™ mass analyzer were set to either 32 or 512 ms to achieve resolving powers of 6,250 or 100,000 at m/z 400, respectively. MS1 spectra were collected at the low-resolution setting. Low- and high-resolution settings were used for MS/MS analysis, each collected in triplicate. The lower resolution setting was used for more accurate relative abundance information because ion abundances can be influenced by the faster decay of larger ions in the Orbitrap analyzer when using long transient lengths.37,38 The higher resolution setting was necessary for automated deconvolution of sequence ions using the Xtract algorithm. At least 300 microscans were averaged for high resolution spectra.
Data Analysis:
High resolution spectra were deconvoluted using the Xtract algorithm with a S/N threshold of 2. Fragments were identified using an algorithm developed in-house (UV-POSIT)39 and ProSight Lite40 with a 10 ppm mass tolerance. Fragments generated from HCD were matched to a, b, and y ions, and those generated by UVPD were matched to a, a+, b, c, x, x+, y, Y, and z ions. Note that current automated methods of matching fragments do not account for internal fragments that arise from secondary dissociation of sequence ions (i.e. ones that do not contain either terminus). Abundances of subunits or subcomplexes (i.e. monomers, dimers, etc.) produced upon UVPD of selected multimeric protein complexes were calculated using a custom script developed in MatLab and normalized to the total ion current (TIC) of each MS/MS spectrum. Total abundance of each subunit or subcomplex was calculated only from charge states that did not overlap significantly with other subunits and subcomplexes. For example, in the case of activation of pentameric protein, CRP, the abundance of the tetramer product was calculated using charge states of 8+ through 11+, 13+, 15+, 17+, 19+, and 21+ and trimer abundance was calculated using charge states of 7+, 8+, 10+, 11+, 13+, 14+, 16+, and 17+ to avoid overlap with monomers and dimers possessing identical m/z values. The weighted average charge (CES) of ejected monomers was calculated from the TIC-normalized intensities and charge states of monomers from MS/MS spectra. Fragment abundances for sequence ion mapping are reported as the natural log of the intensity values (If) normalized to the TIC (ITIC) of the corresponding MS/MS spectrum (i.e. ln).
Result and Discussion
Three different solution compositions (standard native, supercharging, and charge-reducing) resulted in an array of precursor charge states for each protein upon ESI, as illustrated for superoxide dismutase (SOD), a dimeric complex, in Figure 1. For example, 9+ to 11+ charge states were observed for the SOD dimer produced from the solution containing 100 mM ammonium acetate (Figure 1b). A similar distribution of charge states along with the emergence of the 8+ charge state was generated from the charge-reducing solution containing 20 mM triethylammonium acetate and 80 mM ammonium acetate (Figure 1c). The supercharging solution containing 20 mM m-nitrobenzyl alcohol (m-NBA) shifted the distribution of the SOD dimer to the 11+ to 13+ charge states, as shown in Figure 1a. In general, the use of charging additives provided access to a wider range of charge states for subsequent MS/MS analysis of each of the proteins. Similar shifts are observed for streptavidin (SA), transthyretin (TTR), hemoglobin (Hb), and C-reactive protein (CRP) shown in Figures S1-S4, respectively. Interestingly, a notable portion of SOD monomers was produced from the supercharging solution, and a similar change was observed for Hb with an increased abundance of dimers relative to the native tetramers as the charge state distribution increased (Figure S3). An array of both homo- and heterodimers with primarily 0 or 2 heme groups were observed in the ESI mass spectra of Hb in standard and supercharging solution conditions. This unusual array of dimers may suggest some disruption and rearrangement of the native complexes in solution, particularly for the supercharging solution that resulted in an abundance of apo-homodimers of β-Hb (Figure S3a). This outcome for SOD and Hb may arise from an intrinsic shift in the monomer:dimer and dimer:tetramer equilibria for SOD and Hb, respectively, in the supercharging solution owing to disruption of intermolecular forces at the protein-protein interface. Alternatively, the decomposition of tetramers into dimers or dimers into monomers may occur during the ESI process with the enhanced charging causing sufficient Coulombic repulsion of positive charges on the surfaces of the individual subunits to overcome the attractive forces at the dimer interface. The m/z overlap of SOD monomers with dimers inhibited MS/MS analysis of 12+ and 14+ dimers.
Figure 1.
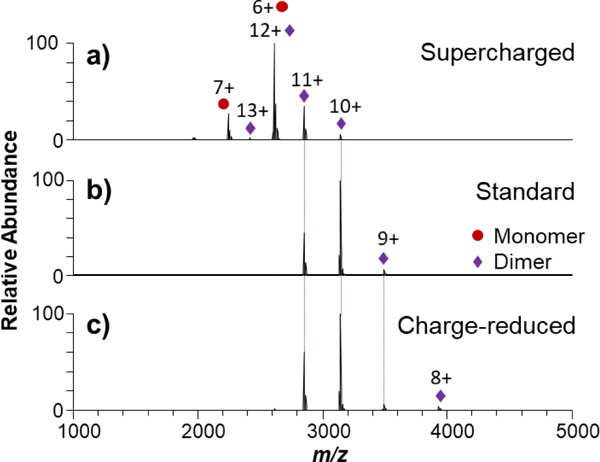
ESI-MS of Cu/Zn superoxide dismutase (SOD) in a) 20 mM m-NBA, 80 mM ammonium acetate solution for supercharging, b) 100 mM ammonium acetate (standard native MS solution), and c) 20 mM triethylammonium acetate, 80 mM ammonium acetate for charge-reducing native solutions.
Each of the charge states generated from ESI of the standard native, supercharging, and charge-reducing solutions was isolated and activated by HCD and UVPD. The abundances of some of the charge states were too low to allow sufficient production and detection of fragment ions and thus were not included in subsequent MS/MS analyses. A summary of the charge states of each precursor that were analyzed is shown in Table S1. The energy deposition from each activation method was varied based on the applied collision energy for HCD or the laser pulse energy for UVPD. Examples of the resulting MS/MS spectra of streptavidin (SA) are shown in Figure 2 and of SOD, TTR, Hb, and CRP in Figures S5-S8.
Figure 2.

MS/MS spectra of streptavidin (SA) in 15+ (top) and 10+ (bottom) charge states using a) a lab frame collision energy of 1 keV for HCD, b) 1 mJ UVPD, and c) 3 mJ UVPD, each using a single laser pulse. The precursor ion is labelled with a star.
