

Abstract

Based on the potent Kv7 agonist RL-81, we prepared new lead structures with greatly improved selectivity for Kv7.2/Kv7.3 over related potassium channels, i.e., Kv7.3/Kv7.5, Kv7.4, and Kv7.4/7.5. RL-36 and RL-12 maintain an agonist EC2x of ca. 1 μM on Kv7.2/Kv7.3 in a high-throughput assay on an automated electrophysiology platform in HEK293 cells but lack activity on Kv7.3/Kv7.5, Kv7.4, and Kv7.4/7.5, resulting in a selectivity index SI > 10. RL-56 is remarkably potent, EC2x 0.11 ± 0.02 μM, and still shows an SI = 2.5. We also identified analogues with significant selectivity for Kv7.4/Kv7.5 over Kv7.2/Kv7.3. The extensive use of fluorine in iterative core structure modifications highlights the versatility of these substituents, including F, CF3, and SF5, to span orders of magnitude of potency and selectivity in medicinal chemistry lead optimizations.
Keywords: Kv7, KCNQ, potassium channel agonist, retigabine, RL-81
Potassium channels represent the most numerous group among the ca. 150 cation channels expressed in the human genome. They trigger a multitude of physiological responses, such as the frequency and duration of action potentials in the brain and heart, muscle contraction, transmitter release, and hormonal secretion. In this realm, the five distinct voltage-dependent Kv7 channel subunits, encoded by the KCNQ genes in humans, are of considerable interest to medicinal chemists. To a significant extent, this is due to their close association with various diseases as well as the tractability of their pharmacological response to small-molecule modifiers.1 Epilepsy, neuropathic pain, tinnitus, migraine, and cardiovascular, metabolic, and psychiatric diseases are frequently associated with defective Kv7 channels.2 The Kv7.1 subtype is primarily located in the cell membranes of cardiac tissue and is critical for the repolarization of the cardiac action potential; aberrations can lead to a prolongation of the QT interval. Kv7.2–Kv7.5 are active in neuronal tissue, and Kv7.4 and Kv7.5 are also expressed in skeletal and smooth muscle cells. The neuronal channels are assembled as homo- or heterotetramers; i.e., they form Kv7.2/Kv7.3, Kv7.3/Kv7.4, Kv7.4/Kv7.5, etc., channel complexes.1,3
Kv7.2/Kv7.3 heterotetramers are slow-activating, voltage-dependent, non-inactivating potassium channels that are open at hyperpolarizing (subthreshold) potentials. Accordingly, they control the subthreshold membrane potential and serve as powerful brakes on neuronal firing activity. Genetic or acquired reductions in the activity of Kv7.2/Kv7.3 heterodimers, underlying native Kv7 currents in neurons,4 are linked with brain disorders that are characterized by neuronal hyperexcitability, including anxiety, mania, tinnitus, and ADHD.5 Consequently, Kv7.2–Kv7.5 channel activators are capable of compensating for a constitutive decline in Kv7 channel activity and thus decrease neuronal excitability and block neurotransmitter release.3 Flupirtine, retigabine, P-retigabine, SF-0034, acrylamide (S)-1, SMB-1, γ-aminobutyric acid (GABA), gabapentine, ICA-27243, diclofenac, BMS-204352, celecoxib, ML-213, NS-1643, and compound 14 are representative structures for the chemical space covered by small-molecule Kv7 activators (Figure 1).3,6−8
Figure 1.

Structures of Kv7 channel activators.
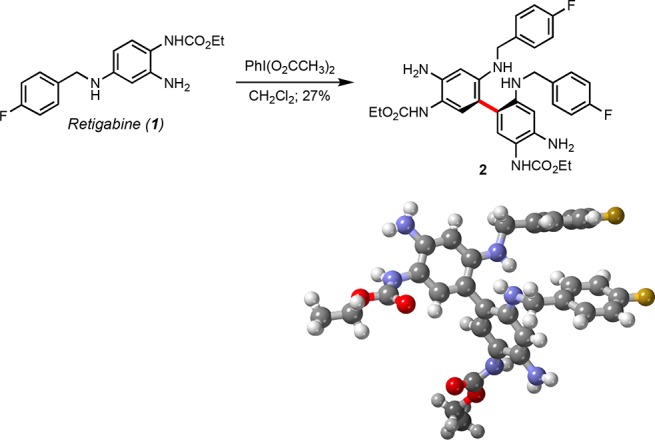
Pharmacological activation of Kv7 channels by retigabine, an FDA-approved antiepileptic drug that serves as a pan-agonist at Kv7.2–Kv7.5 channels, prevents seizures, neuropathic pain, and the development of tinnitus.9,10 In the CNS of vertebrates, Kv7.2/Kv7.3 (KCNQ2/3) propagate the M-current, a muscarinic-inhibited trigger of neuronal excitability. Moreover, natural, nonpharmacologically driven recovery in Kv7.2/Kv7.3 channel activity is linked to resilience to noise-induced tinnitus.11 However, the possibility for severe side effects with retigabine, including urinary retention, blue skin, and retinal discoloration, resulted in a “black box” warning by the FDA for this drug, at first limiting its use to patients who had not responded to alternative treatments, until the drug was discontinued in 2017.12 We hypothesized that these undesirable side effects were due in part to the poor selectivity of retigabine among Kv7.2–Kv7.5 channels as well as the low solubility of its metabolic degradation products. Interestingly, exposure of retigabine to a hypervalent iodine reagent as a model for the oxidative metabolism of xenobiotic substances13 generated the colored, poorly soluble dimer 2, whose structure was confirmed by X-ray analysis (Scheme 1). Dimer 2 has also been identified as an impurity in the retigabine API.14
Scheme 1. Oxidation of Retigabine and X-ray Structure of Symmetrical Dimer 2.
