

Abstract
The development and progression of colorectal cancer (CRC) is closely related to gut microbiome. Here we investigated the impact of lipopolysaccharide (LPS), one of the most prevalent products in the gut microbiome, on CRC immunotherapy. We found that LPS was abundant in orthotopic CRC tissue and was associated with low responses to anti-PD-L1 mAb therapy, and clearance of Gram-negative bacteria from the gut using polymyxin B (PmB), or blockade of Toll-like receptor 4 using TAK-242, would both relieve the immunosuppressive microenvironment and boost T-cell infiltration into the CRC tumor. Further, we designed an engineered LPS-targeting fusion protein and loaded its coding sequence into a lipid-protamine-DNA (LPD) nanoparticle system for selectively expression of LPS trap protein and blocking LPS inside the tumor, and this nano-trapping system significantly relieved the immunosuppressive microenvironment and boosted anti-PD-L1 mAb therapy against CRC tumor. This LPS trap system even attenuated CRC liver metastasis when applied, suggesting the importance of blocking LPS in the gut-liver axis. The strategy applied here may provide a useful new way for treating CRC as well as other epithelial cancers that interact with mucosa microbiome.
Keywords: nanotechnology, cancer therapy, drug delivery, immunotherapy, metastasis
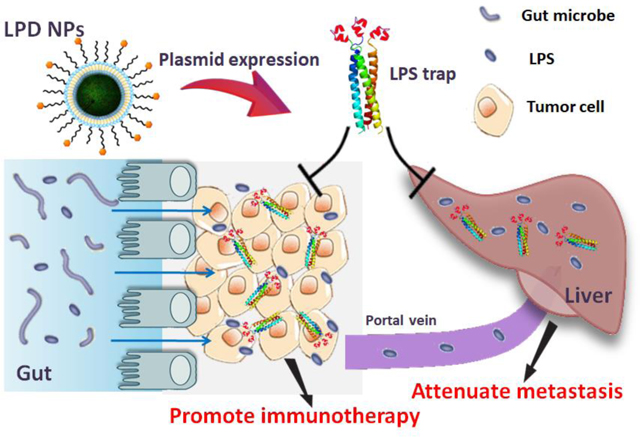
ToC figure
Nanotechnology-based Trapping lipopolysaccharide, a prevalent product of gut microbiome, boosts checkpoint blockade immunotherapy against colorectal cancer and attenuates the liver metastasis.
Cancer immunotherapy has attracted unprecedented attention, based largely on the clinical successes of checkpoint blockade immunotherapy in several cancer types[1]. Unfortunately, this therapy has shown limited efficacy for colorectal cancer (CRC), the third most prevalent cancer and the second leading cause of cancer-related death worldwide[2]. Mismatch-repair (MMR) status is an important intrinsic predictor for the clinical benefit of checkpoint blockade immunotherapy[3]. PD-1/PD-L1 blockade therapy was effective in CRC patients with microsatellite instability-high or MMR-deficient cancers, a type of CRC characterized by high somatic mutations but only represents 5% of the patient population[4]. Recent studies reveal that cyclooxygenase inhibition[5], MAP kinase inhibition[6], or immunogenic chemotherapy[7] promote anti-tumor activity of checkpoint blockade in CRC, suggesting there is value in combining immune therapeutics with pathway-targeted agents or chemotherapies[8]. Moreover, immunotherapeutic efficacy is greatly affected by the complex interplay between the tumor and its surrounding environment, or even host factors including age, obesity and gut microbiome[9]. Recent reports demonstrate that the gut microbiome influences efficacy of PD-1-based immunotherapy against melanoma and epithelial tumors[10]. However, the association between gut microbiome and CRC immunotherapy is still elusive.
CRC develops in close contact with fecal bacteria, and the gut microbiome is known to be closely linked to colorectal carcinogenesis and progression[11]. Both human subjects and animal models with CRC show enhanced intestinal permeability, which results in translocation of microbiota and microbial products into the colorectal tumor and systemic circulation[12]. For example, enrichment of Fusobacterium nucleatum has been viewed as a common feature of human colon cancers and adenomas[13], and studies in diverse experimental models have suggested the role of Fusobacterium in tumor-promotion and chemoresistance[14]. Lipopolysaccharide (LPS) is an important product of gut Gram-negative microbiota. Elevated LPS in the blood, as well as in CRC tissue, is always observed in CRC patients even with early stage adenoma[15]. As an immune-stimulatory ligand, LPS is known for its role in intestinal inflammation and progression of CRC, through activating the Toll-like receptor 4 (TLR4) and nuclear factor NF-κB pathways[16]. LPS was also found to promote CRC metastasis by stimulating TLR4 signaling and increasing β1 integrin-mediated cell adhesion[17], suggesting LPS may be an important target in CRC therapy and CRC liver metastasis.
Here, we investigeated the role of LPS in CRC immunotherapy responses. We began our study by establishing both orthotopic and subcutaneous CRC models with the same murine CRC cell line CT26-FL3, a MMR-proficient CRC cell line with high potential of liver metastasis[18]. When the same anti-PD-L1 mAb therapy was carried out on these two models, distinct responses were observed. Anti-PD-L1 therapy suppressed tumor growth by 53.0% in the subcutaneous CT26-FL3 tumor model but had absolutely no effect on the orthotopic CT26-FL3 tumor (Figure 1a). The unresponsiveness of orthotopic CT26-FL3 tumor to anti-PD-L1 therapy correlates with the clinical status of MMR-proficient CRC in immunotherapy[19]. Immunofluorescence staining of cross sections from these two tumor models showed far less T-cell infiltration in the orthotopic tumor compared to the subcutaneous tumor (Figure 1b). Since the two tumor models were established from the same cell line in the same mouse strain, genetic factors are unlikely to explain the pronounced difference in response. Therefore, environmental factors such as interactions of tumor with the colon may play an important role. Based on our hypothesis that LPS plays a pivotal role in the failure of anti-PD-1/PD-L1 therapy to CRC, we measured the LPS concentration in the orthotopic and subcutaneous tumor tissue. High concentrations of LPS were present in the orthotopic tumor, but its level was negligible in the subcutaneous tumor (Figure 1c).