The effect of charge state on dissociation of protein complexes has been extensively studied using conventional CID, and results obtained here using HCD are consistent with previous observations.15,16,19,20 The characteristic asymmetric charge partitioning of collisional activation is apparent in the HCD spectra of tetrameric streptavidin based on the relatively high charge states of the SA monomers ( 6+) that are ejected from the tetrameric precursor (15+) in Figure 2a. This type of outcome reflects either the heterolytic salt bridge cleavage process described by SaBRe27 or a significant migration of charges to one monomer as it unfolds and detaches prior to disassembly of the complex.15,16,19,20 However, some low-charged (< 6+), presumably compact monomers,15 as well as some dimers were also generated during HCD, indicative of a degree of competitive symmetric dissociation. This atypical behavior has been noted previously for some proteins in higher charge states created using supercharging conditions, such as for avidin, transthyretin, and several other proteins that have few intermolecular salt bridges and higher charge densities.15 The interface properties of a single subunit in each of the complexes are summarized in Table S2, detailing the number of salt bridges and hydrogen bonds that are predicted from the crystal structures using the PISA tool from the European protein data bank (PDBePISA).41 Note that the noncovalent interactions in the gas phase are not expected to be the same as predicted in the PDB, but the values in Table S2 may provide an idea of interfacial strength for each of the complexes studied herein.
The ability of HCD to disassemble protein complexes is suppressed for charge-reduced species, which has been reported previously16,20 and is shown in Figure 2a for the 10+ charge-reduced precursor of SA, which generates very few products when activated by HCD (1000 eV) compared to the 15+ supercharged precursor, the latter of which is completely disassembled using the same lab frame collision energy. The effect of precursor charge on the efficiency of product ion formation is also demonstrated via energy-variable HCD curves in Figure S9a for collision energies spanning 200 – 1000 eV for SA (10+, 11+, 13+, 14+, 15+). The 10+ and 11+ charge-reduced precursors exhibit markedly suppressed formation of products (i.e. lower dissociation efficiencies) across the entire range of collision energies compared to the standard and supercharged precursors (13+ – 15+). Similar trends are observed for TTR, Hb, and CRP in Figure S9b-d, respectively. In essence, complexes in lower charge states appear more resistant to disassembly. Although disruption of the noncovalent subunit interactions is suppressed as the charge state of the precursor decreases, the cleavage of covalent backbone bonds to produce sequence ions has been reported to increase.20 This is supported in the case of SOD, which exclusively generates sequence ions for the charge-reduced precursor (8+) upon activation using 1200 eV lab frame collision energy compared to the dissociation products of the SOD dimer (11+), which are predominantly composed of monomers as shown in Figure S5a. However, although the relative abundances of the sequence ions are greater for charge-reduced SOD, the total number of observed fragments originating from backbone cleavages and the subsequent sequence coverage are greater for the higher charged precursor (from standard native conditions). An increase of sequence coverage with charge state was also observed for Hb and CRP. In addition to enhancement of the disruption of the noncovalent subunit interactions as the charge state of the precursor increases, the cleavage of covalent backbone bonds to produce sequence ions is similarly elevated. Figure 3 demonstrates how the sequence coverage obtained from HCD increases with charge state from 10% (8+) to 21% (11+) for SOD, from 4% (11+) to 15% (17+) for α-Hb, from 2% (11+) to 6% (17+) for β-Hb, and from <1% (17+) to 10% (27+) for CRP. No significant change in sequence coverage was observed for SA and TTR, which each averaged ~8% sequence coverage using HCD.
Figure 3.
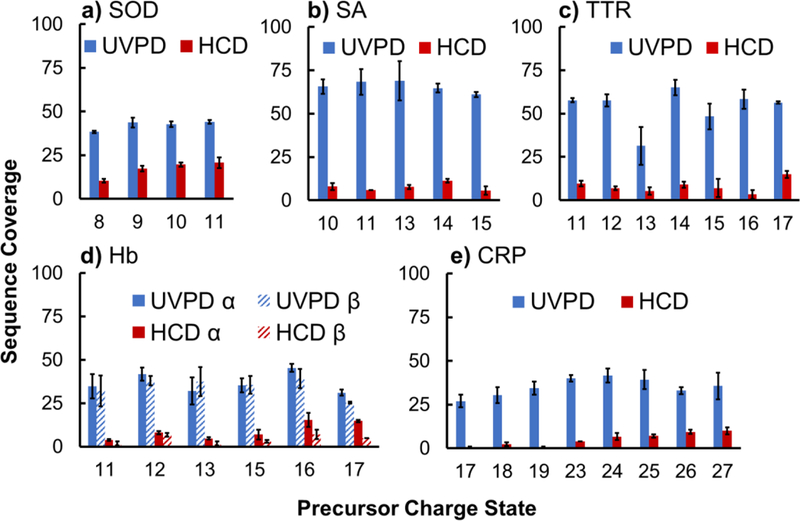
Percent sequence coverage of all of the charge states generated from supercharging, standard, and charge-reducing solutions for a) superoxide dismutase (SOD), b) streptavidin (SA), c) transthyretin (TTR), d) hemoglobin (Hb), e) C-reactive protein (CRP). A single pulse of 3 mJ was used for all UVPD measurements. HCD energy was optimized for each protein and ranged from 1400 to 2500 eV lab frame collision energy. Error bars equal to standard deviation of replicate data. Charge states with insufficient abundances for MS/MS analysis are not included.
Interestingly, when activated by UVPD using relatively low laser pulse energies (Figure 2b), SA (10+ and 15+) demonstrated similar fragmentation behavior as observed for HCD. Both asymmetric and symmetric dissociation schemes were apparent based on the production of highly charged monomers and low-charged monomers/dimers, respectively. The UVPD fragment ion yield (i.e. a measure of dissociation efficiency) was also greatly suppressed for the 10+ charge state of SA relative to the 15+ charge state, consistent with the behavior noted upon HCD, as shown in Figure S9a.
