

Abstract

Methylenetetrahydrofolate dehydrogenase 2 (MTHFD2) plays a key role in one-carbon (1C) metabolism in human mitochondria, and its high expression correlates with poor survival of patients with various types of cancer. An isozyme-selective MTHFD2 inhibitor is highly attractive for potential use in cancer treatment. Herein, we disclose a novel isozyme-selective MTHFD2 inhibitor DS44960156, with a tricyclic coumarin scaffold, which was initially discovered via high-throughput screening (HTS) and improved using structure-based drug design (SBDD). DS44960156 would offer a good starting point for further optimization based on the following features: (1) unprecedented selectivity (>18-fold) for MTHFD2 over MTHFD1, (2) a molecular weight of less than 400, and (3) good ligand efficiency (LE).
Keywords: MTHFD2, inhibitor, isozyme-selective, SBDD, tricyclic coumarin
Methylenetetrahydrofolate dehydrogenase 2 (MTHFD2) is a human mitochondrial enzyme that is known to play a key role in purine and thymidine syntheses by generating one-carbon (1C) units from serine.1 MTHFD2 is a bifunctional enzyme; it catalyzes the dehydrogenation of 5,10-methylene-THF (CH2–THF) with an NAD+ cofactor and cyclohydrolysis of 5,10-methenyl-THF (CH=THF) to yield 10-formyl-THF (CHO−THF), subsequently producing formate as a 1C unit.2 It has recently been revealed that MTHFD2 mRNA and protein are significantly increased in various types of tumors and that patients with high levels of MTHFD2 show a poor prognosis.3−5 On the other hand, most healthy adult tissues do not express MTHFD2, and thus, inhibitors of MTHFD2 could be potential therapeutics for MTHFD2-overexpressing cancers with minimal side effects.6,7
Despite high interest in this target, only a few MTHFD1/2 dual inhibitors have been identified. A folate analog LY345899 (Figure 1) inhibited MTHFD2 (IC50: 663 nM) as well as MTHFD1 (IC50: 96 nM),8,9 and suppressed tumor growth in a mice xenograft model of colorectal cancer following intraperitoneal injection.5 Recently, a nonsubstrate natural product named carolacton, which binds to both MTHFD1 and MTHFD2, with the Ki values in the nanomolar range, was discovered (Figure 1).10 These potent compounds concurrently inhibit MTHFD1 and MTHFD2, likely because the isozymes share the same folding patterns. However, inhibition of MTHFD1 is considered to be undesirable in terms of potential safety, as MTHFD1 is broadly expressed in normal tissues.11 Therefore, a selective inhibitor of MTHFD2 is highly attractive as a lead for drug discovery in this class.
Figure 1.

Reported MTHFD2 inhibitors and HTS hit 1.
Herein, we report the discovery of the first isozyme-selective MTHFD2 inhibitor, DS44960156, with a tricyclic coumarin scaffold. This novel molecule was initially discovered via high-throughput screening (HTS), followed by optimization utilizing a rational structure-based drug design (SBDD). DS44960156 showed more than 18-fold selectivity for MTHFD2 over MTHFD1, with a molecular weight of less than 400.
Through our initial HTS, using a thermal shift assay, we found a novel tetrahydropyrido[4,3-d]pyrimidin-4-one derivative as a series of screening hits. The representative compound 1 possessed inhibitory activity against MTHFD2 dehydrogenation with an IC50 value of 8.3 μM (Figure 1). Interestingly, it did not exhibit inhibitory activity against MTHFD1 (IC50 > 100 μM).
