

It is becoming increasingly clear that chromatin insulators play a key role in the transcriptional control of DNA viruses. The gammaherpesviruses Epstein-Barr virus (EBV) and Kaposi’s sarcoma-associated herpesvirus (KSHV) utilize chromatin insulators to order protein recruitment and dictate the formation of three-dimensional DNA loops that spatially control transcription and latency. The contribution of chromatin insulators in alphaherpesvirus transcriptional control is less well understood. The work presented here begins to bridge that gap in knowledge by showing how one insulator site in HSV-1 modulates lytic gene transcription and heterochromatin deposition as the HSV-1 genome establishes latency.
KEYWORDS: CTCF, CTRL2, HSV-1, chromatin remodeling, epigenetics, insulator, latency, mouse ocular model
ABSTRACT
The cellular insulator protein CTCF plays a role in herpes simplex virus 1 (HSV-1) latency through the establishment and regulation of chromatin boundaries. We previously found that the CTRL2 regulatory element downstream from the latency-associated transcript (LAT) enhancer was bound by CTCF during latency and underwent CTCF eviction at early times postreactivation in mice latently infected with 17syn+ virus. We also showed that CTRL2 was a functional enhancer-blocking insulator in both epithelial and neuronal cell lines. We hypothesized that CTRL2 played a direct role in silencing lytic gene expression during the establishment of HSV-1 latency. To test this hypothesis, we used a recombinant virus with a 135-bp deletion spanning only the core CTRL2 insulator domain (ΔCTRL2) in the 17syn+ background. Deletion of CTRL2 resulted in restricted viral replication in epithelial cells but not neuronal cells. Following ocular infection, mouse survival decreased in the ΔCTRL2-infected cohort, and we found a significant decrease in the number of viral genomes in mouse trigeminal ganglia (TG) infected with ΔCTRL2, indicating that the CTRL2 insulator was required for the efficient establishment of latency. Immediate early (IE) gene expression significantly increased in the number of ganglia infected with ΔCTRL2 by 31 days postinfection relative to the level with 17syn+ infection, indicating that deletion of the CTRL2 insulator disrupted the organization of chromatin domains during HSV-1 latency. Finally, chromatin immunoprecipitation with high-throughput sequencing (ChIP-seq) analyses of TG from ΔCTRL2-infected mice confirmed that the distribution of the repressive H3K27me3 (histone H3 trimethylated at K27) mark on the ΔCTRL2 recombinant genomes was altered compared to that of the wild type, indicating that the CTRL2 site modulates the repression of IE genes during latency.
IMPORTANCE It is becoming increasingly clear that chromatin insulators play a key role in the transcriptional control of DNA viruses. The gammaherpesviruses Epstein-Barr virus (EBV) and Kaposi’s sarcoma-associated herpesvirus (KSHV) utilize chromatin insulators to order protein recruitment and dictate the formation of three-dimensional DNA loops that spatially control transcription and latency. The contribution of chromatin insulators in alphaherpesvirus transcriptional control is less well understood. The work presented here begins to bridge that gap in knowledge by showing how one insulator site in HSV-1 modulates lytic gene transcription and heterochromatin deposition as the HSV-1 genome establishes latency.
INTRODUCTION
Herpes simplex virus 1 (HSV-1) is a significant human pathogen that establishes a lifelong latent infection in sensory neurons (1, 2). Following the establishment of latency, lytic gene transcription is broadly silenced while the latency-associated transcript (LAT), a noncoding RNA that is spliced into two stable introns of 2 kb and 1.5 kb in sensory neurons, is abundantly expressed (3). The latent genome persists as a circular episome associated with histones enriched with posttranslational modifications. The active LAT region of the genome is enriched in the transcriptionally permissive histone marks acetyl H3K9K14 (histone H3 acetylated on K9 and K14) and H3K4me2 (H3 dimethylated on K4) during latency, while the repressed immediate early (IE) lytic genes of ICP0, ICP4, and ICP27 are enriched in repressive heterochromatic histone marks, including the H3K27me3 (H3 trimethylated at K27) mark (4–12). This transcriptional compartmentalization of the latent HSV-1 genome supports a role for chromatin insulator-like elements in the control of viral gene expression during latency.
Chromatin insulators are long-range cis-acting DNA sequence elements and protein complexes that can recruit chromatin-modifying proteins and other transcriptional elements to control gene expression. In the classical sense, chromatin insulators prevent inappropriate signaling from one transcriptional domain to another (13). In mammalian cells, chromatin insulators are comprised of an insulator protein bound to a conserved but complex DNA consensus motif. The most common insulator protein is CCCTC-binding factor (CTCF), an 11-zinc-finger nuclear protein that regulates gene transcription through the formation of higher-order three-dimensional chromatin loops (14). CTCF insulator repression of lytic transcription is an emerging theme in transcriptional control of DNA viruses. Examples include human cytomegalovirus (HCMV) and human papillomavirus (HPV), which have CTCF sites in their major lytic promoters. When these sites were deleted, increased lytic transcription occurred (15, 16). In Kaposi’s sarcoma-associated herpesvirus (KSHV), CTCF binds to an intergenic site between the ORF73 and K14 to repress lytic K14 transcription (17), and in Epstein-Barr virus (EBV) genomes, CTCF binds to a site between the viral origin of replication (OriP) and the C promoter (Cp) to repress transcription of Cp (18). However, the role of CTCF insulators in the control of alphaherpesvirus lytic transcription is less understood. In 2006, Amelio et al. identified seven conserved reiterated CTCF binding motifs in the latent HSV-1 genome (19). The nucleotide positions of these motifs have previously been described and are shown in Fig. 1 (19). Interestingly, while each CTCF binding site was occupied by the insulator protein CTCF during latency in mouse trigeminal ganglion (TG) neurons, this binding was differential, indicating that these potential CTCF insulators may be independently regulated and therefore contribute to the control of viral transcription in a site-specific manner (20). To further illustrate this point, we recently showed that the enhancer-blocking ability of two sites, namely CTRL1 and CTRS3, was dependent on cell type (neuronal versus epithelial) (20). In addition, we showed that only the CTCF sites flanking the immediate early ICP0 and ICP4 genes recruited Suz12, a protein in the polycomb repressive complex 2 (PRC2), indicating that these insulators controlled the repression of IE genes, perhaps through the coordination of chromatin-modifying complexes responsible for the deposition of histone marks that silence transcription (20). With respect to reactivation of HSV-1, we recently showed that depletion of the protein CTCF in neurons led to global HSV-1 reactivation in the rabbit ocular model (21). Collectively, these data indicate that chromatin insulator elements may contribute to the establishment and maintenance of latency.