To generate potent Kv7.2/Kv7.3-selective channel activators, we previously partitioned retigabine’s chemical structure into three distinct zones (Figure 2).15 In these preliminary structure–activity relationship (SAR) studies, we mainly modified zones 1 and 3 and subsequently combined the beneficial modifications found for zones 1 and 3 with a previously known beneficial modification on zone 2, which had also been used in the development of the retigabine analogue SF-0034.10 Our guiding principle for these preliminary SAR studies was to modulate both steric and, in particular, electronic features in zone 1 by introducing fluoride (F), trifluoromethyl (CF3), and pentafluorosulfanyl (SF5), which is considered a “super-trifluoromethyl” substituent.16,17 This strategy led us to the potent and selective benzylamines NR-45, NR-04, and RL-86 (Figure 2).15 Furthermore, by introducing a CF3 group at the 4-position of the benzylamine moiety, combined with a fluorine atom at the 3-position of the aniline ring, we generated RL-81, a new Kv7.2/Kv7.3-specific activator (EC50 = 190 nM) that was ca. 3 times more potent than the most active retigabine analogue, SF-0034 (EC50 = 0.60 μM), at shifting the voltage dependence of Kv7.2/Kv7.3 channels to more hyperpolarized potentials.15 Hence, RL-81 became a promising lead structure for the development of clinical candidates for treating or preventing neurological disorders associated with neuronal hyperexcitability, including tinnitus and epilepsy.
Figure 2.
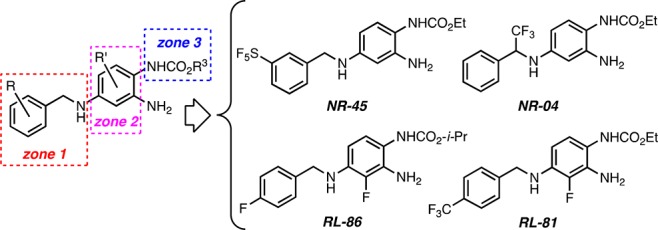
Structures of Kv7 channel activating retigabine analogs.
With RL-81 as a new lead, we focused our attention on zone 2 modifications, in particular the position and the number of fluorine groups on the three unsubstituted carbons of the triaminobenzene moiety, in part motivated by the goal to find potent analogues where the dimerization site in 2 was blocked. We also reoptimized structural features of zone 1 and zone 3 and developed a computational SAR model.
The syntheses of RL-81 and 19 new analogues are summarized in Schemes 2–5. SNAr of benzylamines 3a–3c with fluoroarenes 4a and 4b in the presence of catalytic iodine generated anilines 5a–5d in 63–84% yield (Scheme 2). Reduction of the nitro group and treatment with ethyl chloroformate led to RL-81, RL-73, RL-02, and RL-72 in 47%, 45%, 37%, and 24% yield, respectively. RL-81 was treated with Togni’s trifluoromethylating reagent 6(18) in acetonitrile to obtain the R5 trifluoromethyl derivative RL-073, albeit in a low yield of 17%. For the preparation of the zone 3 modified iso-propyl and cyclo-propyl carbamates RL-32 and RL-56, nitroaniline 5a was reduced with Pd/C under an atmosphere of hydrogen gas in ethanol, and the resulting diamine was selectively acylated with iso-propyl chloroformate and cyclo-propyl chloroformate in 63% and 40% yield, respectively.
Scheme 2. Preparation of RL-81 and 6 Structurally Closely Related Analogues from Monosubstituted Benzylamines 3a–3c.

Scheme 5. Preparation of 3 Analogues Containing the 3-Pyridylmethylamine and the 3-Cyano-2-pyridylmethylamine Moieties in Zone 1.

The use of 2-fluoro-4-trifluoromethylbenzylamine 3d introduced an additional fluorine group into the zone 1 moiety, and in an analogous series of SNAr, zinc reduction, and carbamoylation transformations, ethyl carbamate RL-18 and cyclo-propyl carbamates RL-35, RL-36, RL-46, and RL-50 were obtained in moderate to good yields (Scheme 3). In this series of analogues, we also introduced up to two additional fluorine substituents at zone 2 R5 and R6 positions.
Scheme 3. Preparation of 5 Analogues Containing the 2-Fluoro-4-trifluoromethylbenzylamine Moiety in Zone 1.

A further diversification of the RL-81 lead structure was accomplished by the use of (5-(trifluoromethyl)pyridin-2-yl)methanamine 3e and (3-fluoro-5-(trifluoromethyl)pyridin-2-yl)methanamine 3f (Scheme 4). Reduction of the nitro group in 5i, isolated in 75% yield from the coupling of 3e with 4a, with Pd/C and acylation with ethyl chloroformate provided RL-31 in 43% yield. A zinc reduction and cyclo-propyl chloroformate were used for the preparation of RL-68 in 23% yield from 5i. The penta- and hexa-fluorinated intermediates 5j, 5k, and 5l were obtained from the coupling of 3f with 4a, 4c, and 4d in 45%, 58%, and 37% yield, respectively. Zinc reduction and treatment with cyclo-propyl chloroformate then yielded the corresponding carbamates RL-96, RL-01, and RL-12 in 33%, 37%, and 41%, respectively.