Using a higher laser pulse energy (3 mJ, single pulse) for UVPD resulted in a significant shift in the fragmentation behavior of streptavidin (10+ and 15+), causing ejection of monomers in lower charge states (Figures 2c) and signaling a transition to symmetric dissociation routes. The shift in charge state distribution of ejected monomers due to increasing laser pulse energy can be seen more clearly in Figure S10 for 13+ SA, an intermediate charge state obtained from standard native conditions. These findings support previous observations that showcased the ability of higher-energy activation methods, such as UVPD and SID, to access symmetric dissociation pathways.17,18,22 Increased sequence ion abundance was obtained using the higher laser power (3 mJ, one pulse), yielding enhanced sequence coverages that remained consistently high across all observed charge states (Figure 3). For example, sequence coverages from UVPD averaged 42% for SOD, 66% for SA, 54% for TTR, 37% and 35% for α-Hb and β-Hb, respectively, and 35% for CRP. All values were substantially higher than the coverages garnered from HCD. Sequence coverage information does not consider internal sequence fragments that may result from secondary dissociation. A portion of fragment ions are commonly unassigned, but based on the consistency of coverage across the range of precursor charge states for each protein complex, it is not believed to affect the current scope of analysis. Detailed sequence maps are provided in Figures S11 through S16 and are discussed in more detail later. Note that the decreased sequence coverage for the 13+ TTR tetramer is due to the low abundance of the precursor from both the standard and charge reducing solutions and the subsequent low signal-to-noise of the UVPD fragment ions.
To better assess the balance between asymmetric and symmetric dissociation routes for each of the complexes by the two ion activation methods, the weighted average charge state (CES) of the ejected monomers was calculated as a function of UVPD laser pulse energy ranging from 0.5 to 3.0 mJ per pulse (Figure 4) or HCD lab frame collision energy (Figure S17). SOD was excluded from this type of analysis owing to its dimeric nature; the average charge state of all monomers produced should equal one half of the precursor charge regardless of the symmetry of dissociation, making charge state analysis ineffective. For each of the tetrameric and pentameric complexes, CES decreased as the UVPD laser energy increased. This consistent trend in monomer charge state versus UVPD laser energy recapitulates the increase in symmetric charge partitioning upon greater energy deposition.
Figure 4.
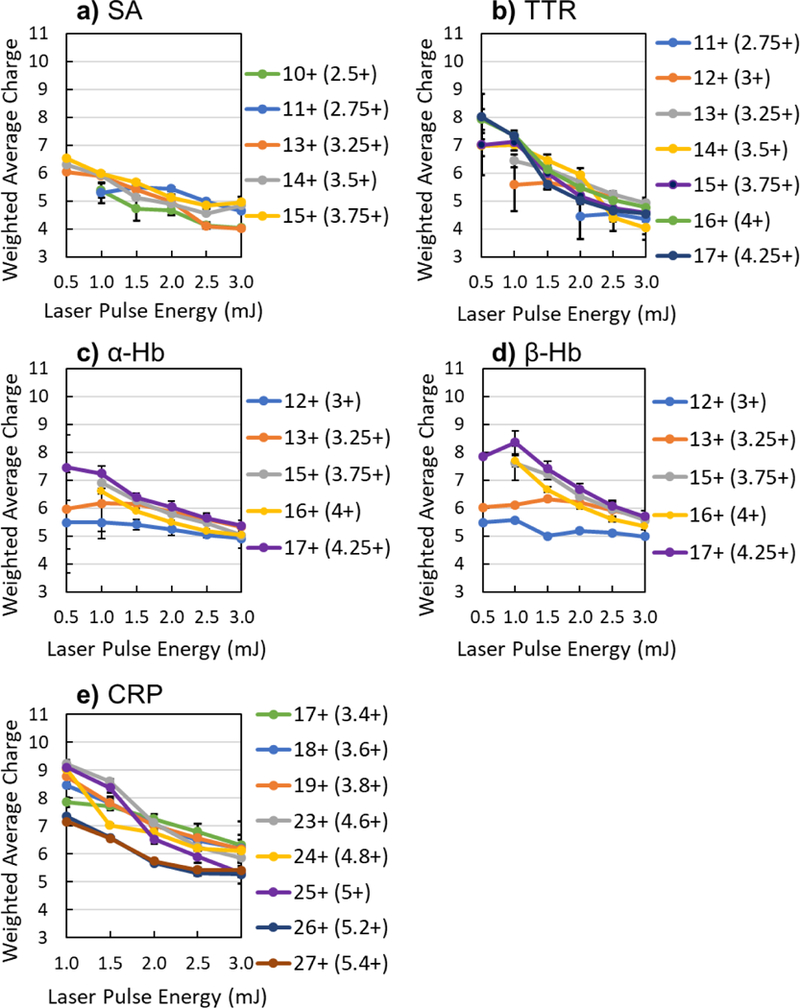
Weighted average charge (CES) of ejected monomers using varying UVPD laser pulse energies for the precursors of a) 10+ to 15+ streptavidin (SA), b) 11+ to 17+ transthyretin (TTR), c,d) 12+ to 17+ hemoglobin (Hb), and e) 17+ to 19+ and 23+ to 27+ C-reactive protein (CRP). Ejected monomers of Hb are shown separately as c) α-subunits and d) β-subunits. Error bars represent the standard deviation of replicate data. Values in parentheses next to each precursor charge indicate the theoretical charge state of each monomer following charge-symmetric partitioning.
Similar graphical analysis of the ejected monomers for HCD (Figure S17) exhibits elevated CES compared to UVPD, reflecting the expected prevalence of asymmetric charge partitioning exhibited by collisional activation. Furthermore, the trends for the CES of ejected monomers remained relatively flat as the collision energy increased for most of the precursor charge states, indicating little dependence on energy deposition. There were some deviations from this trend, particularly for the highest charge states, for which the CES decreased as collision energy increased. This behavior of decreasing weighted average charge state of ejected monomers for both HCD and UVPD can be explained by decreased abundances of highly charged monomers, increased abundances of low charged monomers, or a combination of both.