Figure 1.
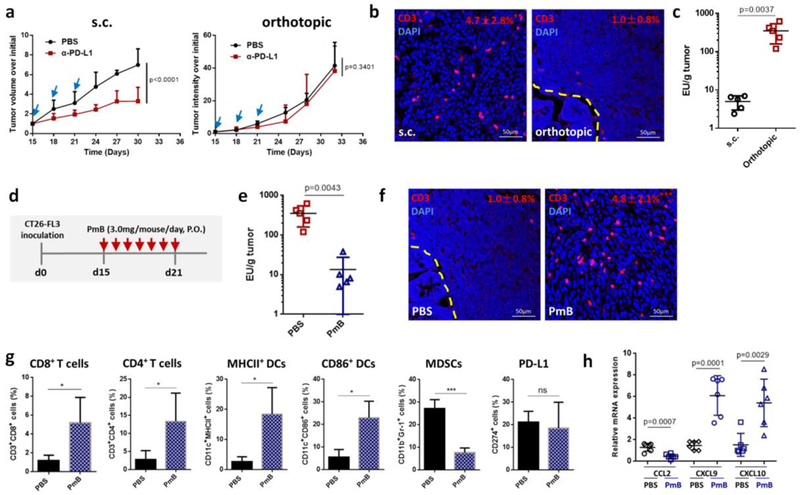
a) Growth curves of s.c. and orthotopic CT26-FL3 tumors treated with PBS or 100 μg/mouse anti-PD-L1 antibody (n = 5 mice per group). b) CD3-immunofluorescence images of s.c. and orthotopic CT26-FL3 tumors. Yellow dashed line indicates the boundary between intestinal mucosa and the orthotopic tumor. * p < 0.05, comparison of CD3+ ratios between s.c. tumor and orthotopic tumor (n = 12 to 15 region of interest (ROI) from four tumors per group). c) Endotoxin test of s.c. CT26-FL3 tumors and orthotopic CT26-FL3 tumors (n = 5). d) Treatment schedule of PmB on orthotopic CT26-FL3 tumor bearing mice. e) Endotoxin test of orthotopic CT26-FL3 tumors in mice treated with PBS and PmB (n = 5). f) CD3-immunofluorescence images of orthotopic CT26-FL3 tumor in mice treated with PBS or PmB (day 22 after tumor inoculation). *** p < 0.001, comparison of CD3+ ratios between PBS and PmB groups (n = 12 to 15 ROIs from four tumors per group). g) Flow cytometry analysis of orthotopic CT26-FL3 tumors in PBS and PmB treated groups (day 22, n = 4). ns, no significant difference, * p<0.05, ***p<0.001. h) Relative mRNA expression of various cytokines and chemokines in the PBS and PmB treated tumors (on day 22, n = 6). Significant differences were assessed in a using two-way ANOVA with multiple comparisons and in others using t test. Results are presented as mean (SD).
To test the effect of LPS on the immunomicroenvironment of the orthotopic tumor, we first used PmB to clear the Gram-negative bacteria from the mouse gut. PmB is a cyclic cationic peptide with potent bactericidal effect on almost all Gram-negative bacilli due to its high affinity to lipid A of LPS[20]. After 7 days of continuous oral gavage of PmB (Figure 1d), most of the Gram-negative bacteria were cleared from the gut (Figure S1, Supporting Information), and the LPS concentration in the orthotopic CT26-FL3 tumor was greatly reduced (Figure 1e). Significantly, while oral PBS had no effect, PmB treatment greatly increased T-cell infiltration inside the tumor (Figure 1f). Further analysis showed that CD8+ and CD4+ T cells, as well as MHCII+ and CD86+ DCs were largely increased in the tumor after PmB treatment, while myeloid-derived suppressor cells (MDSCs) were greatly reduced (Figure 1g). PmB treatment showed partial tumor inhibition effect on the orthotopic CT26-FL3 tumor, while the same treatment had no inhibition effect and even promoted the growth of the subcutaneous tumor (Figure S2, Supporting Information). The expression of the downstream LPS effectors in the orthotopic tumor, including pNF-κB p65, COX-2 and p-STAT3, were all reduced after PmB treatment (Figure S3, Supporting Information), and the expression of the corresponding pro-inflammatory cytokines IL-1β, IL-6, and Ptgs2 were significantly reduced (Figure S4, Supporting Information). In line with these changes, monocyte chemokines CCL2 expression was reduced, while T-cell chemokines CXCL9, and CXCL10 expression was significantly elevated after PmB treatment (Figure 1h). The two chemokines CXCL9 and CXCL10 facilitate chemotactic recruitment of tumor-infiltrating lymphocytes (TILs), and their intratumoral accumulation is a potential way to improve TIL-dependent immune intervention in cancer[21]. Together, these results support that clearance of LPS in orthotopic CRC tumor by clearing the intestinal Gram-negative bacteria would relieve the immunosuppressive microenvironment and elevate immunotherapy for the orthotopic CRC.