The X-ray crystal structure analysis of the MTHFD2–compound 1 complex clearly revealed its binding mode (Figure 2). Compound 1 occupied the folate-binding site of MTHFD2 with a binding mode slightly different from that of LY345899 (Figure 2B). Compound 1 did not occupy the region where the pteridine moiety of LY345899 was found and formed a significant hydrogen bond network, whereas the whole molecule of compound 1, except the terminal benzene ring, shared the same pocket that the rest of the LY345899 molecule occupied. The key interactions of the MTHFD2–compound 1 complex were as follows: (1) four hydrogen bonds (Gln132/Lys88 with C=O of the pyrimidin-4-one; Asn87 with C=O of the linker amide; Gly310 with S=O of the sultam) and (2) a π–π interaction between Tyr84 and the pyrimidin-4-one. These interactions were also observed for the MTHFD2–LY345899 complex, although some of the hydrogen bonds were not strong in the case of the MTHFD2–compound 1 complex, as indicated by their length of >3.0 Å. Particularly, the core pyrimidin-4-one structure is thought to largely contribute to the affinity as it is related to the three key interactions (two hydrogen bonds and one π–π interaction) among the five mentioned above. On the other hand, the C=O bond of the linker amide that interacts with Asn87 is important for isozyme-selectivity. As shown in Figure S2 in the Supporting Information, Asn87 of MTHFD2 corresponds to Val55 of MTHFD1, while the other four residues interacting with 1 are conserved between MTHFD1 and MTHFD2.9 The Val55 side chain of MTHFD1 does not interact with the C=O via hydrogen bonding unlike Asn87 of MTHFD2, and thus, the affinity to MTHFD1 is much weaker than that to MTHFD2. The importance of this C=O in MTHFD2 inhibition was experimentally supported; the deletion of the carbonyl group led to complete loss of the inhibitory activity (see Supporting Information). The terminal benzene ring of compound 1 was surrounded by several hydrophobic residues, such as Leu133, Pro134, and Val205. Considering that this benzene ring occupied the region that was empty for LY345899, it is possible that the hydrophobic interactions between the benzene ring of 1 and the residues partially contributed to the affinity. However, the contribution might be limited because the apparent hydrophobic pocket was not formed and the electron density of the benzene ring was not well established in the region. Based on these structural features, we moved on to the initial derivatization of the hit.
Figure 2.
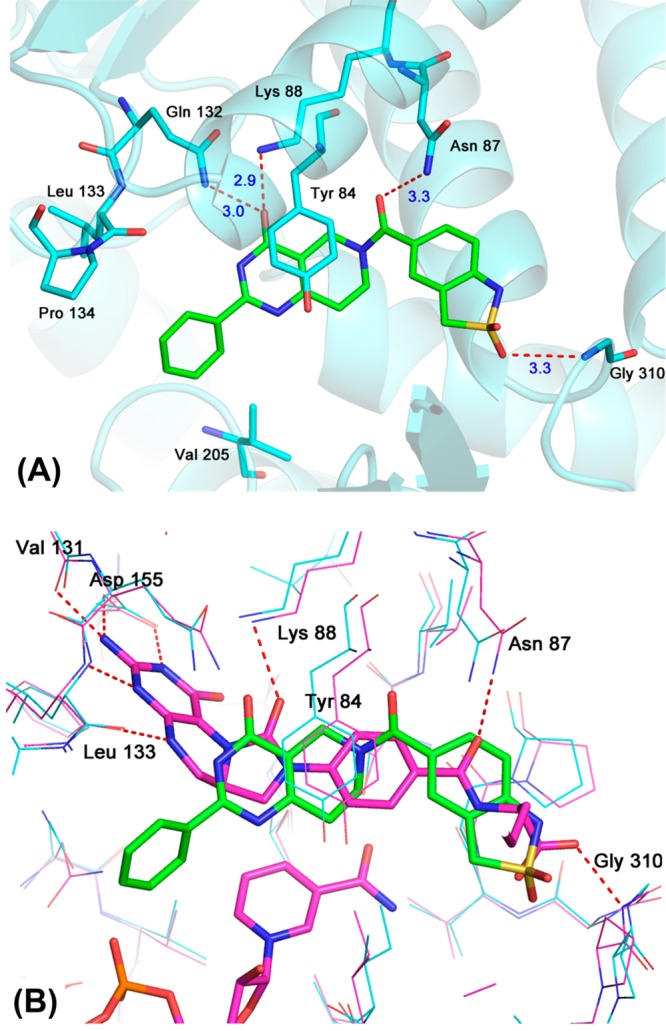
(A) X-ray structure of the MTHFD2–compound 1 complex refined at 2.5 Å resolution (PDB ID: 6JID). (B) Superposition of the MTHFD2–LY345899–NAD complex (PDB ID: 5TC4, magenta) and compound 1 (green).