Scheme 4. Preparation of 5 Analogues Containing the 2-Pyridylmethylamine Moiety in Zone 1.

Finally, we also synthesized analogues with a pyridine in zone 1 by coupling of amine 3g with nitrobenzene 4a, which gave SNAr product 5m in 85% yield (Scheme 5). Reduction and acylation of 5m provided the carbamates RL-24 and RL-67 in 60% and 28% yield, respectively. The 2-pyridyl analogue RL-23, which also contained a cyano group next to the nitrogen of the heterocycle as a chemical replacement mimicking a pyridazine ring,19 was generated in an overall yield of 13% by a reductive amination of pyridine 3h with aniline 4f, followed by zinc reduction and acylation.
The biological evaluation of the new analogues, including RL-81 as a positive standard, was performed in the Lilly OIDD program.20,21 Ion channel agonistic potency was determined in a high-throughput assay using the automated electrophysiology IonWorks Barracuda (IWB) platform in HEK293 cells (Table 1). The resulting 10-point concentration–response curves provide a measure of compound efficacy by determining the agonist concentration that doubles the conductance at the voltage leading to 15% channel activation (EC2x).22 When the EC2x value is lower, the agonist is more potent.
Table 1. Efficacy of RL-81 Analogues in Human Kv7 Channel Conductance Studiesa.
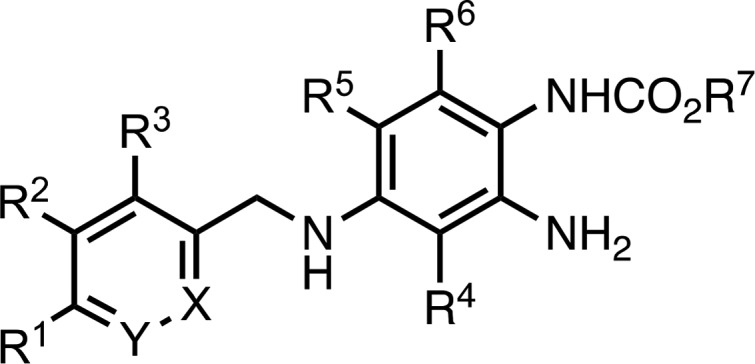
| compd | X | Y | R1 | R2 | R3 | R4 | R5 | R6 | R7 | Kv7.2/3 EC2x ± SD [μM] | Kv7.3/5 EC2x ± SD [μM] | Kv7.4 EC2x ± SD [μM] | Kv7.4/5 EC2x ± SD [μM] | SI |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| FP | CH | CH | F | H | H | Nb | H | H | Et | 2.00 ± 0.69 | 1.58 ± 0.69 | 5.62 ± 1.86 | 3.50 ± 2.68 | 1.8 |
| RG | CH | CH | F | H | H | H | H | H | Et | 0.33 ± 0.08 | 0.34 ± 0.04 | ND | ND | ND |
| RL-81 | CH | CH | CF3 | H | H | F | H | H | Et | 0.26 ± 0.13 | 0.29 ± 0.06 | 0.10 ± 0.04 | 0.09 ± 0.04 | 0.3 |
| RL-73 | CH | CH | H | CF3 | H | F | H | H | Et | 0.14 ± 0.05 | 0.20 ± 0.08 | 0.40 ± 0.13 | 0.66 ± 0.01 | 4.7 |
| RL-02 | CH | CH | H | SF5 | H | F | H | H | Et | 0.36 ± 0.25 | 0.21 ± 0.11 | 0.65 ± 0.39 | 0.65 ± 0.28 | 1.8 |
| RL-72 | CH | CH | CF3 | H | H | H | F | H | Et | 1.26 ± 0.31 | >10 | 4.65 ± 0.95 | >10 | >7 |
| RL-073 | CH | CH | CF3 | H | H | F | CF3 | H | Et | >10 | >10 | >10 | >10 | ND |
| RL-32 | CH | CH | CF3 | H | H | F | H | H | i-Pr | 0.16 ± 0.11 | 0.16 ± 0.10 | 0.19 ± 0.10 | 4.98 ± 1.12 | 31 |
| RL-56 | CH | CH | CF3 | H | H | F | H | H | c-Pr | 0.11 ± 0.02 | 0.23 ± 0.10 | 0.38 ± 0.30 | 0.28 ± 0.03 | 2.5 |
| RL-18 | CF | CH | CF3 | H | H | F | H | H | Et | 0.18 ± 0.11 | 0.34 ± 0.01 | 0.11 ± 0.04 | 0.30 ± 0.26 | 1.7 |
| RL-35 | CF | CH | CF3 | H | H | F | H | H | c-Pr | 0.59 ± 0.08 | 0.47 ± 0.01 | 0.21 ± 0.19 | 0.35 ± 0.17 | 0.6 |
| RL-36 | CF | CH | CF3 | H | H | F | F | H | c-Pr | 0.93 ± 0.21 | >10 | >10 | >10 | >10 |
| RL-46 | CF | CH | CF3 | H | H | F | H | F | c-Pr | 1.47 ± 0.22 | >10 | 0.55 ± 0.14 | >10 | >7 |
| RL-50 | CF | CH | CF3 | H | H | F | F | F | c-Pr | >10 | >10 | >10 | >10 | ND |
| RL-31 | N | CH | CF3 | H | H | F | H | H | Et | 0.66 ± 0.25 | 0.79 ± 0.26 | 0.85 ± 0.14 | 0.59 ± 0.34 | 0.9 |
| RL-68 | N | CH | CF3 | H | H | F | H | H | c-Pr | 0.79 ± 0.03 | 0.72 ± 0.05 | 0.90 ± 0.02 | 0.96 ± 0.05 | 1.2 |
| RL-96 | N | CH | CF3 | H | F | F | H | H | c-Pr | 1.39 ± 0.20 | >10 | >10 | >10 | >7 |
| RL-01 | N | CH | CF3 | H | F | F | F | H | c-Pr | >10 | >10 | >10 | >10 | ND |
| RL-12 | N | CH | CF3 | H | F | F | H | F | c-Pr | 1.00 ± 0.50 | >10 | >10 | >10 | >10 |
| RL-23 | N | CCN | CF3 | H | H | F | H | H | Et | 1.60 ± 0.19 | 1.30 ± 0.16 | 2.17 ± 0.37 | 2.33 ± 0.89 | 1.5 |