Previous studies of the disassembly of protein complexes noted the increase in the sequential losses of monomers as a function of increasing collision energy during which a monomer and (n-2)mer is ejected from a charge-stripped (n-1)mer causing the monomer to bear less charge than if ejected from the intact precursor.4,5,7,14 The charge density of the precursor also influences the dissociation symmetry of protein complexes.14,15,19,20 Notably, collisional activation of complexes with higher charge densities (i.e. supercharged complexes) resulted in ejection of low-charged, compact monomers.15,19 The relatively low CES of ejected monomers at higher collision energies of only the most highly charged precursors is demonstrated in Figure S17. This outcome has been rationalized by two mechanisms. Proponents of the unfolding and monomer ejection mechanism suggest a shift in energy barriers for paths accounting for disruption of the intramolecular noncovalent bonds within subunits that maintain secondary and tertiary structure versus the intermolecular noncovalent bonds between subunits that maintain quaternary structure.15,19 For precursors with higher charge densities, the increased Coulombic repulsion lowers the energy required to disrupt the intermolecular interactions, allowing access to more charge-symmetric dissociation pathways in which minimal subunit unfolding and charge redistribution occurs prior to disassembly.15,19 The second rationalization for symmetric charge partitioning of supercharged precursors stems from the SaBRe mechanism that argues that the inverse correlation of ion pairs and precursor charge state signifies that fewer salt bridges are available to undergo heterolytic, asymmetric cleavage, reducing the overall charge asymmetry of the product ions.27 The absence of salt bridges is also expected to reduce the overall activation barrier for disassembly of the complexes. Both mechanisms justify the generation of low-charged monomers directly from the intact, supercharged complexes. This “atypical” charge-symmetric HCD pattern suggests that increased abundances of low-charged monomers likely outweigh decreased abundances of highly charged monomers as an explanation for the decrease in CES. While this behavior is more akin to unfolding in solution and thus most relevant for MD simulations and structural insight, not all complexes are capable of “atypical” (charge symmetric partitioning) HCD.15,19 Notably, an assembly must be able to sustain high charge densities (e.g. by means of supercharging agents) during ESI. This criterion is not met by SOD and Hb as demonstrated in Figures 1a and S3a, respectively, highlighting the charge-dependent nature of HCD.
The overall symmetry of charge partitioning for each complex upon activation with HCD or UVPD is summarized in Figure 5. Based on Equation 1 shown in the introduction, the ACPF was calculated from the CES of ejected monomers for each native-like, multimeric precursor.24 An ACPF value of 1.0 denotes completely symmetric charge partitioning, which was achieved upon UVPD (3 mJ) of TTR (14+ through 17+, Figure 5b) and CRP (25+ through 27+, Figure 5d). In the case of CRP, the presence of a disulfide bond between residues Cys36 and Cys97 may play a role in the mitigation of unfolding and charge migration or salt bridge rearrangement that leads to asymmetric dissociation. Additionally, the interfacial surface area with neighboring subunits – listed for each complex in Table S2 – is relatively small in CRP, in which each subunit in CRP is only predicted to share 1275 Å2. Smaller interfacial areas may correspond to low activation barriers for subunit ejection, attenuating the charge migration or salt bridge rearrangement that leads to asymmetric dissociation. The subunits in TTR also have relatively small interfacial surface areas (1620 Å2), enhancing access to more symmetric charge partitioning. This dependence on interfacial area would explain the slightly higher overall ACPF values for SA, which has a larger interfacial area (2139 Å2) than TTR. This reasoning, however, does not hold true for Hb, which is a fairly fragile complex with an even smaller interfacial area (1578 Å2). The number of salt bridges and hydrogen bonds between subunits also does not correlate with the ACPF values obtained for the four higher order oligomers (SA, TTR, Hb, and CRP). These results suggest that other characteristics beyond those listed in Table S2 play a role on subunit unfolding and/or salt bridge rearrangement of gas-phase protein complexes.
Figure 5.
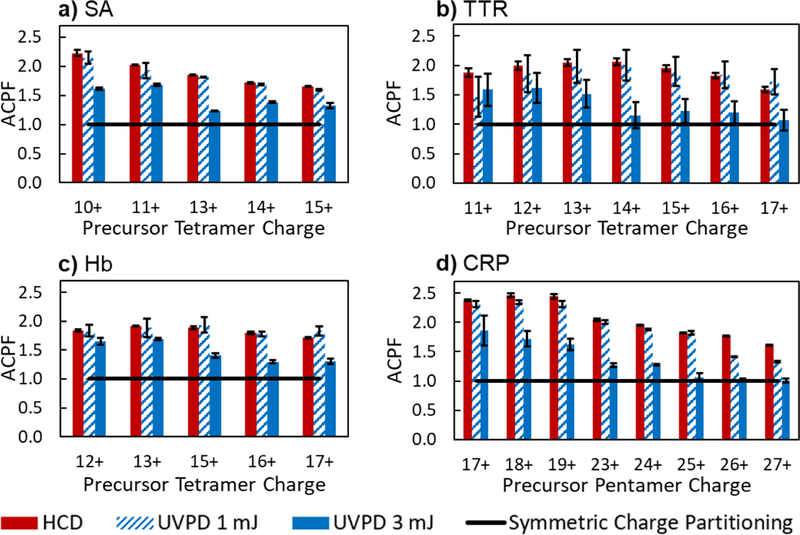
Asymmetric charge partitioning factor of a) 10+ to 15+ streptavidin (SA), b) 11+ to 17+ transthyretin (TTR), c) 12+ to 17+ hemoglobin (Hb), and d) 17+ to 19+ and 23+ to 27+ C-reactive protein (CRP) upon activation with HCD and low- and high-energy UVPD (1 pulse). The ACPF for Hb is shown as the average for α- and β-subunits. The lab frame collision energy was optimized for each precursor and is equal to 1000 eV for SA (a), 1200 eV for TTR and Hb (b and c), and 2000 eV for CRP (d). The ACPF for purely symmetric charge partitioning (1.0) is shown in each graph. Error bars are equal to standard deviation of replicate data.