One strategy to block LPS-mediated signaling pathway is by blocking its receptor. Toll-like receptor 4 (TLR4) is the receptor for Gram-negative LPS and lipid A. We tested the effect of blocking TLR4 on the orthotopic CT26-FL3 tumor using a selective TLR4 inhibitor: TAK242. TAK-242 (resatorvid®) is a drug candidate originally developed to treat sepsis. It is a cell-permeable compound that binds to Cys747 of TLR4 and selectively disrupts its interaction with adaptor molecules TIRAP and TRAM[22]. For in vivo application, TAK-242 was loaded inside a nanoemulsion (NE) system (Figure S5a, Supporting Information). The NE particles had a diameter around 170 nm, and were stable in water for over 25 days (Figure S5b and c, Supporting Information). Similar to what was observed with PmB treatment, partial tumor inhibition and obvious T-cell infiltration was observed inside the orthotopic tumor after TAK-242 treatment (Figure S6, Supporting Information). CD8+ and CD4+ T cells, as well as MHCII+ and CD86+ DCs were largely increased in the tumor after TAK-242 treatment, while the MDSCs were significantly reduced (Figure S7, Supporting Information). The expression of tumor-promoting inflammatory cytokines IL-1β, IL-6 and Ptgs2 were decreased, while the expression of TILs chemokines CXCL9 and CXCL10 were increased (Figure S8, Supporting Information). This is the second piece of evidence that LPS and related pathway contributes to immune suppression in the orthotopic CRC tumor.
To directly test the role of LPS in orthotopic CRC and develop a practical way for LPS clearance, we developed a LPS-binding fusion protein (we call it LPS trap, Sequence S1, Supporting Information) which would selectively block LPS. The uniqueness of this LPS trap is that it has a self-assembly cartilage matrix protein-based trimeric structure that can potently neutralize multiple forms of bacterial LPS endotoxins in a trivalent manner[23], by targeting the relatively conserved lipid A moiety that is present in all the LPS molecules (Figure 2a). The LPS-binding affinity of the trap varies depending on the lipid structure in the LPS molecule, but are in the range from 20 nM to 100 nM when measured by microscale thermophoresis. The detailed biochemcial charcaterization of the trap with different LPS will be published in a separate paper[24]. For tumor-selective delivery of the LPS trap protein, we designed a transient and tumor-selective expression system based on our established lipid-protamine-DNA (LPD) nanoparticle gene delivery sysem35, 36. We coloned the optimized coding sequence for the secreted form of LPS trap protein into the expression vector pcDNA3.1, and then encapsulated the LPS trap plasmid into LPD nanoparticles by complexing the plasmid with protamine and cationic liposome (Figure 2b). The obtained LPD nanoparticles had a hydrodynamic diameter of around 140 nm and surface charge of +40.5 mV (Figure S9, Supporting Information). After injection into orthotopic CT26-FL3 tumor bearing mice through the tail vein, the major LPD accumulation sites were determined to be liver, lung and the tumor (Figure S10, Supporting Information). However, the highest expression of LPS trap protein was observed in the tumor, presumably due to a combination of the enhanced permeability and retention (EPR) effect and the AEAA-mediated targeting effects resulting in more efficient transfection and translation of the LPS trap gene in the tumor tissue than other organs including the liver (Figure 2c). Importantly, the expression of the LPS trap protein is a transient process with high expression from day 1 to day 4 after injection and significantly reduced expression on day 7 (Figure S11, Supporting Information). Similar to what was observed after the PmB treatment, trapping LPS inside the tumor with the LPS trap protein significantly changed the immunosuppressive microenvironment of the orthotopic CT26-FL3 tumor. After three injections at a dose of 50 μg LPS trap plasmid/mouse, partial tumor suppression was observed due to mollification of the tumor-promoting inflammation (Figure S12, Supporting Information). Importantly, the CD8+ and CD4+ T cells, as well as MHCII+ and CD86+ DCs were largely increased in the tumor, while the MDSCs were greatly reduced (Figure 2d, e). Compared to TAK-242, the LPS trap treatment resulted in higher CD3 positive areas and more CD8+ T cell accumulation inside the tumor, presumably due to the continuous and local expression of the LPS trap in the tumor for a period of several days. Further analysis confirmed that the expression of pro-inflammatory cytokines IL-1β, IL-6 and Ptgs2 were decreased (Figure 2f), while the expression of CTL chemokines CXCL9 and CXCL10 were increased (Figure 2g). These results indicate that targeting LPS inside the orthotopic CRC tumor will significantly increase DCs function and T-cell infiltration while reduce the immunosuppressive cell populations, and may provide a good condition for applying immunotherapeutic treatments in CRC.
Figure 2.