| RL-24 | CH | N | CF3 | H | H | F | H | H | Et | 0.61 ± 0.34 | 0.69 ± 0.35 | 0.95 ± 0.13 | 1.27 ± 0.05 | 2.1 |
| RL-67 | CH | N | CF3 | H | H | F | H | H | c-Pr | 0.71 ± 0.23 | 0.69 ± 0.19 | 1.13 ± 0.72 | 1.36 ± 0.70 | 1.9 |
EC2x data were obtained from 10-point concentration curves and 2–5 individual assay determinations, with the exception of FP, for which 6–431 repeats were determined; SD = standard deviation; SI = selectivity index = EC2x(Kv7.4/5)/EC2x(Kv7.2/3); ND = not determined.
FP has a pyridine ring nitrogen at the R4-substituted carbon position.
Compared to the literature standards, flupirtine (FP) and retigabine (RG), our previous lead structure RL-81 demonstrated ca. 8–50 times higher (vs FP) or slightly higher (vs RG) efficacy at all tested Kv7 channels (Table 1) in this assay. Since we were mainly interested in optimizing central (Kv7.2/Kv7.3) over peripheral (Kv7.4/Kv7.5) tissue activities, we also calculated a selectivity index (SI), defined as the inverse ratio of the EC2x values for these two channel types (i.e., SI = EC2x(Kv7.4/Kv7.5)/EC2x(Kv7.2/Kv7.3); when this ratio is higher, the selectivity of the agent for the Kv7.2/Kv7.3 channels is better). Interestingly, RL-81 (SI = 0.3) proved to be considerably more potent at Kv7.4/Kv7.5 than FP (SI = 1.8), suggesting a potentially adverse property profile and setting the stage for further medicinal chemistry optimizations. One of our first modifications was to move the CF3 group in zone 1 in RL-81 from the para- to the meta-position. To our delight, the resulting analogue, RL-73, was twice as potent at Kv7.2/Kv7.3 than RL-81, while substantially decreasing activity at Kv7.4/Kv7.5, resulting in a superior SI = 4.7. These findings were further substantiated by the SF5-containing analogue RL-02, which showed an equivalent channel activity profile to RL-73 and also had a more favorable SI = 1.8. Since structurally the only difference between RL-73 and RL-02 is the switch from a meta-CF3- to a meta-SF5-substituent in zone 1, these assay data further validate the potential for biological mimicry between the trifluoromethyl and the pentafluorosulfanyl groups.16,17 Interestingly, while their Kv7.2/Kv7.3 vs Kv7.4/Kv7.5 selectivity is high, both RL-73 and RL-02 have low EC2x values for Kv7.3/Kv7.5 and Kv7.4 in the 0.2–0.6 μM range, comparable to RL-81’s EC2x of 0.29 and 0.10 μM. Therefore, we continued our structure–activity studies and prepared several zone 2 and zone 3 modifications of RL-81.
Moving the fluorine atom from R4 to R5 in RL-72 had a detrimental effect on the EC2x for Kv7.2/Kv7.3, reducing it 5-fold vs RL-81 to 1.26 μM. However, the selectivity was now superb, with EC2x’s of >10, 4.65, and >10 μM for Kv7.3/Kv7.5, Kv7.4, and Kv7.4/7.5, respectively, accounting for an SI > 7. In contrast, installing a CF3 group at the R5 position in RL-073 completely abrogated activity at all channel types. This position appears to be very sensitive to steric bulk.
Surprisingly, changing the ethyl carbamate in zone 3 to an iso-propyl carbamate slightly increased the potency of the resulting RL-32 at Kv7.2/Kv7.3 but vastly decreased the Kv7.4/7.5 EC2x to 4.98 μM, leading to an SI = 31–two orders of magnitude better than the SI = 0.3 of RL-81. The SI = 2.5 of the corresponding cyclo-propyl carbamate analogue, RL-56, fits this trend, since the steric dimensions of a cyclo-propane are roughly in between those of the iso-propane and the ethane groups. RL-56 proved to be the most active analogue on Kv7.2/Kv7.3 prepared to date, with an EC2x of 0.11 ± 0.02 μM. However, both RL-32 and RL-56 also still showed <0.4 μM EC2x potencies at Kv7.3/Kv7.5 and Kv7.4 channels.
