

Abstract
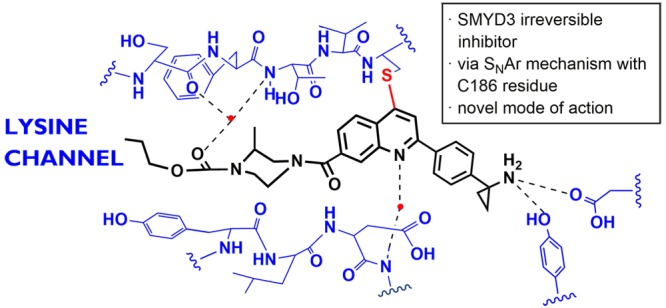
SMYD3 is a histone methyltransferase that regulates gene transcription, and its overexpression is associated with multiple human cancers. A novel class of tetrahydroacridine compounds which inhibit SMYD3 through a covalent mechanism of action is identified. Optimization of these irreversible inhibitors resulted in the discovery of 4-chloroquinolines, a new class of covalent warheads. Tool compound 29 exhibits high potency by inhibiting SMYD3′s enzymatic activity and showing antiproliferative activity against HepG2 in 3D cell culture. Our findings suggest that covalent inhibition of SMYD3 may have an impact on SMYD3 biology by affecting expression levels, and this warrants further exploration.
Keywords: Epigenetics, Covalent, Irreversible, Methyltransferase
SMYD3 (SET and MYND domain containing 3) is a lysine methyltransferase involved in the regulation of cell signaling pathways and gene transcription. Early reports indicated that SMYD3 methylates histones such as histone 3 on lysine 4 (H3K4) and histone 4 on lysine 5 (H4K5),1 while later studies showed that SMYD3 regulates cancer signaling through methylation of MAP3K2 kinase.2 Upon modification at lysine 260, MAP3K2 dissociates from the PP2A phosphatase complex and activates the MEK/ERK signaling pathway, thus suggesting that SMYD3 plays a role in Ras driven cancer progression.2 SMYD3 is found to be overexpressed at high levels in breast, colorectal, hepatocellular, lung, and pancreatic tumors.3 In a more recent report, SMYD3 protein expression was found to be critical for chemically induced liver and colon cancer formation in mice.4
A number of SMYD3 inhibitors have been reported, such as 1 (BCl-121),52 (EPZ031686),6 and 3 (GSK2807)7 (Figure 1). Compound 1 was reported to act by arresting cells at the S/G2 boundary of the cell cycle. Compound 1 was subsequently demonstrated to have an inhibitory effect on H3K4 methylation, including tumor growth inhibition in a HCC mouse xenograft model (HuH7) by Wang et al.8 To the best of our knowledge, this is the first reported evidence of a SMYD3 small molecule inhibitor displaying tumor growth inhibition. Compound 2 is a highly potent SMYD3 inhibitor containing an oxindole-piperidine moiety that is deeply anchored in the lysine channel.6 Unfortunately, it was later shown that 2 did not have any effect against 43 HCC cell lines.9 Compound 3, an analogue of the cofactor S-adenosyl methionine (SAM), suffers from permeability issues.7 More recently, it was shown that silencing SMYD3 gene via CRISPR-Cas 9 leads to no observable effect in the proliferation of cancer cells in cell culture.10 This report is in contrast with early and current literature on SMYD3’s oncogenic role, and the authors objectively concluded that SMYD3′s biology may be more easily uncovered by in vivo studies.
Figure 1.

Published SMYD3 inhibitors and hit 4.
Hit compound 4 was discovered from a high throughput screening campaign of 503 954 compounds. The half-maximal inhibitory concentration (IC50) of 4 was 4.78 μM, which made it a reasonable chemical starting point considering its molecular weight (401.9 g mol–1). Early structure–activity relationship (SAR) was established by varying the length of the carbamate group (Table 1). Shortening the ethyl to a methyl (compound 5) led to a 20-fold loss in potency while lengthening it (compound 6) led to >10-fold improvement. However, lengthening it further did not improve the potency (compound 7). The amide analogue 8 was 4-fold less potent than carbamate 6, suggesting that the oxygen atom is beneficial for the compound’s activity. This acute sensitivity to the carbamate length hinted its close interaction with the lysine channel, prompting us to design 9 and 10. Both compounds carry a primary amine that is capable of mimicking the lysine chain of SMYD3′s substrate. Unfortunately, both compounds turned out to be poor inhibitors.