FIG 1.

CTCF sites in the latent HSV-1 genome. Genomic positions of CTCF binding motifs in the HSV-1 genome are shown. There is an additional CTCF motif (CTUS1) identified by Amelio et al. not shown in this figure (19). The nucleotide position of the core CTCF binding cluster known as CTRL2 and nucleotide location of the deleted sequence used to generate the ΔCTRL2 recombinant virus are indicated. US, unique short region; RL, repeat long region; RS, repeat short region.
To further explore how individual CTCF insulators contribute to or regulate HSV-1 latency, we first targeted the CTRL2 binding site, located downstream of the LAT enhancer element (Fig. 1). Our previous work showed that the CTRL2 site is a potent inhibitor of the LAT enhancer in both epithelial and neuronal cells and that CTCF was rapidly evicted from CTRL2 following reactivation (20, 22). In the experimental data presented in the manuscript, we explored the role of the CTRL2 insulator on the establishment and maintenance of latency in vivo using a recombinant virus with a 135-bp deletion spanning only the core CTRL2 CTCF consensus sequences (ΔCTRL2) in the 17syn+ background. We found that the CTRL2 insulator was required for efficient viral replication in epithelial cells but not in the Neuro 2A cell line, indicating that this element may regulate gene expression in a cell-type-specific manner. Using the mouse ocular model, we found that deletion of the CTRL2 insulator decreased mouse survival and led to the inefficient establishment of latency in the TG. We also found that lytic gene expression increased in the TG of mice infected with the ΔCTRL2 recombinant virus and that the distribution of the repressive H3K27me3 mark on the ΔCTRL2 genomes was altered compared to that of the wild-type (wt) virus, indicating that the CTRL2 insulator contributes to the repression of IE genes, possibly by coordinating the deposition of H3K27me3 on HSV-1 genomes during the establishment of latency. Finally, deletion of the CTRL2 resulted in significant changes in heterochromatin on two HSV-1 elements in proximity to genes UL36 and UL37 that code for tegument proteins, suggesting that alternative chromatin architecture is established when the CTRL2 insulator is deleted.
RESULTS
Deletion of the CTRL2 insulator resulted in a cell-type-specific replication defect.
In order to establish whether the CTRL2 insulator contributes to the ability of HSV-1 to replicate in vitro, we infected rabbit skin (RS), U2OS, 3T3, and Neuro 2A cell lines with both 17syn+ and the recombinant virus ΔCTRL2 at a multiplicity of infection (MOI) of 0.01 and used quantitative PCR (qPCR) to determine the number of genome copies present in each cell type at 8, 16, 24, 30, and 48 h postinfection (p.i.). Following infection of RS, U2OS, and 3T3 cells, there was a 10-fold decrease in the copy numbers of HSV-1 genomes present in the ΔCTRL2-infected cells relative to number in wt 17syn+-infected cells, indicating that the CTRL2 insulator was required for efficient viral replication (Fig. 2A). However, when we infected Neuro 2A cells with the recombinant virus ΔCTRL2, we found a 10-fold increase in viral replication relative to that of wt-infected cells (Fig. 2B). This finding suggests that the CTRL2 insulator is required for lytic replication in epithelial cells but not in cells that are from a neuronal lineage, indicating that the CTRL2 insulator may have diverse roles that are dependent on whether the virus is in a neuronal or nonneuronal cell type.
FIG 2.