First, we transformed the sultam moiety into different functionalities (Table 1), as we found, by HTS, that this moiety affected the inhibitory activity of MTHFD2. We synthesized tetrazole and carboxylic acid variants as bioisosteres.12 The carboxylic acid variant (3) was found to have a 3-fold higher inhibitory activity than 1, while the tetrazole variant (2) was less active. None of the weak acidic moieties examined, i.e. sulfonamides, benzotriazole, and 1,2,4-oxadiazol-5-one (4–7), showed potent inhibitory activity. We then screened the effect of substituents around the benzoic acid moiety. Both the electron-deficient (8) and -donating (9) groups at the ortho-position of the acid tolerated inhibitory activity. In particular, the ortho-Cl analog 8 showed a submicro molar IC50 value. A sterically bulky naphthyl ring (10) instead of benzene ring of 3 was also acceptable, providing a 2-fold increase in the IC50 value (1.4 μM) compared with that of compound 3. Replacement of the benzene ring with pyridine (11, 12) or cyclohexane (13) was not acceptable. Finally, the N-methylated sultam (14) or the nonacidic analog of 3 (the ester 15 or the amide 16) did not show any inhibitory activity against MTHFD2, indicating the importance of acidic functionality at this position.
Table 1. Structure–Activity Relationship of the Sultam Transformationa.
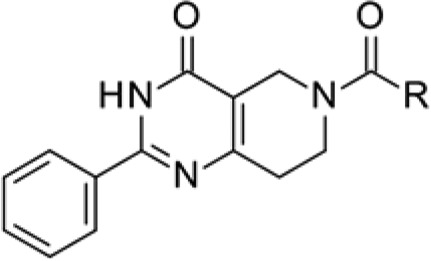

The IC50 values are for the enzymatic assay. The method is described in the Supporting Information.
With the carboxylic acid variants showing promise, we then modified the core scaffold. Cyclic amides, such as pyrimidin-4-one, sometimes show poor physicochemical properties13 that are most likely due to the presence of unmasked weak acidic N–H. As the carboxylic acid moiety of 3 was not easily replaced with other nonacidic groups, as indicated by the results in Table 1, we thought that the removal of the N–H from pyrimidin-4-one would avoid the potential risk of a poor ADME profile. Our initial X-ray analysis of the MTHFD2–compound 1 complex (Figure 2) suggested that the presence of the carbonyl groups and the π–π stacking ability of the core scaffold were required for MTHFD2 inhibition. On the other hand, two nitrogen atoms of the pyrimidin-4-one did not form any key interaction with the protein or water molecules. Moreover, the electron density of the benzene substituent was not apparent, indicating that this benzene was not well packed into the protein pocket and therefore was replaceable with other units. These observations motivated us to demonstrate “structure-based scaffold hopping”14 of the core unit. To obtain the SAR information with simple substituents, we combined a benzoic acid group with each synthesized core.