Table 1. Early SAR of Hit Scaffold.

| compd | Y | R1 | X | SMYD3 IC50 (μM)a |
|---|---|---|---|---|
| 4 | O | Et | Cl | 4.78 ± 0.026 |
| 5 | O | Me | Cl | 123 ± 0.047 |
| 6 | O | nPr | Cl | 0.37 ± 0.044 |
| 7 | O | nBu | Cl | 0.36 ± 0.030 |
| 8 | CH2 | nPr | Cl | 1.52 ± 0.016 |
| 9 | O | –(CH2)2NH2 | Cl | 132.5 ± 0.06 |
| 10 | CH2 | –(CH2)2NH2 | Cl | >750 |
| 11 | O | Et | H | >750 |
| 12 | O | Et | Me | >750 |
Data represent mean value of duplicate experiments.
The chlorine atom in 4 was essential for activity against SMYD3. Removal or replacing the chlorine (11 and 12) led to inactive compounds. While the overall SAR seemed logical, the importance of the chlorine intrigued us to investigate further. MALDI-TOF analysis revealed that a covalent bond was formed between compound 4 and SMYD3. With hindsight, this result is not surprising, as 4-halopyridinium compounds can react with thiols.11 Nevertheless, it is noteworthy that 4-haloquinolines are rarely described or used as electrophilic warheads compared to the more common acrylamide-type Michael acceptors.
Although
we had successfully identified an irreversible SMYD3 inhibitor,
optimizing covalent inhibitors remained a challenge. This is in part
due to the time dependence of the interaction between a covalent inhibitor
and the target protein.12 In fact, the
specificity constant  is normally used to
evaluate the potency
of covalent inhibitors as it takes into account both the initial equilibrium
of the noncovalent complex as well as the rate of inactivation through
covalent bond formation.13 Since we observed
SAR in our preliminary work, we decided to evaluate future compounds
using IC50 values. Other researchers have shown that IC50 value roughly correlates with
is normally used to
evaluate the potency
of covalent inhibitors as it takes into account both the initial equilibrium
of the noncovalent complex as well as the rate of inactivation through
covalent bond formation.13 Since we observed
SAR in our preliminary work, we decided to evaluate future compounds
using IC50 values. Other researchers have shown that IC50 value roughly correlates with  .14
.14
Using compound 6 as reference, >100 analogues were synthesized bearing substituents on the piperazine ring. Some of these included chiral substituents, simple or complex bridged-piperazines, and structurally related piperidines (not shown). None of these showed improvements in IC50’s, except for compound 13, where a simple methyl group is attached (Table 2). The stereochemical orientation of the methyl group proved to be inconsequential. Next, the importance of the cyclohexyl ring in the hit 4 was evaluated by studying compounds 14 and 15. Shrinking or expanding the ring size in both cases were not beneficial.
Table 2. SAR of Alkyl Ring Using Compound 6 as Reference.


Data represent mean value of duplicate experiments.
C7-substitution of the cyclohexyl with either methyl or aminomethyl group gave compounds 16 and 17, respectively, both of which are less active than 6. C8-Substitution with a methyl group also proved ineffective (compound 18). C6-Substitution with a methyl also yielded a less active compound 19, but C6-aminomethyl provided compound 20 possessing the same activity as 6. Encouraged by this result, we synthesized a similar analogue 21, which gave an improved IC50 = 0.067 μM. Gratifyingly, an X-ray cocrystal structure of the ternary complex of 21, SMYD3 and SAM was solved at 2.1 Å resolution (PDB ID: 5YJO, Figure 2). This structure confirmed several important features of 21. The Cys186 residue indeed forms a bond at the C9-position and the propyl carbamate is deeply anchored in the lysine channel (3.7 Å from SAM). The C6-aminomethyl group, on the other hand, did not form a discernible interaction with the protein, but it is clearly oriented toward Asp332 (3.7 Å distance).
Figure 2.
