

Tristetraprolin (TTP) is an anti-inflammatory protein that modulates the stability of certain cytokine/chemokine mRNAs. After initial high-affinity binding to AU-rich elements in 3′ untranslated regions of target mRNAs, mediated through its tandem zinc finger (TZF) domain, TTP promotes the deadenylation and ultimate decay of target transcripts.
KEYWORDS: AU-rich regions, CCR4/NOT, RNA binding proteins, deadenylation, inflammation, tristetraprolin
ABSTRACT
Tristetraprolin (TTP) is an anti-inflammatory protein that modulates the stability of certain cytokine/chemokine mRNAs. After initial high-affinity binding to AU-rich elements in 3′ untranslated regions of target mRNAs, mediated through its tandem zinc finger (TZF) domain, TTP promotes the deadenylation and ultimate decay of target transcripts. These transcripts and their encoded proteins accumulate abnormally in TTP knockout (KO) mice, leading to a severe inflammatory syndrome. To assess the importance of the highly conserved C-terminal CNOT1 binding domain (CNBD) of TTP to the TTP deficiency phenotype in mice, we created a mouse model in which TTP lacked its CNBD. CNBD deletion mice exhibited a less severe phenotype than the complete TTP KO mice. In macrophages, the stabilization of target transcripts seen in KO mice was partially normalized in the CNBD deletion mice. In cell-free experiments, recombinant TTP lacking its CNBD could still activate target mRNA deadenylation by purified recombinant Schizosaccharomyces pombe CCR4/NOT complexes, although to a lesser extent than full-length TTP. Thus, TTP lacking its CNBD can still act to promote target mRNA instability in vitro and in vivo. These data have implications for TTP family members throughout the eukarya, since species from all four kingdoms contain proteins with linked TZF and CNOT1 binding domains.
INTRODUCTION
Members of the tristetraprolin (TTP) family of mRNA binding and destabilizing proteins often contain two linked functional domains, as well as longer regions of apparently low complexity (1). The defining domain, found throughout the eukaryotes, is the 64-amino-acid tandem zinc finger (TZF) domain, which is responsible for high-affinity binding to AU-rich sequences (AREs) in target mRNAs but which by itself is not sufficient to promote mRNA decay (1, 2). The second linked domain is a highly conserved sequence motif that is present at the extreme C termini of the proteins. Early studies by Lykke-Anderson and Wagner (3) suggested a potential mechanism of action for TTP that involved an interaction between an amino-terminal domain of the protein and the CNOT1 component of the CCR4-NOT complex, which includes two deadenylases (4, 5). Later work by Sandler and colleagues (6) suggested that the C-terminal region of TTP interacted with CNOT1, and they concluded “that mRNA decay mediated by binding of TTP alone is clearly dependent on the deadenylase scaffold protein Not1.” More recently, the Sonenberg group solved a crystal structure of a C-terminal fragment of TTP in complex with a central domain of CNOT1 (7), in which the conserved C-terminal peptide of human TTP could form an α-helix that could bind to CNOT1 within a hydrophobic pocket formed by three central helices. Since this C-terminal CNOT1 binding sequence is conserved among proteins from this family throughout the eukaryotes, this interaction between an mRNA binding protein and a complex that includes effector enzymes (deadenylases) raised the possibility of a universal mechanism of action for TTP family proteins in many eukaryotic organisms.
However, when we assayed human TTP activity in a cotransfection assay, we found that deletion of the C-terminal CNOT1 binding domain (CNBD; also called CNOT1-interacting motif [CIM] [7]), significantly inhibited but did not eliminate TTP’s ability to promote instability of AU-rich transcripts (7). We obtained similar data in cotransfection assays using the Drosophila Tis11 protein, which contains typical TZF and CNOT1 binding domains and is very active when expressed in mammalian cells (8). The contribution of the conserved C-terminal CNOT1 binding domain to the activity of members of this family in intact organisms remains undetermined.
Since deadenylation is thought to be the rate-limiting step in mRNA decay in eukaryotes (for reviews, see references 9, to ,11), we addressed the importance of the TTP C-terminal CNOT1 binding domain in vivo by knocking in a deletion of this domain in mice. We used TTP instead of one of the other three mouse TTP family proteins for two main reasons. First, the phenotype of its complete deletion is quite striking and readily assessed in adult mice (12). Second, the three mouse proteins with human orthologues, TTP, ZFP36L1, and ZFP36L2, all have leucine-rich nuclear export sequences (NES) that are responsible for the nuclear export component of their nucleocytoplasmic shuttling behavior (13), but in ZFP36L1 and ZFP36L2, the NES sequences overlap the putative CNOT1 binding domains. Thus, in those two cases, destruction of the CNOT1 binding domain would be expected to prevent nuclear export of the proteins, likely resulting in a severe phenotype because of nuclear sequestration. In the case of TTP, the NES is located instead at the amino terminus of the protein; a deletion of the C-terminal CNOT1 binding domain therefore would not be expected to interfere with nuclear export. Nonetheless, we expect that the results with TTP will reflect the importance of this domain in the other mammalian family members and, indeed, in proteins of this type throughout the eukarya.
Based on our previous results (7, 8), our hypothesis for the present study was that mice expressing TTP that lacked its conserved C-terminal CNOT1 binding domain would have a phenotype intermediate in severity between the complete TTP knockout (KO) and wild-type (WT) phenotypes. We addressed this in two mouse genetic backgrounds, C57BL/6 (B6) and 129, in both sexes, and in both mono- and diallelic comparisons. We also investigated the ability of recombinant full-length and C-terminally truncated TTP to promote deadenylation of target mRNAs by the recombinant CCR4-NOT complex from Schizosaccharomyces pombe (14, 15) and to substitute for the TTP family member Zfs1 in living S. pombe cells (16, 17).
RESULTS
Cotransfection studies.
The TZF domain mutations used in this study (Fig. 1A) can be considered nonbinding mutations. In addition, C-terminal truncations were used that removed the CNOT1 binding domain (CNBD) and the remaining C-terminal amino acids (Fig. 1A).
FIG 1.

Involvement of the TTP CNBD in Mlp-TNF3′ fusion mRNA decay. (A) Sequences of the TZF domains and the C-terminal CNBD from human and mouse TTP, aligned with ClustalW. The locations of the cysteines changed to arginines in the various mutants discussed in this paper are shown. The residues reported to bind to CNOT1 in human TTP (7) are highlighted in orange. Asterisks under the alignments indicate amino acid identity at those sites, a colon indicates amino acid conservation, and a single dot represents less conservation at that site. The numbers at the end of the alignment indicate the numbers of amino acids in the full-length proteins. (B) Results of a coimmunoprecipitation experiment in HEK 293 cells that had been cotransfected with a FLAG-CNOT1800–1020 plasmid and one of three constructs expressing GFP or TTP-GFP fusion proteins: GFP alone (lanes 1 and 2), a C-terminally truncated version of TTP lacking the CNBD (1–313) (lanes 3 and 4), and WT human TTP (lanes 5 and 6). The Western blot shown on the left was blotted with an anti-GFP antibody, and the blot on the right was blotted with an anti-FLAG antibody. In both blots, lanes 1, 3, and 5 show the input extracts (In) that were not incubated with the anti-GFP resin, and lanes 2, 4, and 6 show the proteins that were eluted from the anti-GFP resin (P) after incubation with the indicated extracts. The migration positions of molecular weight standards are shown on the left of the blots, and the migration positions of the fusion proteins (blot on left) or the CNOT1 protein fragment (blot on right) are shown to the right of the blots. (C) Top, Western blot of the various hTTP.tag proteins expressed in HEK 293 cells transfected with WT or mutant constructs, as indicated. The migration positions of molecular weight standards are shown on the left. BS+ indicates cells transfected with the backbone vector alone. Bottom, results of blotting with an anti-β-actin antibody (βAct). (D) Representative Northern blot of the Mlp-TNF3′ fusion mRNA from HEK 293 cells that had been cotransfected with the Mlp-TNF3′ plasmid and one of four constructs expressing GFP or GFP-TTP fusion proteins: a negative control, GFP (lanes 1 and 2); WT human TTP (lanes 3 to 5); TTP 1–313 mutant construct (lanes 6 to 8); and the known inactive TTP mutant construct C124R (lanes 9 and 10). The transfected cells were treated without (ActD 0) or with ActD for the times indicated (0.5 or 1 h). On the right, the migration position of the endogenous Mlp mRNA (endo) is indicated by a line, and that of the fusion Mlp-TNF3′ mRNA is indicated by double arrows. The lower arrow indicates the deadenylated form of the Mlp-TNF3′ transcript. (E) Results of an RNaseH assay using total cellular RNA from cells not treated with ActD. On the right, the migration positions of the endogenous Mlp mRNA (endo) are indicated by two lines, and those of the fusion Mlp-TNF3′ mRNAs are indicated by two arrows. For each mRNA, the upper line or arrow indicates the fully polyadenylated mRNA species, whereas the lower line or arrow indicates the deadenylated form of the mRNA. (F) Relative levels of Mlp-TNF3′ mRNA (mean values ± SD) from four independent experiments. Both bands shown in panel D were used in the phosphorimager quantitation of the Northern blots. Darker bars (ActD 0) represent relative Mlp-TNF3′ transcript values at steady state, before the addition of ActD, and lighter bars (ActD 1 h) represent relative transcript levels after ActD treatment. See the text for calculations and other details.
To investigate whether there was an association between TTP lacking its CNBD and the CNOT1 protein in our cotransfection system, we performed coimmunoprecipitation assays in transfected HEK 293 cells that expressed full-length human WT TTP fused with a green fluorescent protein (GFP) tag or an analogous GFP-tagged TTP protein lacking its 13 C-terminal residues, which contain the TTP CNBD (TTP 1–313, comprising residues 1 to 313). These were coexpressed with the FLAG-tagged central region of human CNOT1, previously shown to interact with the TTP C-terminal domain in cell-free experiments (7). The GFP-tagged WT TTP and the truncated TTP 1–313 version expressed roughly equal levels of protein in the input sample (Fig. 1B), and the anti-GFP resin pulled down both GFP-tagged proteins and GFP itself (Fig. 1B). The anti-GFP resin pulled down the CNOT1 fragment in the presence of the full-length TTP (Fig. 1B) but not in the presence of TTP lacking its CNBD (TTP 1–313) or GFP alone (Fig. 1B). These results mirror exactly those shown by Fabian et al. (7) and support a direct interaction of the CNBD of human TTP with the central region of human CNOT1 in this cellular context. This cellular confirmation is important, since one or both of the serines in the conserved C-terminal regions of TTP (Fig. 1A) have been shown to be phosphorylated in different cell types (18, 19), and these modifications might be expected to influence the CNBD-CNOT1 interaction (7).
To investigate the ability of human TTP and its mutants to promote the decay of an ARE-containing mRNA, we performed experiments at both steady state and after transcription inhibition by actinomycin D (ActD). In transiently transfected HEK 293 cells, full-length or WT TTP, the TTP 1–313 construct, and a TTP TZF domain C124R single point mutant (encoding a change of C to R at position 124) (Fig. 1A) expressed roughly comparable levels of protein (Fig. 1C, lanes 2 to 4). The patterns of immunoreactive bands differed somewhat among the three proteins, due in part to the decreased length of the TTP 1–313 mutant, the possible removal of C-terminal phosphorylation sites (see above), and the presumed structural disruption caused by the cysteine mutation within the RNA binding TZF domain.
We then measured the activity of these proteins in promoting mRNA decay by monitoring levels of a synthetic target transcript, Mlp-TNF3′, expressed from a plasmid (20) that incorporates the promoter, single intron, and protein-coding sequences from the mouse MARCKS-like protein 1 (GenBank accession number NP_034937.1) and 540 bases of the mouse tumor necrosis factor alpha (TNF) mRNA containing the TTP binding sites; transcription of this construct from the MLP promoter is inhibitable by ActD (8). In the presence of WT TTP, before the addition of ActD, there were lower levels of two bands corresponding to the synthetic target Mlp-TNF3′ transcript (Fig. 1D), with the lower band representing the deadenylated transcript species (see below) (21). After ActD treatment, the level of the upper band initially decreased by 30 min, and both bands were further depleted after 1 h (Fig. 1D, compare lanes 2 and 5). In contrast, the TTP 1–313 C-terminal deletion construct did not promote the formation of the characteristic two bands of the Mlp-TNF3′ transcript, and its steady-state level was greater than with WT TTP (Fig. 1D, compare lanes 1, 3, and 6). Decay of the target also appeared to be slower after ActD treatment than it was with WT TTP (Fig. 1D, lanes 3 to 8). As expected, the TZF domain C124R point mutant protein had little or no effect on either the steady-state levels of the target transcript or its decay (Fig. 1D, lanes 9 and 10).