Various pyrimidin-4-one derivatives (Scheme 1, compounds 24–29) were synthesized by condensation of the corresponding amidine (18) and N-Boc-protected β-ketoester (17),15 followed by amidation with the terephthalic acid half ester and hydrolysis. Another quick approach was to use t-Bu terephthalic amide intermediate (22), prepared in one step from commercially available reagents, for condensation. The bicyclic pyrazolin-3-one system (32–34) was also accessible using the same intermediate via reactions with hydrazine derivatives.16 For 4-pyridone derivatives, N-benzylpiperidin-4-one (35) was converted to enamine17 and reacted with β-ketoester to give a bicyclic pyran-4-one intermediate (36).18 Substitution with methylamine resulted in the corresponding 4-pyridone core (37).19 Subsequent debenzylation, amidation, and deprotection resulted in the target compound 38.
Scheme 1. Synthesis of Compounds with Various Core Scaffolds.

Reagents and conditions: (a) K2CO3, EtOH, 35%–75%; (b) HCl, 1,4-dioxane or TFA, CH2Cl2, 24%–quant.; (c) carboxylic acid, WSCI-HCl, HOBt, DMF, or CH2Cl2, 65%–97%; (d) 1 M NaOH, THF, rt, 67%–95%; (e) EtOH, Et3N, 61%–100%; (f) morpholine, toluene, reflux, quant.; (g) ethyl benzoylacetate, xylenes, reflux, 26%; (h) MeNH2, MeOH, 80 °C–100 °C, 87%; (i) Pd(OH)2, H2, 1 M HCl, MeOH, rt.
The tricyclic coumarin intermediate 39 was prepared using a previously reported Pechmann condensation.20 We followed the protocol for the first batch; however, the reaction was not efficient (∼10% yield) due to the low reactivity of phenol. Subsequently, we developed a new stepwise protocol for tricyclic coumarin construction (Scheme 2). The pinacol borate intermediate 42 was prepared by triflation of β-ketoester,21 followed by Miyaura borylation.22 Under Suzuki coupling conditions between o-bromophenol and 42, Pd-catalyzed coupling and the following 6-membered lactone formation simultaneously proceeded. This tandem reaction and the subsequent deprotection yielded the tricyclic coumarin intermediate 39 as a single product. The total yield of intermediate 39 from 17 was 26%. As a further benefit, this sequential protocol enabled the construction of the amide variant (45),23 by replacing o-bromophenol with o-bromoaniline.
Scheme 2. Synthesis of Tricyclic Coumarin Derivatives.

Reagents and conditions: (a) 4-(tert-butoxycarbonyl)benzoic acid, WSCI-HCl, HOBt, CH2Cl2 or DMT-MM, MeOH, 86%–94%; (b) HCl, 1,4-dioxane or TFA, CH2Cl2, 31%–quant.; (c) Pd(dppf)Cl2, o-bromophenol or o-bromoaniline, NaHCO3, THF-H2O, 45%–57%.
Table 2 shows the results of scaffold hopping. Our strategy was to change the heteroaromatic system of the bicyclic pyrimidin-4-one moiety without deleting the essential carbonyl groups and π–π stacking ability. Deletion of phenyl substitution (24) reduced the inhibitory activity, suggesting that the benzene ring contributed to the binding, likely via interaction with the surrounding hydrophobic residues, and therefore was important. Ring expansion of piperidine (25) or the introduction of methyl groups (26, 27) did not result in inhibitory activity. We then removed the free N–H of the pyrimidin-4-one to minimize the potential risk of poor physicochemical properties. As mentioned earlier, this N–H did not form any significant hydrogen bonds or interactions. As a result, N-methylated (28) and cyclized compounds (29) possessed moderate inhibitory activity, although the IC50 values were 9- and 3-fold lower than that of the parent compound 3, respectively. Replacement of pyrimidin-4-one with a 4-pyridone core (38) resulted in an IC50 value comparable to that of compound 3. The fused pyrazolin-3-one system was then investigated; we found that the bulkier substituents increased the inhibitory activity (32–34). This result is consistent with the significant increase in potency due to phenyl substitution at the 2-position of pyrimidin-4-one (3 vs 24). Compound 34 showed the highest IC50 value in this series. Finally, the tricyclic coumarin scaffold was examined. Compound 41 (DS44960156) was found to be more potent than the other derivatives examined in Table 2, including the original pyrimidin-4-one. The amide variant (45) was not promising compared to DS44960156. It is worth noting that the IC50 value against MTHFD1 of all compounds examined was >30 μM.