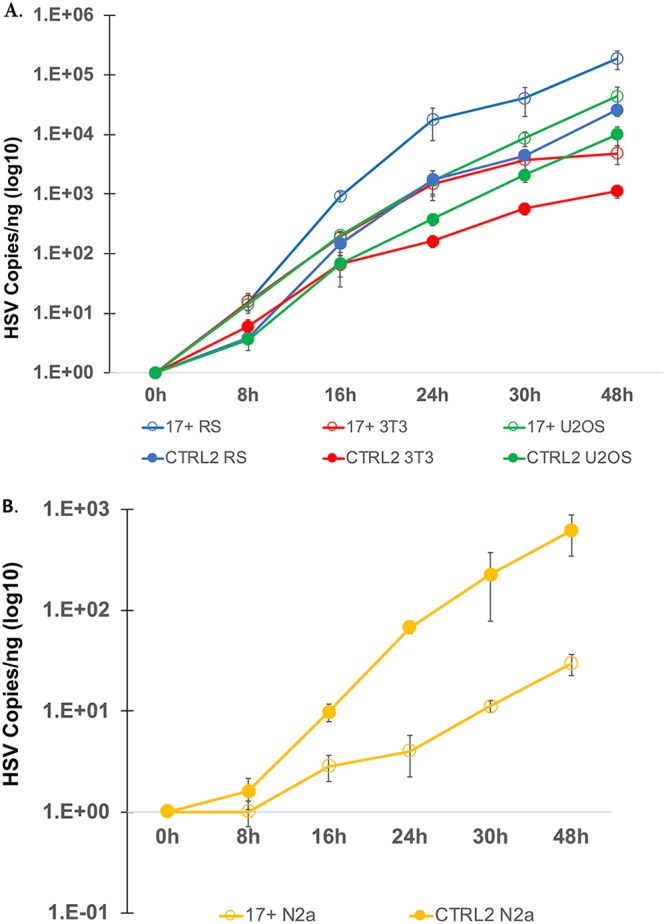
HSV-1 growth curves in cell culture. (A) Growth curves were done using RS, mouse 3T3, and human U2OS cells as epithelial cell models. Cells were plated at 150,000 cells per well and infected at an MOI of 0.01 with either wild-type 17syn+ or the ΔCTRL2 recombinant. Cells were harvested at the times indicated on the x axis, DNA was extracted, and qPCR was done to measure the HSV-1 DNA polymerase gene, which was quantified using a standard curve generated from purified 17syn+. The ΔCTRL2 recombinant had a replication defect in all epithelial cells used in the quantification. (B) Growth curves using the Neuro 2A (N2a) cell line were done as described above. No replication defect was observed for the ΔCTRL2 recombinant in Neuro 2A cells.
Deletion of the CTRL2 insulator increased virulence and attenuated the establishment of latency in vivo.
To characterize the phenotype of the recombinant ΔCTRL2 virus in mice following ocular infection, 120 mice were inoculated with the ΔCTRL2 virus, and 125 were infected with 17syn+. Infections were confirmed by slit lamp on postinfection day 3, and mice were monitored daily for weight loss, hunched posture, hair loss, and other signs of morbidity for 28 days following inoculation. Surviving mice were counted each day, and the survival rates for each cohort are described in Table 1. We observed a significant decrease in survival rates in mice following corneal infection with the ΔCTRL2 virus compared to rates in the wild-type-infected animals (P < 0.003). In summary, 47.5% of mice infected with the ΔCTRL2 recombinant died in contrast to 29.6% of mice infected with 17syn+ (Table 1). Further, the deaths of mice infected with the ΔCTRL2 virus occurred over an extended period of time, with the majority of deaths occurring after postinfection day 11 and extending through p.i. day 21 (Fig. 3). In contrast, in wild-type-infected animals, most mortalities occurred between days 7 and 14. No deaths in either group occurred post-21 days of infection. These data were also consistent with our findings in Neuro 2A cells, where ΔCTRL2 replicated to significantly higher levels than wt virus. Collectively, our data suggested that deletion of the CTRL2 insulator of HSV-1 resulted in increased lytic replication in murine sensory ganglia.
TABLE 1.
Mortality rates in mice infected with the ΔCTRL2 mutant
| Virus | No. of infected animals | No. of deaths | % Mortality |
|---|---|---|---|
| ΔCTRL2 | 120 | 57 | 47.5a |
| 17syn+ | 125 | 37 | 29.6 |
P < 0.0005 by the exact version of chi-square analysis for both pairwise comparisons with 17syn+.
FIG 3.
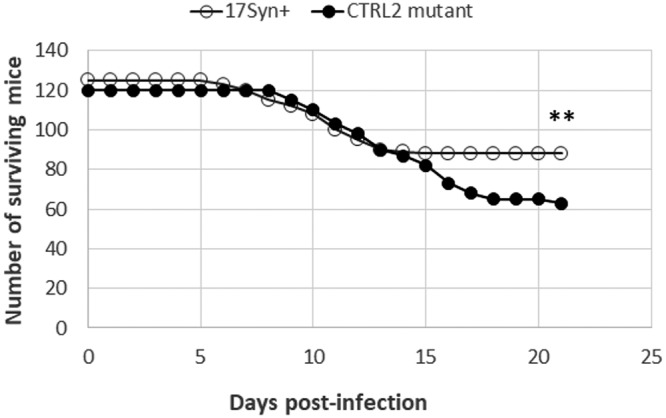
Survival rates of mice infected with the 17syn+ and ΔCTRL2 strains. A total of 125 mice were ocularly inoculated with 17syn+, and 120 mice were ocularly inoculated with ΔCTRL2. Mice were monitored daily through postinfection day 28 for signs of morbidity and for mortalities. Statistical analysis was done with SigmaPlot, version 12.5, using chi-square analysis for the number of surviving mice expected versus the number observed with 17syn+ infection (**, P < 0.005).
The significant decrease in survival and extended period of mortality of mice infected with ΔCTRL2 suggested that latency might not be established as efficiently in ΔCTRL2-infected mice. To test this, we harvested TG from mice infected with either ΔCTRL2 or wt 17syn+ virus at 31 days postinfection, and isolated DNA from the TG. qPCR was performed for the HSV-1 DNA polymerase (Pol) gene (UL30) in order to quantify the number of viral genomes per ganglion. All values were normalized to the level of a host control, the mouse Aprt gene. We found significantly fewer viral genomes per ganglion for the mice infected with the ΔCTRL2 virus than for those infected with the wt 17syn+ at 31 days postinfection, suggesting that the CTRL2 insulator was required for the efficient establishment of latency in vivo (Fig. 4).
