

Abstract
Therapeutic target characterization involves many components, including accurate molecular weight (MW) determination. Knowledge of the accurate MW allows one to detect the presence of post-translational modifications, proteolytic cleavages, and importantly, if the correct construct has been generated and purified. Denaturing liquid chromatography-mass spectrometry (LC-MS) can be an attractive method for obtaining this information. However, membrane protein LC-MS methodology has remained relatively under-explored and under-incorporated in comparison to methods for soluble proteins. Here, systematic investigation of multiple gradients and column chemistries has led to development of a 5-minute denaturing LC-MS method for acquiring membrane protein accurate MW measurements. Conditions were interrogated with membrane proteins such as GPCRs and ion channels, as well as bispecific antibody constructs of variable sizes with the aim to provide the community with rapid LC-MS methods necessary to obtain chromatographic and accurate MW measurements in a medium-to high-throughput manner. The 5-minute method detailed has successfully produced MW measurements for hydrophobic proteins with a wide size range (17.5 to 105 kDa) and provided evidence that some constructs indeed contain unexpected modifications or sequence clipping. This rapid LC-MS method is also capable of baseline separating formylated and non-formylated aquaporinZ membrane protein.
For TOC only:
Membrane proteins are responsible for multiple cellular functions1 and have been linked to many disease states such as cystic fibrosis, multiple cancers, and retinitis pigmentosa,2–4 making them prime therapeutic targets.5,6 Having a rapid method for determining their accurate molecular weight (MW) would be of great utility to the pharmaceutical industry for drug design and development. However, their characterization via traditional biophysical techniques, such as SPR or NMR, is often difficult7–11 due to the challenges associated with expression, purification, and solubilization, as well as overall sample amounts.12 Native mass spectrometry (native-MS) has been an enabling technique for investigation of numerous membrane protein systems13 as it allows for the study of intact membrane protein complex stoichiometry and structure, as well as interactions with various lipids and small molecules, using relatively small amounts of material.14–16 However, membrane protein complexes can afford native-MS spectra that may include multiple non-volatile adducts, bound ligands and/or protein stoichiometries9, which can complicate accurate MW determination. Accurate MW measurements can provide insight into potential proteolytic cleavage(s) and/or low MW modifications, and importantly, whether the correct construct has been purified. Denaturing liquid chromatography-mass spectrometry (LC-MS) is an attractive technique to attain this information as it involves the removal of non-covalently bound lipids or small molecules that may remain using other techniques. Also, it provides dissociation of membrane protein complexes, avoiding potential spectral interpretation issues.
Nevertheless, a widely incorporated high-throughput denaturing LC-MS method for membrane proteins has remained somewhat elusive throughout the literature. Early methods (1983–1998) utilized C8, C18 or polystyrene-divinyl benzene copolymer (PLRP/S) columns.17–19 These methods involve long gradients (40+ minutes) and pre-injection sample acidification in formic acid (FA) or trifluoroacetic acid (TFA). Methods from the early 2000s also employ PLRP/S columns.7,20 These methods utilized lower concentrations of TFA or FA, however still require lengthy gradients (55+ minutes), pre-injection sample acidification and, in some cases, required a second elution with 60% FA to elute larger subunits.20 The column particle size employed for these methods is 5 μm. The most current methodology, to our knowledge, employs ZORBAX 300SB-C3 columns in multiple lengths (achieved by tandemly linking multiple 50 mm columns).21 These gradients are 12–40 minutes and use 0.1% FA as the mobile phase additive. Here, the protein stock is diluted in 1% FA prior to injection. While the column particle size used for this work is not reported, only 3.5 or 5 μm particle size is commercially available from the manufacturer for the column dimensions reported in the manuscript.21
Many of these gradients employ solvents such as acetonitrile, 2-propanol and methanol with varying concentrations of ion pairing agents. It has also been reported that denaturing LC-MS and non-native size exclusion chromatography (SEC) have been used prior to top-down MS of membrane proteins.21–24 These methods employ unique solvents, such as mixtures of chloroform and methanol.24,25 While all of these methods have provided quality data, they require extensive gradient lengths and/or pre-injection sample handling. Additionally, most of these methods are quite extensive, and their main purpose was the chromatographic separation of complex mixtures or separation of membrane protein complex subunits. The need to rapidly screen membrane protein constructs for MW determination or confirmation does not require such extensive separation. Instead, a more high-throughput method is optimal for this type of analysis, especially in a biopharmaceutical environment.
To extend denaturing LC-MS of membrane proteins and other hydrophobic proteins to an industrial platform method where high-throughput analysis is highly desirable, we systematically interrogated conditions to reduce method length, eliminate pre-injection sample handling and extend column lifetime. Here, we present a medium-to high-throughput denaturing rapid LC-MS method for membrane protein accurate MW determination. Development of this 5-minute method involved investigation of several column chemistries, mobile phases, different ion pairing agents, and gradients (all investigated without pre-injection sample handling). Furthermore, we have also explored bispecific antibody constructs to assess this method with other hydrophobic molecules. The goal of this work was to provide a rapid method for screening the quality of prepared and purified protein via accurate MW determination, not the separation of the protein from multiple solution contaminants (as the protein constructs analyzed herein are previously purified). For brevity, only the optimal chromatography conditions will be discussed. Refer to the Supporting Information for details regarding the other chromatography conditions investigated.
Experimental
Membrane protein preparation for rapid denaturing LC-MS.