Table 2. Scaffold Hopping of the Corea.
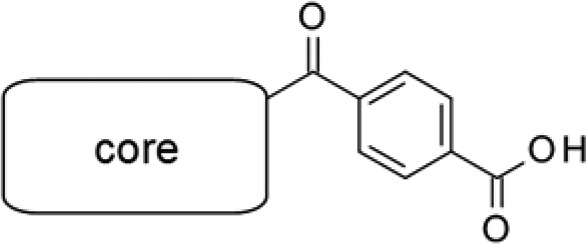

The IC50 values are for the enzymatic assay. The method is described in the Supporting Information.
The structure of the MTHFD2–DS44960156 complex determined by X-ray crystallography is shown in Figure 3. The binding mode was almost the same as that of compound 1. All the key features (the four hydrogen bonds and the π–π stacking) still existed, and the terminal carboxylate strongly interacted with the main chain N–H of Gly310 via hydrogen bonding, as observed in the sultam of 1. Both the oxygen atoms of the carboxylate of DS44960156 were located approximately 3 Å from the N–H of Gly310 and appeared to contribute to the binding, whereas one of the oxygen atoms of the sultam of 1 was oriented differently and approximately 4.4 Å away from the same N–H. This might be one of the reasons for the enhanced potency of DS44960156.
Figure 3.
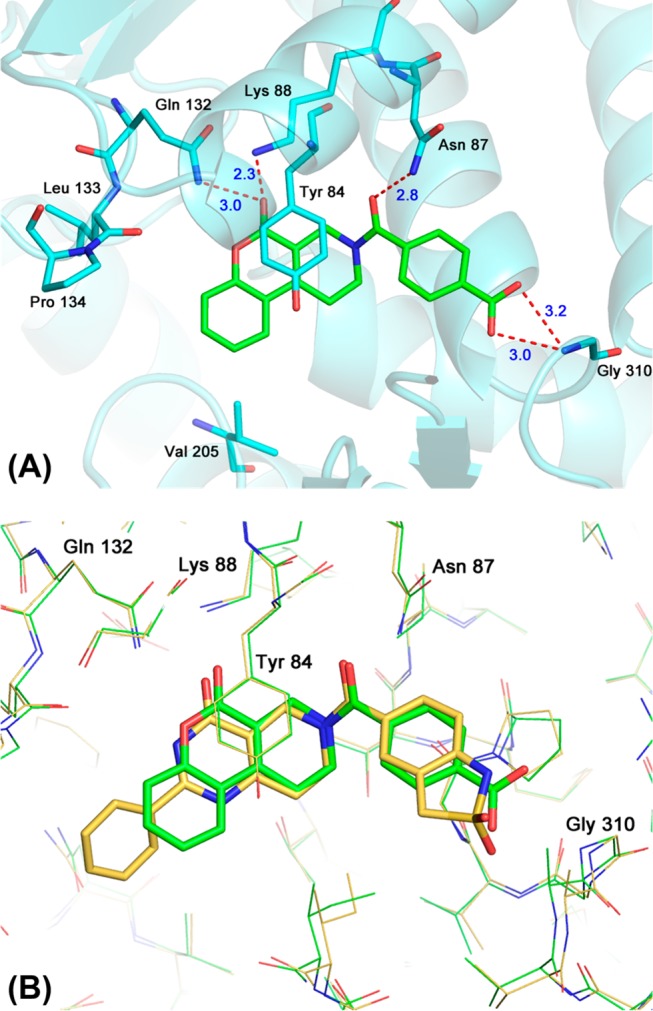
(A) X-ray analysis of the MTHFD2–DS44960156 complex refined at 2.25 Å resolution (PDB ID: 6JIB). (B) Superposition of DS44960156 (green) and compound 1 (yellow).