FIG 4.
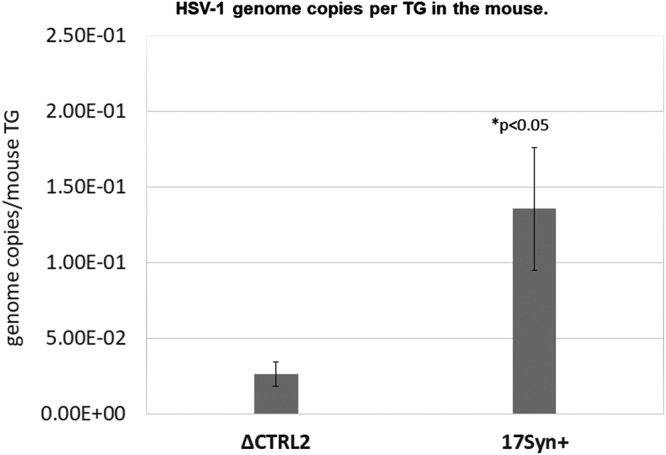
HSV-1 genomes in latent mice TG. TG were harvested at 31 days postinfection, and DNA was extracted. Data are presented as the ratio of HSV-1 DNA polymerase/APRT. One-way ANOVA showed a significant difference in the number of HSV-1 copies between the viruses (n = 6).
Deletion of the CTRL2 insulator disrupted repression of IE regions during latency.
The CTRL2 site is an enhancer-blocking insulator element that binds and is enriched in the cellular protein CTCF (19, 22). Because we observed a decrease in mouse survival combined with a decrease in the number of genomes per ganglion at >28 days postinfection with the ΔCTRL2 strain, we hypothesized that the CTRL2 region controlled the silencing of lytic genes as the virus established latency in sensory neurons and that the ΔCTRL2 virus could not as efficiently repress lytic transcription to establish latency. To test this, we isolated RNA from the TG of mice infected with either ΔCTRL2 or 17syn+ at 31 days postinfection and performed quantitative reverse transcription-PCR (qRT-PCR) using primers and probes specific for the viral targets for the LAT intron, ICP0, ICP4, ICP27, and VP16 (Table 2). We found significantly increased expression of the IE genes ICP0 and ICP27 genes (P < 0.05) and, while not statistically significant by one-way analysis of variance (ANOVA) of statistical means, a 2-fold increase in ICP4 expression in ganglia infected with ΔCTRL2 relative to the level with wt infection (Fig. 5). In contrast, we observed no increase in expression of VP16 (UL48) (Fig. 5E), indicating that only the regions proximal to the CTRL2 binding domain are controlled by CTRL2 while lytic transcripts farther away remained unaffected. Interestingly, we also found a significant 3-fold increase in the expression of the LAT intron at 31 days postinfection, consistent with previously reported findings from Lee et al. using a CTRL2 recombinant virus from the KOS parent background (23). This was an interesting finding since the expression of the LAT has long been considered a hallmark of latency, and latency establishment appears to be disrupted in the ΔCTRL2 virus. Nonetheless, the increased expression levels of the LAT, ICP0, ICP4, and ICP27 regions in the CTRL2 recombinant suggest that the chromatin domains that are established and maintained during HSV-1 latency were disrupted in the ΔCTRL2 virus, further indicating that the insulator site plays a role in the maintenance of the latent transcriptional domains.
TABLE 2.
Primer/probe sequences used for qRT-PCR
| Target and primer or probe name | Primer sequence | Nucleotide positiona |
|---|---|---|
| DNA Pol | 65880–65953 | |
| Forward | 5′-AGA GGG ACA TCC AGG ACT TTG T-3′ | |
| Reverse | 5′-CAG GCG CTT GTT GGT GTA C-3′ | |
| Probe | 5′-ACC GCC GAA CTG AGC A-3′ | |
| VP16 | 103946–104191 | |
| Forward | 5′-CCT CGA TGG TAG ACC CGT AA-3′ | |
| Reverse | 5′-ACA TTC GCG AGC ACC TTA AC-3′ | |
| Probe | 5′-CAT AAA GTA CCC AGA GGC-3′ | |
| ICP27 | 113945–114034 | |
| Forward | 5′-GCC CGT CTC GTC CAG AAG-3′ | |
| Reverse | 5′-GCG CTG GTT GAG GAT CGT T-3′ | |
| Probe | 5′-CAG CAC CCA GAC GCC-3′ | |
| LAT Intron | 119721–119795 | |
| Forward | 5′-ACC CAC GTA CTC CAA GAA GGC-3′ | |
| Reverse | 5′-TAA GAC CCA AGC ATA GAG AGC CA-3′ | |
| Probe | 5′-TCC CAC CCC GCC TGT GTT TTT-3′ | |
| ICP0 | 121385–121453 | |
| Forward | 5′-GGC CGA GGG AGG TTT CC-3′ | |
| Reverse | 5′-CCG CTT CCG CCT C-3′ | |
| Probe | 5′-CTC CCA GGG CAC CGA C-3′ | |
| ICP4 | 130224–130292 | |
| Forward | 5′-CAC GGG CCG CTT CAC-3′ | |
| Reverse | 5′-GCG ATA GCG CGC GTA GA-3′ | |
| Probe | 5′-CGA CGC GAC CTC C-3′ |
Position according to GenBank accession no. NC_001806.