To demonstrate that the lower band shown in Fig. 1D, lanes 3 and 4, was the deadenylated species of the target transcript, we removed the poly(A) tails from mRNA with oligo(dT)12–18 plus RNase H, as described previously (22) (Fig. 1E). This experiment provides evidence that the lower bands seen with both the endogenous Mlp mRNA and the fusion Mlp-TNF3′ transcript were the deadenylated transcript species (Fig. 1E).
When the results of four independent cotransfection experiments of this type were analyzed (Fig. 1F), both WT TTP and the TTP 1–313 mutant were shown to have caused decreased levels of the Mlp-TNF3′ transcript in the steady state compared to the results for either GFP or the C124R mutant (Fig. 1F). When the steady-state levels of the Mlp-TNF3′ transcripts were compared statistically, the P values were as follows: WT versus TTP 1–313 mutant, P = 0.024; WT versus C124R mutant, P < 0.0001; and TTP 1–313 mutant versus C124R mutant, P = 0.002. When the target mRNA levels after 1 h of ActD incubation were compared to the steady-state levels (Fig. 1F, ActD 0 versus ActD 1 h), the P values for each pair were as follows: GFP, 0.1223; WT TTP, 0.002; TTP 1–313 mutant, 0.128; and C124R mutant, 0.539. These comparisons indicate that the TTP 1–313 mutant protein was not as effective as WT TTP in promoting the decay of the Mlp-TNF3′ transcript in this assay but that it still retained some measure of TTP activity compared to that in the inactive controls.
In separate experiments, we confirmed that the TTP 1–313 mutant was cytoplasmic when fused to GFP (data not shown), as expected given the N-terminal location of the nuclear export sequence in this protein but not in other mouse members of the family (13, 23).
Cell-free deadenylation assays.
Full-length mouse TTP partially complements a deficiency in the TTP family member Zfs1 in S. pombe (24). Therefore, we examined the activities of recombinant WT and mutant forms of human TTP in a deadenylation assay involving purified, recombinant CCR4-NOT complexes from S. pombe that were expressed in and purified from insect cells (14, 15). Full-length, purified recombinant WT TTP, in the form of an N-terminal fusion with maltose binding protein (MBP), stimulated the deadenylation activity of the CCR4-NOT complex when tested against a synthetic mRNA target that contained a single ARE (Fig. 2A). These assays used protein concentrations such that WT TTP retained activity but a zinc finger mutant form of TTP (C118R mutant, with a change of C to R at position 118), the TTP TZF domain alone, and MBP alone all had negligible activity (Fig. 2A). Under these conditions, TTP lacking the CNBD (ΔCNBD) still retained some ability to stimulate deadenylation but was less active than WT TTP (Fig. 2A). In both cases, the TTP-activated complex not only completely removed the poly(A) tail of the target mRNA but it continued to exert 3′-to-5′ exonucleolytic activity toward the RNA until it reached the site of TTP binding (Fig. 2A), similar to what was shown recently with the S. pombe TTP family member Zfs1 (25).
FIG 2.

TTP-stimulated deadenylation of recombinant S. pombe CCR4-NOT complexes in vitro, and TTP-stimulated mRNA decay in S. pombe in vivo. (A) Two independent replicate deadenylation assays (1 and 2). The ARE-containing RNA probe and the purified recombinant CCR4-NOT complexes were incubated for the times indicated with no additions (−) or in the presence of identical concentrations of various purified recombinant human TTP proteins, including the WT TTP-MBP fusion protein, the ΔCNBD-MBP fusion protein, representing the human version of the mouse TTPΔCNBD mutant, the C118R-MBP fusion protein, representing the non-RNA binding TZF domain mutant of human TTP, TZF (the human TTP TZF domain alone), and MBP (MBP protein alone). Numbers on the right indicate the poly(A) tail lengths, and negative numbers indicate the numbers of nucleotides removed immediately 5′ of the poly(A) tail. The asterisk indicates the removal of 6 nucleotides (−6) between the TTP binding site sequence and the poly(A) tail. (B) Schematic representation of the S. pombe zfs1 locus, in which the protein coding domain of zfs1 was replaced by either an epitope-tagged version of Zfs1 or the indicated full-length mouse TTP or its mutant protein coding sequences (CDS), in all cases epitope tagged. Note that in all cases, the endogenous zfs1 promoter region and 3′ UTR remained intact. (C) Western blot in which each lane contained identical concentrations of protein extracts from cells expressing the mouse proteins described in panel B, with the first lane containing an extract from the zfs1Δ cells. (D) Results of a NanoString assay of Arz1 (SPCC1494.03) mRNA levels, with the first bar labeled WT representing RNA from S. pombe cells expressing full-length, FLAG-tagged Zfs1, as shown schematically in panel B, and the second bar labeled WT representing RNA from cells expressing WT mouse TTP in place of Zfs1. Each bar represents the mean value ± SD of data from 4 independent isolates. The results were analyzed by one-way ANOVA with Tukey’s multiple comparison test. The asterisks indicate adjusted P values of <0.0001 in all comparisons.
Complementation assays in S. pombe.
We then tested the ability of the analogous mutant mouse proteins to complement the molecular phenotype of Zfs1 deficiency in S. pombe, in which several dozen ARE-containing transcripts accumulate in the absence of Zfs1 (16, 17). A schematic representation of the WT and mutant mouse proteins inserted into the S. pombe zfs1 locus is depicted in Fig. 2B, and a Western blot analysis demonstrating roughly equal expression of each of the three mouse proteins is shown in Fig. 2C. As a readout of TTP family member activity, we measured the accumulation of Arz1 mRNA, a prominent Zfs1 target transcript (16, 17). As shown by the results in Fig. 2D, Arz1 mRNA levels were increased by nearly 5-fold in the Zfs1-deficient cells compared to the levels in WT cells. As shown previously (24), the expression of WT mouse TTP largely complemented this defect; however, the zinc finger mutant protein had no effect, even though it was expressed at comparable levels (Fig. 2C), resulting in Arz1 mRNA levels that were similar to those seen in the Zfs1-deficient strain (Fig. 2D). When the C-terminally truncated mutant of mouse TTP was expressed, again at comparable levels (Fig. 2C), a level of complementation was seen that was intermediate between the results for the inactive zinc finger mutant and the WT protein (Fig. 2D).
Phenotype of mice with a C-terminal TTP deletion.
To better understand the contribution of the TTP C-terminal CNBD to the function of TTP in an intact animal, we created a knock-in (KI) mouse model in which the DNA sequence encoding the TTP CNBD was removed, using standard targeting techniques (Fig. 3). In this model, TTP is expressed from its normal locus and under normal regulatory control, but the mutant allele expresses a protein missing the C-terminal 12 amino acids of the sequence with GenBank accession number NP_035886.1, comprising the CNBD.
FIG 3.

Scheme of Zfp36 gene targeting in mice. (A) Schematic representation of the mouse Zfp36 locus, the targeting vector, the homologously recombined features in the mutant allele, and after the Flp-mediated deletion of the PGK-Neo positive-selection cassette. The blue or pink arrows indicate the sizes of the SpeI- or PvuII-digested fragments from WT Zfp36 and the correctly recombined mutant locus. The gray arrows indicate the sizes of the left or right homologous recombination arms in the targeting vector. The half-headed red arrows represent the genotyping primers. Sp, SpeI; Pv, PvuII; Xb, XbaI; 1, exon 1; 2, exon 2; CNBD, CNOT1 binding domain; Frt, flippase recognition site; DTA, MC1-DTA negative-selection marker. (B) Sample Southern blots show SpeI- or PvuII-digested fragments of genomic DNA from five ES cell clones, including one (lanes 1) with the correctly recombined Zfp36 mutant locus, showing hybridization with the 5′ and 3′ probes, respectively. Lanes 3 represent a clone with apparent partial integration at the Zfp36 locus.
The phenotypes of the original TTP KO mice (12) and the recently described TTP TZF domain mutant mice (26) are essentially identical and quite complex, with failure of weight gain and cachexia, arthritis, myeloid hyperplasia, autoimmunity, and bone erosions with generalized osteopenia. The most readily observable external signs of this phenotype are failure to gain weight over time, beginning several weeks after birth, and peripheral joint arthritis, as manifested by redness and swelling. In the current studies, we monitored body weight and peripheral joint appearance as signs of the “TTP deficiency syndrome”; these were supplemented by molecular studies of TTP-dependent mRNA stability, as described below.
We analyzed mice of two different genotypes on two genetic backgrounds and in both sexes. In what we refer to here as the “mixed heterozygote comparison,” we compared mice with one normal Zfp36 allele and one CNBD deletion mutant allele (Zfp36+/ΔCNBD; referred to here as TTP+/ΔCNBD mice) to mice derived from the original KO strain that had one normal Zfp36 allele and one KO allele (Zfp36+/−; referred to here as TTP+/−) and mice with one KO allele and one mutant allele (Zfp36−/ΔCNBD; referred to here as TTP−/ΔCNBD). In general, the TTP+/− heterozygotes have no obvious phenotype in terms of weight loss or arthritis. We also performed a homozygote comparison, in which we compared the homozygous WT mice (Zfp36+/+; referred to here as WT) with their homozygous CNBD deletion KI counterparts (Zfp36ΔCNBD/ΔCNBD; referred to here as TTPΔCNBD/ΔCNBD mice).
Body weights.
By the end of the 24-week observation period, there were substantial decreases in body weight in the TTPΔCNBD/− mice compared to the weights of the TTP+/− mice, when comparing mice from both sexes and both genetic backgrounds (Fig. 4A). The average weights of the TTPΔCNBD/− mice ranged from 76.2% to 80.3% of the weights of the TTP+/− mice, with an overall average decrease to 78.8% of the weights for the control mice. This should be compared with an overall average for the original TTP KO mice of about 52.2% of WT weights after 24 weeks; however, many KO mice do not survive until 24 weeks, and the nonsurvivors often have much lower body weights at the time of death.
FIG 4.