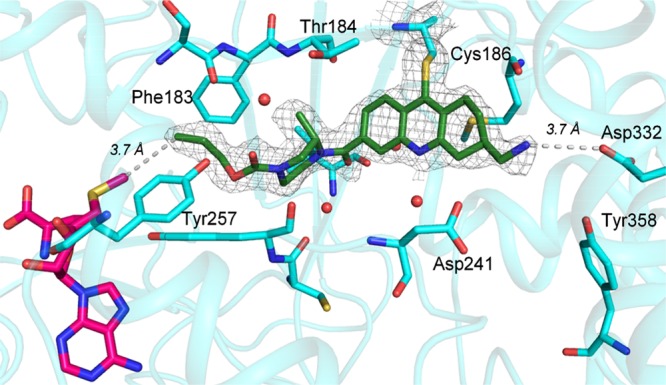
Structure of compound 21 (green) and SAM (pink) bound to SMYD3 (cyan) (PDB ID: 5YJO). SMYD3 residues 3.5 Å away from 21 are shown in stick representation. Tyr358 and Asp332 are at 3.7 and 5.1 Å distance from 21 respectively. The electron density collected cannot detect the absolute configuration of the C6-aminomethyl group and the preferred isomer shown based on the model.
At this point, our curiosity toward the reactivity of hit scaffold led us to perform kinetic studies using glutathione as an external nucleophile.13 Using the known Michael acceptor 22, its rate of consumption was monitored in the presence of excess glutathione over time (Table 3). Using the same protocol to study compound 13, it was evident that 13 is much less reactive than 22 toward glutathione with a half-life of 14.4 h. The higher solubility imparted by the free amine in 21 gave an excellent R2 value for the best-fit line. A T1/2 value of 23 h was obtained signifying that both 13 and 21 are equally unreactive, and the gain in SMYD3 inhibitory potency is not due to an increase in kinact but an optimized KI.
Table 3. Comparison of Reactivity of Compounds Using Glutathione as External Nucleophile.

Values were measured in parallel runs.
Poor R2 value was obtained.
In order to further evaluate these compounds, they were tested for permeability in the Caco-2 transport assay and microsomal stability (Table 4). Compound (S)-13 exhibited intermediate permeability with a Papp of 10.35 × 10–6 cm s–1 (A–B) with no efflux. However, compound (S)-13 displayed low microsomal stability, with T1/2’s of <3 min in both human and mouse liver microsomes (H/MLM). Initial hit 4 also displayed poor microsomal stability with a T1/2 < 9 min, strongly suggesting that the pharmacophore had metabolic stability issues. Introducing an amine group (20, 21) led to compounds with poor permeability.
Table 4. In Vitro Pharmacokinetics of Selected Compounds.
| HLM |
MLM |
||||
|---|---|---|---|---|---|
| compd | Caco-2 Papp (10–6 cm s–1) | T1/2 (min) | CL′int (μL/min/mL) | T1/2 (min) | CL′int (μL/min/mL) |
| 4 | n.d. | 9 | 107 | 4 | 245 |
| (S)-13 | 10.35 (A–B), 8.29 (B–A) | 3 | 304 | 1 | 858 |
| 20 | 4.43 (A–B), 16.20 (B–A) | n.d. | n.d. | n.d. | n.d. |
| 21 | 0 (A–B), 2.19 (B–A) | n.d. | n.d. | n.d. | n.d. |
In an attempt to identify the “metabolic hotspot”, a series of quinoline derivatives 23–29 were synthesized (Table 5). However, 23 is 10-fold less active than 6. While the HLM stability improved (T1/2 = 40 min), the compound was still unstable against MLM. We found that attachment of a C2-substituent such as a phenyl group as in 24, was crucial in maintaining the activity against SMYD3 (IC50 = 0.3 μM). But again, the compound had good HLM stability (T1/2 > 30 min), but poor-to-moderate MLM stability. Further improvement in metabolic stability was achieved when a substituent was added to block the 4-position of the phenyl ring (26–29). The successful optimization of both the potency and metabolic stability of quinoline-compounds 23–29 was guided by X-ray crystallography. Based on the structural data of 21 (Figure 2), residues Asp332 and Tyr358 are potential sites for salt bridge or hydrogen bond interactions. By changing the scaffold to a quinoline core, we were now able to expand our molecules outward from the C2-position. Consequently, the improved IC50 observed for 29 suggests that a significant interaction has been made with the protein. An X-ray crystal structure of compound 29 with SMYD3 was successfully solved at 2.35 Å resolution (PDB ID: 6ILJ, Figure 3A). This structure shares similar features to that with 21, specifically the covalent bond between Cys186 and the C4 position of the quinoline ring and the deep anchoring of the propyl carbamate in the lysine channel. This structure differs in the orientation of the piperazine, since the methyl group is on a different position. The most distinct feature of this structure is the interaction between cyclopropylamine of 29 and Asp332 and Tyr358, showing an intimate distance of 2.9 and 3.3 Å respectively. An overlay of the structures of 21 and 29 in Figure 3B depicts the amine in 29 is in much closer proximity to the acidic amino acid residues.