Protein stock preparation for all membrane proteins is detailed in the Supporting Information. Concentrations were determined using A280 measurements. For denaturing UPLC-MS experiments, the bacteriorhodopsin (bR) mutants L111A, T47A, P50A and P50A/T46A were diluted for 10 μg injections. Proteolytic digestion conditions and instrument settings for bR P50A are detailed in the Supporting Information. Wild type (WT) and W14A aquaporinZ (AqpZ) were diluted for 2 μg monomer injections. Both WT and W14A AqpZ were also analyzed with covalent green fluorescent protein (GFP) tags. AqpZ-GFP samples were diluted for 1.5 μg injections of both the WT and W14A monomers. All AqpZ and bR samples were diluted in 200 mM ammonium acetate/ 1.1% (w/v) n-octyl-β-D-glucoside (OG). Coq10 was analyzed as received at 6 μg per injection. The ammonia transporter AmtB fused with maltose-binding periplasmic protein (AmtB-MBP) was received at 9 mg/mL26 and was diluted in 200 mM ammonium acetate/ 0.017% (w/v) n-dodecyl-β-D-maltopyranoside (DDM) for 6 μg of monomer per injection. AmtB was also analyzed at 6 μg monomer after TEV (91636; Invitrogen; Carlsbad, CA) cleavage to remove MBP. This cleavage required overnight incubation of AmtB-MBP and TEV in a 100:1 ratio with 5 μM β-mercaptoethanol at 4 °C. KcsA-Kv1.3 (K-K) was analyzed, as prepared, in n-decyl-β-D-maltopyranoside (DM), at 4 μg monomer per injection after removal of the N-terminal His-tag by caspase3 (made in house) cleavage. This cleavage was executed by incubation of 350 μg of K-K with 6 μg of caspase3 at 4 °C for 6–8 hours. Completion of the cleavage reactions was confirmed via the LC-MS method described herein. Optimization of starting % B was achieved with injections of WT AqpZ (polyphenol column) and caspase3 cleaved K-K (C3 column) under increasing starting % B from 5 to 40%. To demonstrate reproducibility, WT AqpZ-GFP was measured 10 additional times. Polyphenol column limit of detection studies were performed with K-K (0.2–15 μg on column). Limit of detection studies on the C3 column were performed with both bR P50A T46A (0.2–15.2 μg) and K-K (0.075–20 μg on column). Five Fc-provided bispecific antibody constructs (D6Q, K8T, M3X, X2W, and J8R) were analyzed at 10 μg on column. A non-Fc-provided bispecific antibody construct (Construct A), was analyzed at 20 μg on column at 5% and 30% B. All constructs and concentrations were analyzed in triplicate in positive ionization mode.
Rapid Denaturing LC-MS conditions.
An Acquity UPLC (Waters MS-Technologies; Manchester, UK) with a 450 Å, 2.7 μm, 2.1 × 50 mm BioResolve RP mAb Polyphenol column (186008944; Waters; Milford, MA) which is compatible with both UPLC and HPLC systems, was employed. Classically, the term UPLC refers to sub-2 μm particle size columns.27 As stated above, the particle size used in this method is 2.7 μm. However, according to manufacturer specifications, this column is compatible with UPLC systems. Additionally, this method is run on an Acquity UPLC system operated at typical UPLC pressure (>6000 psi). A 5-minute method (including gradient and re-equilibration) was applied using a mix of 0.1% (v/v) FA / 0.1% (v/v) TFA in water as mobile phase A and in 90% n-propanol as mobile phase B. The gradient was maintained at 30% B from 0–1 minutes, increased to 95% B from 1–2.5 minutes where it was held for 1 minute. The gradient was then reduced to 30% B from 3.5–4 minutes and held for an additional minute at 30% B. The flow rate was 500 μL/min with a column temperature of 65 °C. The same method was also employed on a 2.1 × 50 mm, 1.8 μm ZORBAX RRHD 300SB-C3 column (857750–909; Agilent; Santa Clara CA), and these results are discussed in the Supporting Information. This C3 column is representative of a standard UPLC particle size column.
The Acquity UPLC was run in-line with a Xevo Q-ToF mass spectrometer (Waters MS-Technologies). MS data was collected over the m/z 500–5000 range. For instrument tune settings and pressures refer to the Supporting Information. Zero-charge accurate mass measurements were determined using MaxEnt28 in the MassLynx software (MassLynx 4.1, Waters MS-Technologies). MaxEnt parameters can be found in the Supporting Information. Limit of detection studies used the total ion counts from the area centered deconvoluted spectra.
Results and Discussion
The amphipathic nature of membrane proteins (hydrophilic-extracellular regions and hydrophobic-transmembrane regions) makes them intrinsically difficult to characterize by traditional UPLC-MS methods. Bispecific antibody constructs represent another class of hydrophobic molecules difficult to characterize by traditional UPLC methods. A bispecific antibody construct is composed of two single-chain variable fragments (scFv’s) joined using short protein linkers.29,30 One example of a bispecific antibody construct is Blinatumomab, a second-line treatment for Philadelphia chromosome-negative relapsed or refractory acute lymphoblastic leukemia. Blinatumomab consists of two fused scFv’s against CD19 and CD3.30 Bispecific antibody constructs are roughly one third the MW of traditional monoclonal antibodies, however they are significantly more hydrophobic and have the propensity to aggregate.29,30 Due to the similarity of hydrophobic characteristics, we felt that bispecific antibody constructs could also be used to assess this rapid LC-MS method meant for molecules exhibiting significant hydrophobic characteristics.
Reversed-phase HP(UP)LC commonly employs a C18 bonded silica stationary phase for small molecules.31 However, membrane proteins would likely be retained during a short gradient given their hydrophobic character, and thus C18 columns were not tested during this study and are not recommended. During method development, C3, C4, C8, Cyano (CN) and polyphenol columns were investigated. Only the C8 column proved to be too retentive for the proteins under all of the gradient conditions investigated, which was apparent from lack of protein signal and increasing column back pressure. Investigation of the C3, C4, CN and polyphenol columns, which share similar short chain bonding, revealed that multiple gradient conditions resulted in protein elution. Thus it can be inferred that short chain bonding is an ideal column chemistry for hydrophobic protein denaturing LC-MS; this is a logical inference considering minimization of column interactions with short chain lengths, thus decreasing unwanted retention. Based on the results, the polyphenol column operated at UPLC pressures at 65 °C (column temperature limited to 65 °C by manufacturer) was determined to provided optimal chromatography. (See the Supporting Information for discussion of the other column chemistries.)