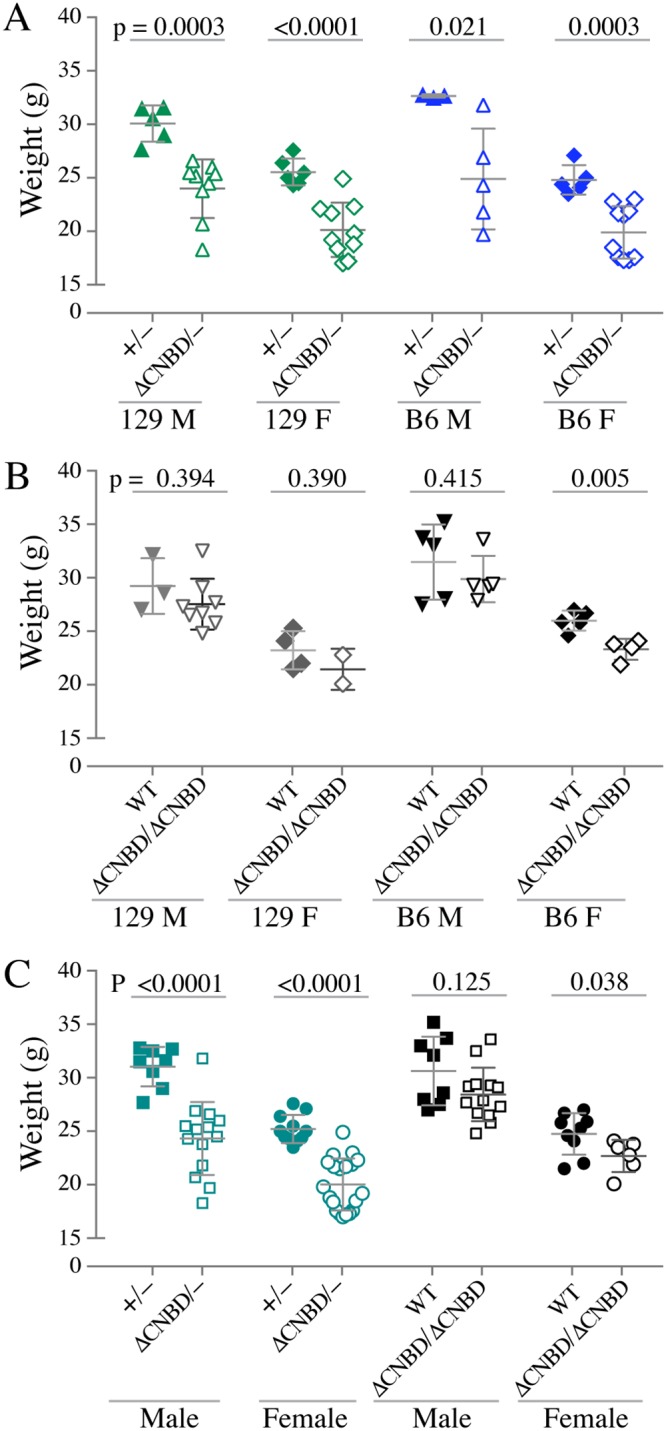
Body weights at 24 weeks of age. Body weight measurements for mice of all genotypes and both sexes at 24 weeks of age are shown as scatter plots, in which each dot represents the weight of an individual mouse. Also shown are the mean values ± SD, as well as the P value for each comparison, calculated from two-tailed, unpaired t tests with Welch’s correction. (A) Comparisons between TTP+/− and TTPΔCNBD/− mice of the indicated sex and background. (B) Comparisons between WT and TTPΔCNBD/ΔCNBD mice of the indicated sex and background. (C) Comparisons between mice of the same pairs of genotypes when weights from mice of each sex of both backgrounds were combined.
The average weights of the TTPΔCNBD/ΔCNBD mice at 24 weeks were decreased to 89.7% to 94.9% of the WT weights, with an overall average decrease to 92.8% (Fig. 4B). In only one case was the P value of this average decrease less than 0.05 (Fig. 4B). When data from mice of both genetic backgrounds were combined, the weights of the TTPΔCNBD/− mice of both sexes were found to be significantly lower than those of the TTP+/− mice (Fig. 4C), whereas there was no significant difference between the combined weights of the TTPΔCNBD/ΔCNBD and WT male mice, and weights for female mice were only slightly lower (P = 0.038) (Fig. 4C).
These comparisons demonstrated that the TTPΔCNBD/ΔCNBD mice were much less severely affected than the KO mice. The increased severity of the TTPΔCNBD/− body weight phenotype compared to that of the TTPΔCNBD/ΔCNBD mice indicates that the ΔCNBD allele represents a hypomorphic allele, based on the classical definition that involves increased phenotype severity when placed in trans from a null allele (27).
Arthritis and dactylitis.
In the original TTP KO (TTP−/−) mice, as well as the C116R KI mice, a universal aspect of the phenotype is severe peripheral joint arthritis within the first several months of life, with the front paws generally more severely affected than the rear paws (12, 26). The arthritis is characterized externally by redness and swelling of the paws and histologically by invasion of the inflammatory synovium into the joint, with destruction of bone and cartilage, neutrophil infiltration into soft tissues, and myeloid hyperplasia. Micro-computed tomography (micro-CT) of the paws revealed extensive bone destruction, and staining indicated increased osteoclast activity.
In the present study, the TTPΔCNBD/ΔCNBD mice on the B6 background generally exhibited only slight involvement of the front paws, with mild swelling and redness that was limited to one to a few digits (dactylitis) and was often transient (Table 1). Mice on the 129 background had higher frequencies of dactylitis (Table 1), and there was occasional rear limb involvement. The dactylitis, illustrated in Fig. 5B (arrowhead), was often accompanied by leukocyte infiltration into the soft tissues of the affected digits (see Fig. 6 and 7 below), but there were no instances of larger joint involvement. No instances of dactylitis were observed in the WT, TTP+/−, or TTPΔCNBD/+ mice monitored in parallel.
TABLE 1.
Overall frequencies of paw or digit swelling in mice of the TTPΔCNBD/ΔCNBD and TTPΔCNBD/− genotypes
| Parameter | % of mice with indicated genotype on indicated genetic background |
|||||||
|---|---|---|---|---|---|---|---|---|
| TTPΔCNBD/ΔCNBD |
TTPΔCNBD/− |
|||||||
| B6 |
129 |
B6 |
129 |
|||||
| Male | Female | Male | Female | Male | Female | Male | Female | |
| Swellinga | 50 | 73 | 71 | 82 | 93 | 65 | 95 | 100 |
| Resolvedb | 29 | 55 | 30 | 22 | 54 | 36 | 0 | 0 |
| Improvedc | 29 | 36 | 0 | 0 | 8 | 0 | 0 | 0 |
Percentage of mice that exhibited any swelling of digits or paws that occurred at any time during the 24-week observation period, even if transient.
Represents a subgroup of the “Swelling” group and refers to mice with complete resolution of swelling by the end of the observation period, i.e., no swelling of any digits at the end of the experiment.
Refers to mice with partial resolution of swelling at the end of the observation period, either less swelling of affected digits or fewer affected digits.
FIG 5.
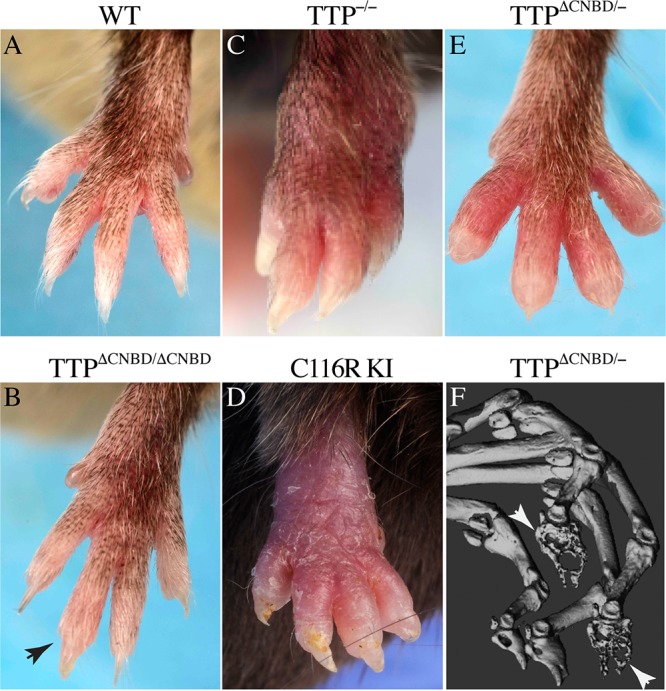
(A to E) External phenotypes of the front paws. Shown are representative paws from WT (A), TTPΔCNBD/ΔCNBD (B), TTP−/− (C), C116R KI (D), and TTPΔCNBD/− (E) mice. All mice were males. The images in panels A, B, E, and F were from mice on a 129 background, and the images in panels C and D were from mice on a C57BL/6 background. (B) The arrowhead indicates a single swollen terminal digit in the TTPΔCNBD/ΔCNBD mouse. (F) Part of a micro-CT image from a TTPΔCNBD/− mouse paw; the arrows indicate two of the distal phalanges that were affected by bony erosions, whereas the other two appear to be unaffected.
FIG 6.

Paw immunohistochemistry with an antibody to Ly6G. Shown are images from sections of WT, TTPΔCNBD/−, and TTPΔCNBD/ΔCNBD paws. Row I shows low-power sections that were treated the same as the rest of the sections in the figure during the immunostaining process but were incubated with the purified rat IgG2a isotype control instead of the Ly6G antibody. Row II shows comparable low-power sections incubated with the Ly6G antibody. Rows III and IV show magnified areas from the sections shown in row II, with row III showing synovial spaces and row IV soft tissues. Arrows in panels F, G, and J point to neutrophil infiltration in soft tissues, and arrowheads in panels F and G indicate Ly6G-positive cells in bone marrow.
FIG 7.

Paw immunohistochemistry with an antibody to CD45. Shown are images from sections of WT, TTPΔCNBD/−, and TTPΔCNBD/ΔCNBD paws. All sections were incubated with the CD45 antibody. The images in panels A, C, and E show synovial spaces. The images in panels C, D, and F show cells in bone marrow cavities, with immunoreactive cells present in the sections shown in panels D and F.
In the mixed heterozygote TTPΔCNBD/− mice, the arthritis phenotype was more severe and more frequent than in the TTPΔCNBD/ΔCNBD mice, but it was still less frequent and less severe than that seen in the TTP−/− mice (Fig. 5C) or the TZF domain C116R mutation KI mice (Fig. 5D). In addition, there was occasional involvement of larger joints. Dactylitis was more common and more severe than in the TTPΔCNBD/ΔCNBD mice (Table 1; Fig. 5E). Swelling and redness of the front digits was seen in most animals on the B6 background and in almost all mice on the 129 background (Table 1). More digits were involved in each animal, and the digit swelling was generally more severe than that seen in the homozygous TTPΔCNBD/ΔCNBD mice (Fig. 5E compared with Fig. 5B) and was still more severe on the 129 background. Swelling and redness of the digits of the rear limbs were observed in almost all mice on the 129 background by the end of the study, and there was no resolution or improvement of either front or rear digits (Table 1). When some of the dactylitic digits from the TTPΔCNBD/− mice were evaluated by micro-CT, they showed evidence of bone erosions (Fig. 5F, arrows) that were often limited to the terminal phalanges, whereas in the C116R KI mice, the erosions involved almost all bones of the paws (26).
Histologically, the TTPΔCNBD/− arthritis/dactylitis was associated with swelling of the soft tissues and increased infiltration of the soft tissues by neutrophils (Fig. 6F and G, arrows; Fig. 6H shows a more detailed view) but little or no synovitis or destruction of the bone and cartilage of the synovial joints (Fig. 6G). There were marked increases in Ly6G-positive cells (granulocytes) in the TTPΔCNBD/− mouse bone marrow (Fig. 6F and G, arrowheads), indicating myeloid hyperplasia, a universal characteristic of the TTP KO and C116R KI mice (12, 26). In the homozygote comparisons, soft tissue infiltration by neutrophils was observed in a few areas of the TTPΔCNBD/ΔCNBD paws (Fig. 6J, arrows; Fig. 6L is a detailed view), but there was less bone marrow infiltration of Ly6G-positive cells in the TTPΔCNBD/ΔCNBD mouse paws (Fig. 6K).
In both the TTPΔCNBD/− and TTPΔCNBD/ΔCNBD mice, there were evident increases in leukocyte infiltration of soft tissues, as seen histologically with CD45-positive cells (Fig. 7C to F), but with little sign of synovial inflammation in either case (Fig. 7C and E).
Response to acute LPS challenge.
Myeloid cell-specific TTP KO mice (28) also had a mild external phenotype, but low-dose lipopolysaccharide (LPS) injections resulted in signs of severe endotoxemia and extremely high serum levels of TNF, consistent with the absence of TTP in LPS-responsive leukocytes (28). We therefore investigated the response of the TTPΔCNBD/ΔCNBD mice to intraperitoneal injections of LPS, at two different doses of LPS. After injections of LPS at 2 mg/kg of body weight, there were statistically significant genotype influences on the increases of TNF, granulocyte-macrophage colony-stimulating factor (GM-CSF), interleukin-6 (IL-6), CXCL1, CXCL2, and CCL3 (Fig. 8A) in the serum, as well as granulocyte colony-stimulating factor (G-CSF) (P = 0.008) (data not shown). When we analyzed the responses from the two different sexes, we found that only the increases of CCL3 were significantly influenced by genotype in both males (P = 0.046) and females (P = 0.009), while only in females were the increases of GM-CSF (P = 0.029), IL-6 (P = 0.003), CXCL2 (P = 0.046), and G-CSF (P = 0.029) significantly influenced by genotype. In the hours after the LPS injections, there were body temperature decreases in response to LPS in both the WT and TTPΔCNBD/ΔCNBD mice, with a significant genotype influence (P = 0.0141) in the body temperature changes (Fig. 8B).