Table 5. Optimization of Quinoline Compounds 23–29.


Data represent mean value of duplicate experiments (see the Supporting Information).
Figure 3.
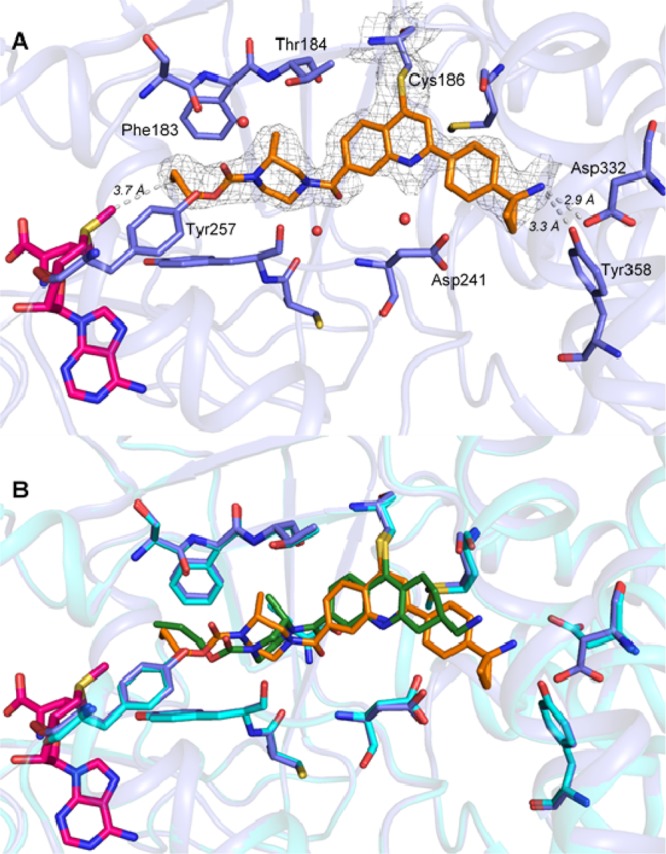
(A) Structure of compound 29 (orange) and SAM (pink) bound to SMYD3 (slate) (PDB ID: 6ILJ). SMYD3 residues 3.5 Å away from 29 are shown in stick representation. (B) Overlay of structures of compound 21 and 29 bound to SMYD3.
Three compounds 24, 28, and 29 were selected for 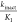 measurement (Table 5). Both compounds 24 and 28 have similar IC50 and
measurement (Table 5). Both compounds 24 and 28 have similar IC50 and 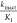 values. Viewing the kinact or KI values
individually
suggests that both molecules are just as reactive as they are capable
at reversibly binding SMYD3 before the covalent reaction. Compound 29, on the other hand, has a
values. Viewing the kinact or KI values
individually
suggests that both molecules are just as reactive as they are capable
at reversibly binding SMYD3 before the covalent reaction. Compound 29, on the other hand, has a 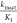 value that
is larger, and it is clear that
the main contributor is the almost 10-fold lower KI value, indicating that we have optimized the initial
reversible binding to SMYD3 with 29.
value that
is larger, and it is clear that
the main contributor is the almost 10-fold lower KI value, indicating that we have optimized the initial
reversible binding to SMYD3 with 29.