Optimization of Chromatography Conditions.
Historically, denaturing LC-MS methods for membrane proteins involved high concentrations of acid in the mobile phases (isopropanol, CHCl3/methanol, methanol) and/or long gradients. Some gradients employed TFA in place of FA, however remained quite lengthy (40+ minutes). Additionally, no direct comparison of TFA and FA chromatography for the same membrane protein has been performed. While recent efforts have reduced the mobile phase FA concentration,21 the multiple methods presented in that work run upwards of 12 minutes. Here, method development was aimed at decreasing the method length and minimizing pre-injection sample handling through investigation of various FA and TFA concentrations, mobile phases and column chemistries (Supporting Information), similar to previous HPLC-MS method develoment for monoclonal antibody (mAb) characterization.32,33
Representative chromatograms and spectra obtained with the final optimized method conditions using 90% n-propanol are shown in Figure 1. Acetonitrile, methanol, isopropanol and 90% n-propanol were evaluated with both TFA and FA as ion pairing agents. Methanol and isopropanol resulted in higher back pressure, poor chromatography and/or low protein signal intensity. While contrary to previous reports,21 employing acetonitrile with either 0.1% or 1% FA produced high quality chromatography and protein signal (data not shown). However, extreme carryover issues were observed with as little as 0.25 μg of protein. Replacing acetonitrile with 90% n-propanol, which is used for antibody reversed-phase chromatography, fixed the carryover issues without compromising protein signal.33 Method development for mAb characterization reported by Dillon et al. demonstrated that employment of n-propanol for mobile phase B reduced peak tailing and increased resolution in comparison to acetonitrile, methanol and isopropanol.33 This was attributed to the high eluotropic coefficient of n-propanol, as compared to the other solvents, which helped to reduce column interactions.33 Here, employment of 90% n-propanol as mobile phase B for membrane protein UPLC-MS analysis, as expected, aided in reducing column interactions, decreasing peak tailing, and eliminating carryover as compared to other organic solvents investigated.33
Figure 1.
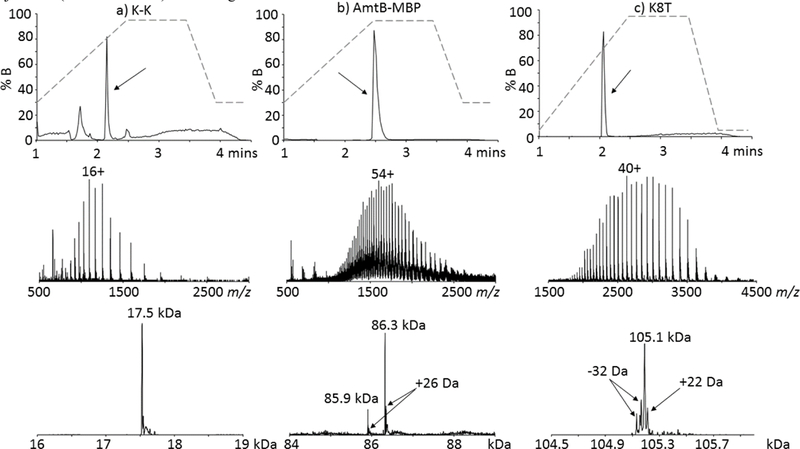
Representative reconstructed UPLC ion chromatograms (RICs) (upper), MS spectra (middle) and deconvoluted MS spectra (lower) of the membrane proteins (a) K-K (~17.5 kDa); (b) AmtB-MBP (~86.3 kDa) and (c) Fc-provided antibody construct K8T (~105.1 kDa). The black arrow in each chromatogram denotes where the protein elutes from the polyphenol column. The Y-axis for each chromatogram is the % B gradient profile (dashed line), starting at 30% B for the membrane protein samples and at 5% B for the bispecific antibody constructs. The most intense charge state is labeled in each MS spectrum. The 85.9 kDa species observed for AmtB-MBP corresponds to an N-terminal clip, vide infra. The +26 Da and +22 Da species present in the deconvoluted spectra for AmtB-MBP and K8T are presumably salt adducts. The Y-axis for the spectra represents intensity with arbitrary units.
Addition of 0.1% TFA to both mobile phases (water and 90% n-propanol) resulted in good chromatography with no observable carryover. TFA is a commonly utilized solvent additive for reversed-phase HP(UP)LC because it is an excellent ion pairing agent, increases the surface tension of water, and improves peak shape.34,35 However, it is not preferred for LC-MS as electrospray of solutions with high surface tension and conductivity is difficult and ultimately results in analyte signal suppression.35,36 Thus, as expected, protein ionization during these experiments was inferior when compared to those performed with FA.35 Use of FA as a solvent additive is generally not recommended for HP(UP)LC of proteins due to potential O-formylation of amino acids during exposure.17 However, protein exposure to FA during the experiments detailed within was limited to the 5-minute method. Additionally, only the four AqpZ constructs analyzed displayed N-terminal formylation (vide infra), which was previously detected by native-MS and LC-MS/MS for the WT construct.37
Adding 1% FA to both mobile phases resulted in good chromatography and MS performance, but significant protein carryover was observed in blank runs between protein injections (data not shown). Lowering to 0.1% FA decreased the carryover while maintaining the strong ion signal. However, optimal results were achieved with a mix of 0.1% FA and 0.1% TFA. Upon mixing, superior chromatography was observed on the polyphenol column with the separation of the non-formylated and formylated AqpZ monomers (Figures 2 and S1). For all AqpZ constructs, the formylated construct elutes an average of 0.05 minutes after the non-formylated construct on the 5-minute gradient. Though some separation was clearly achieved within 5 minutes, improved separation was attained with a 7 and 15 minute gradients (Figure S2). Increased separation of the two species in the 0.1% FA mobile phases was also achieved with the same 7 and 15 minute gradients (Figure S2). Chromatographic separation of a non-formylated and formylated protein has been previously reported for human S100b, with a retention time shift of < 0.5 minutes.38 Additionally, a shift in retention time (mean value of 2.8 minutes) upon N-terminal formylation has also been reported for small peptides.39 Formylation alters the N-terminal amine to an amide, causing the loss of a chargeable basic site, which could account for the retention time shift. All runs were performed at a column temperature of 65 °C, which minimized baseline fluctuation and protein carryover as compared to 40 °C (data not shown). Previous work done by Dillon et al demonstrated that increased column temperature (60 to 75 °C) decreased column fouling and peak tailing while improving resolution and recovery.33,40 Additionally, mixing TFA and FA decreased the level of signal suppresion observed on both the polyphenol and C3 columns with TFA alone (Figures S1 and S3, respectively).