FIG 8.

Serum cytokine concentrations and body temperatures after LPS challenge. (A and C) Levels of cytokines and chemokines (mean values ± SD) in the sera of WT or TTPΔCNBD/ΔCNBD mice before and after intraperitoneal injection of LPS at either 2 mg/kg of body weight (A) or 10 mg/kg (C). The P values beneath the protein name indicate the levels of significance of the differences in response between the two genotypes (see Materials and Methods for their determination). (B and D) Body temperatures (mean values ± SD) measured before and after injection of LPS at 2 mg/kg (B) or 10 mg/kg (D).
Higher-dose injections (10 mg/kg) gave similar results (Fig. 8C), except that there was no significant genotype effect on levels of CXCL1. When the results were broken down by sex, only the increases in GM-CSF were significantly influenced by genotype in both males (P = 0.004) and females (P = 0.023); only in females were the increases in TNF (P = 0.036), and only in males were the increases in CXCL2 (P = 0.003) and CCL3 (P = 0.025) significantly influenced by genotype. The body temperature decreases were more dramatic with this dose of LPS (Fig. 8D), but there was no influence of genotype in this case.
Before the LPS injections, there were no discernible differences in serum cytokine levels between the two genotypes of mice. This is in contrast to the results obtained in similar experiments in the TZF C116R point mutant KI mice, which exhibited significant increases in the basal levels of TNF (P = 0.0224, unpaired t test with Welch’s correction) and CXCL1 (P = 0.0085).
Analysis of mRNA stability in primary BMDM.
We first compared levels of TTP protein expression in bone marrow-derived macrophages (BMDM) from WT, TTP+/−, and TTPΔCNBD/− mice. A Western blot analysis of unstimulated cells and cells stimulated for 3 h with LPS confirmed the decreased expression of TTP protein in the TTP+/− cells (Fig. 9A), but the multiple bands characteristic of this protein under these circumstances, reflecting multisite phosphorylation, were present in their normal configuration. In the TTPΔCNBD/− cells, there was approximately the same level of protein expression as seen in the TTP+/− cells, but the banding pattern was slightly modified, reflecting the decreased size of the protein and perhaps loss of one or more C-terminal phosphorylation sites (Fig. 9A).
FIG 9.

TTP protein expression and TNF or TTP mRNA decay in TTP+/− and TTPΔCNBD/− BMDM after LPS stimulation. BMDM from WT, TTP+/−, and TTPΔCNBD/− mice were treated with LPS for various times, as indicated. (A) Western blot demonstrating TTP protein expression in cells of the three different genotypes. Top, extracts from cells under control conditions (lanes 1, 3, and 5) and from cells treated with LPS for 3 h (lanes 2, 4, and 6). Bottom, results of blotting gels containing the same extracts with an anti-β-actin antibody (βAct). (B) Representative Northern blots showing TNF (top) and TTP (bottom) mRNA decay after ActD treatment. BMDM were stimulated with LPS for 1 h, followed by an ActD decay time course assay. Total RNA samples from TTP+/− cells were loaded into lanes 1 to 9, and total RNA samples from TTPΔCNBD/− cells were loaded into lanes 10 to 18. Migration positions of TNF and TTP mRNA are indicated on the right, and a line at the right of the bottom panel indicates the migration position of a TTP-neo fusion mRNA from the TTP null allele. (C and D) Decay curves (mean values ± SD) from five independent experiments of the type shown in panel B are shown for TNF mRNA decay (C) and TTP mRNA decay (D). As in the experiment described for panel B, cells were stimulated with LPS for 1 h, followed by ActD treatment of WT, TTP+/−, and TTPΔCNBD/− cells, as indicated.
Complete TTP deficiency, as well as a non-RNA binding TZF domain mutation in TTP, has major effects on the stability of several cytokine mRNAs that have been shown to be direct targets of TTP binding, deadenylation, and destabilization (for reviews, see references 29, to ,31). In a previous study in BMDM from C116R KI mice, we found that treatment with ActD following 1 h of LPS stimulation provided a greater contrast between TNF mRNA levels and decay rates in cells of the WT and mutant genotypes than ActD treatment at either 3 or 6 h after LPS treatment. We therefore compared BMDM of TTP+/− and TTPΔCNBD/− mice after treatment with LPS for 1 h, using Northern blotting to evaluate effects on TNF mRNA expression and decay. As the example in Fig. 9B shows, there was a slower decay rate of TNF mRNA in the TTPΔCNBD/− cells than in the TTP+/− cells. When data from five different experiments of this type were averaged, with cells derived from five groups of mice, there was a minor but statistically significant delay in TNF mRNA decay in the TTPΔCNBD/− cells compared to that observed in the WT cells, while there were no differences between WT and TTP+/− cells (Fig. 9C). The average times to decay (time at which mRNA decreased to 50% of its original level [t50s]) (mean values ± standard deviation [SD]) were similar in the WT (20 ± 1 min) and in the TTP+/− (23 ± 6 min) cells, and the average t50 in the TTPΔCNBD/− cells was 34 ± 11 min (Table 2). For comparison, there was a much more marked delay in TNF mRNA decay in a previous comparison of WT versus KO cells (26) (Table 2). There were no apparent differences in the TTP mRNA decay profiles for WT, TTPΔCNBD/−, and TTP+/− cells (P = 0.092) (Fig. 9D), in contrast to the behavior of TTP mRNA in the C116R point mutant cells, in which it was significantly stabilized in the mutant cells (Table 2) (26).
TABLE 2.
Time for the TNF mRNA to decay to 50% of its original level in cells of the various genotypes
| Genotype | t50 (min) (mean ± SD)a | 95% confidence interval | P valueb |
|---|---|---|---|
| WT | 20 ± 1 | 18–22 | 0.045 |
| TTPΔCNBD/− | 34 ± 11 | 21–47 | |
| WT | 15 ± 2 | 11–18 | 0.024 |
| TTPΔCNBD/ΔCNBD | 33 ± 9 | 19–48 | |
| WTc | 17 ± 2 | 14–21 | 0.003 |
| TTP–/–c | 59 ± 15 | 41–78 | |
| WTc | 18 ± 3 | 15–22 | <0.0001 |
| TTPC116R/C116Rc | 69 ± 15 | 51–88 |
Data are from cells derived from 5 mice in each group, except for the WT versus TTPΔCNBD/ΔCNBD comparison, in which there were data and cells from 4 mice in each group.
Determined with unpaired, two-tailed t tests with Welch’s correction.
Data from analogous experiments with cells derived from the original TTP KO mice and from the TTPC116R/C116R mice were taken from a previous paper (26) and are shown here for comparison.
We then performed similar analyses on BMDM from the WT and TTPΔCNBD/ΔCNBD mice. A Western blot analysis of TTP showed comparable levels of induced protein after 1 or 3 h of LPS stimulation (Fig. 10A). The slight size decrease and the change in the protein banding pattern were similar to those noted above for the TTPΔCNBD/− cells (Fig. 9A).
FIG 10.

TTP protein expression and TNF and other mRNA decay in WT and TTPΔCNBD/ΔCNBD BMDM after LPS stimulation. BMDM from WT and TTPΔCNBD/ΔCNBD mice were treated with LPS for the times indicated. (A) Top, Western blot demonstrating TTP protein expression in cells of the indicated genotypes under control conditions (lanes 1and 4) and from cells treated with LPS for 1 h (lanes 2 and 5) or for 3 h (lanes 3 and 6). Bottom, results of blotting the same extracts with an anti-β-actin antibody (βAct). (B) Representative Northern blots illustrating TNF (top) and TTP (bottom) mRNA decay after 1 h of LPS stimulation followed by an ActD time course assay. Total RNA from WT cells was loaded into lanes 1 to 9, and from TTPΔCNBD/ΔCNBD cells into lanes 10 to 18. Migration positions of TNF and TTP mRNA are indicated on the right. (C) NanoString analysis of BMDM mRNA decay. Total RNA samples from four decay experiments were performed, using cells from four pairs of mice of the two indicated genotypes. Data are shown as fractions of the original mRNA remaining (mean values ± SD; n = 4) during the ActD time course assay, with a value of 1 set for the samples taken before ActD was added. The numbers in parentheses following the name of the mRNA indicate the numbers of core TTP binding sites (UAUUUAU) found in the mRNA 3′ UTR. The P values beneath the mRNA names refer to statistical comparisons between the two genotypes; see Materials and Methods for their determination. In the panel labeled TNF, the red lines represent mean values from a previous experiment (26), in which cells from mice with a cysteine point mutation in the TZF domain (open circles) were compared to cells from WT mice (closed circles). (D) Original mRNA levels, determined by NanoString assays, from total RNA prepared after 1 h of LPS stimulation but before adding ActD. The data shown are normalized counts (mean values ± SD).
The decay patterns of mRNA after 1 h of LPS treatment followed by ActD incubation indicated a modest delay in the decay of TNF mRNA in the TTPΔCNBD/ΔCNBD cells, as shown by Northern blotting (Fig. 10B, top). The average t50s of TNF mRNA decay of four independent experiments using BMDM from four pairs of mice showed a 2-fold increase in average t50 of the TNF mRNA in the TTPΔCNBD/ΔCNBD cells compared to the results for their WT counterparts (Table 2). The delay was similar in magnitude to that observed in the WT versus TTPΔCNBD/− comparison (Table 2). The decay of TTP mRNA was similar in both WT and TTPΔCNBD/ΔCNBD cells (Fig. 10B, bottom). The average t50s of TTP mRNA decay were similar (P = 0.955) in the WT (14 ± 3 min) and the TTPΔCNBD/ΔCNBD cells (14 ± 2 min).
Using NanoString assays (32), we examined the effects of the TTPΔCNBD/ΔCNBD genotype on the decay of several additional TTP target mRNAs and several internal controls (26). These assays validated the effectiveness of the ActD treatment; specifically, the curves from known rapidly decaying internal controls, Dusp2 and Socs3 mRNAs, were very rapidly degraded after ActD addition, with superimposable curves from cells of the two genotypes (Fig. 10C). Similarly, there were no apparent differences in the decay curves for TTP mRNA itself (Fig. 10C). We observed comparable results for these control transcripts and for TTP mRNA when RNA samples from TTP+/− and TTPΔCNBD/− cells were tested in the same assays (not shown).
The decay of TNF mRNA was significantly slowed in the TTPΔCNBD/ΔCNBD cells compared to its decay in the WT cells (Fig. 10C; Table 2). This decreased rate of decay was not as great as that seen in the point mutant cells (26); the average values from those decay curves are shown in red in Fig. 10C for comparison. Using the same type of analysis, a significant genotype influence on TNF mRNA decay was also observed with the TTP+/− versus TTPΔCNBD/− comparison (P = 0.002). Significant effects of the TTPΔCNBD/ΔCNBD genotype were also found in the decay curves of Cxcl1, Cxcl2, and Ptge2 mRNAs (Fig. 10C), transcripts previously identified as TTP targets (26, 33–35); in these cases, the decreased decay after ActD treatment was more similar to that seen previously in the cells from the zinc finger mutant mice, but t50 values could not be calculated for those transcripts because of the unusual decay patterns. There were minor genotype influences of the TTPΔCNBD/ΔCNBD genotype on the decay of Myd116 and CD274 mRNAs; these were not significantly different in this experiment.
When the initial values of these mRNAs (before ActD was added) were compared (Fig. 10D, LPS 1 h), TNF was the only one found to be present at significantly higher levels in the TTPΔCNBD/ΔCNBD group (P = 0.037).
Predictions of disordered regions in human and mouse TTP.