FIG 5.

Gene expression increases in ΔCTRL2-infected mice ganglia. Real-time PCRs using the primers and probes listed in Table 2 were done to quantitate HSV-1 LAT, ICP0, ICP4, ICP27, and VP16 expression at 31 days postinfection. Mouse TG were isolated and placed in RNAlater and stored according to the manufacturer’s specifications. RNA was extracted by removing RNAlater from samples and adding TRIzol reagent (Sigma-Aldrich) to each sample. RNA was precipitated and DNase treated, and reverse transcription was done using a high-capacity One Step cDNA reverse transcription kit (ABI), according to the manufacturer’s instructions. In the case of ICP0, a 10-μl aliquot of purified RNA was used with the strand-specific primer for the ICP0 transcript (LAT I-1, GACACGGATTGGCTGGTGTAGTGGG; nucleotides 120797 to 120820) (37). Controls for qRT-PCRs included no-template and no-reverse transcriptase wells for each plate for each gene analyzed. One-way ANOVA was used to determine statistical relevance of the expression data (n = 6 to 8). **, P < 0.05.
CTRL2 controlled the deposition of H3K27me3 on the latent genome.
To determine the impact of the deletion of the CTRL2 insulator on latent HSV-1 chromatin composition, TG from mice latently infected with wild-type 17syn+ or ΔCTRL2 virus were used for triplicate H3K27me3 (histone H3 trimethylated at K27) chromatin immunoprecipitation with high-throughput sequencing (ChIP-seq) experiments. Enriched regions of H3K27me3 (normalized over input) across the HSV-1 genome were determined, and differential enrichment analysis was performed. In general, the patterns of H3K27me3 were similar for the two viruses; however, the ΔCTRL2 virus exhibited a significantly lower level of H3K27me3 at distinct genomic regions (Fig. 6). In particular, when CTRL2 was deleted, H3K27me3 enrichment was decreased 2- to 4-fold at sites mapping back to the LAT 5′ exon (Fig. 6A and C) (nucleotides [nt] 5000 to 7500 and 119000 to 121000), ICP27 (Fig. 6B) (nt 113000 to 115000), and ICP0 (Fig. 6C) (nt 125000 to 127000) regions in the ΔCTRL2 virus compared to levels for wild-type 17syn+. In addition, there was a 2-fold enrichment decrease in H3K27me3 at the CTRS3′ element upstream of the ICP4 promoter region of the HSV-1 genome (Fig. 6D) (nt 150000 to 152000), indicating that the status of transcriptional domains in latent HSV-1 were disrupted with the deletion of the CTRL2 site. These decreases in H3K27me3 enrichments also correspond to the observed increased transcription of ICP0, ICP4, LAT, and ICP27 that we quantitated by qRT-PCR. Interestingly, ChIP-seq also showed significant changes in H3K27me3 enrichment at two other regions of the genome in the ΔCTRL2 virus mapping back to genomic sites in HSV-1 that correspond to the inner tegument proteins known as UL36 and UL37 (Fig. 6, top schematic, gray bar indicating nucleotide positions 70000 to 90000). The implications of these findings for latent HSV-1 biology are under investigation.
FIG 6.

ΔCTRL2 affects H3K27me3 occupancy. The schematic at the top of the figure represents ChIP-seq signals of merged experimental replicates (n = 6). H3K27me3 wild-type 17syn+ and ΔCTRL2 signals from TG of latently infected mice were quantified. Both signal tracks were calculated using all 6 inputs combined for either the wild-type 17syn+ or the ΔCTRL2 viruses. The patterns of H3K27me3 signals were similar between the wild-type 17syn+ and ΔCTRL2 viruses; however, there was differential H3K27me3 enrichment at distinct genomic regions. There was 2- to 4-fold H3K27me3 enrichment at the LAT 5′ exon and ICP0 (RL2) regions (A and C) compared to the level for the wild-type 17syn+ and a slight decrease (less than a 1-fold change) in the ICP27 (UL54) region (C). In addition, there was a 2-fold decrease in H3K27me3 enrichment at the CTRS3′ element upstream of the ICP4 promoter region (RS1) (D) and an increase (0.5- to 2-fold) of H3K27me3 enrichment in the ΔCTRL2 virus at the UL36 and UL37 sites located between nt 70000 and 90000 (in the schematic at the top, the horizontal line below the sequence denotes where the histone marks differ between the two viruses). Data in panels represent merged experiments using 3 mice and 6 TG each (n = 6). Data are plotted as fold enrichment over input for all panels.
DISCUSSION
Recent experimental evidence from several labs has shown that CTCF binding domains of HSV-1 play an important role in the establishment and maintenance of latency through the coordinated binding of the insulator protein CTCF to the HSV-1 genome. Importantly, seven individual CTCF binding domains have been identified in the latent genome, and it is becoming increasingly clear that each CTCF binding domain is differentially regulated, recruits different chromatin-modifying protein complexes, and likely has cell-type-specific roles in lytic infections and during latency (19–23). The disruption of CTCF binding to sites in the latent genome evokes reactivation in vivo, suggesting not only that CTCF binding domains contribute to the deposition of chromatin marks on the HSV-1 genome but also that their presence and function as insulators are critical to the maintenance of latency in vivo (21).