Figure 2.
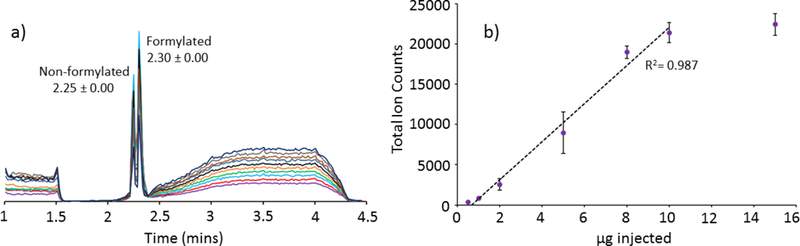
Reproducibility and limit of detection experiments. Reproducibility tests (a) with WT AqpZ-GFP at 1.6 μg/injection. Each run is represented with a different color and show an overall reproducibility of retention time of 2.25 ± 0.00 and 2.30 ± 0.00 minutes for the non-formylated and formylated WT AqpZ-GFP, respectively. The Y-axis for the spectra represents intensity with arbitrary units. Limit of detection studies (b) with K-K, injection amounts based on A280 readings ranged from 0.2 to 15 μg for K-K. Outliers existed at 0.2 and 15 μg. Total ion counts on the y-axis were obtained from MaxEnt deconvolution of the LC integrated peak area.
In general, these proteins are very amphiphilic and some constructs could be retained longer than others. Thus regardless of the gradient conditions tested, some carryover was always observed in blank injections run post protein injection (<10% carryover). However, carryover was never observed in proteins analyzed back-to-back. It has been previously reported that some membrane proteins elute with limited efficiency, thus yielding “ghost peaks” in subsequent runs.22,41 Conversely, with optimization of injection concentration, these “ghost peaks” were not detected throughout these experiments. Additionally, column backing pressure remained stable throughout this entire work, indicating minimal protein retention over time. Since membrane proteins can be challenging to express and purify in high quantities, minimization of the protein concentration necessary to obtain quality chromatographic and accurate mass measurements was a key part of method development.
During method development it was observed that chromatographic separation between the detergent and the protein for DDM solubilized proteins decreased as the method length was decreased to 5 minutes. In cases of detergent signal interference, a 7 or 9 minute method detailed in the Supporting Information is suggested. Additionally, it is worth noting that proteins are often diluted in mobile phase A for denaturing HPLC-MS injections.31 However, injections of bR mutants from protein stock diluted in mobile phase A resulted in very low chromatographic protein signal, thus none of the samples were diluted in mobile phase A prior to injection. Therefore, none of the membrane proteins were denatured when injected, and instead existed in their “native-state” detergent micelle enviroment; this suggests that the proteins fully denatured only when subjected to the LC-MS gradient.
Initial 5-minute method experiments at 5% B showed the non-formylated and formylated AqpZ partially resolved, however were highly overpowered by detergent signal. Optimization of starting % B was performed by increasing it from 5% to 50% B to determine the starting % needed to maximize chromatographic resolution. As shown in Figures 2 and S4, the chromatographic resolution of the two AqpZ species was easily observed at ≥ 30% B due to decreased chromatographic detergent signal. This separation was not observed on the C3 column so optimization of starting % B (Figure S5) was performed with membrane protein K-K because it eluted near the detergent signal and multiple chromatographic peaks containing various low MW species were detected. Both columns demonstrated that 30% B provided optimal chromatography for resolution of modified constructs, separation from detergent peaks and decreasing the chromatographic and spectral intensity of co-eluting species. Hence 30% B was used for membrane protein analysis. Chromatograms and spectra for K-K, AmtB-MBP and the Fc-provided antibody construct K8T are shown in Figure 1. The gradient profiles have been overlaid on these respective chromatograms demonstrating the difference in starting % B for the membrane protein samples as compared to the bispecific antibody constructs. Representative chromatograms and spectra for the remaining membrane protein samples on the polyphenol and C3 columns are shown in Figures S6 and S7, respectively. Bispecific antibody constructs were run only on the polyphenol column (Figure S8).
It should be noted that the bR mutants provided a higher intensity MS signal on the C3 column as opposed to the polyphenol column (compare Figures S6 and S7). On the polyphenol column, monitoring protein elution by UV absorbance clearly showed that each of the bR mutants eluted at 2.4 minutes. Furthermore, Coq10 eluted at 30% from the polyphenol and C3 columns, however, optimal chromatography was at 25% or 20% B for the polyphenol and C3 column, respectively. Results for Coq10 at 25 and 20% B are shown in Figures S5 and S6, respectively. Data acquired at 30% B on both columns is provided in Figure S9. If a protein is not chromatographically separating from the detergent peak, reducing the starting % organic mobile phase is suggested to rectify the issue.