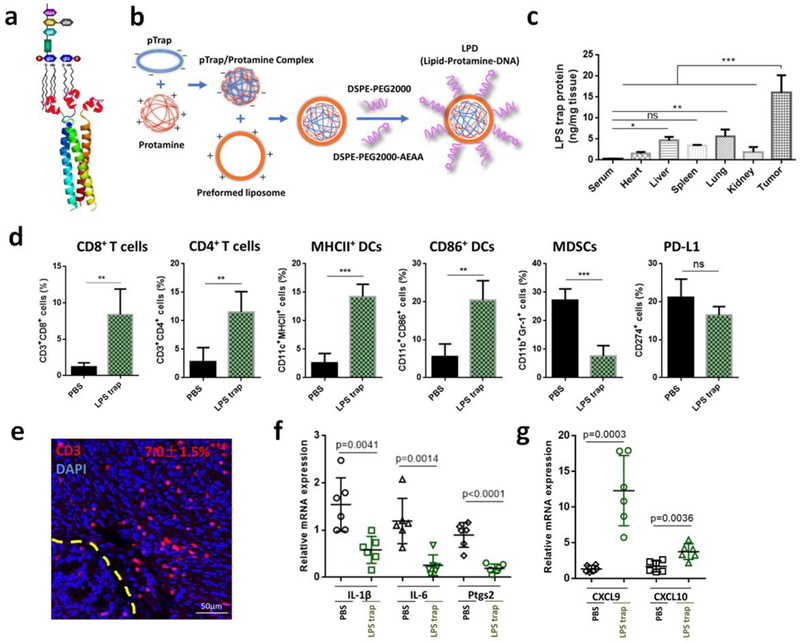
a) The schematic of the trimeric LPS trap and multivalent interaction of the LPS trap with the lipid A region of LPS. The red region is the LPS-binding moiety. b) Preparation of LPS trap plasmid (pTrap) loaded LPD nanoparticles. c) LPS trap protein expression in serum, major organs and CT26-FL3 tumor. LPS trap plasmid loaded LPD was given on day 0, and the LPS trap expression was measured using ELISA by targeting the His (6×)-tag engineered at the C-terminus of the LPS trap on day 2 (n = 3). *** p < 0.001. d) Flow cytometry analysis of orthotopic CT26-FL3 tumor after PBS and LPS trap treatment (on day 22, n = 4). ns, no significant difference, ** p<0.01, ***p<0.001. e) CD3-immunofluorescence images of orthotopic CT26-FL3 tumor after LPS trap treatment (day 22). Yellow dashed line indicates the boundary between intestinal mucosa and the orthotopic tumor. *** p < 0.001, comparison of CD3+ ratios between PBS and LPS trap groups (n = 12 to 15 ROIs from four tumors per group). f-g) Relative mRNA expression of various cytokines or chemokines in the PBS and LPS trap treated tumors (n = 6). Significant differences were assessed using t test. Results are presented as mean (SD).
Trapping LPS with the LPS trap system did not change much on the PD-L1 ratios in the orthotopic CT26-FL3 tumors (Figure 2d). The high ratio of PD-L1+ cells inside tumor may be attributed to tumor cells, DCs, macrophages, as well as many other lymphocytes, and justified the necessity for combining the LPS trap with anti-PD-L1 treatment. In addition, the abundant T-cell infiltration induced by LPS trap treatment is an criterion in determining whether the tumor is suitable or not for PD-1-based checkpoint blockade immunotherapy[25]. Therefore, we combined LPS trap with anti-PD-L1 mAb for the orthotopic CT26-FL3 tumor therapy (Figure 3a). The orthotopic CT26-FL3 tumors were unresponsive to anti-PD-L1 therapy alone; however, the combination therapy resulted in significant tumor growth suppression over other groups (Figure 3b, c). It should be noted that the combination therapy prolonged the survival time (~47 days) of the tumor bearing mice in comparison with the anti-PD-L1 therapy alone (~32 days) or LPS trap therapy alone (~40.5 days) (Figure 3d). Further analysis showed that the combination treatment resulted in more CD8+ and CD4+ T cells inside the tumor (Figure 3e). The tumor inhibition effect of the combination treatment was obviously CD8+ T-cell dependent, as depletion of CD8+ T cells significantly impaired the tumor inhibition efficiency (Figure 3f).
Figure 3.
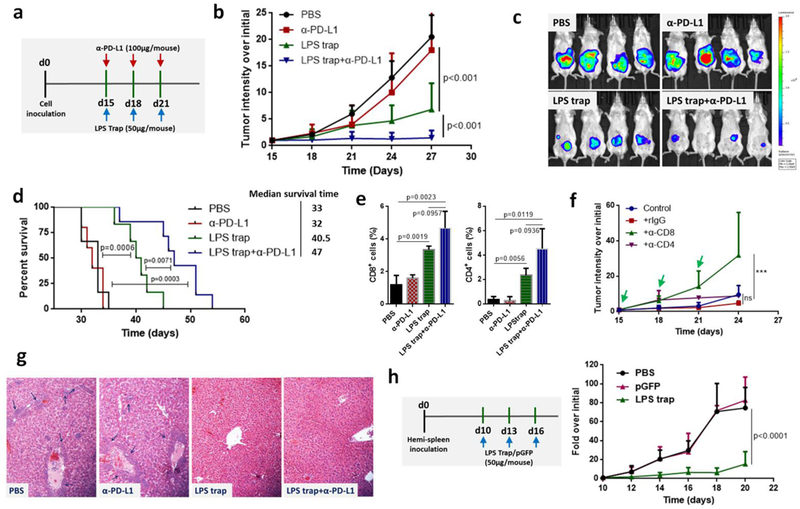
a) Treatment scheme of orthotopic CT26-FL3 tumor. b) Growth curves of orthotopic CT26-FL3 tumors in PBS, α-PD-L1, LPS trap plasmid and LPS trap+α-PD-L1 treated groups (n = 6–10 mice per group). c) Representative bioluminescence imaging of tumor burden, applying a maximum luminescence threshold of 2 × 109. d) Kaplan-Meier survival curves; the difference between different groups is significant by log-rank test. e) Flow cytometry analysis of orthotopic CT26-FL3 tumor after various treatments (on day 27, n = 4). f) CD4+ and CD8+ T cell depletion test of the LPS trap+α-PD-L1 treatment (n = 5 mice per group). Rat IgG, α-CD8 or α-CD4 (200 μg/mouse, i.p. injection) together with LPS trap+α-PD-L1 were given on day 15, 18 and 21. ns, no significant difference, *** p<0.001. g) Pathological analyses of liver after various treatments. The regions pointed by blue arrows are metastasis sites. The photos were taken at 10× magnification. h) Treatment scheme and tumor growth curves of CT26-FL3 liver metastatic tumors (n = 5 mice per group). Significant differences were assessed in b, f, h using two-way ANOVA with multiple comparisons and in e using t test. Results are presented as mean (SD).