We hypothesized that individual CTCF binding domains in HSV-1 contribute to the establishment and maintenance of latency differentially. To begin to dissect the role of individual sites, we focused on the CTRL2 site. The CTRL2 binding motif is positioned downstream of the LAT promoter/enhancer complex, a complex region of the genome that is critical for efficient reactivation from latency (10). Further, the CTRL2 site has already been characterized as an insulator element that has potent enhancer-blocking and -silencing capabilities, regardless of the cell type (19, 20). Previous work showed that the LAT region of the genome is populated with transcriptionally permissive marks, while the juxtaposed IE gene regions, specifically ICP0, ICP4, and ICP27, are populated with transcriptionally repressive histone marks during latency (7, 10, 24, 25). The position of the CTRL2 insulator within the context of these distinct transcriptional domains indicates a fundamental role for this element in the preservation of a latent genome conformation. Our findings support this, and in addition we show that the CTRL2 site promotes efficient replication in epithelial cells (deletion of CTRL2 results in a replication defect); but this defect was not observed in neuronal cell lines infected with ΔCTRL2. This, combined with our findings that the ΔCTRL2 virus is more virulent and does not establish latency in mouse TG as efficiently as wt virus, suggests that the CTRL2 insulator plays a critical role in establishment of latency and prevents inappropriate signaling from the LAT enhancer to nearby immediate early gene regions. In the absence of the CTRL2 insulator, lytic genes are not silenced, as evidenced by the observation that ICP0, ICP27, and ICP4 are all expressed in the TG of mice infected with the ΔCTRL2 recombinant virus at a time point consistent with a latent infection (31 days postinfection). Interestingly, LAT expression was significantly higher (3-fold) in TG harvested from mice infected with ΔCTRL2, suggesting that LAT may be expressed at different levels in a cell-type-dependent manner during lytic infection. This is currently being explored.
The CTRL2 site has silencer capabilities (19), suggesting that it plays a role in the recruitment of corepressive proteins to silence the IE regions of HSV-1 as the virus establishes latency. This would be consistent with our findings that deletion of this site disrupted the establishment of latency. We previously showed that two other sites, namely the CTRS1/2 and CTRS3 sites in HSV-1 were also insulators that likely recruited the PRC2. CTCF insulators directly interact with the protein Suz12 in mammalian cells, and since the PRC2 is responsible for the trimethylation of H3K27 to silence transcription (26), CTCF recruitment of PRC2 through a Suz12 interaction in HSV-1 was consistent with the H3K27Me3 status of lytic regions during latency. We previously found that Suz12 colocalized primarily to CTCF binding domains that flank the ICP0 and ICP4 gene regions of the genome but not to the CTRL2 insulator, suggesting an alternative role for the CTRL2 insulator in latency establishment. This is supported by our findings that the CTRL2 recombinant virus has an altered H3K27me3 profile compared to that of wild-type input in vivo. We found a consistent decrease in the enrichment of H3K27me3 on the ICP0, ICP4, and ICP27 regions of HSV-1 in the absence of the CTRL2 insulator, suggesting that this site controls heterochromatin deposition to the latent HSV-1 genome.
It should be noted that overall our interpretations of data are consistent with the findings of experimental data presented in a recent manuscript from Lee et al. (23); however, there are some key differences in our findings that should be discussed. In the manuscript from Lee et al., a 370-bp deletion of the CTRL2 site plus flanking sequences was generated in the background of the KOS wild-type strain of HSV-1 (23). Here, the authors showed that deletion of CTRL2 resulted in the encroachment of heterochromatin onto the LAT region of the genome to disrupt the establishment of latency, and they provided additional data that indicated that mice infected with the KOS/ΔCTRL2 recombinant had lower survival rates, similar to our findings. However, Lee et al. also showed that their ΔCTRL2 recombinant was capable of establishing an equivalent latent infection in the mouse TG and displayed no replication defect during the lytic infection (in vivo). While both parent viruses are commonly used in HSV-1 research, there are well-documented strain-specific differences between the KOS and 17syn+ viruses with respect to histone marks and other epigenetic factors that may control aspects of latency and reactivation (7, 24). Given the observed differences between the two recombinant viruses, we propose that there are strain-specific differences that influence latency and reactivation.
We also analyzed latently infected mouse TG by ChIP-seq to survey the entire HSV-1 genome for altered H3K27me3 enrichment following the deletion of the CTRL2 insulator in vivo. Our findings with respect to the H3K27me3 population on the genome were consistent with our evaluation of IE gene expression. We observed increased expression of ICP0, ICP4, and ICP27 and a parallel decrease in the enrichment of the heterochromatic marker near these genomic sites in the CTRL2 recombinant virus. This suggests that, at a minimum, the CTRL2 site is responsible for the deposition of heterochromatin on genomic sites adjacent to the CTRL2 insulator. However, surprisingly, we found that ΔCTRL2 exhibited an enrichment in the H3K27me3 repressive marker on two genomic regions that were near the genes that code the inner tegument proteins UL36 and UL37. This heterochromatic enrichment was not observed in wild-type viral input from our ChIP-seq experiments. Cytosolic capsids associate with the inner tegument proteins UL36 and UL37 for their intracellular transport along microtubules to cytoplasmic membranes where they meet other tegument and viral membrane proteins for secondary envelopment and virion formation. These proteins are essential for efficient capsid envelopment and fusion for the release of infectious particles and are involved in larger protein complexes that together are involved in axonal targeting for viral spread during reactivation (27, 28). Interestingly, Lee et al. reported that in mice infected with the ΔCTRL2 strain in the KOS background, virus failed to reactivate efficiently in the explant model (23). In addition, we have preliminary data using the ΔCTRL2 virus in the 17syn+ background indicating that reactivation of our ΔCTRL2 recombinant was also attenuated in the rabbit model of infection and reactivation (unpublished data), and therefore the chromatinization of these two gene regions of HSV-1 is consistent with these observed phenotypes.