Analysis of the bispecific antibody constructs revealed that Construct A was not retained on the column at 30% B (data not shown). Exploration of lower starting % B yielded data for the non Fc-provided construct A and Fc-provided bispecific antibody constructs at under 15% B, with optimal chromatography at 5% B. These observations suggest that while these species have been reported to exhibit hydrophobic characteristics, they may not be as hydrophobic as membrane proteins, which provide optimal chromatography at higher starting % B. However, additional non Fc-provided bispecific antibody constructs will need to be tested to further support this inference. The Fc-provided bispecific antibody constructs all displayed similar chromatographic results to Construct A.
Reproducibility and Limit of Detection.
Triplicate injections of each membrane protein showed minimal retention time variability (Table 1a–c). To further demonstrate reproducibility, ten additional WT AqpZ-GFP injections were run, showing negligible rentention time variability (Figure 2). Additionally, with a single blank between each WT AqpZ-GFP injection, only low levels of carryover were detected (<5%). Injection amounts were optimized to maintain observable protein signal in the presence of high chromatographic signals observed from the solubilization detergents. In general, it is known that detergents will preferentially ionize by ESI in comparison to proteins, thus high detergent concentration can suppress analyte ionization.31 Limit of detection studies with caspase3 cleaved K-K showed detection to be as low as 0.2 μg on column. However, both the chromatographic resolution and ion signal were optimal at or above 2 μg. As shown in Figure 2, saturation occurred around 20,000 total ion counts (10 μg of K-K). C3 column reproducibility, with WT AqpZ-GFP, and limit of detection studies, with bR P50A T46A and K-K, are provided in Figure S10.
Table 1. Average Mass Accuracies of Membrane Proteins and Bispecific Antibody Constructs.
Average MWs, mass accuracy (PPM), retention times and monomer transmembrane (TM) regions for the 12 measured proteins. Theoretical MWs are the expected protein average MW and account for any protein modifications known prior to the work herein. The theoretical MWs for the bR constructs consider the N-terminal pyroglutamate and the loss of aspartate from the C terminus. The theoretical MW of construct A considered an N-terminal pyroglutamate and 4 disulfide bonds. Each MW measurement is an average of triplicate runs. The formylated and non-formylated AqpZ constructs (a); the retinal bound and unbound bR constructs (b); and the clipped and unclipped constructs (c). Bispecific antibody construct mass accuracy and retention time data is reported in (d).
| 1a) Protein |
Non-formylated | Formylated | Retention Time (min) |
Monomer TM Regions |
||||
|---|---|---|---|---|---|---|---|---|
| Theoretical Mass (Da) |
Measured Mass (Da) |
PPM | Theoretical Mass (Da) |
Measured Mass (Da) |
PPM | |||
| WT AqpZ | 24,268.56 | 24,268.30 ± 0.09 | 10.53 | 24,296.57 | 24,296.05 ± 0.25 | 21.53 | 2.29 ± 0.01 2.35 ± 0.01 |
6^ |
| W14A AqpZ | 24,153.43 | 24,153.14 ± 0.13 | 11.81 | 24,181.44 | 24,180.65 ± 0.30 | 32.52 | 2.28 ± 0.01 2.34 ± 0.01 |
6^ |
| WT AqpZ-GFP | 49,861.42 | 49,862.29 ± 0.36 | −17.35 | 49,889.43 | 49,890.45 ± 0.42 | −20.45 | 2.25 ± 0.00 2.30 ± 0.00 |
6^ |
| W14A AqpZ-GFP | 49,746.29 | 49,747.20 ± 0.07 | −18.42 | 49,774.30 | 49,774.93 ± 0.27 | −12.76 | 2.22 ± 0.00 2.27 ± 0.00 |
6^ |
| 1b) Protein |
Retinal Unbound | Retinal Bound | Retention Time (min) |
Monomer TM Regions |
||||
|---|---|---|---|---|---|---|---|---|
| Theoretical Mass (Da) |
Measured Mass (Da) |
PPM | Theoretical Mass (Da) |
Measured Mass (Da) |
PPM | |||
| bR L111A | 26,741.53 | 26,740.40 ± 0.49 | 42.25 | 27,007.96 | 27,007.66 ± 0.83 | 11.15 | 2.40 ± 0.00 | 7 |
| bR T47A | 26,753.59 | 26,753.44 ± 1.05 | 5.44 | 27,020.02 | 27,019.28 ± 0.28 | 27.24 | 2.42 ± 0.00 | 7 |
| bR P50A | 26,757.57 | 26,756.36 ± 0.64 | 45.54 | 27,024.00 | 27,023.39 ± 0.18 | 43.19 | 2.42 ± 0.00 | 7 |
| bR P50A T46A | 26,727.55 | 26,727.01 ± 0.29 | 20.16 | 26,993.98 | 26,992.94 ± 0.55 | 38.35 | 2.42 ± 0.00 | 7 |
| 1c) Protein |
Clipped | Unclipped | Retention Time (min) |
Monomer TM Regions |
||||
|---|---|---|---|---|---|---|---|---|
| Theoretical Mass (Da) |
Measured Mass (Da) |
PPM | Theoretical Mass (Da) |
Measured Mass (Da) |
PPM | |||
| Coq10* | 18,307.43 | 18,306.96 ± 0.11 | 25.55 | 20,302.61 | 20,302.30 ± 0.09 | 15.42 | 1.79 ± 0.00 | Unknown |
| K-K | 17,528.23 | 17,527.78 ± 0.12 | 25.64 | 2^ | ||||
| AmtB-MBP | 85,905.27 | 85,905.23± 0.44 | 0.42 | 86,321.77 | 86,321.28 ± 0.91 | 5.68 | 2.51 ± 0.00 | 11 |
| AmtB | 42,263.89 | 42,263.58 ± 0.61 | 7.29 | 2.54 ± 0.00 | 11 | |||
| 1d) Bispecific Anti body Constructs |
Theoretical Mass (Da) | Measured Mass (Da) | PPM | Retention Time (min) |
|---|---|---|---|---|
| D6Q | 105,123.77 | 105,123.21 ± 1.04 | 5.31 | 2.05 ± 0.01 |
| K8T | 105,190.86 | 105,191.10 ± 1.39 | −2.20 | 2.04 ± 0.01 |
| M3X | 105,193.86 | 105,191.68 ± 1.09 | 20.79 | 2.05 ± 0.00 |
| X2W | 105,065.73 | 105,065.31 ± 1.89 | 4.07 | 2.06 ± 0.00 |
| V2T | 105,332.08 | 105,332.72 ± 1.00 | −6.04 | 2.04 ± 0.01 |
| Construct A | 54,635.20 | 54,634.61 ± 0.80 | 10.88 | 2.05 ± 0.01 |
Reported retention time and mass accuracy for Coq 10* is from 25% B data.
denotes two additional half-membrane or intramembrane regions. An overall RMS error of 22.17 PPM (0.80 Da) was measured for all constructs.