For the next SAR iteration, we introduced a 2-fluoro-4-trifluoromethylbenzylamine moiety in zone 1 and systematically varied fluorinations in zone 2. The parent compound in this series, RL-18, was both potent, EC2x = 0.18 ± 0.11 μM, and moderately selective, SI = 1.7 (Table 1). Changing the ethyl to the cyclo-propyl carbamate decreased potency and SI (i.e., RL-35), but as previously found, adding fluorine at R5 or R6 recovered a high selectivity, generating an SI > 10 and SI > 7, respectively, for RL-36 and RL-46. In agreement with the data found for RL-72, the R5-fluorinated RL-36 also lacked agonist activity at Kv7.3/Kv7.5, Kv7.4, and Kv7.4/7.5, and due to its remaining sub-micromolar EC2x = 0.93 at Kv7.2/Kv7.3, RL-36 therefore represents an overall significant improvement over RL-81. In contrast, the hepta-fluorinated RL-50 lost all activity at these channels.
In a final round of SAR investigations, we replaced the trifluorobenzene in zone 1 with trifluoromethylated pyridines. Initially, we focused on the 2-pyridyl analogues RL-31, RL-68, RL-96, RL-01, RL-12, and RL-23. With the exception of the R3-fluorinated RL-96 and RL-12, these analogues demonstrated either low selectivity or low potency. The R3,R4,R6-trifluorinated RL-12, in particular, maintained a respectable 1.00 μM EC2x at Kv7.2/Kv7.3, with an SI > 10 and no detectable agonist activity at other channels. Compared to their 2-pyridyl isomers RL-31 and RL-68, the 3-pyridyl analogues RL-24 and RL-67 showed slightly increased selectivity but essentially equivalent potency, demonstrating that, among the studied chemotypes, the fluorination pattern was the most significant determinant of selectivity and activity.
Since substitution with fluorine has profound effects on the electron distribution in conjugated π-systems, we examined the electrostatically encoded electron-density surfaces of RL-81 vs RL-50, which has additional fluorinations at the benzylamine moiety and, especially, at R5 and R6 of the triaminobenzene, rendering it inactive at all Kv7 channel types (Figure 3). Most significantly, the electron density at the carbamate oxygen and the ortho-aniline nitrogen is significantly decreased as a result of the two additional fluorinations at the triaminobenzene moiety in RL-50. In comparison, RL-46, which only has one additional fluorine atom at R6 vs RL-81 and is still quite active, shows an intermediate electron density at the carbamate oxygen and the ortho-aniline nitrogen. We briefly also explored the possibility that a hindered rotation at the zone 1 benzylamine moiety could be responsible for the lack of activity of the o,o′-disubstituted aniline RL-50. However, a potential energy analysis (HF-6-31G*) suggested a barrier of only 1.5–2.5 kcal/mol for the C–NH–CH2–C dihedral angle for both RL-50 and the o-monosubstituted reference compound RL-46 and geometrically similar energy minima.21 Accordingly, we hypothesize that Kv7.2–5 channel potency is largely due to the electrostatic/H-bonding interactions at the carbamate oxygen and the ortho-aniline, with decreased electron density being detrimental. In contrast, steric and conformational effects at the carbamate and the benzylamine moieties appear to influence channel selectivity. This rationalization is also supported by literature evidence. On the basis of mutation and modeling studies, the tryptophan residue W236 in Kv7.2 (or, analogously and respectively, W265, W242, and W235 in Kv7.3, Kv7.4, and Kv7.5) is thought to be critical for binding and participates in hydrogen bonding interactions with carbamate or amide groups of small-molecule agonists.23,24 In contrast, Kv7.1, which lacks a corresponding W residue, is not activated by retigabine-type molecules.
Figure 3.
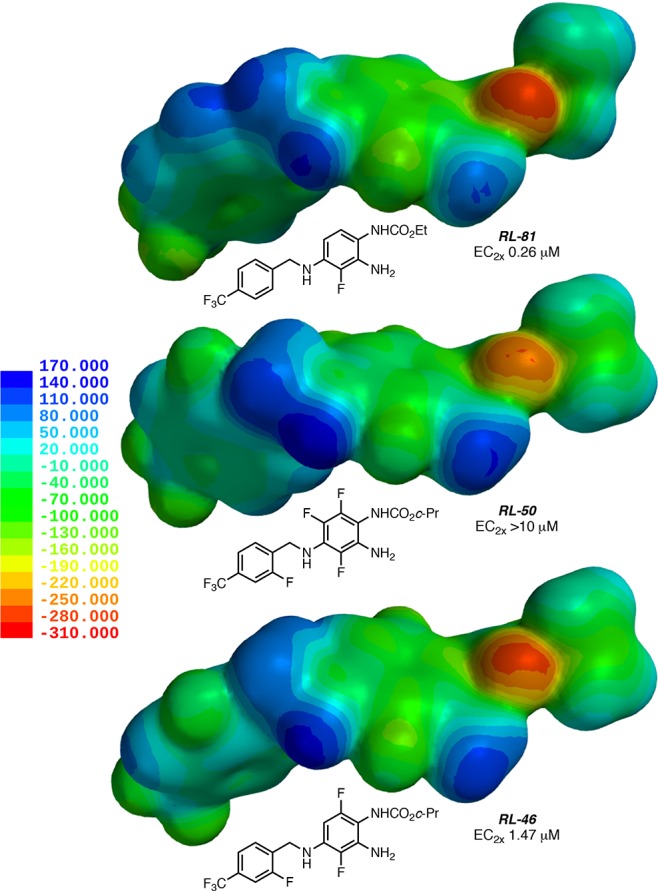
Maps of the electron-density surface encoded with electrostatic potential for RL-81 (top), RL-50 (middle), and RL-46 (bottom). Colors reflect a property range from +170 kJ/mol (blue) to −310 kJ/mol (red).25
An alternative, but possibly complementary, interpretation of the differential selectivities observed for fluorinated and heterocyclic analogues of retigabine and RL-81 could be their preference for different binding sites on the channels and hence different mechanisms of action. While retigabine binds to the pore domain on Kv7.2–Kv7.5, other compounds have been suggested to target the voltage-sensing domain.26 In the future, we plan to perform experimental and computational studies to further investigate this hypothesis on our lead compounds.