Table 3 shows a brief comparison of the profiles of compounds 1 and DS44960156. DS44960156 presented an IC50 value higher than that of compound 1 (1.6 vs 8.3 μM) and had a lower molecular weight (349 vs 422, respectively). Ligand efficiency (LE) is now widely used as a simple metric of binder, where LE = free energy of ligand binding/heavy atom count.24 A ligand of LE ≥ 0.3 is considered to be a good binder for further optimization.25 Although the HTS hit 1 (LE = 0.24) had a lower LE than the standard value, DS44960156 possessed an LE of 0.31, which is a commonly accepted value, similar to that of carolacton (LE = 0.35). Overall, DS44960156 has an acceptable profile for an initial lead and would offer a good starting point for further optimization.
Table 3. Profiles of Compounds 1 and 41 (DS44960156).

| Compound | MTHFD2 IC50 (μM) | Mw | LE |
|---|---|---|---|
| 1 | 8.3 | 422 | 0.24 |
| 41 (DS44960156) | 1.6 | 349 | 0.31 |
In summary, we discovered DS44960156, a novel small molecule MTHFD2 inhibitor with a tricyclic coumarin scaffold. Design of this scaffold, assisted by SBDD, enabled us to transform the HTS hit into a promising compound. It is noteworthy that the revised synthetic protocols made it possible to derivatize the initial lead. DS44960156 has >18-fold selectivity for MTHFD2 over MTHFD1, with a low molecular weight and a good LE. Evaluation of the biological roles of MTHFD2 with the derivatized molecule, and its further optimization to achieve greater potency, are ongoing.
Acknowledgments
We appreciate Drs. Takashi Asahi, Shinji Tsutsumi, and Yasuhide Hirota for their contribution and helpful support in pharmacology. We thank the members of the Protein Production Research Group for protein purification, Dr. Hiroyuki Hanzawa for support in SBDD, and Mr. Toru Taniguchi for his generous help in manuscript preparation. We also thank Drs. Naohiro Matsugaki and Noriyuki Igarashi for their help with the data collection at the Tsukuba Photon factory.
Glossary
Abbreviations
- THF
tetrahydrofolate or tetrahydrofuran
- HTS
high-throughput screening
- SBDD
structure-based drug design
- Boc
tert-butoxycarbonyl
- t-Bu
tert-butyl
- EtOH
ethanol
- TFA
trifluoroacetic acid
- WSCI
1-ethyl-3-(3-(dimethylamino)propyl)carbodiimide
- HOBt
1-hydroxybenzotriazole
- DMF
N,N-dimethylformamide
- MeOH
methanol
- DMT-MM
4-(4,6-dimethoxy-1,3,5-triazin-2-yl)-4-methylmorpholinium chloride
- Mw
molecular weight
- LE
ligand efficiency
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.9b00069.