Next, we investigated the SMYD3 expression levels in a panel of human cell lines (see the Supporting Information). Due to our interest in HCC, further cellular studies were conducted using HepG2 cells. Endogenous methylation of MAP3K2 was observed in HepG2 cells when tested using our customized anti-MAP3K2-Me2/3 antibody (Figure 4A). Depletion of SMYD3 expression levels was performed using CRISPR-Cas 9 whereby 4 HepG2 clones were generated (including control), effecting different extents of SMYD3 knockdown (Figure 4B). Of the two full-knockout clones 3 and 4, clone 4 showed a significantly lower level of MAP3K2-Me2/3 when compared against the control clone. In all cases, we did not observe full inhibition of methylation of MAP3K2. The growth rate of these clones was monitored over a 7 day period in a 2D monolayer cell culture and was found to have no significant difference from the parental HepG2 cells. This observation is consistent with the findings in the publication by Thomenius et al.10 Cell-based assays using traditional 2D cell cultures have demonstrated to be a valuable method for proliferation studies, but its limitations have been increasingly documented.15 The 3D cell assays are known to stimulate tumor microenvironment better in comparison and may lead to closer predictions to in vivo behavior.16−18 Thus, when cultured in an anchorage-independent condition (3D soft agar assay), HepG2 clones 2–4 showed reduced proliferation corresponding to the degree of SMYD3 gene silencing, with significant inhibition of growth for clone 4 (Figure 4C). In fact, such colony formation inhibition by SMYD3 knockdown has been demonstrated in other instances on human cancer cell lines.2,8,19,20
Figure 4.

(A) Detection of SMYD3 and MAP3K2-me2/me3 in HepG2 cells using Western blot analysis. (B) Reduction of MAP3K2-me2/me3 using different HepG2 clones with SMYD3 knockout. (C) Inhibition of proliferation of HepG2 cells in anchorage-independent condition.
Next, 24–29 were tested rapidly using the CellTiter-Glo luminescent cell viability assay on HepG2 cells. Consistent with our knockout studies, we did not see a significant antiproliferative activity against HepG2 cells with the exception of 25 and 29 (Table 6). Based on these values, 25 and 29 were tested with HepG2 cells in the 3D soft agar assay and both compounds did have an effect on the proliferation of HepG2 colonies. Compounds 25 and 29 showed similar activities with GI50’s of 0.79 and 1.04 μM, respectively, although 29 is the more potent compound. Compound 30 which is a close noncovalent analogue did not have antiproliferative activity.
Table 6. Effect of Compounds 4, 24−25, and 27−30 on HepG2.
| HepG2
GI50 (μM) |
|||
|---|---|---|---|
| compd | SMYD3 IC50 (μM) | 2D | 3D |
| 4 | 4.78 ± 0.026 | >100 | n.d. |
| 24 | 0.3 ± 0.017 | >100 | n.d. |
| 25 | 0.17 ± 0.014 | 24.36 | 0.79 |
| 27 | 0.26 ± 0.026 | >100 | n.d. |
| 28 | 0.25 ± 0.015 | >100 | n.d. |
| 29 | 0.0117 ± 0.0109 | 17.69 | 1.04 |
| 30 | >250 | 60.77 | >10 |
The inhibition of cellular MAP3K2 methylation was quantified using the capillary isoelectric focusing (cIEF) separation technique in the nanofluidic proteomic immunoassay.21 Using this assay, ∼40% inhibition of MAP3K2-me2/me3 was observed with SMYD3-KO HepG2 clone 4 (Figure 5A). Since full inhibition of MAP3K2 methylation was not observed previously using Western blot analysis, we were not too surprised by this result, which might be due to a limitation of the customized antibody. HepG2 cells treated with 25 showed 55% inhibition of MAP3K2 methylation, similar to the SMYD3-KO clone (Figure 5B). This observed cellular potency can be attributed to the covalent mechanism of action of our compounds via the irreversible inhibition of SMYD3. This is further verified when the noncovalent analogue 30 did not inhibit cellular MAP3K2 methylation. Lastly, 29 showed an improved 66% inhibition of MAP3K2 methylation, possibly due to a combination of its covalent mechanism and optimized molecular recognition (Figure 5C).
Figure 5.

(A) Relative levels of MAP3K2-me2/me3 in wild type HepG2 vs SMYD3 KO HepG2 clone 4. (B) Relative levels of MAP3K2-me2/me3 in HepG2 when treated with DMSO and compounds 25 and 30 at 20 μM for 24 h. (C) Relative levels of MAP3K2-me2/me3 in HepG2 when treated with DMSO and compounds 25 and 29 at 20 μM for 24 h. (D) Chemical structure of 30.