In conclusion, we have prepared and characterized analogues of the potent Kv7 agonist RL-81 and obtained several new lead structures with greatly improved selectivity for Kv7.2/Kv7.3 over the other tested potassium channels, i.e., Kv7.3/Kv7.5, Kv7.4, and Kv7.4/7.5. Specifically, RL-36 and RL-12 maintain an agonist EC2x of ca. 1 μM on Kv7.2/Kv7.3 in a high-throughput assay on the automated IWB platform in HEK293 cells but lack activity on Kv7.3/Kv7.5, Kv7.4, and Kv7.4/7.5, resulting in a selectivity index SI > 10. We have also identified an analogue of RL-81, i.e., RL-56, with a ca. 3 times more potent EC2x of 0.11 ± 0.02 μM. Furthermore, RL-56 also shows an SI = 2.5 for Kv7.2/Kv7.3 over Kv7.4/Kv7.5; i.e., it is almost an order of magnitude more selective than RL-81 (SI = 0.3). Accordingly, RL-56, RL-36, and RL-12 represent promising new lead structures for the therapeutic use of selective potassium channel agonists in epilepsy, neuropathic pain, anxiety, mania, ADHD, depression, migraines, and tinnitus. It is also worth mentioning that the R5 substitution on RL-36 should prevent the oxidative dimerization observed for RG; further studies to test this hypothesis, including in vivo metabolite identification, are planned. In a preliminary experiment, subjecting RL-36 and the R5,R6-difluorinated RL-50 to the oxidative conditions from Scheme 1 did not produce a corresponding dimer product by LCMS analysis, and starting materials were recovered in 43% and 51% yield, respectively, based on NMR integration of the crude reaction mixture in the presence of an internal standard. A metabolic stability analysis of RG, RL-81, and eight analogues in pooled human (HLM) and male mouse (MLM) liver microsomes suggested that the pyridine-containing RL-24 (HLM t1/2 = 406 min) and the iso-propyl carbamate RL-32 (HLM t1/2 = 451 min) have longer half-lives than RL-81 (HLM t1/2 = 344 min). Only the cyclo-propyl carbamate RL-36 (HLM t1/2 = 76 min) has a substantially shorter half-live than RL-81, but the microsomal degradation of all tested analogues is significantly faster than RG (HLM t1/2 = 970 min), which we hypothesize will prove advantageous in overcoming RG’s undesired toxicities (see Tables S1 and S2 in the Supporting Information for comprehensive HLM and MLM data).
Our SAR analysis demonstrates the ability of fluorine substituents to substantially alter the potency and selectivity of pharmaceutical candidates. While the utility of fluorinated drug candidates has been highlighted in particular for their increases in metabolic stability, conformational effects, and pKa modulation and for applications as bioisosteric replacements, fewer studies have focused on their effects on specific target affinity.27 The extensive use of fluorine as the guiding principle in iterative core structure modifications in this work further supports the versatility of fluorine-containing substituents, including F, CF3, and SF5, to span orders of magnitude of potency and selectivity in lead optimization.
While our primary goal was the preparation of a Kv7 agonist with a high selectivity for Kv7.2/Kv7.3 over Kv7.4/Kv7.5, we note that this work also identified several analogues with significant selectivity for Kv7.4/Kv7.5 over Kv7.2/Kv7.3, i.e., compounds with an SI < 1, such as RL-81 and RL-35. Kv7.4 is the primary potassium channel in the smooth muscle of the bladder, where it serves to regulate contractility.28 Activation of Kv7.4 leads to membrane hyperpolarization and a resultant loss of contractile function, a possible etiology for the urinary retention side effect of RG. While currently there is no clinical precedence for selective agonists of Kv7.4 and Kv7.5, other medicinal chemistry efforts toward this goal have been reported.29 Therefore, RL-81 and RL-35 could form a foundation for the development of much more selective Kv7.4/Kv7.5 activators, with potential applications for the treatment of visceral smooth-muscle-related diseases.
Acknowledgments
OIDD screening data are supplied courtesy of Eli Lilly and Company—used with Lilly’s permission, and gratefully acknowledged. The authors thank T. S. Maskrey (University of Pittsburgh) for project management and technical assistance, S. J. Geib (University of Pittsburgh) for the X-ray analysis of 2, and J. C. Burnett and R. Cao (University of Pittsburgh) for preliminary Kv7 channel modeling studies.
Glossary
Abbreviations
- KCNQ
potassium channel, voltage-gated, KQT-like subfamily
- ADHD
attention deficit hyperactivity disorder
- HEK293
human embryonic kidney 293
- SAR
structure–activity relationship.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.9b00097.
Experimental details and 1H and 13C NMR spectra for new synthetic intermediates and products, assay information from the Eli Lilly OIDD program, and details of dihedral angle conformational analysis (PDF)
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
This work was supported in part by DOD Award W81XWH-18-1-0623 (to T.T. and P.W.).
The authors declare no competing financial interest.