The ACPF values calculated from HCD and low-energy UVPD (1 mJ) are virtually identical across all charge states of the protein complexes and were consistently higher (indicating more asymmetric charge partitioning) than the values calculated from higher energy UVPD (3 mJ). Even in the case of CRP that is structurally constrained by the disulfide bond, ACPF values of greater than 2.0, indicating charge retention of more than double what is expected for symmetric partitioning, are obtained for charge state ≤ 23+ of the pentameric precursor activated by HCD and low-energy UVPD. For each activation method, the highest ACPF value (most asymmetric) was typically obtained for the lowest charged precursors: 10+ and 11+ SA, 12+ and 13+ Hb, and 17+ through 19+ CRP. TTR deviated from this trend and instead, the highest ACPF values for HCD and low-energy UVPD were obtained from the mid-charge state 14+ precursor. The higher ACPF values for the lower charged precursors are rationalized because as precursor charge decreases, the CES of the native-like precursors converge around the same charge state (± 0.5) for the whole range of charge states as illustrated in Figure 4 for 3 mJ UVPD of each protein complex. It would be expected that CES would decrease with decreasing precursor charge. ACPF values greater than 1.0 may support the unfolding and ejection mechanism of asymmetric dissociation; however, it is possible that the monomers ejected from activation using higher energy UVPD (3 mJ) are compact even when they carry a disproportionate amount of charge. Previously reported ion mobility (IM) results, showed that low-charged, collisionally-ejected monomers from TTR (≤6+) or from CRP (≤7+) retain a compact structure.15,16 Additionally, other IM studies reported similar cross sections for identically charged proteins, regardless of their origin.42,43 These findings have also been rationalized by the SaBRe mechanism of dissociation that justifies the compact cross section of monomers that bear a disproportionate amount of precursor charge based the rearrangement of salt bridges within the protein complex.27 The present results suggest that monomers ejected using high energy UVPD (3 mJ) may have compact, potentially-folded conformations, regardless of precursor charge state, and the charge partitioning of protein complex disassembly is not necessarily indicative of the extent of unfolding during activation.
To confirm that low-charged, presumably folded monomers are being ejected during higher energy UVPD, the possibility of secondary dissociation of highly charged monomers upon UVPD, a process that would artificially deplete the population of the monomers representative of asymmetric charge partitioning, was evaluated as an alternative explanation using energy-variable UVPD (variation of laser pulse energy). The distribution of high-charged monomers, low-charged monomers, and sequence ions was mapped as a function of laser pulse energy for each protein studied (Figures S18-S22), again excluding SOD owing to its dimeric nature. The set of curves for each charge state demonstrates a similar trend: at the onset of fragmentation, the higher charged monomers that carry a disproportionate amount of precursor charge are abundant and the lower charged, presumably compact monomers are low abundance or nonexistent. As the laser pulse energy is increased, the highly charged monomers diminish and the abundances of the low-charged monomers increase. One explanation for this behavior originates from the likelihood of multiphoton absorption during the activation period. As the laser power increases, the probability of multiphoton absorption increases, resulting in higher energy deposition and faster dissociation. Multiphoton absorption may increase the propensity for direct fragmentation from an excited electronic state.31 This type of direct fragmentation would lead to cleavage of covalent bonds and production of sequence ions directly from intact multimers or subcomplexes thereof. Additionally, the energy absorbed from each photon may be converted to vibrational energy, which may favor disruption of noncovalent bonds.31 This tremendous molecular heating from photon absorption and intramolecular vibrational energy redistribution (IVR) occurs on the femtosecond to picosecond timescale similar to SID activation.31 This type of fragmentation supersedes unfolding, which occurs on the micro- to millisecond timescale in the gas phase,44 and would favor symmetric dissociation and lead to production of monomers in lower charge states.22 Post-UVPD ion mobility measurements would be required to verify production of compact monomers. Both direct fragmentation of covalent bonds and IVR contribute to the enhanced sequence coverage and symmetric dissociation observed with UVPD, as a single covalent cleavage would likely be insufficient to liberate a sequence-type fragment from a complex owing to the number of retained noncovalent interactions in the gas phase.31
As laser power is increased further (Figures S18-S22), the abundances of low-charged monomers also begin to diminish and sequence ions dominate. The wealth of sequence ions obtained using higher laser pulse energies may originate from secondary dissociation of ejected subunits/subcomplexes or direct fragmentation of the multimeric precursor. The origin of sequence ions, i.e., whether they are generated from secondary dissociation of ejected monomers or directly from the multimeric precursor, is probed by monitoring the abundances of trimers from the tetrameric precursors or tetramers from pentameric precursors (Figures S23-S25). If monomers were directly dissociating into sequence ions, presumably the abundances of the monomers would decay more rapidly than the corresponding trimers (from tetrameric SA and TTR) or tetramers (from pentameric CRP). With few exceptions, the abundances of monomers and trimers from SA and TTR (Figures S23 and S24, respectively) grow and decay in parallel for all precursor charge states. Although this does not exclude the possibility of secondary dissociation from both monomeric and multimeric products, it suggests that if secondary dissociation is occurring, it is happening for both monomers and multimeric species. In the case of pentameric CRP (Figure S25), the population of tetrameric products decays more rapidly than the population of monomers, potentially signifying greater propensity of the tetramers to further dissociate by production of sequence ions or by ejection of another monomer to generate a trimeric product. The trimer population is also displayed in Figure S25 to provide further insight. Although the trends in Figures S25a-e are somewhat inconclusive, the trends in Figures S25f-h have characteristics similar to what one might envision if a portion of the trimer population were being generated from ejected tetramers: the decay of trimers parallels the tetramers as the laser power is increased from 1.0 to 2.0 mJ, but as the laser power is increased to 2.5 mJ or beyond, the trimer abundance increases slightly as the tetramer population continues to diminish. However, the monomer population also begins to decay more steeply than the trimer, indicating that the plateau in trimer abundance is not solely related to secondary dissociation of tetramers to monomers and trimers. Instead, the increased trimer abundance may again stem from the shift in symmetry of dissociation from mostly asymmetric (pentamer → monomer + tetramer) to mostly symmetric (pentamer → trimer + dimer) owing to multiphoton absorption. Tracking the dimer population would provide considerably more insight into this issue, but quantification of dimer abundance is largely prohibited by overlap with other subcomplexes (e.g. identical m/z values for 4+ monomer, 8+ dimer, 12+ trimer, 16+ tetramer). The relatively sharp decrease in observed monomer population from 2.0 to 3.0 mJ laser pulse energy in Figures S25f-h generally coincides with the increase of sequence ions, either directly from the pentameric precursor or from any of the ejected subcomplexes, thus accounting for the erosion of intact monomers. Essentially, we believe that the enormous amount of energy absorbed from multiple photons contributing to direct covalent fragmentation and IVR leads to dissociation of the intact complex directly into sequence ions and subcomplexes without excessive charge migration or unfolding. Monomers are observed as the predominant subcomplex in the higher energy (3 mJ) UVPD spectra of each of the complex precursors in lieu of higher order subcomplexes (i.e. dimer, trimers, tetramers). Detection of the higher order subcomplexes is convoluted by the presence of truncated subcomplexes that are missing one or more sequence fragments. These truncated subcomplexes are suspected to overlap in the higher m/z range as seen by the density of fragments around and to the right of the precursor in Figures S5c, S6c, S7c, and S8c.