Experimental procedures, analytical data, and information on additional studies (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Ducker G. S.; Chen L.; Morscher R. J.; Ghergurovich J. M.; Esposito M.; Teng X.; Kang Y.; Rabinowitz J. D. Reversal of Cytosolic One-Carbon Flux Compensates for Loss of the Mitochondrial Folate Pathway. Cell Metab. 2016, 23, 1140–1153. 10.1016/j.cmet.2016.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allaire M.; Li Y.; MacKenzie R. E.; Cygler M. The 3-D Structure of a Folate-Dependent Dehydrogenase/ Cyclohydrolase Bifunctional Enzyme at 1.5 Å Resolution. Structure 1998, 6, 173–182. 10.1016/S0969-2126(98)00019-7. [DOI] [PubMed] [Google Scholar]
- Nilsson R.; Jain M.; Madhusudhan N.; Sheppard N. G.; Strittmatter L.; Kampf C.; Huang J.; Asplund A.; Mootha V. K. Metabolic Enzyme Expression Highlights a Key Role for MTHFD2 and the Mitochondrial Folate Pathway in Cancer. Nat. Commun. 2014, 5, 3128. 10.1038/ncomms4128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu F.; Liu Y.; He C.; Tao L.; He X.; Song H.; Zhang G. Increased MTHFD2 Expression Is Associated with Poor Prognosis in Breast Cancer. Tumor Biol. 2014, 35, 8685–8690. 10.1007/s13277-014-2111-x. [DOI] [PubMed] [Google Scholar]
- Ju H.-Q.; Lu Y.-X.; Chen D.-L.; Zuo Z.-X.; Liu Z.-X.; Wu Q.-N.; Mo H.-Y.; Wang Z.-X.; Wang D.-S.; Pu H.-Y.; et al. Modulation of Redox Homeostasis by Inhibition of MTHFD2 in Colorectal Cancer: Mechanisms and Therapeutic Implications. JNCI J. Natl. Cancer Inst., published online December 8, 2018; 10.1093/jnci/djy160. [DOI] [PMC free article] [PubMed]
- Tedeschi P. M.; Vazquez A.; Kerrigan J. E.; Bertino J. R. Mitochondrial Methylenetetrahydrofolate Dehydrogenase (MTHFD2) Overexpression Is Associated with Tumor Cell Proliferation and Is a Novel Target for Drug Development. Mol. Cancer Res. 2015, 13, 1361–1366. 10.1158/1541-7786.MCR-15-0117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinberg S. E.; Chandel N. S. Targeting Mitochondria Metabolism for Cancer Therapy. Nat. Chem. Biol. 2015, 11, 9–15. 10.1038/nchembio.1712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt A.; Wu H.; Mackenzie R. E.; Chen V. J.; Bewly J. R.; Ray J. E.; Toth J. E.; Cygler M. Structures of Three Inhibitor Complexes Provide Insight into the Reaction Mechanism of the Human Methylenetetrahydrofolate Dehydrogenase/Cyclohydrolase. Biochemistry 2000, 39, 6325–6335. 10.1021/bi992734y. [DOI] [PubMed] [Google Scholar]
- Gustafsson R.; Jemth A. S.; Gustafsson N. M. S.; Farnegardh K.; Lose-va O.; Wiita E.; Bonagas N.; Dahllund L.; Llona-Minguez S.; Haggblad M.; et al. Crystal Structure of the Emerging Cancer Target MTHFD2 in Complex with a Substrate-Based Inhibitor. Cancer Res. 2017, 77, 937–948. 10.1158/0008-5472.CAN-16-1476. [DOI] [PubMed] [Google Scholar]
- Fu C.; Sikandar A.; Donner J.; Zaburannyi N.; Herrmann J.; Reck M.; Wagner-Döbler I.; Koehnke J.; Müller R. The Natural Product Carolacton Inhibits Folate-Dependent C1Metabolism by Targeting FolD/MTHFD. Nat. Commun. 2017, 8, 1529. 10.1038/s41467-017-01671-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tissue expression of MTHFD1 - Summary - The Human Protein Atlas. www.proteinatlas.org/ENSG00000100714-MTHFD1/tissue (accessed January 30, 2019).