Because recent experiments have demonstrated that Zfs1, the single TTP family member expressed in S. pombe, can interact with the S. pombe CCR4/NOT complex through unstructured domains in Zfs1 that are N-terminal to the C-terminal TZF domain in that protein (25), we analyzed the human and mouse TTP protein sequences for the presence of predicted disordered regions. As shown by the data in Fig. 11, the disorder prediction program DISOPRED3 (36) predicts that there are large unstructured regions in both the N- and C-terminal halves of both mouse and human proteins that might interact with the mammalian CCR4/NOT complex in similar ways.
FIG 11.

Disordered protein predictions for human and mouse TTP. The protein sequences for human and mouse TTP (hTTP and mTTP, respectively) were subjected to disordered protein segment predictions using DISOPRED3 (36), using default parameters. The domain structures of the two proteins are illustrated schematically at the top of the figure. NES, nuclear export signal; TZF, tandem zinc finger domain; CNBD, CNOT1 binding domain. Note that the TZF domain and CNBD especially are predicted to be ordered, with large regions of disorder predicted for large stretches of the N- and C-terminal halves of the proteins. Predicted disordered regions are indicated by blue lines, and predicted protein binding site locations by orange lines.
DISCUSSION
Early studies of cells derived from TTP KO mice established that TTP could promote decay of mRNAs containing specific AU-rich sequences in their 3′ untranslated regions (3′ UTRs) (26, 35, 37, 38). This process was initiated by high-affinity binding of TTP to the AU-rich sequences, followed by the stimulation of mRNA deadenylation (22, 39), thought to be the rate-limiting step in eukaryotic mRNA decay (for reviews, see references 9 to 11). It was clear that the defining characteristic of TTP family proteins, the TZF domain, was responsible for the RNA binding; however, the nature of the interactions with deadenylases remained obscure.
A second conserved sequence motif that is found in TTP family members throughout the eukaryotes is generally found at the extreme C terminus (1, 7). This short motif in human TTP was found to interact directly with the human CNOT1 protein (7), and a crystal structure of this interaction demonstrated key residues in the TTP C terminus that have been conserved throughout eukaryotic evolution. Since CNOT1 is a component of the CCR4-NOT complex, a multiprotein complex that contains two 3′-to-5′ exonucleases, or deadenylases, this provided an attractive model for the activity of TTP and its family members: after binding to mRNA through the TZF domain, the C-terminal domain would recruit the CCR4-NOT complex to the targeted mRNA, resulting in its accelerated deadenylation and ultimate decay (7, 40).
However, there are some problems with this model. First, in the cell transfection studies with both human TTP and Drosophila Tis11, the CNBD deletions did not fully destroy the ability of the proteins to promote the decay of ARE-containing mRNAs (7, 8). Second, many widely used model organisms express TTP family members, as defined by the presence of typical TZF domains, that appear to lack any sequences analogous to the C-terminal CNBD described above (1). Examples of these include the yeasts Saccharomyces cerevisiae, S. pombe, and Candida albicans and the eudicot plant Arabidopsis thaliana. In many of these cases, the TTP family proteins end shortly after the TZF domain, without an obvious C-terminal CNBD. This is particularly striking given that certain primitive fungi and many primitive plants contain more typical metazoan-style proteins with linked central TZF domains and C-terminal CNBD (1, 41). Despite the apparent absence of these domains in yeasts, the TTP family members from those species can still bind to AU-rich-element-containing mRNAs and promote their decay.
The present experiments attempted to determine the importance of the conserved C-terminal CNBD to the function of TTP in the intact mouse, using knock-in technology to remove this portion of the protein while leaving the Zfp36 gene otherwise intact and subject to its normal regulation. Based on our previous cotransfection studies with human TTP and Drosophila TIS11 (7, 8), our hypothesis was that this C-terminal truncation would result in a phenotype intermediate in severity between the phenotypes of normal mice and TTP KO mice. As mentioned above, we chose TTP over the other family members expressed in mouse because of its severe and well-described phenotype in adult mice and because of the overlap of the CNBD of the two other widely expressed TTP family members, ZFP36L1 and ZFP36L2, with their experimentally verified nuclear export sequences (13).
The experiments reported here support this hypothesis. In fact, the mutant gene encoding the C-terminally truncated protein behaved like a hypomorphic allele, with a phenotype that was more severe when it was in trans with a null allele (27). As in the case of mice heterozygous for a point mutation in the TZF domain that abrogates RNA binding, we saw no evidence of a dominant-negative effect of the mutant allele (26). We conclude from these experiments that TTP lacking this conserved C-terminal domain is less active than its full-length counterpart in mice but still retains considerable activity in the intact organism.
These findings were supported by three very different types of experiments. In mammalian cell transfection studies, the C-terminally truncated version of TTP was clearly less active than WT TTP at promoting deadenylation and decay of a target transcript but more active than either an unrelated protein (GFP) or a TZF domain cysteine mutant that is incapable of binding RNA. This was despite the facts that, in transfection experiments in the same cell type, full-length human TTP was able to pull down the central TTP binding domain of CNOT1 but the C-terminally truncated protein was not, exactly mirroring the association results using recombinant proteins expressed in and purified from Escherichia coli (7). These data indicate that the phosphorylation of one or both serines in the C-terminal fragment, which has been observed in other cell types (18, 19), might be expected to interfere with association with CNOT1 (7). Similarly, in cell-free experiments with recombinant human TTP and its mutants and recombinant CCR4-NOT complexes from S. pombe, the C-terminally truncated mutant of TTP retained some ability to promote the deadenylation of ARE-containing target RNAs, under conditions in which WT TTP was active and a TZF domain point mutant, the TZF domain alone, and MBP were all inactive. Finally, the C-terminally truncated form of mouse TTP was clearly less active than the full-length protein in an S. pombe Zfs1 complementation assay, but it was more active than the zinc finger point mutant protein. Taken together, these data suggest that the removal of the conserved C-terminal sequence from TTP decreases its activity in vitro and in vivo but does not abolish it.
In general, the mouse phenotype seen when only one copy of TTPΔCNBD was expressed, in trans with a null allele, was considerably less severe than the phenotype seen in the complete TTP KO mice (12) or in mice with a knocked-in point mutation in one of the zinc-coordinating cysteines within the TZF domain (26). The phenotype in the homozygous mice expressing two copies of the TTPΔCNBD mutant was still less severe, in terms of weight gain and peripheral evidence of arthritis, as would be expected with an apparent hypomorphic allele (27). However, despite the nearly normal weight gain in the TTPΔCNBD/ΔCNBD mice, there were some abnormal phenotypes that raise the possibility that specific actions of TTP are differentially affected. For example, both the TTPΔCNBD/− and many of the TTPΔCNBD/ΔCNBD mice developed dactylitis, with soft tissue swelling and leukocyte infiltration in the digits and bony erosions that were most severe in the terminal phalanges. This dactylitis was actually transient in some of the TTPΔCNBD/ΔCNBD mice, i.e., they seemed to recover completely, at least in terms of external physical examination. This kind of transient arthritis is apparently not uncommon in certain human rheumatologic diseases, such as “palindromic rheumatism” (42, 43), and may reflect less severe arthritogenic stimuli than found complete in TTP-deficient mice. Both types of C-terminus mutant mice had no or minimal involvement of the larger joints that are so severely affected in the TTP KO and TZF domain point mutant mice (12, 26). Dactylitis is frequently seen in human psoriatic arthritis (44), and it can be used to distinguish this condition from other types of inflammatory arthritis. We also found that myeloid hyperplasia, a prominent part of the original TTP deficiency syndrome, was evident in both the TTPΔCNBD/− and the TTPΔCNBD/ΔCNBD mice. Macrophages from the TTPΔCNBD/ΔCNBD mice exhibited stabilization of Cxcl1 and Cxcl2 mRNA that was very similar to that seen in the more severe forms of TTP deficiency (26), even though TNF and TTP mRNAs were stabilized to a lesser extent. It is possible that certain aspects of the phenotype that differ from the complete KO phenotype are the result of specific effects of the mutant protein on certain transcripts, the nature of which remain to be determined.
Recently, Zhang et al. (45) described an interesting mouse model in which endogenous TTP was tagged with a C-terminal V5 tag. Based on the targeting method used, the encoded protein should lack the C-terminal seven amino acids of mouse TTP. Based on the results of Fabian et al. (7), this truncation should decrease or eliminate binding of the C-terminal domain to CNOT1. To our knowledge, these mice do not have an evident phenotype, but as in the current study, it may be necessary to put the mutant allele in trans with a null allele to elicit a high-frequency inflammatory phenotype of the kind seen with the TTPΔCNBD/− mice.
Many possibilities remain for how C-terminally truncated TTP can still promote deadenylation of its targets. These include binding of other regions of TTP to CNOT1 or to other members of the CCR4-NOT complex, as suggested by the experiments in the present paper with purified CCR4-NOT complexes from S. pombe and from previous work (3, 46), binding to other deadenylases, either directly or through intermediary proteins, and other possibilities. Regardless of the mechanism of this residual activity, the present results demonstrate that removal of the conserved C-terminal domain of TTP has deleterious effects on the function of the protein, suggesting that other family members throughout the eukarya also may rely on this domain for full activity.
MATERIALS AND METHODS
Animal procedures, genotyping, and sequence analysis of the ΔCNBD allele.
The targeting scheme was designed to remove the CNOT1 binding domain (CNBD)-coding sequence from exon 2 of the Zfp36 locus, the gene encoding TTP (Fig. 3). A 9.1-kb fragment spanning the Zfp36 locus and flanking sequence was shuttled from bacterial artificial chromosome (BAC) RP23-204B7 (Children’s Hospital Oakland Research Institute, Oakland, CA), using standard BAC recombineering techniques, into a plasmid containing the MC1-DTA negative-selection marker. Site-directed mutagenesis was used to remove the coding sequence of the CNBD (5′-CGTCTCCCCATCTTCAATCGTATCTCTGTCTCTGAG-3′), coding for the extreme C terminus of mouse TTP (GenBank accession number NP_003398.2): RLPIFNRISVSE. This is analogous to the deletion used to eliminate CNOT1 binding to human TTP in the work of Fabian et al. (7). An Frt-flanked neomycin resistance cassette from pL451 (NCI, Frederick, MD) was then subcloned into the XbaI site of the single intron of Zfp36. The linearized targeting vector was electroporated into 129S6-derived embryonic stem (ES) cells (AB2.2). Properly targeted clones were identified by 5′ and 3′ Southern blot analysis utilizing probes generated by PCR of 129 mouse genomic DNA (TTP-5′Fwd, 5′-AACCACGTGACTACCCAACC-3′; TTP-5′Rev, 5′-GCAAGTGGCCTCTGGAGAAT-3′; TTP-3′Fwd, 5′-CAGATTCTCACGCCTCCTGG-3′; and TTP-3′Rev, 5′-ACTGGCTAACTCCTCGGTCT-3′). Proper integration of the CNOT1 binding site-deficient exon 2 was confirmed by sequencing. Correctly targeted ES cell clones were microinjected into albino C57BL/6 blastocysts to generate chimeric founders. Germ line-transmitting chimeras were bred to FLP recombinase-expressing mice (129S4, JAX stock number 003946; The Jackson Laboratory) to excise the FLP recombination target (FRT)-flanked neomycin resistance cassette to generate the final Zfp36 CNOT1BD allele (Zfp36+/ΔCNBD).
Heterozygous Zfp36+/ΔCNBD mice were bred with C57BL/6 mice to remove the Cre transgene and then bred with the original TTP+/− mice (12) on 129 and C57BL/6 genetic backgrounds to generate either mixed heterozygote mice with one TTP-KO allele and one CNBD deletion mutant allele in the Zfp36 locus (Zfp36−/ΔCNBD) or homozygous CNBD deletion (Zfp36ΔCNBD/ΔCNBD) mice. Offspring were genotyped by Transnetyx Genotyping Services.
The expression of TTP mRNA lacking the CNBD was verified individually in cells from four Zfp36ΔCNBD/ΔCNBD mice with reverse transcription (RT)-PCR, using total RNA isolated from bone marrow-derived macrophages (BMDM; see below). The sequence of the 5′ PCR primer was within the TTP protein-coding region, 5′-CGACAAAGCATCAGCTTCTC-3′, and the sequence of the 3′ primer was located in the 3′ UTR of TTP mRNA, 5′-TGCCCTCTCTTTGAGATCTAG-3′. The resulting 455-bp cDNA was sequenced, and this confirmed that the coding sequence for RLPIFNRISVSE at the extreme C terminus of TTP was indeed missing.