As mentioned previously and displayed in Figure 3, the sequence coverage of each protein via UVPD of the intact complexes significantly exceeds that obtained by conventional collisional activation (HCD). Sequence coverages of SOD and CRP were inhibited by intramolecular disulfide bonds, which are apparent in the sequence maps in Fig. S11 with the disulfide occurring between residues Cys55 and Cys144 in SOD and between residues Cys36 and Cys97 in CRP. The impact of the intramolecular disulfide bonds are also demonstrated in Figures S11a,f as well as Figure S12 for SOD and Figure S16 for CRP, which show significant gaps in coverage between the cysteine residues. The ability to generate extensive sequence ions of intact proteins using photodissociation has been well established.18,22,34 The charge-independence of UVPD has also been noted for peptides and proteins of various sizes and composition34,35 and can now be extended to multimeric complexes based on the consistency in sequence coverage for all charge states of the proteins studied herein.
Further evidence of the lack of significant charge state dependence of UVPD on extensive production of sequence ions is demonstrated in Figure S11 in which the locations and abundances of sequence ions are plotted along the primary sequence of each protein. Not only is the total coverage consistent for each charge state (Figure 3), but so also are the trends in the locations of backbone cleavages for all charge states. Figures S12 through S16 show the fragment maps for each of the protein complexes. The location of backbone cleavages that result in sequence ions generated from UVPD as well as the fragment type (a/x green, b/y blue, c/z red) are displayed. Also illustrated in Figures S12-S16 are the crystal structures of the proteins colorized to reflect the abundances of sequence fragments from UVPD based on cleavages of the backbone throughout the protein. The origins of sequence ions with respect to the positions of backbone cleavages are of particular interest because structural insight can be obtained from the fragmentation patterns of native-like proteins or complexes.45–49
Conclusions
In this work, the charge-independent nature of UVPD to yield exceptional sequence coverage has been extended to an array of intact protein complexes. As originally reported, UVPD demonstrates the ability to provide access to symmetric dissociation pathways at higher laser powers (higher photon flux leading to multiphoton absorption).18,22 Direct dissociation from excited electronic states prior to protein unfolding establishes the opportunity for further structural analysis of protein assemblies.35,45,46 Results for SOD, SA, and TTR are in agreement with previous reports of similar UVPD analyses.18,22 In the present study, UVPD analysis was expanded to a greater number of native charge states for a more comprehensive understanding of the charge-dependence of UVPD for multimeric complexes. At lower laser powers, greater abundances of unfolded monomers and charge-stripped subcomplexes are generated, likely caused by the prevalence of intramolecular vibrational redistribution following photon absorption. Although some structural characterization is impeded by this asymmetric dissociation pathway, subunit stoichiometry can be readily determined and relative interfacial strength can be inferred based on access to ‘atypical’ (charge symmetric) fragmentation patterns from both HCD and low-energy UVPD. However, while dissociation efficiency for low-energy UVPD (1 mJ per pulse) and HCD of protein complexes decreases with decreasing precursor charge state, high-energy UVPD (3 mJ per pulse) characterize protein complexes across a range of charge states with fragmentation that is consistently more charge-symmetric.
Supplementary Material
ACKNOWLEDGEMENTS
Funding from the NIH (R01GM121714) and the Robert A. Welch Foundation (F-1155) is acknowledged. The authors thank Drs. Alexander Makarov, Maria Reinhardt-Szyba, and Kyle Fort for their valuable insight and feedback on technical aspects of the prototype UHMR Orbitrap.
REFERENCES
- (1).Fenn JB; Mann M; Meng CK; Wong SF; Whitehouse CM Electrospray Ionization for Mass Spectrometry of Large Biomolecules. Science 1989, 246 (4926), 64–71. 10.2307/1703916. [DOI] [PubMed] [Google Scholar]
- (2).Bolla JR; Sauer JB; Wu D; Mehmood S; Allison TM; Robinson CV Direct Observation of the Influence of Cardiolipin and Antibiotics on Lipid II Binding to MurJ. Nat. Chem. 2018. 10.1038/nchem.2919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Barrera NP; Bartolo ND; Booth PJ; Robinson CV Micelles Protect Membrane Complexes from Solution to Vacuum. Science 2008, 321 (5886), 243–246. 10.1126/science.1159292. [DOI] [PubMed] [Google Scholar]
- (4).Liko I; Allison TM; Hopper JT; Robinson CV Mass Spectrometry Guided Structural Biology. Curr. Opin. Struct. Biol. 2016, 40, 136–144. 10.1016/j.sbi.2016.09.008. [DOI] [PubMed] [Google Scholar]
- (5).Zhou M; Jones CM; Wysocki VH Dissecting the Large Noncovalent Protein Complex GroEL with Surface-Induced Dissociation and Ion Mobility-Mass Spectrometry. Anal. Chem. 2013, 85 (17), 8262–8267. 10.1021/ac401497c. [DOI] [PubMed] [Google Scholar]
- (6).Li H; Nguyen HH; Loo RRO; Campuzano IDG; Loo JA An Integrated Native Mass Spectrometry and Top-down Proteomics Method That Connects Sequence to Structure and Function of Macromolecular Complexes. Nat. Chem. 2018, 10, 139–148. 10.1038/nchem.2908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).van Duijn E; Simmons DA; van den Heuvel RHH; Bakkes PJ; van Heerikhuizen H; Heeren RMA; Robinson CV; van der Vies SM; Heck AJR Tandem Mass Spectrometry of Intact GroEL−Substrate Complexes Reveals Substrate-Specific Conformational Changes in the Trans Ring. J. Am. Chem. Soc. 2006, 128 (14), 4694–4702. 10.1021/ja056756l. [DOI] [PubMed] [Google Scholar]