Notes
CCDC 1899172 contains the supplementary crystallographic data for this paper. These data can be obtained free of charge via www.ccdc.cam.ac.uk/data_request/cif, or by emailing data_request@ccdc.cam.ac.uk, or by contacting The Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ, U.K.; fax: + 44 1223 336033.
Supplementary Material
References
- Grunnet M.; Stroebaek D.; Hougaard C.; Christophersen P. Kv7 Channels as Targets for Anti-Epileptic and Psychiatric Drug-Development. Eur. J. Pharmacol. 2014, 726, 133–137. 10.1016/j.ejphar.2014.01.017. [DOI] [PubMed] [Google Scholar]
- Barrese V.; Stott J. B.; Greenwood I. A. KCNQ-Encoded Potassium Channels as Therapeutic Targets. Annu. Rev. Pharmacol. Toxicol. 2018, 58, 625–648. 10.1146/annurev-pharmtox-010617-052912. [DOI] [PubMed] [Google Scholar]
- Miceli F.; Soldovieri M. V.; Ambrosino P.; Manocchio L.; Mosca I.; Taglialatela M. Pharmacological Targeting of Neuronal Kv7.2/3 Channels: A Focus on Chemotypes and Receptor Sites. Curr. Med. Chem. 2018, 25 (23), 2637–2660. 10.2174/0929867324666171012122852. [DOI] [PubMed] [Google Scholar]
- Jentsch T. J. Neuronal KCNQ Potassium Channels: Physiology and Role in Disease. Nat. Rev. Neurosci. 2000, 1, 21–30. 10.1038/35036198. [DOI] [PubMed] [Google Scholar]
- Li S.; Choi V.; Tzounopoulos T. Pathogenic Plasticity of Kv7.2/3 Channel Activity Is Essential for the Induction of Tinnitus. Proc. Natl. Acad. Sci. U. S. A. 2013, 110, 9980–9985. 10.1073/pnas.1302770110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manville R. W.; Abbott G. W. Gabapentin Is a Potent Activator of KCNQ3 and KCNQ5 Potassium Channels. Mol. Pharmacol. 2018, 94, 1155–1163. 10.1124/mol.118.112953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davoren J. E.; Claffey M. M.; Snow S. L.; Reese M. R.; Arora G.; Butler C. R.; Boscoe B. P.; Chenard L.; DeNinno S. L.; Drozda S. E.; Duplantier A. J.; Moine L.; Rogers B. N.; Rong S.; Schuyten K.; Wright A. S.; Zhang L.; Serpa K. A.; Weber M. L.; Stolyar P.; Whisman T. L.; Baker K.; Tse K.; Clark A. J.; Rong H.; Mather R. J.; Lowe J. A. III Discovery of a Novel Kv7 Channel Opener as a Treatment for Epilepsy. Bioorg. Med. Chem. Lett. 2015, 25, 4941–4944. 10.1016/j.bmcl.2015.04.074. [DOI] [PubMed] [Google Scholar]
- Seefeld M. A.; Lin H.; Holenz J.; Downie D.; Donovan B.; Fu T.; Pasikanti K.; Zhen W.; Cato M.; Chaudhary K. W.; Brady P.; Bakshi T.; Morrow D.; Rajagopal S.; Samanta S. K.; Madhyastha N.; Kuppusamy B. M.; Dougherty R. W.; Bhamidipati R.; Mohd Z.; Higgins G. A.; Chapman M.; Rouget C.; Lluel P.; Matsuoka Y. Novel Kv7 Ion Channel Openers for the Treatment of Epilepsy and Implications for Detrusor Tissue Contraction. Bioorg. Med. Chem. Lett. 2018, 28, 3793–3797. 10.1016/j.bmcl.2018.09.036. [DOI] [PubMed] [Google Scholar]
- Wu C.; Gopal K. V.; Lukas T. J.; Gross G. W.; Moore E. J. Pharmacodynamics of Potassium Channel Openers in Cultured Neuronal Networks. Eur. J. Pharmacol. 2014, 732, 68–75. 10.1016/j.ejphar.2014.03.017. [DOI] [PubMed] [Google Scholar]
- Kalappa B. I.; Soh H.; Duignan K. M.; Furuya T.; Edwards S.; Tzingounis A. V.; Tzounopoulos T. Potent KCNQ2/3-Specific Channel Activator Suppresses in Vivo Epileptic Activity and Prevents the Development of Tinnitus. J. Neurosci. 2015, 35, 8829–8842. 10.1523/JNEUROSCI.5176-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S.; Kalappa B. I.; Tzounopoulos T. Noise-Induced Plasticity of KCNQ2/3 and HCN Channels Underlies Vulnerability and Resilience to Tinnitus. eLife 2015, 4, e07242. 10.7554/eLife.07242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eskioglou E.; Perrenoud M. P.; Ryvlin P.; Novy J. Novel Treatment and New Drugs in Epilepsy Treatment. Curr. Pharm. Des. 2017, 23, 6389–6398. 10.2174/1381612823666171024143541. [DOI] [PubMed] [Google Scholar]
- Xu D.; Penning T. M.; Blair I. A.; Harvey R. G. Synthesis of Phenol and Quinone Metabolites of Benzo[a]Pyrene, a Carcinogenic Component of Tobacco Smoke Implicated in Lung Cancer. J. Org. Chem. 2009, 74, 597–604. 10.1021/jo801864m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dousa M.; Srbek J.; Radl S.; Cerny J.; Klecan O.; Havlicek J.; Tkadlecova M.; Pekarek T.; Gibala P.; Novakova L. Identification, Characterization, Synthesis and HPLC Quantification of New Process-Related Impurities and Degradation Products in Retigabine. J. Pharm. Biomed. Anal. 2014, 94, 71–76. 10.1016/j.jpba.2014.01.042. [DOI] [PubMed] [Google Scholar]