All animal procedures were performed according to the U.S. Public Health Service policy on the humane care and use of laboratory animals, and the National Institute of Environmental Health Sciences Institutional Animal Care and Use Committee approved all animal procedures used in this study.
Body weights were measured weekly, from 6 weeks after birth until 24 weeks of age. We compared possible differences in body weights at age 24 weeks between the two pairs of genotypes using the unpaired, two-tailed t test with Welch’s correction.
Micro-CT.
Front paws from WT, TTP−/ΔCNBD, and TTPΔCNBD/ΔCNBD mice were prepared and analyzed using a microcomputed tomography system (micro-CT) as described previously (26, 47, 48). Briefly, the isolated paws were fixed in 10% neutral buffered formalin for 24 h and then stored in 70% ethanol. For micro-CT scanning, the fixed paws were immobilized in 2% agarose gel, vertically set in the tubes, and measured in the micro-CT system (μCT40; Scanco Medical AG, Zurich, Switzerland). The scanned images were analyzed, and three-dimensional (3-D) images were remodeled.
Histology and immunohistochemistry.
Animals were euthanized between 4 and 5 months of age, and front paws were fixed in 10% neutral buffered formalin, embedded in paraffin, and sectioned (5-μm sections). To perform immunohistochemistry, the sections were deparaffined and rehydrated, and antigens were retrieved by heat and pressure in a decloaking chamber. For Ly6G antibody staining, the antigen retrieval solution used was 1× antigen decloaker citrate buffer, pH 6.0 (Biocare Medical, Concord, CA). Hydrogen peroxide (3%) was applied to block endogenous peroxidase. The sections were then incubated with Dako protein-blocking reagent (Dakocytomation Corporation, Carpinteria, CA) for 10 min, followed by the avidin-biotin blocking reagent (Vector Laboratories, Burlingame, CA). The sections were incubated with a Ly6G IgG2a (catalog number 127602, lot number B192830; Biolegend, Inc., San Diego, CA) primary antibody at a 1:500 dilution for 30 min, or for negative-control sections, the incubation was with an equivalent dilution of purified rat IgG2a (BD Biosciences San Jose, CA) isotype control. The sections were then incubated for 30 min with a biotinylated rabbit anti-rat secondary antibody (Vector Laboratories, Burlingame, CA) at a 1:500 dilution, followed by incubation with the R.T.U. Vectastain elite ABC reagent (Vector Laboratories, Burlingame, CA) for 30 min. The antigen-antibody complex was visualized using diaminobenzidine (DAB; Dako, Carpinteria, CA) for 6 min. The sections were counterstained with hematoxylin for 10 s, dehydrated, cleared, and coverslip mounted. For CD45 antibody staining, the antigen retrieval solution used was 1× EDTA decloaker retrieval solution (Biocare Medical, Concord, CA). Hydrogen peroxide (3%) was applied to block endogenous peroxidase. The sections were then incubated with 2.5% normal goat serum (ImmPRESS anti-rat Ig kit; Vector Laboratories, Burlingame, CA) for 20 min. The sections were incubated with a CD45 monoclonal IgG2b (catalog number 550539, lot number 3122745; BD Biosciences, Inc., San Jose, CA) primary antibody at a 1:150 dilution for 60 min, or for negative-control sections, the incubation was with an equivalent dilution of purified rat IgG2a isotype control (BD Biosciences San Jose, CA). The sections were then incubated with anti-rat Ig polymer (ImmPRESS anti-rat Ig kit; Vector Laboratories, Burlingame, CA) for 30 min. Antigen-antibody complexes were visualized using DAB (Dako Corp., Carpinteria, CA) for 6 min. The sections were counterstained with hematoxylin, dehydrated, cleared, and coverslip mounted.
Endotoxin challenge, clinical pathology, and cytokine/chemokine multiplex assay.
For the low-dose test (2 mg/kg body weight), four groups (n = 5 in each) of 8- to 13-week-old male or female mice, WT or TTPΔCNBD/ΔCNBD, were used for the endotoxin challenge study. Body temperatures were determined and peripheral blood samples were collected before intraperitoneal injection of a single dose of lipopolysaccharide (LPS 0111:B4 in normal saline, product number L2630; Sigma) or normal saline alone. One mouse from each group was injected with normal saline alone. During the whole testing period, the four saline-injected mice showed stable body temperatures, and their serum cytokines remained at basal levels. For the high-dose test (10 mg/kg body weight), 9 mice (5 males and 4 females) of each genotype at 8 to 13 weeks old were used. Body temperature measurements and peripheral blood collections were repeated at 1.5 h and 3 h after the LPS injection. Peripheral blood was collected via retro-orbital bleeding into tubes containing serum gel/clotting activator (Sarstedt, Inc., Newton, NC). After clotting, the tubes were centrifuged for 5 min at 10,000 × g at room temperature. The resulting sera were analyzed with a mouse cytokine/chemokine magnetic bead panel (Milliplex MAP; EMD Millipore) on a Luminex 200.
Transfection assays.
A human TTP (GenBank accession number NP_003398.1) expression construct, CMV.hTTP.tag, its zinc finger mutant, and its C terminus deletion constructs have been described previously (7, 21). These constructs were transfected into HEK 293 cells by the calcium phosphate precipitation method as described previously (49) to express TTP protein (0.5 μg DNA and 4.5 μg of vector BS+ DNA per 100-mm dish) for Western blotting with a hemagglutinin (HA) antibody (HA probe [F-7]-horseradish peroxidase [HRP], sc7392; Santa Cruz). The RNA-destabilizing activity of TTP expressed by these constructs (5 ng DNA/dish) was assessed with HEK 293 cell cotransfection assays (49), using a reporter construct, Mlp-TNF3′ (3 μg DNA/dish), that expressed a fusion mRNA containing 540 bases of the TNF 3′ UTR (20). Forty-eight hours after transfection of the constructs, the cells were treated without or with ActD (5 μg/ml, product number A4262; Sigma). Dishes were rinsed in cold phosphate-buffered saline (PBS) and lysed in lysis solution (illustra RNAspin minikit; GE Healthcare). The lysates from the 3 plates from the same treatment time point were pooled, and cellular RNA was prepared according to the manufacturer’s protocol.
The ability of TTP expressed by these constructs to degrade the Mlp-TNF3′ fusion mRNA was visualized and quantitated by Northern blotting with an Mlp cDNA probe, as described previously (8, 20). The Mlp-TNF3′ fusion mRNA and the endogenous Mlp mRNA expression level on each gel lane were quantitated using a PhosphorImager and mRNA probe bound volume (volume quantitation by ImageQuant; Molecular Dynamics). The probe bound volume of Mlp-TNF3′ fusion mRNA was corrected by the bound volume of endogenous Mlp mRNA, which served as a loading control. For comparison, the steady-state (without ActD treatment) levels of Mlp-TNF3′ mRNA in the RNA from cells transfected with the TTP expression constructs were compared to those from cells transfected with the control construct CMV.EGFP at steady state, and the state of Mlp-TNF3′ mRNA decay after 1 h in ActD in the presence of the TTP expression constructs was compared to that in the presence of the CMV.EGFP control after ActD treatment of the cells.
RNase H assays were performed as described previously (8, 21, 22). Briefly, total cellular RNA samples from cells (without ActD treatment) cotransfected with Mlp-TNF3′ and various TTP or GFP expression constructs were incubated at 55°C in 50 mM KCl with or without 0.6 μg of oligo(dT)12–18 for 5 min, followed by another 20 min at 25°C. All reaction mixtures included 10 μg of RNA, except for the GFP and C124R mutant samples incubated with RNase H, which included 5 μg. Reaction buffer containing RNase H (1 U per reaction sample) was added to samples annealed with oligo(dT)12–18, or reaction buffer without the enzyme was added to samples without oligo(dT)12–18, followed by incubation at 37°C for 1 h. After precipitating the RNA, the samples were used for Northern blotting as described above.
Coimmunoprecipitations of TTP and its mutants with a CNOT1 peptide were performed using extracts of HEK 293 cells cotransfected with a mixture of Lipofectamine 2000 (Invitrogen) and 2.5 μg of a CMV.FLAG.hCNOT1800–1020 construct that expressed the middle part of the human CNOT1 protein (GenBank accession no. NP_996882.1, amino acids 800 to 1020) that was previously shown to bind to the C terminus of TTP (7), together with control plasmid CMV.EGFP or the CMV.hTTP.EGFP constructs of WT TTP or of TTP 1–313 that lacks the C-terminal CNBD. One day after transfection, the cells were lysed with 1 ml RNP lysis buffer (50 mM Tris-HCl, pH 7.5, 1 mM MgCl2, 150 mM NaCl, 50 mM NaF, 1% [vol/vol] IGEPAL CA-630, 10% [vol/vol] glycerol) supplemented with 1× cOmplete protease inhibitor cocktail (Roche, without EDTA), 1 mM dithiothreitol (DTT), and 1 mM sodium orthovanadate. The cleared lysates were used in immunoprecipitation reactions that were performed with anti-GFP antibodies coupled to an agarose resin (54) (NIEHS, Structural Biology Core Laboratory, Protein Expression Facility). In addition, 250 units of RNase I (Ambion; Invitrogen) was added to each immunoprecipitation reaction mixture, and the reaction mixtures constantly mixed at 4°C for 2 h. The agarose resin-bound complexes were next washed 4 times with ice-cold RNP lysis buffer and then boiled at 90°C for 4 min in a mixture of 1× Laemmli buffer with 80 mM DTT to both liberate and denature the bound proteins on the anti-GFP resin. Twenty percent (by volume) of the denatured, immunoprecipitated proteins and 10 μg of total protein from the input lysate (also denatured using the conditions described above) were subjected to SDS-PAGE, followed by transfer of the proteins from the polyacrylamide gel to a nitrocellulose membrane and then immunoblotting the membrane with either anti-GFP (B-2)-HRP antibody (Santa Cruz Biotechnology) or anti-FLAG M2-HRP antibody (Sigma-Aldrich).
Macrophage cultures, cell treatments, RNA preparation, and Northern blotting.
BMDM were obtained by culturing bone marrow cells in macrophage growth medium (RPMI 1640 medium with 10% fetal bovine serum [FBS], 30% L929 cell culture supernatant, 100 U/ml penicillin, 100 μg/ml streptomycin, and 6 mM l-glutamine) in 10-cm tissue culture plates as described previously (26). To prepare for experiments, the 70% to 80% confluent BMDM monolayers in 10-cm plates were incubated in RPMI 1640 medium (10 ml each plate) supplemented with 1% FBS and the antibiotics for 16 to 24 h.
The mRNA decay assays were performed as described previously (26). For each experiment, 4 plates of cells of each genotype were used as controls (untreated), and 4 plates were treated with LPS (product number L6529; Sigma) alone as references. At the end of the 1-h pretreatment period, these reference plates were quickly rinsed with cold PBS and the cells in monolayers were lysed in lysis solution. Thereafter, three plates of cells were combined for use for each mRNA decay point. ActD was added to these plates precisely at the end of the indicated LPS treatment period to stop mRNA transcription, and the cells were further incubated for the indicated times, followed by cold PBS rinses, cell lysis, and cellular RNA extraction as described above.
For Northern blotting, each gel lane was loaded with 8 μg of total RNA. Northern blots were hybridized with the α-32P-labeled probes for mouse TNF and TTP as described previously (50, 51). The blots were quantitated using a phosphorimager, and the probe-bound mRNA volumes were obtained using Volume Quantification (ImageQuant; Molecular Dynamics).
NanoString RNA analysis.