While the heterochromatinization of UL36 and UL37 was a surprising finding and one that we are currently exploring with respect to HSV-1 biology, one provocative explanation for heterochromatinization of the UL36 and UL37 gene regions lies within the potential for CTCF insulators to dimerize and establish higher-order chromatin structures for the regulation of gene expression. CTCF can self-dimerize on insulators to form higher-order chromatin structures, known as chromatin loops, to spatially orient distance-separated transcriptional elements to regulate gene expression (29–31). CTCF insulators in mammalian cells regulate the local balance of active and repressive histone marks within chromatin loops through the recruitment of corepressors, and there is evidence that support roles for chromatin loops in the regulation of other herpesviruses such as KSHV and EBV (32, 33). In KSHV, CTCF and cohesion proteins colocalize to mediate DNA looping (34). Conventional chromosome conformation capture (3C) assays in cell culture have also identified a chromatin loop that likely mediates type I and type III latency in EBV (35). Considering the growing evidence for CTCF-mediated chromatin loops in other DNA viruses, it is possible that the HSV-1 CTCF insulator, CTRL2, may regulate lytic gene expression during latency and reactivation through spatial proximity of the LAT enhancer with lytic promoters. Conversely, if the CTRL2 insulator is involved in the formation of higher-order chromatin structures, then deletion of this site would likely have impact on the chromatin organization of distally separated gene regions, potentially even the UL36 and UL37 regions of HSV-1. This hypothesis is currently being explored. Nonetheless, here we provide evidence indicating that the CTCF insulator CTRL2 contributes to the establishment and maintenance of latency through the organization of heterochromatic domains in HSV-1.
MATERIALS AND METHODS
Viruses and cells.
All experiments were performed using HSV-1 strain 17syn+ or the recombinant ΔCTRL2 (constructed in the 17syn+ background). The ΔCTRL2 virus was constructed by homologous recombination, as previously reported, with a 135-bp deletion spanning nucleotides 120500 to 120635 (GenBank accession number NC_001806) (36) (Fig. 1). High-throughput sequencing (HTS) of the ΔCTRL2 virus was done, and the data obtained were compared to the sequence of our lab stock of 17syn+. The results of this sequence analysis revealed no indels within coding regions of genes, and only one single nucleotide base change in UL36, which was synonymous. There were four additional single nucleotide base substitutions and one two-nucleotide deletion that occurred in noncoding regions (mainly in reiterated regions in the repeats) (36). Considering these sequencing data, it is unlikely that any sequence changes resulted in changes to the fitness of the 17ΔCTRL2 recombinant. All viruses were amplified and titrated on rabbit skin (RS) cells (ATCC CCL-68) using Eagle's minimal essential medium (MEM; Life Technologies) supplemented with 5% calf serum, 250 U of penicillin/ml, 250 μg of streptomycin/ml, and 292 μg of l-glutamine/ml (Life Technologies).
HSV-1 growth curves.
Rabbit skin (RS) cells were cultured in MEM (catalog no. 10-010; Corning) plus 5% bovine serum, human U2OS cells were cultured in McCoy’s 5A medium (catalog no. 10-050; Corning) plus 10% fetal bovine serum (FBS), murine 3T3 fibroblasts were cultured in Dulbecco’s modified Eagle’s medium (DMEM) (catalog no. 15-013; Corning) plus 10% FBS, and undifferentiated murine Neuro 2A cells were cultured in MEM plus 10% FBS with addition of MEM nonessential amino acids (catalog no. 25-025-Cl; Corning). Cells were plated at 150,000 cells per well of 24-well plates and the next day infected at an MOI of 0.01 with HSV-1 17syn+ or ΔCTRL2. Virus inoculum (50 μl) was adsorbed onto cells for 1 h (0-h time point p.i.) at 37°C in a humidified incubator with 5% CO2, at which time virus was removed, and cells were overlaid with medium. Cells were harvested at the times indicated in the x axis of Fig. 2, and DNA was extracted with DNAzol (catalog no. 10503-027; ThermoFisher Scientific) according to the manufacturer’s recommendation. For analysis of virus replication, 20 ng of total DNA was used in qPCR reactions containing 1× master mix (catalog no. 4352042; ABI) along with primers and probe corresponding to the HSV-1 DNA polymerase region (Table 2). Quantification was performed using a standard curve generated from 10-fold serial dilutions of purified 17syn+ genomes. Replication of genomes was determined following normalization of each time point to the level at 0 h postinfection (incoming genomes).
Mouse ocular infections.
BALB/c mice 4 to 6 weeks of age (Taconic) were anesthetized using intramuscular injections of ketamine and xylazine (200 mg/kg/ and 10 mg/kg body weight). Using a 27-gauge needle, a light 2-by-2 crosshatch pattern was made on the corneal epithelium. Equal amounts of virus, as determined by standard plaque assay, were applied to both eyes of each mouse. Mice were monitored daily through postinfection day 28 for signs of morbidity and for mortalities. Animals were considered latent at >28 days postinfection.
Ethics statement.
The animal studies were approved by the Institutional Animal Care and Use Committee of the Louisiana State University Health Sciences Center, New Orleans (institutional animal welfare assurance no. A3094-01 and LSUHSC IACUC approval).
qPCR for genome copies/ganglia.