Mass Accuracy.
Overall mass accuracy for the 12 membrane proteins and the 6 bispecific antibody constructs are shown in Table 1, with an overall measured RMS error of 22.17 ppm (0.80 Da). Several proteins investigated showed readily observable post-translational modifications or proteolytic cleavages. For example, during previous WT AqpZ experiments,37 N-terminal methionine formylation was detected, which was observed for each of the AqpZ constructs herein (Table 1a). N-terminal formylation of AqpZ was observed due to expression of this protein in E. coli, however, the non-formylated species was observed because of incomplete post-translational removal of the formyl group. Additionally, when expressed in its native Halobacterium host, bR is known to undergo proteolytic cleavage on both the N-and C-termini.24 Proteolytic digestion of bR mutants with chymotrypsin followed by LC-MS/MS collision-induced dissociation (CID) (Supporting Information) verified both N-terminal pyroglutamate formation and a loss of aspartate from the C-terminus. Loss of aspartate had occurred in roughly 95% of the individual construct (Figure S11). These findings are accounted for in the theoretical MWs reported in Table 1b. Additionally, the Schiff-linked retinal (+266.43 Da) species was also observed with each of the mutants. Roughly <20% of each mutant did not retain the Schiff-linked retinal.
Furthermore, Coq10 manifested two main species, one of which correlated to within 15.42 ppm (0.31 Da) of its intact theoretical mass (Figure S11). The second species corresponded to a clipped sequence, which based on MW could have been either an N-or C-terminal clip (Table 1c). In the deconvoluted spectrum (Figure S12), several species were observed corresponding to additions of ~510 Da, which were determined to be DDM adducts. This was the only protein to show detergent adduction upon deconvolution. Through Glu-C proteolytic digestion followed by LC-MS/MS (Supporting Information) it was determined to be N-terminal cleavage (Figure S13). Also, initial deconvoluted spectra for K-K, revealed several species. To reduce heterogeneity, caspase3 cleavage was employed to remove the His-tag used for purification. Post caspase3 cleavage revealed only two species: the fully cleaved (most abundant) and another cleaved product, mis-cleaved by a single amino acid (less abundant). Of the two, the mis-cleaved product provided poorer mass accuracy. For AmtB-MBP, the clipped sequence corresponds to an N-terminal clip from the MBP of –MDIG. The Fc-provided bispecific antibody constructs are known to be processed and include N-terminal pyroglutamate formation as well as C-terminal lysine cleavage. These sequence characteristics were considered in the reported theoretical masses.
Conclusions
The ability to obtain accurate MW measurements of membrane proteins is an important aspect of construct characterization. Here, we have demonstrated that this is possible with a 5-minute denaturing LC-MS method. This 5-minute method (including column re-equilibration) employs a mix of 0.1% (v/v) TFA / 0.1% (v/v) FA in water as mobile phase A and in 90% n-propanol as mobile phase B with a flow rate of 500 μL/min and a column temperature of 65 °C. Using this rapid LC-MS method we are able to derive a MW measurement RMS error of 22.17 ppm (0.80 Da) for 12 membrane proteins and six bispecific antibody constructs, ranging in MW 17.5 to 105 kDa on a time-of-flight MS system operating at a modest resolution setting (10,000 FWHM)
Additionally we demonstrate chromatographic separation of non-formylated and formylated AqpZ constructs. Minimal testing of a C8 column yielded no protein elution, which given the hydrophobicity of these multiple transmembrane region containing proteins is not surprising. Thus avoidance of column chemistries similar to C8 and C18 for development of other denaturing UPLC-MS and rapidLC-MS methods for membrane and hydrophobic proteins is recommended. However, with some optimization, C3, C4 and CN columns similar to those investigated here could be employed for high-throughput denaturing HPLC and UPLC-MS of membrane and other hydrophobic proteins. Although not investigated in this work, monolithic columns may also be useful for rapid denaturing LC-MS of hydrophobic proteins as the large pore size of these columns would help minimize unwanted column retention. Contrary to previous opinions regarding the hydrophobicity of non Fc-provided bispecific antibody constructs, thought to be similarly hydrophobic to membrane proteins, they did not exhibit similar chromatographic properties when analyzed by rapid LC-MS. The observation that chromatography of bispecific antibody constructs showed drastic improvement when decreasing mobile phase B from 30% to 5% supports a hypothesis that these molecules are, in general, not as hydrophobic as membrane proteins. Again, more non Fc-provided bispecific antibody constructs would need to be analyzed to further support this hypothesis.
Furthermore, the authors acknowledge the considerable interest in identifying co-purified lipids in membrane protein purification reactions. While low MW species were detected in the chromatograms presented here, none of the species were pursued for further identification as the focus of this method development was to provide a rapid screening method for accurate MW determination.
Furthermore, the optimized method requires no pre-injection sample acidification or resuspension. The polyphenol column method development was performed using a single column, over 150 runs in total, with no loss in column viability. Based on the results presented herein using a short 5-minute method on a commercially available polyphenol column, we expect that denaturing LC-MS of membrane proteins can indeed be a high-throughput characterization tool within both the industrial and academic-research setting. Additionally, the authors recognize that 2.7 μm particle size is not standard for UPLC-MS analysis. However, this column is not yet available in 1.7 μm particle size. If made available, improved chromatographic separation and potentially even shorter method times could be achieved.