A Western blot analysis was conducted to probe the effect of this series of inhibitors on SMYD3 protein levels in HepG2 cells (see the Supporting Information). Interestingly, compound 29 showed significant reduction of SMYD3 protein levels up to 50% reduction compared to DMSO control when tested at 5 μM with no further difference at 20 μM. Such small molecule induced-protein degradation phenotype in a non-PROTAC context has been described in the literature and in some cases, the extent of protein degradation correlates with antiproliferative activity.22 Since our earlier experiments with SMYD3-knockout showed that SMYD3 is essential for HepG2 cell proliferation in a 3D assay, and our covalent inhibitors 25 and 29 that reduced SMYD3 protein levels also show antiproliferative activity, these evidence suggest that the SMYD3 protein level (and not enzymatic activity alone) is likely to be critical to the viability of these cells. Lastly, compound 29 displayed high selectivity when tested against a panel of methyltransferases, namely, SMYD1-2, G9a, PRDM9, and PRMT5 (see the Supporting Information).
In conclusion, we have designed a new class of inhibitors that covalently modify SMYD3 via a nucleophilic aromatic substitution reaction. These irreversible inhibitors potently inhibit SMYD3 and are chemically less reactive than classical covalent warheads. While studying our compounds in vitro, we found our compounds show an effect on the growth of HepG2 colonies. SMYD3 promotes the proliferation and invasion of HCC cells in vitro and contributes to tumorigenicity and metastasis in vivo.8 A 3D cell culture system better simulates the tumor microenvironment and is useful for assessment of these properties. Lastly, the covalent mode of action of our inhibitors has a profound effect on the biology of SMYD3, which we hope could provide further insight for future studies.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.9b00170.
Synthesis and data for compounds described, crystallography methods and data, biochemical assay conditions, kinetics data, and cell-based assay conditions (PDF)
Author Contributions
† C.H. and S.S.L. contributed equally.
The authors declare no competing financial interest.
Supplementary Material
References
- Foreman K. W.; Brown M.; Park F.; Emtage S.; Harriss J.; Das C.; Zhu L.; Crew A.; Arnold L.; Shaaban S.; Tucker P. Structural and functional profiling of the human histone methyltransferase SMYD3. PLoS One 2011, 6, e22290 10.1371/journal.pone.0022290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazur P. K.; Reynoird N.; Khatri P.; Jansen P. W.; Wilkinson A. W.; Liu S.; Barbash O.; Van Aller G. S.; Huddleston M.; Dhanak D.; Tummino P. J.; Kruger R. G.; Garcia B. A.; Butte A. J.; Vermeulen M.; Sage J.; Gozani O. SMYD3 links lysine methylation of MAP3K2 to Ras-driven cancer. Nature 2014, 510, 283–287. 10.1038/nature13320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamamoto R.; Silva F. P.; Tsuge M.; Nishidate T.; Katagiri T.; Nakamura Y.; Furukawa Y. Enhanced SMYD3 expression is essential for the growth of breast cancer cells. Cancer Sci. 2006, 97, 113–118. 10.1111/j.1349-7006.2006.00146.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarris M. E.; Moulos P.; Haroniti A.; Giakountis A.; Talianidis I. Smyd3 is a Transcriptional Potentiator of Multiple Cancer-Promoting Genese and Required for Liver and Colon Cancer Development. Cancer Cell 2016, 29, 354–366. 10.1016/j.ccell.2016.01.013. [DOI] [PubMed] [Google Scholar]