- Meanwell N. A. Synopsis of Some Recent Tactical Application of Bioisosteres in Drug Design. J. Med. Chem. 2011, 54, 2529–2591. 10.1021/jm1013693. [DOI] [PubMed] [Google Scholar]
- Nakajima H.; Kakui N.; Ohkuma K.; Ishikawa M.; Hasegawa T. A. Newly Synthesized Poly(ADP-Ribose) Polymerase Inhibitor, DR2313 [2-Methyl-3,5,7,8-Tetrahydrothiopyrano[4,3-d]-Pyrimidine-4-One]: Pharmacological Profiles, Neuroprotective Effects, and Therapeutic Time Window in Cerebral Ischemia in Rats. J. Pharmacol. Exp. Ther. 2004, 312, 472–481. 10.1124/jpet.104.075465. [DOI] [PubMed] [Google Scholar]
- Böhm H. J.; Flohr A.; Stahl M. Scaffold Hopping. Drug Discovery Today: Technol. 2004, 1, 217–224. 10.1016/j.ddtec.2004.10.009. [DOI] [PubMed] [Google Scholar]
- Breslin M. J.; Coleman P. J.; Cox C. D.; Raheem I. T.; Schreier J. D.. Alkoxy Tetrahydro-Pyridopyrimidine PDE10 Inhibitors. WO2010/138430.
- Moreno-Manas M.; Perez M.; Pleixats R.. Palladium-Catalyzed Allylation of 3-Hydroxyisoxazole, 5-Isoxazolone and 5-Pyrazolone Systems. Tetrahedron 1994, 50, 515–528. 10.1016/S0040-4020(01)80773-0 [DOI] [Google Scholar]
- Asai F.; Sugidachi A.; Ikeda T.; Koike H.; Inoue T.; Takata K.; Iwamura R.; Kita J.; Yoneda K.. Cyclic Amine Derivatives. US6087379.
- Cordonnier G.; Sliwa H. A New Route to 6-Azachromones. An Improved Synthesis of 2-Azaxanthone. J. Heterocycl. Chem. 1987, 24, 111–115. 10.1002/jhet.5570240123. [DOI] [Google Scholar]
- Ma Y.; Luo W.; Quinn P. J.; Liu Z.; Hider R. C. Design, Synthesis, Physicochemical Properties, and Evaluation of Novel Iron Chelators with Fluorescent Sensors. J. Med. Chem. 2004, 47, 6349–6362. 10.1021/jm049751s. [DOI] [PubMed] [Google Scholar]
- Brown R. E.; Shavel J. J.. Substituted Benzopyrano[3,4-c]pyridines. US3946008.
- Zheng G.; Smith A. M.; Huang X.; Subramanian K. L.; Siripurapu K. B.; Deaciuc A.; Zhan C. G.; Dwoskin L. P. Structural Modifications to Tetrahydropyridine-3-Carboxylate Esters En Route to the Discovery of M5-Preferring Muscarinic Receptor Orthosteric Antagonists. J. Med. Chem. 2013, 56, 1693–1703. 10.1021/jm301774u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campeau L. C.; Dolman S. J.; Gauvreau D.; Corley E.; Liu J.; Guidry E. N.; Ouellet S. G.; Steinhuebel D.; Weisel M.; O’Shea P. D. Convergent Kilo-Scale Synthesis of a Potent Renin Inhibitor for the Treatment of Hypertension. Org. Process Res. Dev. 2011, 15, 1138–1148. 10.1021/op2001063. [DOI] [Google Scholar]
- Bommagani S.; Lee N. R.; Zhang X.; Dwoskin L. P.; Zheng G. Synthesis of O- and N-Alkylated Products of 1,2,3,4-Tetrahydrobenzo[c][2,7]Naphthyrin-5(6H)-One. Tetrahedron Lett. 2015, 56, 6472–6474. 10.1016/j.tetlet.2015.09.156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hopkins A. L.; Groom C. R.; Alex A. Ligand Efficiency: A Useful Metric for Lead Selection. Drug Discovery Today 2004, 9, 430–431. 10.1016/S1359-6446(04)03069-7. [DOI] [PubMed] [Google Scholar]
- Shultz M. D. Setting Expectations in Molecular Optimizations: Strengths and Limitations of Commonly Used Composite Parameters. Bioorg. Med. Chem. Lett. 2013, 23, 5980–5991. 10.1016/j.bmcl.2013.08.029. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.