- (8).Fort KL; Waterbeemd M van de; Boll D; Reinhardt-Szyba M; Belov ME; Sasaki E; Zschoche R; Hilvert D; Makarov AA; Heck AJR Expanding the Structural Analysis Capabilities on an Orbitrap-Based Mass Spectrometer for Large Macromolecular Complexes. Analyst 2017, 143 (1), 100–105. 10.1039/C7AN01629H. [DOI] [PubMed] [Google Scholar]
- (9).van de Waterbeemd M; Fort KL; Boll D; Reinhardt-Szyba M; Routh A; Makarov A; Heck AJR High-Fidelity Mass Analysis Unveils Heterogeneity in Intact Ribosomal Particles. Nat. Methods 2017, 14 (3), 283–286. 10.1038/nmeth.4147. [DOI] [PubMed] [Google Scholar]
- (10).Baldwin AJ; Kay LE NMR Spectroscopy Brings Invisible Protein States into Focus. Nat. Chem. Biol. 2009, 5 (11), 808 10.1038/nchembio.238. [DOI] [PubMed] [Google Scholar]
- (11).Acharya KR; Lloyd MD The Advantages and Limitations of Protein Crystal Structures. Trends Pharmacol. Sci. 2005, 26 (1), 10–14. 10.1016/j.tips.2004.10.011. [DOI] [PubMed] [Google Scholar]
- (12).Yu H Extending the Size Limit of Protein Nuclear Magnetic Resonance. Proc. Natl. Acad. Sci. U. S. A. 1999, 96 (2), 332–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Shen H-B; Chou K-C QuatIdent: A Web Server for Identifying Protein Quaternary Structural Attribute by Fusing Functional Domain and Sequential Evolution Information. J. Proteome Res. 2009, 8 (3), 1577–1584. 10.1021/pr800957q. [DOI] [PubMed] [Google Scholar]
- (14).Benesch JLP; Aquilina JA; Ruotolo BT; Sobott F; Robinson CV Tandem Mass Spectrometry Reveals the Quaternary Organization of Macromolecular Assemblie. Chem. Biol. 2006, No. 13, 597–605. [DOI] [PubMed] [Google Scholar]
- (15).Hall Z; Hernández H; Marsh JA; Teichmann SA; Robinson CV The Role of Salt Bridges, Charge Density, and Subunit Flexibility in Determining Disassembly Routes of Protein Complexes. Structure 2013, 21 (8), 1325–1337. 10.1016/j.str.2013.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Zhou M; Dagan S; Wysocki VH Impact of Charge State on Gas-Phase Behaviors of Noncovalent Protein Complexes in Collision Induced Dissociation and Surface Induced Dissociation. Analyst 2012, No. 138, 1353–1362. [DOI] [PubMed] [Google Scholar]
- (17).Quintyn RS; Yan J; Wysocki VH Surface-Induced Dissociation of Homotetramers with D2 Symmetry Yields Their Assembly Pathways and Characterizes the Effect of Ligand Binding. Chem. Biol. 2015, 22 (5), 583–592. 10.1016/j.chembiol.2015.03.019. [DOI] [PubMed] [Google Scholar]
- (18).Tamara S; Dyachenko A; Fort KL; Makarov AA; Scheltema RA; Heck AJR Symmetry of Charge Partitioning in Collisional and UV Photon-Induced Dissociation of Protein Assemblies. J. Am. Chem. Soc. 2016, 138 (34), 10860–10868. 10.1021/jacs.6b05147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Hall Z; Politis A; Bush MF; Smith LJ; Robinson CV Charge-State Dependent Compaction and Dissociation of Protein Complexes: Insights from Ion Mobility and Molecular Dynamics. J. Am. Chem. Soc. 2012, 134 (7), 3429–3438. 10.1021/ja2096859. [DOI] [PubMed] [Google Scholar]
- (20).Pagel K; Hyung S-J; Ruotolo BT; Robinson CV Alternate Dissociation Pathways Identified in Charge-Reduced Protein Complex Ions. Anal. Chem. 2010, 82 (12), 5363–5372. 10.1021/ac101121r. [DOI] [PubMed] [Google Scholar]
- (21).Lermyte F; Sobott F Electron Transfer Dissociation Provides Higher-Order Structural Information of Native and Partially Unfolded Protein Complexes. PROTEOMICS 2015, 15 (16), 2813–2822. 10.1002/pmic.201400516. [DOI] [PubMed] [Google Scholar]
- (22).Morrison LJ; Brodbelt JS 193 Nm Ultraviolet Photodissociation Mass Spectrometry of Tetrameric Protein Complexes Provides Insight into Quaternary and Secondary Protein Topology. J. Am. Chem. Soc. 2016, No. 138, 10849–10859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Zhang H; Cui W; Wen J; Blankenship RE; Gross ML Native Electrospray and Electron-Capture Dissociation FTICR Mass Spectrometry for Top-Down Studies of Protein Assemblies. Anal. Chem. 2011, 83 (14), 5598–5606. 10.1021/ac200695d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Compton PD; Fornelli L; Kelleher NL; Skinner OS Probing Asymmetric Charge Partitioning of Protein Oligomers during Tandem Mass Spectrometry. Int. J. Mass Spectrom. 2015, 390, 132–136. 10.1016/j.ijms.2015.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Sciuto SV; Liu J; Konermann L An Electrostatic Charge Partitioning Model for the Dissociation of Protein Complexes in the Gas Phase. J. Am. Soc. Mass Spectrom. 2011, 22 (10), 1679 10.1007/s13361-011-0205-x. [DOI] [PubMed] [Google Scholar]
- (26).Jurchen JC; Williams ER Origin of Asymmetric Charge Partitioning in the Dissociation of Gas-Phase Protein Homodimers. J. Am. Chem. Soc. 2003, 125 (9), 2817–2826. 10.1021/ja0211508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Loo RRO; Loo JA Salt Bridge Rearrangement (SaBRe) Explains the Dissociation Behavior of Noncovalent Complexes. J. Am. Soc. Mass Spectrom. 2016, 27 (6), 975–990. 10.1007/s13361-016-1375-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Harvey SR; Yan J; Brown JM; Hoyes E; Wysocki VH Extended Gas-Phase Trapping Followed by Surface-Induced Dissociation of Noncovalent Protein Complexes. Anal. Chem. 2016, 88 (2), 1218–1221. 10.1021/acs.analchem.5b03479. [DOI] [PubMed] [Google Scholar]