In further pathological analysis, we observed that the LPS trap and the combination treatments greatly inhibited the spontaneous liver metastasis of the orthotopic CT26-FL3 tumor (Figure 3g, Figure S13, Supporting Information). Blood chemistry analysis confirmed that the LPS trap and combination treated groups had normal levels of serum aspartate aminotransferase (AST) and alanine aminotransferase (ALT), in contrast to the much elevated levels in the PBS and anti-PD-L1 treated groups (Figure S14, Supporting Information). Liver is the primary metastatic site for CRC, and an important reason for CRC related death is liver metastasis. Our results showed that the LPS trap treatment would not only promote immunotherapy for the primary CRC, but also prevent liver metastasis happen. Some recent reports may explain the effect of trapping LPS in preventing CRC liver metastatis. It was observed that LPS promoted migratory ability of CRC cells by activation of the SDF-1α/CXCR4 axis and epithelial-mesenchymal transition[26], and systemic inflammation caused by elevated LPS levels in the enteric cavity and portal venous blood of patients with CRC was shown to increase hepatic recruitment of cancer cells[27]. In addition, LPS also exerts a direct effect on promoting hepatocarcinogensis through the interaction with TLR4[28]. To clarify the effect of LPS trap on the growth of CT26-FL3 tumor cells already metastasized into the liver, we established an experimental liver metastasis model by hemi-splenic inoculation of CT26-FL3 cells[29]. Similar to what was observed in other liver diseases[30], metastatic tumor in liver resulted in increased translocation of microbial products from the gut to the liver through portal circulation and increased levels of LPS in liver (Figure S15, Supporting Information). Significantly, when we administered three injections of LPS trap nanoparticles to the mice, CT26-FL3 tumor cell growth in the liver was significantly inhibited (Figure 3h, Figure S16, Supporting Information). The dramatic inhibition effect was confirmed by the histological analysis and serum chemistry analysis (Figure S17, Supporting Information). Elevated LPS trap protein was detected in the liver with tumor metastasis, presumably via direct transfection of the tumor cells in the liver (Figure S18, Supporting Information). The above results highlight the potential of the LPS trap system in CRC therapy, by not only promote immunotherapy against the primary tumor, but also attenuate CRC liver metastasis.
In summary, we have demonstrated the prevalent role of LPS in CRC immunotherapy, and developed a nanoparticle-based LPS trap system for promoting PD-1-based immunotherapy for CRC and inhibiting liver metastasis. We showed that nano-trapping of LPS inside the orthotopic CRC tumor promoted T-cell infiltration into tumor and boosted checkpoint blockade therapy in the otherwise anti-PD-1/PD-L1 unresponsive CRC. Trapping LPS also reduced liver metastasis of primary CRC and attenuated metastasized tumor growth in the liver, suggesting the prevalent role of LPS in the metastasis of CRC through the gut-liver axis.
Our results highlight the possibility of designing further means for neutralizing LPS from gut microbiome or microbiota modulation as potential treatments for CRC. Clearance of Gram-negative bacteria from the gut using antibiotic cocktails is a direct way to do so, however, one concern is the negative effect of the broad-spectrum antibiotics on the healthy intestinal microbiota. Intra-tumor expression of the LPS trap protein used in this study is a safe and tumor-specific way for clearance of LPS from the tumor. Along with this concept, one would expect to develop genetically-engineered bacteria that secrete LPS trap or even use Bacillus polymyxa to directly produce PmB. These bacteria could be introduced to the host gut in proper way to scavenge LPS and modulate the CRC microenvironment.
The majority of the bacteria found in the body are localized in the gut; therefore, the influence of gut microbiota on disease is important to understand. However, bacteria also exist on the surface or deep layer of the skin, mouth, vagina, bladder, and so on. Important questions raised by our findings include whether cancers occurring at these sites are also abundant with LPS, and whether LPS will modulate the responses of these tumors to immunotherapy. Emerging evidence supports that the microorganisms inhabiting many sites of the body have a role beyond infection[31]. Our results provide a strong foundation for pursuing targeted approaches to mucosa-related cancer treatment directed against LPS and other key constituents of the cancer microbiota.
Supplementary Material
Acknowledgements
This work was supported by NIH grants CA198999 (to L.H.) and CA157738 (to R.L.) and a grant from Eshelman Institute for Innovation (to L.H. and R. L.). It was also supported by National Natural Science Foundation of China (Project 51673185) and China Scholarship Council. We gratefully appreciate Dr. Aaron Anselmo from UNC Eshelman School of Pharmacy for his discussion on the use of PmB, Nikki McCoy and Prof. Tope Keku from UNC Center for Functional GI & Motility Disorders for discussion on this project, and Dr. Ashutosh Tripathy at UNC Macromolecular Interactions Facility for assistance in biophysical analysis.
Footnotes
Supporting Information
Supporting Information is available from the Wiley Online Library or from the author.
Competing financial interests: R.L. and L.H. are co-founders of OncoTrap, Inc. which has licensed the trap technology.