- Peserico A.; Germani A.; Sanese P.; Barbosa A. J.; di Virgilio V.; Fittipaldi R.; Fabini E.; Bertucci C.; Varchi G.; Moyer M. P.; Caretti G.; del Rio A.; Simone C. A SMYD3 Small- Molecule Inhibitor Impairing Cancer Cell Growth. J. Cell. Physiol. 2015, 230, 2447–2460. 10.1002/jcp.24975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell L. H.; Boriack-Sjodin P. A.; Smith S.; Thomenius M.; Rioux N.; Munchhof M.; Mills J. E.; Klaus C.; Totman J.; Riera T. V.; Raimondi A.; Jacques S. L.; West K.; Foley M.; Waters N. J.; Kuntz K. W.; Wigle T. J.; Scott M. P.; Copeland R. A.; Smith J. J.; Chesworth R. Novel Oxindole Sulfonamides and Sulfamides: EPZ031686, the First Orally Bioavailable Small Molecule SMYD3 Inhibitor. ACS Med. Chem. Lett. 2016, 7, 134–138. 10.1021/acsmedchemlett.5b00272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Aller G. S.; Graves A. P.; Elkins P. A.; Bonnette W. G.; McDevitt P. J.; Zappacosta F.; Annan R. S.; Dean T. W.; Su D. S.; Carpenter C. L.; Mohammad H. P.; Kruger R. G. Structure-Based Design of a Novel SMYD3 Inhibitor that Bridges the SAM-and MEKK2-Binding Pockets. Structure 2016, 24, 774–781. 10.1016/j.str.2016.03.010. [DOI] [PubMed] [Google Scholar]
- Wang Y.; Xie B.-H.; Lin W.-H.; Huang Y.-H.; Ni J.-Y.; Hu J.; Cui W.; Zhou J.; Shen L.; Xu L.-F.; Lian F.; Li H.-P. Amplification of SMYD3 promotes tumorigenicity and intrahepatic metastasis of hepatocellular carcinoma via upregulation of CDK2 and MMP2. Oncogene 2019, 10.1038/s41388-019-0766-x. [DOI] [PubMed] [Google Scholar]
- Mitchell L. H.; Boriack-Sjodin P. A.; Smith S.; Thomenius M.; Rioux N.; Munchhof M.; Mills J. E.; Klaus C.; Totman J.; Riera T. V.; Raimondi A.; Jacques S. L.; West K.; Foley M.; Waters N. J.; Kuntz K. W.; Wigle T. J.; Scott M. P.; Copeland R. A.; Smith J. J.; Chesworth R. Identification of a Novel Potent Selective SMYD3 Inhibitor with Oral Bioavailability. AACR Poster Presentation. Mol. Cancer Ther. 2015, 10.1158/1535-7163.TARG-15-C85. [DOI] [Google Scholar]
- Thomenius M. J.; Totman J.; Harvey D.; Mitchell L. H.; Riera T. V.; Cosmopoulos K.; Grassian A. R.; Klaus C.; Foley M.; Admirand E. A.; Jahic H.; Majer C.; Wigle T.; Jacques S. L.; Gureasko J.; Brach D.; Lingaraj T.; West K.; Smith S.; Rioux N.; Waters N. J.; Tang C.; Raimondi A.; Munchhof M.; Mills J. E.; Ribich S.; Porter Scott M.; Kuntz K. W.; Janzen W. P.; Moyer M.; Smith J. J.; Chesworth R.; Copeland R. A.; Boriack-Sjodin P. A. Small molecule inhibitors and CRISPR/Cas9 mutagenesis demonstrate that SMYD2 and SMYD3 activity are dispensible for autonomous cancer cell proliferation. PLoS One 2018, 13, e0197372 10.1371/journal.pone.0197372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson C. M.; Linsky T. W.; Yoon D. W.; Person M. D.; Fast W. Discovery of halopyridines as quiescent affinity labels: inactivation of dimethylarginine dimethylaminohydrolase. J. Am. Chem. Soc. 2011, 133, 1553–1562. 10.1021/ja109207m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serafimova I. M.; Pufall M. A.; Krishnan S.; Duda K.; Cohen M. S.; Maglathlin R. L.; McFarland J. M.; Miller R. M.; Frodin M.; Taunton J. Reversible targeting of noncatalytic cysteines with chemically tuned electrophiles. Nat. Chem. Biol. 2012, 8, 471–476. 10.1038/nchembio.925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flanagan M. E.; Abramite J. A.; Anderson D. P.; Aulabaugh A.; Dahal U. P.; Gilbert A. M.; Li C.; Montgomery J.; Oppenheimer S. R.; Ryder T.; Schuff B. P.; Uccello D. P.; Walker G. S.; Wu Y.; Brown M. F.; Chen J. M.; Hayward M. M.; Noe M. C.; Obach R. S.; Philippe L.; Shanmugasundaram V.; Shapiro M. J.; Starr J.; Stroh J.; Che Y. Chemical and computational methods for the characterization of covalent reactive groups for the prospective design of irreversible inhibitors. J. Med. Chem. 2014, 57, 10072–10079. 10.1021/jm501412a. [DOI] [PubMed] [Google Scholar]