- (29).Quintyn RS; Harvey SR; Wysocki VH Illustration of SID-IM-SID (Surface-Induced Dissociation-Ion Mobility-SID) Mass Spectrometry: Homo and Hetero Model Protein Complexes. Analyst 2015, 140 (20), 7012–7019. 10.1039/C5AN01095K. [DOI] [PubMed] [Google Scholar]
- (30).VanAernum ZL; Gilbert JD; Belov ME; Makarov AA; Horning SR; Wysocki VH Surface-Induced Dissociation of Noncovalent Protein Complexes in an Extended Mass Range Orbitrap Mass Spectrometer. Anal. Chem. 2019. 10.1021/acs.analchem.8b05605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Julian RR The Mechanism Behind Top-Down UVPD Experiments: Making Sense of Apparent Contradictions. J. Am. Soc. Mass Spectrom. 2017, 28 (9), 1823–1826. 10.1007/s13361-017-1721-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Yin S; Loo JA Top-down Mass Spectrometry of Supercharged Native Protein–Ligand Complexes. Int. J. Mass Spectrom. 2011, 300 (2), 118–122. 10.1016/j.ijms.2010.06.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Zenaidee AM; Donald AW Electron Capture Dissociation of Extremely Supercharged Protein Ions Formed by Electrospray Ionisation. Anal. Methods 2015, 7 (17), 7132–7139. 10.1039/C5AY00710K. [DOI] [Google Scholar]
- (34).Shaw JB; Li W; Holden DD; Zhang Y; Griep-Raming J; Fellers RT; Early BP; Thomas PM; Kelleher NL; Brodbelt JS Complete Protein Characterization Using Top-Down Mass Spectrometry and Ultraviolet Photodissociation. J. Am. Chem. Soc. 2013, 135 (34), 12646–12651. 10.1021/ja4029654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).O’Brien JP; Li W; Zhang Y; Brodbelt JS Characterization of Native Protein Complexes Using Ultraviolet Photodissociation Mass Spectrometry. J. Am. Chem. Soc. 2014, 136 (37), 12920–12928. 10.1021/ja505217w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Mehaffey MR; Sanders JD; Holden DD; Nilsson CL; Brodbelt JS Multistage Ultraviolet Photodissociation Mass Spectrometry To Characterize Single Amino Acid Variants of Human Mitochondrial BCAT2. Anal. Chem. 2018, 90 (16), 9904–9911. 10.1021/acs.analchem.8b02099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Sanders JD; Grinfeld D; Aizikov K; Makarov A; Holden DD; Brodbelt JS Determination of Collision Cross-Sections of Protein Ions in an Orbitrap Mass Analyzer. Anal. Chem. 2018, 90 (9), 5896–5902. 10.1021/acs.analchem.8b00724. [DOI] [PubMed] [Google Scholar]
- (38).Makarov A; Denisov E Dynamics of Ions of Intact Proteins in the Orbitrap Mass Analyzer. J. Am. Soc. Mass Spectrom. 2009, 20 (8), 1486–1495. 10.1016/j.jasms.2009.03.024. [DOI] [PubMed] [Google Scholar]
- (39).Rosenberg J; Parker WR; Cammarata MB; Brodbelt JS UV-POSIT: Web-Based Tools for Rapid and Facile Structural Interpretation of Ultraviolet Photodissociation (UVPD) Mass Spectra. J. Am. Soc. Mass Spectrom. 2018, 29 (6), 1323–1326. 10.1007/s13361-018-1918-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Fellers RT; Greer JB; Early BP; Yu X; LeDuc RD; Kelleher NL; Thomas PM ProSight Lite: Graphical Software to Analyze Top-down Mass Spectrometry Data. PROTEOMICS 2015, 15 (7), 1235–1238. 10.1002/pmic.201400313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Krissinel E; Henrick K Inference of Macromolecular Assemblies from Crystalline State. J. Mol. Biol. 2007, 372 (3), 774–797. 10.1016/j.jmb.2007.05.022. [DOI] [PubMed] [Google Scholar]
- (42).Laszlo KJ; Buckner JH; Munger EB; Bush MF Native-Like and Denatured Cytochrome c Ions Yield Cation-to-Anion Proton Transfer Reaction Products with Similar Collision Cross-Sections. J. Am. Soc. Mass Spectrom. 2017, 28 (7), 1382–1391. 10.1007/s13361-017-1620-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Jhingree RJ; Barran PE Ion-Ion Interactions Affect (Re)Folding Dynamics of Ubiquitin Ions in the Gas Phase. Distinct Folding Intermediates Formed via Charge-Reduction Pathways. 10.26434/chemrxiv.5678872.v1. [DOI] [Google Scholar]
- (44).Breuker K; McLafferty FW Stepwise Evolution of Protein Native Structure with Electrospray into the Gas Phase, 10−12 to 102 s. Proc. Natl. Acad. Sci. 2008, 105 (47), 18145–18152. 10.1073/pnas.0807005105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Mehaffey MR; Cammarata MB; Brodbelt JS Tracking the Catalytic Cycle of Adenylate Kinase by Ultraviolet Photodissociation Mass Spectrometry. Anal. Chem. 2018, 90 (1), 839–846. 10.1021/acs.analchem.7b03591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Cammarata MB; Brodbelt JS Structural Characterization of Holo- and Apo-Myoglobin in the Gas Phase by Ultraviolet Photodissociation Mass Spectrometry. Chem. Sci. 2015, 6 (2), 1324–1333. 10.1039/C4SC03200D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Cammarata MB; Thyer R; Rosenberg J; Ellington A; Brodbelt JS Structural Characterization of Dihydrofolate Reductase Complexes by Top-Down Ultraviolet Photodissociation Mass Spectrometry. J. Am. Chem. Soc. 2015, 137 (28), 9128–9135. 10.1021/jacs.5b04628. [DOI] [PubMed] [Google Scholar]
- (48).Theisen A; Black R; Corinti D; Brown JM; Bellina B; Barran PE Initial Protein Unfolding Events in Ubiquitin, Cytochrome c and Myoglobin Are Revealed with the Use of 213 Nm UVPD Coupled to IM-MS. J. Am. Soc. Mass Spectrom. 2019, 30 (1), 24–33. 10.1007/s13361-018-1992-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Theisen A; Yan B; Brown JM; Morris M; Bellina B; Barran PE Use of Ultraviolet Photodissociation Coupled with Ion Mobility Mass Spectrometry To Determine Structure and Sequence from Drift Time Selected Peptides and Proteins. Anal. Chem. 2016, 88 (20), 9964–9971. 10.1021/acs.analchem.6b01705. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.