Contributor Information
Wantong Song, Division of Pharmacoengineering and Molecular Pharmaceutics, Eshelman School of Pharmacy, University of North Carolina at Chapel Hill, Chapel Hill, NC 27599, USA; Key Laboratory of Polymer Ecomaterials, Jilin Biomedical Polymers Engineering Laboratory, Changchun Institute of Applied Chemistry, Chinese Academy of Sciences, Changchun, 130022, P. R. China.
Karthik Tiruthani, Division of Chemical Biology and Medicinal Chemistry, Eshelman School of Pharmacy, University of North Carolina at Chapel Hill, Chapel Hill, NC 27599, USA.
Ying Wang, Division of Pharmacoengineering and Molecular Pharmaceutics, Eshelman School of Pharmacy, University of North Carolina at Chapel Hill, Chapel Hill, NC 27599, USA; Division of Chemical Biology and Medicinal Chemistry, Eshelman School of Pharmacy, University of North Carolina at Chapel Hill, Chapel Hill, NC 27599, USA.
Limei Shen, Division of Pharmacoengineering and Molecular Pharmaceutics, Eshelman School of Pharmacy, University of North Carolina at Chapel Hill, Chapel Hill, NC 27599, USA.
Mengying Hu, Division of Pharmacoengineering and Molecular Pharmaceutics, Eshelman School of Pharmacy, University of North Carolina at Chapel Hill, Chapel Hill, NC 27599, USA.
Oleksandra Dorosheva, Division of Chemical Biology and Medicinal Chemistry, Eshelman School of Pharmacy, University of North Carolina at Chapel Hill, Chapel Hill, NC 27599, USA.
Kunyu Qiu, Division of Pharmacoengineering and Molecular Pharmaceutics, Eshelman School of Pharmacy, University of North Carolina at Chapel Hill, Chapel Hill, NC 27599, USA.
Karina A. Kinghorn, Division of Pharmacoengineering and Molecular Pharmaceutics, Eshelman School of Pharmacy, University of North Carolina at Chapel Hill, Chapel Hill, NC 27599, USA
Rihe Liu, Division of Chemical Biology and Medicinal Chemistry, Eshelman School of Pharmacy, University of North Carolina at Chapel Hill, Chapel Hill, NC 27599, USA; Carolina Center for Genome Sciences, University of North Carolina at Chapel Hill, Chapel Hill, NC 27599, USA..
Leaf Huang, Division of Pharmacoengineering and Molecular Pharmaceutics, Eshelman School of Pharmacy, University of North Carolina at Chapel Hill, Chapel Hill, NC 27599, USA.
References
- [1].Mellman I, Coukos G, Dranoff G, Nature 2011, 480, 480; [DOI] [PMC free article] [PubMed] [Google Scholar]; Hoos A, Nature reviews. Drug discovery 2016, 15, 235. [DOI] [PubMed] [Google Scholar]
- [2].Kuipers EJ, Grady WM, Lieberman D, Seufferlein T, Sung JJ, Boelens PG, van de Velde CJH, Watanabe T, Nature Reviews Disease Primers 2015, 1, 15065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Le DT, Durham JN, Smith KN, Wang H, Bartlett BR, Aulakh LK, Lu S, Kemberling H, Wilt C, Luber BS, Wong F, Azad NS, Rucki AA, Laheru D, Donehower R, Zaheer A, Fisher GA, Crocenzi TS, Lee JJ, Greten TF, Duffy AG, Ciombor KK, Eyring AD, Lam BH, Joe A, Kang SP, Holdhoff M, Danilova L, Cope L, Meyer C, Zhou S, Goldberg RM, Armstrong DK, Bever KM, Fader AN, Taube J, Housseau F, Spetzler D, Xiao N, Pardoll DM, Papadopoulos N, Kinzler KW, Eshleman JR, Vogelstein B, Anders RA, Diaz LA, Science (New York, N.Y.) 2017, 357, 409; [DOI] [PMC free article] [PubMed] [Google Scholar]; Zou W, Wolchok JD, Chen L, Sci Transl Med 2016, 8, 328rv4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Le DT, Uram JN, Wang H, Bartlett BR, Kemberling H, Eyring AD, Skora AD, Luber BS, Azad NS, Laheru D, Biedrzycki B, Donehower RC, Zaheer A, Fisher GA, Crocenzi TS, Lee JJ, Duffy SM, Goldberg RM, de la Chapelle A, Koshiji M, Bhaijee F, Huebner T, Hruban RH, Wood LD, Cuka N, Pardoll DM, Papadopoulos N, Kinzler KW, Zhou S, Cornish TC, Taube JM, Anders RA, Eshleman JR, Vogelstein B, Diaz LAJ, New England Journal of Medicine 2015, 372, 2509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Zelenay S, van der Veen Annemarthe G., Böttcher Jan P., Snelgrove Kathryn J., Rogers N, Acton Sophie E., Chakravarty P, Girotti Maria R., Marais R, Quezada Sergio A., Sahai E, Reis e Sousa C, Cell 2015, 162, 1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Ebert PJR, Cheung J, Yang Y, McNamara E, Hong R, Moskalenko M, Gould SE, Maecker H, Irving BA, Kim JM, Belvin M, Mellman I, Immunity 2016, 44, 609. [DOI] [PubMed] [Google Scholar]