- Prime M. E.; Andersen O. A.; Barker J. J.; Brooks M. A.; Cheng R. K.; Toogood-Johnson I.; Courtney S. M.; Brookfield F. A.; Yarnold C. J.; Marston R. W.; Johnson P. D.; Johnsen S. F.; Palfrey J. J.; Vaidya D.; Erfan S.; Ichihara O.; Felicetti B.; Palan S.; Pedret-Dunn A.; Schaertl S.; Sternberger I.; Ebneth A.; Scheel A.; Winkler D.; Toledo-Sherman L.; Beconi M.; Macdonald D.; Munoz-Sanjuan I.; Dominguez C.; Wityak J. Discovery and structure-activity relationship of potent and selective covalent inhibitors of transglutaminase 2 for Huntington’s disease. J. Med. Chem. 2012, 55, 1021–1046. 10.1021/jm201310y. [DOI] [PubMed] [Google Scholar]
- Imamura Y.; Mukohara T.; Shimono Y.; Funakoshi Y.; Chayahara N.; Toyoda M.; Kiyota N.; Takao S.; Kono S.; Nakatsura T.; Minami H. Comparison of 2D- and 3D-culture models as drug-testing platforms in breast cancer. Oncol. Rep. 2015, 33, 1837–1843. 10.3892/or.2015.3767. [DOI] [PubMed] [Google Scholar]
- Breslin S.; O’Driscoll L. Three-dimensional cell culture: the missing link in drug discovery. Drug Discovery Today 2013, 18, 240–249. 10.1016/j.drudis.2012.10.003. [DOI] [PubMed] [Google Scholar]
- Lovitt C. J.; Shelper T. B.; Avery V. M. Advanced cell culture techniques for cancer drug discovery. Biology 2014, 3, 345–367. 10.3390/biology3020345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weigelt B.; Ghajar C. M.; Bissell M. J. The need for complex 3D culture models to unravel novel pathways and identify accurate biomarkers in breast cancer. Adv. Drug Delivery Rev. 2014, 69–70, 42–51. 10.1016/j.addr.2014.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S.-Z.; Luo X.-G.; Shen J.; Zou J.-N.; Lu Y.-H.; Xi T. Knockdown of SMYD3by RNA interference inhibits cervical carcinoma cell growth and invasion in vitro. BMB Rep. 2008, 41, 294–299. 10.5483/BMBRep.2008.41.4.294. [DOI] [PubMed] [Google Scholar]
- Cock-Rada A. M.; Medjkane S.; Janski N.; Yousfi N.; Perichon M.; Chaussepied M.; Chluba J.; Langsley G.; Weitzman J. B. SMYD3 Promotes Cancer Invasion by Epigenetic Upregulation of the Metalloproteinase MMP-9. Cancer Res. 2012, 72, 810–820. 10.1158/0008-5472.CAN-11-1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Neill R. A.; Bhamidipati A.; Bi X.; Deb-Basu D.; Cahill L.; Ferrante J.; Gentalen E.; Glazer M.; Gossett J.; Hacker K.; Kirby C.; Knittle J.; Loder R.; Mastroieni C.; MacLaren M.; Mills T.; Nguyen U.; Parker N.; Rice A.; Roach D.; Suich D.; Voehringer D.; Voss K.; Yang J.; Yang T.; Vander Horn P. B. Isoelectric focusing technology quantifies protein signaling in 25 cells. Proc. Natl. Acad. Sci. U. S. A. 2006, 103, 16153–16158. 10.1073/pnas.0607973103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerres N.; Steurer S.; Schlager S.; Bader G.; Berger H.; Caligiuri M.; Dank C.; Engen J. R.; Ettmayer P.; Fisherauer B.; Flotzinger G.; Gerlach D.; Gerstberger T.; Gmaschitz T.; Greb P.; Han B.; Heyes E.; Iacob R. E.; Kessler D.; Köller H.; Lamarre L.; Lancia D. R.; Lucas S.; Mayer M.; Mayr K.; Mischerikow N.; Mück K.; Peinsipp C.; Petermann O.; Reiser U.; Rudolph D.; Rumpel K.; Salomon C.; Scharn D.; Schnitzer R.; Schrenk A.; Schweifer N.; Thompson D.; Traxler E.; Varecka R.; Voss T.; Weiss-Puxhaum A.; Winkler S.; Zheng X.; Zoephel A.; Kraut N.; McConnell D.; Pearson M.; Koegl M. Chemically Induced Degradation of the Oncogenic Transcription Factor BCL6. Cell Rep. 2017, 20, 2860–2875. and references therein 10.1016/j.celrep.2017.08.081. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.