Total cellular RNA from BMDM used in the RNA decay assays was analyzed using the NanoString nCounter method (32) for a specific group of mRNAs. The observed counts were normalized by a method described previously (26). The internal housekeeping controls for normalizing the counts were Shfm1 (NM_009169.1), Tbca (NM_009321.2), Pbrm1 (NM_001081251.1), Gnai2 (NM_008138.4), Psma7 (NM_011969.1), Cox8a (NM_007750.2), and Mom2 (NM_007505.2). GenBank accession numbers for each transcript presented in Fig. 10 are as follows: Cd274 (NM_021893.3), Cxcl1 (NM_008176.1), Cxcl2 (NM_009140.2), Dusp2 (NM_010090.2), Myd116 (NM_008654.2), Ptgs2 (NM_011198.4), Socs3 (NM_007707.2), Tnf (NM_011638.4), and Zfp36 (TTP) (NM_011756.3).
In vitro deadenylation assays.
(i) Plasmid constructs. An E. coli expression plasmid encoding human TTP (RefSeq accession number NP_003398.1), with an N-terminal maltose binding protein (MBP) tag and a C-terminal 6-residue histidine tag, was a kind gift from Marc R. Fabian, McGill University, and was described previously (7). The C118R single point mutant, mutated within the TZF domain, and the C-terminal deletion mutant, lacking amino acid residues 315 to 326 that were previously reported to comprise the CNOT1 binding site (7), were generated by PCR mutagenesis using the vector encoding human TTP as the template. The TZF domain of human TTP (residues 103 to 170, based on the sequence with RefSeq accession number NP_003398.1), was cloned using the SspI site of a modified pET28/30 vector, as described in reference 24. The modified pMAL-c5x vector encoding MBP was a kind gift from Lars Pederson, NIEHS (52).
(ii) Protein expression and purification. Human full-length TTP, its full-length C118R mutant, and its C-terminal deletion mutant proteins fused to MBP were expressed in Rosetta-2(DE3) cells (Millipore-Sigma) after induction with 0.1 mM IPTG (isopropyl-β-d-1-thiogalactopyranoside) for 16 h at 20°C. Cells were lysed by sonication, followed by centrifugation at 35,000 rpm at 4°C for 30 min. Supernatants were applied to HisTrap columns (GE Healthcare Life Sciences) and eluted with 40 mM imidazole. Proteins were subsequently loaded onto a Superdex S200 size exclusion column (GE Healthcare Life Sciences) in 20 mM HEPES, pH 7.5, 100 mM KCl, 25 μM ZnSO4, 2 mM 2-mercaptoethanol, 5% glycerol, and 10 mM maltose. Fractions containing purified TTP fusion proteins were pooled and concentrated using a 10,000 molecular-weight-cutoff (MWCO) Vivaspin 20 centrifugal filtering device (GE Healthcare Life Sciences) and stored at −80°C in buffer with 10% glycerol (final) after flash freezing in liquid nitrogen.
The human TTP TZF domain fusion peptide was expressed in BL21(DE3) cells (Millipore-Sigma) after induction with 0.1 mM IPTG for 16 h at 20°C and was purified as described above for full-length and mutant TTP proteins, with the exception that the affinity tag was removed following elution from the HisTrap column by incubation with tobacco etch virus (TEV) protease (vector kindly supplied by Traci T. Hall, NIEHS) for 16 h at 4°C in the presence of 2 mM DTT. Isolated TZF domains were further purified using a Superdex S200 column in 20 mM HEPES, pH 7.5, 100 mM KCl, 25 μM ZnSO4, 2 mM 2-mercaptoethanol, and 5% glycerol. After concentration using a 3,000 MWCO Vivaspin 20 centrifugal device, purified protein was flash frozen in liquid nitrogen and stored at −80°C.
MBP was overexpressed in BL21(DE3) cells after induction with 0.3 mM IPTG for 16 h at 20°C. Cells were lysed as described above, and supernatants were incubated with amylose resin (New England Biolabs) and eluted with 40 mM maltose. Fractions containing MBP were subsequently applied to a Superdex S200 column. After concentration using a 10,000 MWCO Vivaspin 20 centrifugal device, protein was stored at −80°C in a final buffer containing 20 mM HEPES, pH 7.5, 100 mM KCl, 10 mM maltose, and 10% glycerol after flash freezing in liquid nitrogen.
Cell-free deadenylation assays were performed as described previously (15, 25), using a target mRNA sequence at a final concentration of 200 nM that contained a single TTP family member binding site, 5′-[6FAM/fluorescein]-AAUCAUCCUUAUUUAUUACCAUUAAAAAAAAAAAAAAAAAAAAAAAAAAAAAA-3′ (the binding site is underlined). Recombinant purified CCR4-NOT complexes from S. pombe were incubated in the presence or absence of identical concentrations (60 nM) of the following recombinant MBP fusion proteins: wild-type full-length human TTP, its C-terminally truncated mutant (ΔCNDB), its nonbinding C118R zinc finger mutant, its TZF domain alone, or the MBP protein alone. Preliminary experiments had determined that this concentration of reactants resulted in accelerated deadenylation in the presence of WT TTP but not in the presence of the C118R mutant. Deadenylation was initiated by the addition of 20 nM recombinant CCR4-NOT complex. Aliquots of the reaction mixture were removed at the times indicated and added to tubes containing the denaturing loading dye to stop the reactions. The RNA products were resolved by urea-TBE (Tris-borate-EDTA) polyacrylamide gel electrophoresis.
S. pombe complementation experiments.
These experiments were performed as described previously (24), except that a zinc finger cysteine mutant and a C-terminally truncated mutant of mouse TTP were also knocked into the zfs1 locus, as illustrated in Fig. 2B. Western blotting of epitope-tagged mouse TTP and NanoString analysis of Arz1 mRNA levels were conducted as described previously (24).
Antibodies and Western blotting.
At the end of the indicated reagent treatment periods, BMDM cell protein extracts were prepared as described previously (26). Equal amounts of protein (100 μg/lane) were resolved on 12% SDS-PAGE gels and then transferred onto nitrocellulose membranes (GE Healthcare). The membranes were preincubated in TBS-T (Tris-buffered saline and 0.5% Tween 20) with 5% dry milk for 1 h, followed by incubation with an antiserum against mouse TTP (53) at a 1:10,000 dilution in TBS-T–5% dry milk at 4°C overnight. Blots were rinsed with TBS-T and incubated with a secondary antibody conjugated to horseradish peroxidase (Bio-Rad) at a 1:50,000 dilution in TBS-T–5% dry milk for 60 min at room temperature. After rinsing with TBS-T, the blots were developed using enhanced chemiluminescence (ECL; Thermo Scientific). Protein loading controls were visualized by incubation of the membrane with an anti-β-actin antibody (1:25,000; Abcam) followed by incubation with a secondary antibody conjugated to horseradish peroxidase (goat anti-mouse IgG–HRP; Bio-Rad) at a 1:50,000 dilution.
Statistical analysis.
Data are generally presented as mean values ± standard deviations (SD). In the BMDM RNA decay experiments, the value obtained after 1 h of LPS stimulation (i.e., pre-ActD treatment reference value) was set at 1, with data from the ActD treatment time course assay expressed as a fraction of that original value. The time to decay (t50) of an assayed mRNA was defined as the time at which the mRNA decreased to its 50% level during the ActD treatment time course, as analyzed by interpolation using nonlinear regression (i.e., finding an X value [ActD min] for Y at 0.5) (Prism 7; GraphPad). The differences in t50 values between the TTPΔCNBD/ΔCNBD or the TTPΔCNBD/− groups and the WT groups were compared (Prism 7; GraphPad) by the two-tailed unpaired Student’s t test (with Welch’s correction) on the data from 5 individual experiments (unless otherwise indicated); the difference between two groups was considered significant when the test P value was <0.05. In analysis of the NanoString assay results, a 2-way analysis of variance (ANOVA) with Sidak’s multiple comparison test was used to determine how an mRNA decay response was affected by these two factors: (i) the two different genotypes and (ii) the treatment by LPS followed by an ActD time course assay. The difference in responses of the two genotypes was considered significant when the test P value was <0.05. Similarly, in the analysis of cytokine expression in blood after LPS injection, we used a 2-way ANOVA with Sidak’s multiple comparison test to determine how cytokine accumulation was affected by (i) the two different genotypes and (ii) the time after LPS injection. The difference in responses of the two genotypes was considered significant when the test P value was <0.05.
ACKNOWLEDGMENTS
We are grateful to Dori Germolec and Don Cook for constructive comments on the manuscript and Fred Miller for helpful discussions. We also thank the Comparative Medicine Branch, NIEHS, for mouse care and assistance with the endotoxin challenge study, Sukhdev Brar of the Cellular & Molecular Pathology Branch, NIEHS, for cytokine assays, the NIEHS Molecular Genomics Core Laboratory for the NanoString analyses, the NIEHS histology facility for tissue processing, immunostaining, and photography, and James A. W. Stowell (MRC LMB) for providing purified S. pombe CCR4/NOT complexes.
This research was supported by the Intramural Research Program of the National Institute of Environmental Health Sciences, NIH, by NIH grants R01 AR053628 and AR066551 and Shriners Hospital grant 85100 to R.F., by Medical Research Council (MRC) grant number MC_U105192715, and under the European Union’s Horizon 2020 research and innovation programme (ERC consolidator grant agreement no. 725685 to L.A.P.).