- [7].Song W, Shen L, Wang Y, Liu Q, Goodwin TJ, Li J, Dorosheva O, Liu T, Liu R, Huang L, Nature Communications 2018, 9, 2237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Sharma P, Allison JP, Cell 2015, 161, 205; [DOI] [PMC free article] [PubMed] [Google Scholar]; Adams JL, Smothers J, Srinivasan R, Hoos A, Nature reviews. Drug discovery 2015, 14, 603. [DOI] [PubMed] [Google Scholar]
- [9].Klevorn LE, Teague RM, Trends in immunology 2016, 37, 354; [DOI] [PMC free article] [PubMed] [Google Scholar]; Iida N, Dzutsev A, Stewart CA, Smith L, Bouladoux N, Weingarten RA, Molina DA, Salcedo R, Back T, Cramer S, Dai R-M, Kiu H, Cardone M, Naik S, Patri AK, Wang E, Marincola FM, Frank KM, Belkaid Y, Trinchieri G, Goldszmid RS, Science (New York, N.Y.) 2013, 342, 967; [DOI] [PMC free article] [PubMed] [Google Scholar]; Sivan A, Corrales L, Hubert N, Williams JB, Aquino-Michaels K, Earley ZM, Benyamin FW, Man Lei Y, Jabri B, Alegre M-L, Chang EB, Gajewski TF, Science (New York, N.Y.) 2015, 350, 1084; [DOI] [PMC free article] [PubMed] [Google Scholar]; Vétizou M, Pitt JM, Daillère R, Lepage P, Waldschmitt N, Flament C, Rusakiewicz S, Routy B, Roberti MP, Duong CPM, Poirier-Colame V, Roux A, Becharef S, Formenti S, Golden E, Cording S, Eberl G, Schlitzer A, Ginhoux F, Mani S, Yamazaki T, Jacquelot N, Enot DP, Bérard M, Nigou J, Opolon P, Eggermont A, Woerther P-L, Chachaty E, Chaput N, Robert C, Mateus C, Kroemer G, Raoult D, Boneca IG, Carbonnel F, Chamaillard M, Zitvogel L, Science (New York, N.Y.) 2015, 350, 1079; [DOI] [PMC free article] [PubMed] [Google Scholar]; Viaud S, Saccheri F, Mignot G, Yamazaki T, Daillère R, Hannani D, Enot DP, Pfirschke C, Engblom C, Pittet MJ, Schlitzer A, Ginhoux F, Apetoh L, Chachaty E, Woerther P-L, Eberl G, Bérard M, Ecobichon C, Clermont D, Bizet C, Gaboriau-Routhiau V, Cerf-Bensussan N, Opolon P, Yessaad N, Vivier E, Ryffel B, Elson CO, Doré J, Kroemer G, Lepage P, Boneca IG, Ghiringhelli F, Zitvogel L, Science (New York, N.Y.) 2013, 342, 971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Gopalakrishnan V, Spencer CN, Science (New York, N.Y.) 2018, 359, 97; [DOI] [PMC free article] [PubMed] [Google Scholar]; Matson V, Fessler J, Bao R, Science (New York, N.Y.) 2018, 359, 104; [DOI] [PMC free article] [PubMed] [Google Scholar]; Routy B, Le Chatelier E, Science (New York, N.Y.) 2018, 359, 91. [DOI] [PubMed] [Google Scholar]
- [11].Sears CL, Garrett WS, Cell host & microbe 2014, 15, 317; [DOI] [PMC free article] [PubMed] [Google Scholar]; Arthur JC, Perez-Chanona E, Muhlbauer M, Tomkovich S, Uronis JM, Fan TJ, Campbell BJ, Abujamel T, Dogan B, Rogers AB, Rhodes JM, Stintzi A, Simpson KW, Hansen JJ, Keku TO, Fodor AA, Jobin C, Science (New York, N.Y.) 2012, 338, 120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Brenchley JM, Douek DC, Annual review of immunology 2012, 30, 149; [DOI] [PMC free article] [PubMed] [Google Scholar]; Grivennikov SI, Wang K, Mucida D, Stewart CA, Schnabl B, Jauch D, Taniguchi K, Yu GY, Osterreicher CH, Hung KE, Datz C, Feng Y, Fearon ER, Oukka M, Tessarollo L, Coppola V, Yarovinsky F, Cheroutre H, Eckmann L, Trinchieri G, Karin M, Nature 2012, 491, 254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Castellarin M, Warren RL, Freeman JD, Dreolini L, Krzywinski M, Strauss J, Barnes R, Watson P, Allen-Vercoe E, Moore RA, Holt RA, Genome research 2012, 22, 299; [DOI] [PMC free article] [PubMed] [Google Scholar]; Li Y-Y, Ge Q-X, Cao J, Zhou Y-J, Du Y-L, Shen B, Wan Y-JY, Nie Y-Q, World Journal of Gastroenterology 2016, 22, 3227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Bullman S, Pedamallu CS, Sicinska E, Clancy TE, Zhang X, Cai D, Neuberg D, Science (New York, N.Y.) 2017, 358, 1443; [DOI] [PMC free article] [PubMed] [Google Scholar]; Geller LT, Barzily-Rokni M, Science (New York, N.Y.) 2017, 357, 1156; [DOI] [PMC free article] [PubMed] [Google Scholar]; Yu T, Guo F, Yu Y, Sun T, Ma D, Han J, Qian Y, Kryczek I, Sun D, Nagarsheth N, Chen Y, Chen H, Hong J, Zou W, Fang JY, Cell 2017, 170, 548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Kang M, Edmundson P, Araujo-Perez F, McCoy AN, Galanko J, Keku TO, BMC Cancer 2013, 13, 91; [DOI] [PMC free article] [PubMed] [Google Scholar]; Zhu G, Huang Q, Huang Y, Zheng W, Hua J, Yang S, Zhuang J, Wang J, Ye J, Oncotarget 2016, 7, 73711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Killeen SD, Wang JH, Andrews EJ, Redmond HP, British journal of cancer 2009, 100, 1589; [DOI] [PMC free article] [PubMed] [Google Scholar]; Fukata M, Chen A, Vamadevan AS, Cohen J, Breglio K, Krishnareddy S, Hsu D, Xu R, Harpaz N, Dannenberg AJ, Subbaramaiah K, Cooper HS, Itzkowitz SH, Abreu MT, Gastroenterology 2007, 133, 1869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Hsu RYC, Chan CHF, Spicer JD, Rousseau MC, Giannias B, Rousseau S, Ferri LE, Cancer research 2011, 71, 1989. [DOI] [PubMed] [Google Scholar]