Supplementary Material
Acknowledgments
Support from the US National Institutes of Health (R01GM103479) and the US Department of Energy (UCLA/DOE Institute for Genomics and Proteomics; DE-FC03–02ER63421) to JAL is acknowledged. M.T.M. and J.E.K. are supported by the Bisgrove Scholar Award from Science Foundation Arizona and the National Institute of General Medical Sciences and National Institutes of Health under Award Number R35 GM128624 to M.T.M. The authors would like to acknowledge Drs. James Bowie and Nicholas Woodall (UCLA) for their generous gift of the bacteriorhodopsin constructs, and Hui S. Tsui and Dr. Catherine F. Clarke (UCLA) for the gift of the Coq10 protein. Furthermore, the authors thank Tawnya Flick (Amgen) and Anthony Reed (Amgen) for the initial discussions regarding the analysis of bispecific antibody constructs and N-terminal formylation, respectively. Patrick Hoffmann (Amgen ARM Research and Development) and Will Hamouda (Amgen Pre-Pivotal Attribute Sciences) are acknowledged for providing the bispecific antibody constructs. Furthermore, the authors thank Mengjie Lu and Qiang Zhao (SIMM) and Dandan Zhang and Yingli Ma (Amgen Shanghai) for their willingness to provide additional membrane proteins for method testing. The Amgen postdoctoral program is acknowledged for its support of this project.
ASSOCIATED CONTENT
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website.
REFERENCES
- 1.Almén MS, Nordström KJ, Fredriksson R & Schiöth HB Mapping the human membrane proteome: a majority of the human membrane proteins can be classified according to function and evolutionary origin. BMC Biology 7, 50, doi: 10.1186/1741-7007-7-50 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kampen KR Membrane Proteins: The Key Players of a Cancer Cell. The Journal of Membrane Biology 242, 69–74, doi: 10.1007/s00232-011-9381-7 (2011). [DOI] [PubMed] [Google Scholar]
- 3.Sanders CR & Nagy JK Misfolding of membrane proteins in health and disease: the lady or the tiger? Current Opinion in Structural Biology 10, 438–442 (2000). [DOI] [PubMed] [Google Scholar]
- 4.Sanders CR & Myers JK Disease-Related Misassembly of Membrane Proteins. Annual Reviews Biomolecular Structure 33, 25–51 (2004). [DOI] [PubMed] [Google Scholar]
- 5.Hopkins AL & Groom CR The druggable genome. Nature Reviews Drug Discovery 1, 727–730, doi: 10.1038/nrd892 (2002). [DOI] [PubMed] [Google Scholar]
- 6.Arinaminpathy Y, Khurana E, Engelman DM & Gerstein MB Computational analysis of membrane proteins: the largest class of drug targets. Drug Discovery Today 14, 1130–1135, doi: 10.1016/j.drudis.2009.08.006 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.le Coutre J et al. Proteomics on Full-Length Membrane Proteins Using Mass Spectrometry. Biochemistry 39, 4237–4242, doi: 10.1021/bi000150m (2000). [DOI] [PubMed] [Google Scholar]
- 8.Savage DF, Egea PF, Robles-Colmenares Y, O’Connell JD III & Stroud RM Architecture and Selectivity in Aquaporins 2.5: Α X-Ray Structure of Aquaporin Z. PLOS Biology 1, 334–340, doi: 10.1371/journal/pbio.0000072 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang SC et al. Ion Mobility Mass Spectrometry of Two Tetrameric Membrane Protein Complexes Reveals Compact Structures and Differences in Stability and Packing. Journal of the American Chemical Society 132, 15468–15470, doi: 10.1021/ja104312e (2010). [DOI] [PubMed] [Google Scholar]
- 10.Chang Y-C & Bowie JU Measuring membrane protein stability under native conditions. Proceedings of the National Academy of Sciences 111, 219–224, doi: 10.1073/pnas.1318576111 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fisette O et al. Hydration Dynamics of a Peripheral Membrane Protein. Journal of the American Chemical Society 138, 11526–11535, doi: 10.1021/jacs.6b07005 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Barrera NP & Robinson CV Advances in the Mass Spectrometry of Membrane Proteins: From Individual Proteins to Intact Complexes The Annual Review of Biochemistry 80, 247–271, doi: 10.1146/annurev-biochem-062309-093307 (2011). [DOI] [PubMed] [Google Scholar]
- 13.Marcoux J & Robinson CV Twenty Years of Gas Phase Structural Biology. Structure 21, 1541–1550 (2013). [DOI] [PubMed] [Google Scholar]
- 14.Laganowsky A, Reading E, Hopper JTS & Robinson CV Mass Spectrometry of intact membrane protein complexes. Nature Protocols 8, 639–651, doi: 10.1038/nprot.2013.024 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Laganowsky A et al. Membrane proteins bind lipids selectively to modulate their structure and function. Nature 510, 172–175, doi: 10.1038/nature13419 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gault J et al. High-resolution mass spectrometry of small molecules bound to membrane proteins. Nature Methods 13, 333–336, doi: 10.1038/NMETH.3771 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tarr GE & Crabb JW Reverse-Phase High-Performance Liquid Chromatography of Hydrophobic Proteins and Fragments Thereof. Analytical Biochemistry 131, 99–107 (1983). [DOI] [PubMed] [Google Scholar]
- 18.Musatov A & Robinson NC Subunit Analysis of Bovine Cytochrome bc1 by Reverse-Phase HPLC and Determination of the Subunit Masses by Electrospray Ionization Mass Spectrometry. Biochemistry 33, 10561–10567 (1994). [DOI] [PubMed] [Google Scholar]