- Kumar M.; Reed N.; Liu R.; Aizenman E.; Wipf P.; Tzounopoulos T. Synthesis and Evaluation of Potent KCNQ2/3-Specific Channel Activators. Mol. Pharmacol. 2016, 89, 667–677. 10.1124/mol.115.103200. [DOI] [PubMed] [Google Scholar]
- Wipf P.; Mo T.; Geib S. J.; Caridha D.; Dow G. S.; Gerena L.; Roncal N.; Milner E. E. Synthesis and Biological Evaluation of the First Pentafluorosulfanyl Analogs of Mefloquine. Org. Biomol. Chem. 2009, 7, 4163–4165. 10.1039/b911483a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alverez C.; Arkin M. R.; Bulfer S. L.; Colombo R.; Kovaliov M.; Laporte M. G.; Lim C.; Liang M.; Moore W. J.; Neitz R. J.; Yan Y.; Yue Z.; Huryn D. M.; Wipf P. Structure-Activity Study of Bioisosteric Trifluoromethyl and Pentafluorosulfanyl Indole Inhibitors of the AAA ATPase p97. ACS Med. Chem. Lett. 2015, 6, 1225–1230. 10.1021/acsmedchemlett.5b00364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisenberger P.; Gischig S.; Togni A. Novel 10-I-3 Hypervalent Iodine-Based Compounds for Electrophilic Trifluoromethylation. Chem. - Eur. J. 2006, 12, 2579–2586. 10.1002/chem.200501052. [DOI] [PubMed] [Google Scholar]
- Southall N. T.; Ajay Kinase Patent Space Visualization Using Chemical Replacements. J. Med. Chem. 2006, 49, 2103–2109. 10.1021/jm051201m. [DOI] [PubMed] [Google Scholar]
- For information on the, as of 2/1/2019 discontinued, OIDD program, please see https://openinnovation.lilly.com/dd/login.jsp.
- Physicochemical properties of agonists were calculated with Instant JChem 18.13.0 (ChemAxon, Cambridge, MA). A HF-6-31G* dihedral angle/conformational energy analysis was performed with Spartan 18 v. 1.3.0 (Wavefunction, Inc., Irvine, CA).
- For a related, but antagonist assay set up, see:; Priest B. T.; Cerne R.; Krambis M. J.; Schmalhofer W. A.; Wakulchik M.; Wilenkin B.; Burris K. D.. Automated Electrophysiology Assays. In Assay Guidance Manual; Sittampalam S., Coussens N. P., Brimacombe K., Grossman A., Arkin M., Auld D., Austin C., Baell J., Bejcek B., Caaveiro J. M. M., Chung T. D. Y., Dahlin J. L., Devanaryan V., Foley T. L., Glicksman M., Hall M. D., Haas J. V., Inglese J., Iversen P. W., Kahl S. D., Kales S. C., Lal-Nag M., Li Z., McGee J., McManus O., Riss T., Trask O. J., Weidner J. R., Wildey M. J., Xia M., Xu X., Eds. Eli Lilly & Company and the National Center for Advancing Translational Sciences: Bethesda, MD, 2018; pp 493–543; https://www.ncbi.nlm.nih.gov/books/NBK53196/pdf/Bookshelf_NBK53196.pdf. [Google Scholar]
- Schenzer A.; Friedrich T.; Pusch M.; Saftig P.; Jentsch T. J.; Groetzinger J.; Schwake M. Molecular Determinants of KCNQ (Kv7) K+ Channel Sensitivity to the Anticonvulsant Retigabine. J. Neurosci. 2005, 25, 5051–5060. 10.1523/JNEUROSCI.0128-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim R. Y.; Yau M. C.; Kurata H. T.; Galpin J. D.; Ahern C. A.; Seebohm G.; Pless S. A. Atomic Basis for Therapeutic Activation of Neuronal Potassium Channels. Nat. Commun. 2015, 6, 8116. 10.1038/ncomms9116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spartan’10; Wavefunction, Inc.: Irvine CA. Calculations performed using the PM6 semiempirical parametrization set.
- Wang A. W.; Yang R.; Kurata H. T. Sequence Determinants of Subtype-Specific Actions of KCNQ Channel Openers. J. Physiol. 2017, 595, 663–676. 10.1113/JP272762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meanwell N. A. Fluorine and Fluorinated Motifs in the Design and Application of Bioisosteres for Drug Design. J. Med. Chem. 2018, 61, 5822–5880. 10.1021/acs.jmedchem.7b01788. [DOI] [PubMed] [Google Scholar]
- Greenwood I. A.; Ohya S. New Tricks for Old Dogs: KCNQ Expression and Role in Smooth Muscle. Br. J. Pharmacol. 2009, 156, 1196–1203. 10.1111/j.1476-5381.2009.00131.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L.; Qiao G.-H.; Hu H.-N.; Gao Z.-B.; Nan F.-J. Discovery of Novel Retigabine Derivatives as Potent KCNQ4 and KCNQ5 Channel Agonists with Improved Specificity. ACS Med. Chem. Lett. 2019, 10, 27–33. 10.1021/acsmedchemlett.8b00315. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.