REFERENCES
- 1.Wells ML, Perera L, Blackshear PJ. 2017. An ancient family of RNA-binding proteins: still important! Trends Biochem Sci 42:285–296. doi: 10.1016/j.tibs.2016.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Carrick DM, Lai WS, Blackshear PJ. 2004. The tandem CCCH zinc finger protein tristetraprolin and its relevance to cytokine mRNA turnover and arthritis. Arthritis Res Ther 6:248–264. doi: 10.1186/ar1441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lykke-Andersen J, Wagner E. 2005. Recruitment and activation of mRNA decay enzymes by two ARE-mediated decay activation domains in the proteins TTP and BRF-1. Genes Dev 19:351–361. doi: 10.1101/gad.1282305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Collart MA. 2016. The Ccr4-Not complex is a key regulator of eukaryotic gene expression. Wiley Interdiscip Rev RNA 7:438–454. doi: 10.1002/wrna.1332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Miller JE, Reese JC. 2012. Ccr4-Not complex: the control freak of eukaryotic cells. Crit Rev Biochem Mol Biol 47:315–333. doi: 10.3109/10409238.2012.667214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sandler H, Kreth J, Timmers HT, Stoecklin G. 2011. Not1 mediates recruitment of the deadenylase Caf1 to mRNAs targeted for degradation by tristetraprolin. Nucleic Acids Res 39:4373–4386. doi: 10.1093/nar/gkr011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fabian MR, Frank F, Rouya C, Siddiqui N, Lai WS, Karetnikov A, Blackshear PJ, Nagar B, Sonenberg N. 2013. Structural basis for the recruitment of the human CCR4-NOT deadenylase complex by tristetraprolin. Nat Struct Mol Biol 20:735–739. doi: 10.1038/nsmb.2572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Choi YJ, Lai WS, Fedic R, Stumpo DJ, Huang W, Li L, Perera L, Brewer BY, Wilson GM, Mason JM, Blackshear PJ. 2014. The Drosophila Tis11 protein and its effects on mRNA expression in flies. J Biol Chem 289:35042–35060. doi: 10.1074/jbc.M114.593491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Goldstrohm AC, Wickens M. 2008. Multifunctional deadenylase complexes diversify mRNA control. Nat Rev Mol Cell Biol 9:337–344. doi: 10.1038/nrm2370. [DOI] [PubMed] [Google Scholar]
- 10.Schott J, Stoecklin G. 2010. Networks controlling mRNA decay in the immune system. Wiley Interdiscip Rev RNA 1:432–456. doi: 10.1002/wrna.13. [DOI] [PubMed] [Google Scholar]
- 11.Yan YB. 2014. Deadenylation: enzymes, regulation, and functional implications. Wiley Interdiscip Rev RNA 5:421–443. doi: 10.1002/wrna.1221. [DOI] [PubMed] [Google Scholar]
- 12.Taylor GA, Carballo E, Lee DM, Lai WS, Thompson MJ, Patel DD, Schenkman DI, Gilkeson GS, Broxmeyer HE, Haynes BF, Blackshear PJ. 1996. A pathogenetic role for TNF alpha in the syndrome of cachexia, arthritis, and autoimmunity resulting from tristetraprolin (TTP) deficiency. Immunity 4:445–454. doi: 10.1016/S1074-7613(00)80411-2. [DOI] [PubMed] [Google Scholar]
- 13.Phillips RS, Ramos SBV, Blackshear PJ. 2002. Members of the tristetraprolin family of tandem CCCH zinc finger proteins exhibit CRM1-dependent nucleocytoplasmic shuttling. J Biol Chem 277:11606–11613. doi: 10.1074/jbc.M111457200. [DOI] [PubMed] [Google Scholar]
- 14.Stowell JAW, Webster MW, Kogel A, Wolf J, Shelley KL, Passmore LA. 2016. Reconstitution of targeted deadenylation by the Ccr4-Not complex and the YTH domain protein Mmi1. Cell Rep 17:1978–1989. doi: 10.1016/j.celrep.2016.10.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Webster MW, Stowell JAW, Tang TTL, Passmore LA. 2017. Analysis of mRNA deadenylation by multi-protein complexes. Methods 126:95–104. doi: 10.1016/j.ymeth.2017.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cuthbertson BJ, Liao Y, Birnbaumer L, Blackshear PJ. 2008. Characterization of Zfs1 as an mRNA-binding and -destabilizing protein in Schizosaccharomyces pombe. J Biol Chem 283:2586–2594. doi: 10.1074/jbc.M707154200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wells ML, Huang W, Li L, Gerrish KE, Fargo DC, Ozsolak F, Blackshear PJ. 2012. Posttranscriptional regulation of cell-cell interaction protein-encoding transcripts by Zfs1p in Schizosaccharomyces pombe. Mol Cell Biol 32:4206–4214. doi: 10.1128/MCB.00325-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chrestensen C, Schroeder M, Shabanowitz J, Hunt D, Pelo J, Worthington M, Sturgill T. 2004. MAPKAP kinase 2 phosphorylates tristetraprolin on in vivo sites including Ser178, a site required for 14-3-3 binding. J Biol Chem 279:10176–10184. doi: 10.1074/jbc.M310486200. [DOI] [PubMed] [Google Scholar]
- 19.Zhou H, Di Palma S, Preisinger C, Peng M, Polat AN, Heck AJ, Mohammed S. 2013. Toward a comprehensive characterization of a human cancer cell phosphoproteome. J Proteome Res 12:260–271. doi: 10.1021/pr300630k. [DOI] [PubMed] [Google Scholar]
- 20.Blackshear PJ, Phillips RS, Ghosh S, Ramos SBV, Ramos SVB, Richfield EK, Lai WS. 2005. Zfp36l3, a rodent X chromosome gene encoding a placenta-specific member of the Tristetraprolin family of CCCH tandem zinc finger proteins. Biol Reprod 73:297–307. doi: 10.1095/biolreprod.105.040527. [DOI] [PubMed] [Google Scholar]
- 21.Lai WS, Carballo E, Strum JR, Kennington EA, Phillips RS, Blackshear PJ. 1999. Evidence that tristetraprolin binds to AU-rich elements and promotes the deadenylation and destabilization of tumor necrosis factor alpha mRNA. Mol Cell Biol 19:4311–4323. doi: 10.1128/MCB.19.6.4311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Carballo E, Lai WS, Blackshear PJ. 2000. Evidence that tristetraprolin is a physiological regulator of granulocyte-macrophage colony-stimulating factor messenger RNA deadenylation and stability. Blood 95:1891–1899. [PubMed] [Google Scholar]
- 23.Frederick ED, Ramos SB, Blackshear PJ. 2008. A unique C-terminal repeat domain maintains the cytosolic localization of the placenta-specific tristetraprolin family member ZFP36L3. J Biol Chem 283:14792–14800. doi: 10.1074/jbc.M801234200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wells ML, Hicks SN, Perera L, Blackshear PJ. 2015. Functional equivalence of an evolutionarily conserved RNA binding module. J Biol Chem 290:24413–24423. doi: 10.1074/jbc.M115.673012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Webster MW, Stowell JA, Passmore LA. 2019. RNA-binding proteins distinguish between similar sequence motifs to promote targeted deadenylation by Ccr4-Not. Elife 8:e40670. doi: 10.7554/eLife.40670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lai WS, Stumpo DJ, Qiu L, Faccio R, Blackshear PJ. 2018. A knock-in tristetraprolin (TTP) zinc finger point mutation in mice: comparison with complete TTP deficiency. Mol Cell Biol 38:e00488-17. doi: 10.1128/MCB.00488-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wilkie AO. 1994. The molecular basis of genetic dominance. J Med Genet 31:89–98. doi: 10.1136/jmg.31.2.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Qiu LQ, Stumpo DJ, Blackshear PJ. 2012. Myeloid-specific tristetraprolin deficiency in mice results in extreme lipopolysaccharide sensitivity in an otherwise minimal phenotype. J Immunol 188:5150–5159. doi: 10.4049/jimmunol.1103700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Khabar KS. 2014. Post-transcriptional control of cytokine gene expression in health and disease. J Interferon Cytokine Res 34:215–219. doi: 10.1089/jir.2013.0151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Maeda K, Akira S. 2017. Regulation of mRNA stability by CCCH-type zinc-finger proteins in immune cells. Int Immunol 29:149–155. doi: 10.1093/intimm/dxx015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Stumpo DJ, Lai WS, Blackshear PJ. 2010. Inflammation: cytokines and RNA-based regulation. Wiley Interdiscip Rev RNA 1:60–80. doi: 10.1002/wrna.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fortina P, Surrey S. 2008. Digital mRNA profiling. Nat Biotechnol 26:293–294. doi: 10.1038/nbt0308-293. [DOI] [PubMed] [Google Scholar]
- 33.Datta S, Biswas R, Novotny M, Pavicic PG Jr, Herjan T, Mandal P, Hamilton TA. 2008. Tristetraprolin regulates CXCL1 (KC) mRNA stability. J Immunol 180:2545–2552. doi: 10.4049/jimmunol.180.4.2545. [DOI] [PubMed] [Google Scholar]
- 34.Kratochvill F, Machacek C, Vogl C, Ebner F, Sedlyarov V, Gruber AR, Hartweger H, Vielnascher R, Karaghiosoff M, Rulicke T, Muller M, Hofacker I, Lang R, Kovarik P. 2011. Tristetraprolin-driven regulatory circuit controls quality and timing of mRNA decay in inflammation. Mol Syst Biol 7:560. doi: 10.1038/msb.2011.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Qiu LQ, Lai WS, Bradbury A, Zeldin DC, Blackshear PJ. 2015. Tristetraprolin (TTP) coordinately regulates primary and secondary cellular responses to proinflammatory stimuli. J Leukoc Biol 97:723–736. doi: 10.1189/jlb.3A0214-106R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jones DT, Cozzetto D. 2015. DISOPRED3: precise disordered region predictions with annotated protein-binding activity. Bioinformatics 31:857–863. doi: 10.1093/bioinformatics/btu744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Carballo E, Lai WS, Blackshear PJ. 1998. Feedback inhibition of macrophage tumor necrosis factor-alpha production by tristetraprolin. Science 281:1001–1005. doi: 10.1126/science.281.5379.1001. [DOI] [PubMed] [Google Scholar]
- 38.Lai WS, Parker JS, Grissom SF, Stumpo DJ, Blackshear PJ. 2006. Novel mRNA targets for tristetraprolin (TTP) identified by global analysis of stabilized transcripts in TTP-deficient fibroblasts. Mol Cell Biol 26:9196–9208. doi: 10.1128/MCB.00945-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lai WS, Kennington EA, Blackshear PJ. 2003. Tristetraprolin and its family members can promote the cell-free deadenylation of AU-rich element-containing mRNAs by poly(A) ribonuclease. Mol Cell Biol 23:3798–3812. doi: 10.1128/MCB.23.11.3798-3812.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Patial S, Blackshear PJ. 2016. Tristetraprolin as a therapeutic target in inflammatory disease. Trends Pharmacol Sci 37:811–821. doi: 10.1016/j.tips.2016.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Blackshear PJ, Perera L. 2014. Phylogenetic distribution and evolution of the linked RNA-binding and NOT1-binding domains in the tristetraprolin family of tandem CCCH zinc finger proteins. J Interferon Cytokine Res 34:297–306. doi: 10.1089/jir.2013.0150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hardo HG. 1981. Palindromic rheumatism: a review. J R Soc Med 74:521–524. doi: 10.1177/014107688107400713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mankia K, Emery P. 2017. What can palindromic rheumatism tell us? Best Pract Res Clin Rheumatol 31:90–98. doi: 10.1016/j.berh.2017.09.014. [DOI] [PubMed] [Google Scholar]
- 44.Rothschild BM, Pingitore C, Eaton M. 1998. Dactylitis: implications for clinical practice. Semin Arthritis Rheum 28:41–47. doi: 10.1016/S0049-0172(98)80027-9. [DOI] [PubMed] [Google Scholar]
- 45.Zhang X, Chen X, Liu Q, Zhang S, Hu W. 2017. Translation repression via modulation of the cytoplasmic poly(A)-binding protein in the inflammatory response. Elife 6:e27786. doi: 10.7554/eLife.27786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bulbrook D, Brazier H, Mahajan P, Kliszczak M, Fedorov O, Marchese FP, Aubareda A, Chalk R, Picaud S, Strain–Damerell C, Filippakopoulos P, Gileadi O, Clark AR, Yue WW, Burgess-Brown NA, Dean J. 2018. Tryptophan-mediated interactions between tristetraprolin and the CNOT9 subunit are required for CCR4-NOT deadenylase complex recruitment. J Mol Biol 430:722–736. doi: 10.1016/j.jmb.2017.12.018. [DOI] [PubMed] [Google Scholar]
- 47.Cremasco V, Decker CE, Stumpo D, Blackshear PJ, Nakayama KI, Nakayama K, Lupu TS, Graham DB, Novack DV, Faccio R. 2012. Protein kinase C-delta deficiency perturbs bone homeostasis by selective uncoupling of cathepsin K secretion and ruffled border formation in osteoclasts. J Bone Miner Res 27:2452–2463. doi: 10.1002/jbmr.1701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zamani A, Decker C, Cremasco V, Hughes L, Novack DV, Faccio R. 2015. Diacylglycerol kinase zeta (DGKzeta) is a critical regulator of bone homeostasis via modulation of c-Fos levels in osteoclasts. J Bone Miner Res 30:1852–1863. doi: 10.1002/jbmr.2533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lai WS, Kennington EA, Blackshear PJ. 2002. Interactions of CCCH zinc finger proteins with mRNA: non-binding tristetraprolin mutants exert an inhibitory effect on degradation of AU-rich element-containing mRNAs. J Biol Chem 277:9606–9613. doi: 10.1074/jbc.M110395200. [DOI] [PubMed] [Google Scholar]
- 50.Lai WS, Carballo E, Thorn JM, Kennington EA, Blackshear PJ. 2000. Interactions of CCCH zinc finger proteins with mRNA. Binding of tristetraprolin-related zinc finger proteins to AU-rich elements and destabilization of mRNA. J Biol Chem 275:17827–17837. doi: 10.1074/jbc.M001696200. [DOI] [PubMed] [Google Scholar]
- 51.Lai WS, Stumpo DJ, Blackshear PJ. 1990. Rapid insulin-stimulated accumulation of an mRNA encoding a proline-rich protein. J Biol Chem 265:16556–16563. [PubMed] [Google Scholar]
- 52.Moon AF, Mueller GA, Zhong X, Pedersen LC. 2010. A synergistic approach to protein crystallization: combination of a fixed-arm carrier with surface entropy reduction. Protein Sci 19:901–913. doi: 10.1002/pro.368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cao H, Tuttle JS, Blackshear PJ. 2004. Immunological characterization of tristetraprolin as a low abundance, inducible, stable cytosolic protein. J Biol Chem 279:21489–21499. doi: 10.1074/jbc.M400900200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Schellenberg MJ, Petrovich RM, Malone CC, Williams RS. 2018. Selectable high-yield recombinant protein production in human cells using a GFP/YFP nanobody affinity support. Protein Sci 27:1083–1092. doi: 10.1002/pro.3409. [DOI] [PMC free article] [PubMed] [Google Scholar]