- 19.Whitelegge JP, Gundersen CB & Faull KF Electrospray-ionization mass spectrometry of intact intrinsic membrane proteins. Protein Science 7, 1423–1430 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Whitelegge JP, Zhang H, Aguilera R, Taylor RM & Cramer WA Full Subunit Coverage Liquid Chromatography Electrospray Ionization Mass Spectrometry (LCMS) of an Oligomeric Membrane Protein: Cytochrome b6f complex from spinach and the cyanobacterium mastigocladus laminosus. Molecular & Cellular Proteomics 1, 816–827 (2002). [DOI] [PubMed] [Google Scholar]
- 21.Berridge G et al. High-performance liquid chromatography separation and intact mass analysis of detergent-solubilized integral membrane proteins. Analytical Biochemistry 410, 272–280, doi: 10.1061/j.ab.2010.11.006 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Whitelegge JP, Halgand F, Souda P & Zabrouskov V Top-down mass spectrometry of integral membrane proteins. Expert Rev. Proteomics 3, 585–596 (2006). [DOI] [PubMed] [Google Scholar]
- 23.Zabrouskov V & Whitelegge JP Increased Coverage in the Transmembrane Domain with Activated-Ion Electron Capture Dissociation for Top-Down Fourier-Transform Mass Spectrometry of Integral Membrane Proteins. Journal of Proteome Research 6, 2205–2210 (2007). [DOI] [PubMed] [Google Scholar]
- 24.Ryan CM et al. Post-translational Modifications of Integral Membrane Proteins Resolved by Top-down Fourier Transform Mass Spectrometry with Collisionally Activated Dissociation. Molecular & Cellular Proteomics 9 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Whitelegge JP et al. Toward the bilayer proteome, electrospray ionization-mass spectrometry of large, intact transmembrane proteins. PNAS 96, 10695–10698 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Reid DJ et al. Engineering Nanodisc Scaffold Proteins for Native Mass Spectrometry. Analytical Chemistry 89, 11189–11192 (2017). [DOI] [PubMed] [Google Scholar]
- 27.Plumb R et al. Ultra-performance liquid chromatography coupled to quadrupole-orthogonal time-of-flight mass spectrometry. Rapid Commun. Mass Spectrom. 18, 2331–2337, doi: 10.1002/rcm.1627 (2004). [DOI] [PubMed] [Google Scholar]
- 28.Ferrige AG et al. Disentangling electrospray spectra with maximum entropy. Rapid Communications in Mass Spectrometry 6, 707–711, doi: 10.1002/rcm.129061115 (1992). [DOI] [Google Scholar]
- 29.Bannas P, Hambach J & Koch-Nolte F Nanobodies and Nanobody-Based Human Heavy Chain Antibodies As Antitumor Therapeutics. Frontiers in Immunology 8, 1–13, doi: 10.3389/fimmu.2017.01603 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Goyon A et al. Characterization of 30 therapeutic antibodies and related products by size exclusion chromatography: Feasibility assessment for future mass spectrometry hypenation. Journal of Chromatography B 1065-1066, 35–43 (2017). [DOI] [PubMed] [Google Scholar]
- 31.Guzzetta A Reverse Phase HPLC Basics for LC/MS: An IonSource Tutorial, http://www.ionsource.com/tutorial/chromatography/rphplc.htm> (2001).
- 32.Dillon TM et al. LC/MS method of analyzing high molecular weight proteins. United States of America patent (2005). [Google Scholar]
- 33.Dillon TM et al. Optimization of a reversed-phase high-performance liquid chromatography-mass spectrometry method for characterizing recombinant antibody heterogeneity and stability. Journal of Chromatography A 1120, 112–120, doi: 10.1016/j.chroma.2006.01.016 (2006). [DOI] [PubMed] [Google Scholar]
- 34.Apffel A, Fischer S, Goldberg G, Goodley PC & Kuhlmann FE Enhanced sensitivity for peptide mapping with electrospray liquid chromatography-mass spectrometry in the presence of signal suppression due to trifluoroacetic acid-containing mobile phases. Journal of Chromatography A 712, 177–190 (1995). [DOI] [PubMed] [Google Scholar]
- 35.Annesley TM Ion Suppresion in Mass Spectrometry. Clinical Chemistry 49, 1041–1044, doi:10.1373/49.1041 (2003). [DOI] [PubMed] [Google Scholar]
- 36.Eshraghi J & Chowdhury SK Factors Affecting Electrospray Ionization of Effluents Containing Trifluoroacetic Acid for High-Performance Liquid Chromatography/Mass Spectrometry. Analytical Chemistry 65, 3528–3533 (1993). [DOI] [PubMed] [Google Scholar]
- 37.Lippens JL et al. Fourier Transform-Ion Cyclotron Resonance Mass Spectrometry as a Platform for Characterizing Multimeric Membrane Protein Complexes. Journal of The American Society for Mass Spectrometry 29, 183–193, doi: 10.1007/s13361-017-1799-4 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Smith SP, Barber KR & Shaw GS Identification and structural influence of a differentially modified N-terminal methionine in human S100b. Protein Science 6, 1110–1113 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lengqvist J et al. Observed peptide pI and retention time shifts as a result of post-translational modifications in multidimentional separations using narrow-range IPG-IEF. Amino Acids 40, 697–711 (2011). [DOI] [PubMed] [Google Scholar]
- 40.Dillon TM, Bondarenko PV & Ricci MS Development of an analytical reversed-phase high-performance liquid chromatography-electrospray ionization mass spectrometry method for characterization of recombinant antibodies. Journal of Chromatography A 1053, 299–305, doi: 10.1016/j.chroma.2004.08.058 (2004). [DOI] [PubMed] [Google Scholar]
- 41.Souda P, Ryan CM, Cramer WA & Whitelegge JP Profiling of integral membrane proteins and their post translational modifications using high-resolution mass spectrometry. Methods 55, 330–336 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.