- [18].Zhang Y, Davis C, Ryan J, Janney C, Pena MM, Clinical & experimental metastasis 2013, 30, 903; [DOI] [PMC free article] [PubMed] [Google Scholar]; Castle JC, Loewer M, Boegel S, de Graaf J, Bender C, Tadmor AD, Boisguerin V, Bukur T, Sorn P, Paret C, Diken M, Kreiter S, Türeci Ö, Sahin U, BMC Genomics 2014, 15, 190; [DOI] [PMC free article] [PubMed] [Google Scholar]; Castle JC, Loewer M, Boegel S, de Graaf J, Bender C, Tadmor AD, Boisguerin V, Bukur T, Sorn P, Paret C, Diken M, Kreiter S, Tureci O, Sahin U, BMC Genomics 2014, 15, 190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Le DT, Uram JN, Wang H, Bartlett BR, Kemberling H, Eyring AD, Skora AD, Luber BS, Azad NS, Laheru D, Biedrzycki B, Donehower RC, Zaheer A, Fisher GA, Crocenzi TS, Lee JJ, Duffy SM, Goldberg RM, de la Chapelle A, Koshiji M, Bhaijee F, Huebner T, Hruban RH, Wood LD, Cuka N, Pardoll DM, Papadopoulos N, Kinzler KW, Zhou S, Cornish TC, Taube JM, Anders RA, Eshleman JR, Vogelstein B, Diaz LA, New England Journal of Medicine 2015, 372, 2509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Emmelot CH, van der Waaij D, The Journal of hygiene 1980, 84, 331; [DOI] [PMC free article] [PubMed] [Google Scholar]; Domingues MM, Inacio RG, Raimundo JM, Martins M, Castanho MA, Santos NC, Biopolymers 2012, 98, 338. [DOI] [PubMed] [Google Scholar]
- [21].Bronger H, Singer J, Windmuller C, Reuning U, Zech D, Delbridge C, Dorn J, Kiechle M, Schmalfeldt B, Schmitt M, Avril S, British journal of cancer 2016, 115, 553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Matsunaga N, Tsuchimori N, Matsumoto T, Ii M, Molecular pharmacology 2011, 79, 34. [DOI] [PubMed] [Google Scholar]
- [23].Kim D, Kim SK, Valencia CA, Liu R, Biochemistry 2013, 52, 10.1021/bi400716w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Jingjing. Li, Karthik Tituthani, Olekasandra Dorosheva, Rihe Liu, In submission 2018.
- [25].Tang H, Wang Y, Chlewicki LK, Zhang Y, Guo J, Liang W, Wang J, Wang X, Fu Y-X, Cancer Cell 2016, 29, 285; [DOI] [PMC free article] [PubMed] [Google Scholar]; Pfirschke C, Engblom C, Rickelt S, Cortez-Retamozo V, Garris C, Pucci F, Yamazaki T, Poirier-Colame V, Newton A, Redouane Y, Lin Y-J, Wojtkiewicz G, Iwamoto Y, Mino-Kenudson M, Huynh TG, Hynes RO, Freeman GJ, Kroemer G, Zitvogel L, Weissleder R, Pittet MJ, Immunity 2016, 44, 343; [DOI] [PMC free article] [PubMed] [Google Scholar]; Rosenbaum MW, Bledsoe JR, Morales-Oyarvide V, Huynh TG, Mino-Kenudson M, Modern pathology : an official journal of the United States and Canadian Academy of Pathology, Inc 2016, 29, 1104. [DOI] [PubMed] [Google Scholar]
- [26].Liu W-T, Jing Y-Y, Yan F, Han Z-P, Lai F-B, Zeng J-X, Yu G-F, Fan Q-M, Li R, Zhao Q-D, Wu M-C, Wei L-X, Cell Adhesion & Migration 2017, 11, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].McDonald B, Spicer J, Giannais B, Fallavollita L, Brodt P, Ferri LE, International journal of cancer 2009, 125, 1298. [DOI] [PubMed] [Google Scholar]
- [28].Dianne H Dapito A Mencin G-Y Gwak J-P Pradere M-K Jang I Mederacke, Jorge M Caviglia H Khiabanian A Adeyemi R Bataller, Jay H Lefkowitch M Bower R Friedman RB Sartor R Rabadan, Robert F Schwabe, Cancer Cell 2012, 21, 504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Goodwin TJ, Zhou Y, Musetti SN, Liu R, Huang L, Science Translational Medicine 2016, 8, 364ra153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Szabo G, Gastroenterology 2015, 148, 30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Schwabe RF, Jobin C, Nature reviews. Cancer 2013, 13, 800; [DOI] [PMC free article] [PubMed] [Google Scholar]; Whiteside SA, Razvi H, Dave S, Reid G, Burton JP, Nature reviews. Urology 2015, 12, 81. [DOI] [PubMed] [Google Scholar]
- [32].Kasuya H, Kuruppu DK, Donahue JM, Choi EW, Kawasaki H, Tanabe KK, Cancer research 2005, 65, 3823. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.