The numbers of genome copies per ganglion were measured by quantitative real-time PCR using primers and a probe specific for HSV-1 DNA polymerase (Table 2). All real-time PCR experiments were performed using TaqMan universal PCR master mix and no AmpErase uracil N-glycosylase on a single-color qTower PCR instrument (Analytik Jena) using custom-designed primer-probe mixes (Life Technologies) with the general protocol as follows: 95°C for 10 min (1×) and then 45 cycles of 95°C for 15 s followed by 60°C for 1 min. Threshold values used for PCR analyses were set within the linear range of PCR target amplification based on a standard curve generated for each plate. The relative HSV-1 copy numbers for each sample were normalized to the copy numbers of the host control mouse adenine phosphoribosyltransferase (APRT). Primer probe sequences for mouse APRT were the following: forward, CTCAAGAAATCTAACCCCTGACTCA; reverse, GCGGGACAGGCTGAGA; probe, CCCCACACACACCTC.
qRT-PCR analysis.
Mouse TG were isolated and placed in RNAlater and stored according to the manufacturer’s specifications. RNA was extracted by removing RNAlater from samples and adding TRIzol reagent (Sigma-Aldrich) to each sample. Briefly, each TG was homogenized in 1.2 ml of TRIzol, and following the addition of a 0.2 volume of chloroform, samples were centrifuged for phase separation. RNA was precipitated from the aqueous phase using a 0.7 volume of isopropanol, followed by DNase treatment using DNA-free (Ambion), according to the manufacturer’s directions. One-step RT-PCR was done using a SuperScript III One-Step RT-PCR system with Platinum Taq DNA polymerase according to the manufacturer’s protocol (Life Technologies). In the case of ICP0, a 10-μl aliquot of purified RNA was used with the strand-specific primer for the ICP0 transcript (LAT I-1, GACACGGATTGGCTGGTGTAGTGGG; nucleotides 120797 to 120820) (37). Real-time PCRs were performed on cDNA according to the above-described procedures, and protocols were performed using the primers and probes listed in Table 2.
ChIP-seq assays.
Chromatin immunoprecipitation (ChIP) assays were performed as previously described for mice using the specific antibody anti-H3K27me3 (Millipore) (22). Each ChIP assay contained pooled TG of three mice (6 TG). All solutions and buffers were maintained at 4°C and treated with HALT protease inhibitor (ThermoFisher) until de-cross-linking. The TG were rapidly removed following euthanasia, the ganglia were homogenized in phosphate-buffered saline, and the chromatin was cross-linked in 1% formaldehyde (Sigma-Aldrich) for 10 min at room temperature. Cross-linking was quenched by glycine (final concentration of 0.125 M), and cells were pelleted, washed, and resuspended in sodium dodecyl sulfate lysis solution for 30 min. The cell lysate was sonicated using a Bioruptor sonicator (Diagenode) using a total of 36 cycles on high power to shear the chromatin into fragments between 300 and 800 bp. Fragment size following sonication was confirmed by agarose gel electrophoresis using a 1.5% gel. Ten percent of the lysate was saved to for the input controls, while the remaining fraction was incubated with 10 mg/μl of H3K27me3 (07-449; Millipore) or with IgG as a nonspecific antibody binding control overnight at 4°C. Following incubation, the unbound fraction was saved for normalization purposes. Antibody complexes were captured by MagnaChIP protein A/G magnetic beads (Millipore) and washed in accordance with Millipore’s protocol. Following immunoprecipitation, samples were eluted from beads using elution buffer (final concentration of 0.1% sodium dodecyl sulfate and 0.1 M NaHCO3 at 65°C) and de-cross-linked with NaCl (final concentration of 0.2 M) at 55°C for 4 h. Eluates were treated with RNase A and proteinase K and purified with a QiaQuick PCR purification column (Qiagen). ChIPs were validated using real-time PCR using TaqMan Universal PCR Master Mix (Applied Biosystems) and a StepOne Plus qPCR instrument (Applied Biosystems). The same reagents were also used to determine enrichment over levels of viral genes. Prior to Illumina library construction, HSV-1 sequences were enriched by hybridization capture-based target enrichment. Briefly, eight PCR cycle-indexed libraries were pooled for selection using a SureSelectXT high sensitivity (HS) system with custom probes spanning the HSV-1 genome (Roche-Nimblegen). This enrichment platform consists of custom-biotinylated DNA fragments between 50 and 100 bp in length that bind specifically to the DNA of HSV-1 strain 17 (GenBank accession no. NC_001806). Any sequences within the viral genome that also matched mouse or human sequences were removed from the array design. Each region of the viral genome was covered by multiple unique and overlapping baits for redundancy. Following enrichment, samples were subjected to four additional PCR cycles and used for 100-bp paired-end Illumina sequencing on a HiSeq platform. Sequencing data quality was assessed using FASTQC (38), and H3K27me3 broad peak enrichment analysis was performed using MACS2 (39) using a standardized input control merged from six experimental replicates with either the wt or the ΔCTRL2 virus. Fold enrichment signal tracks were generated using MACS2 using merged H3K27me3 ChIP data sets for wt virus and the ΔCTRL2 experiments against the standardized input control and visualized using the seqsetvis Bioconductor package (40). Reproducibility of data sets was checked using principal-component analysis (PCA) and clustering using the DiffBind Bioconductor package (41). Significant changes in ChIP-seq data sets were also identified using DiffBind, whereby the analysis was performed using the triplicate H3K27me3 data sets derived from either wt or ΔCTRL2 virus infection; a false discovery rate (FDR) of <0.05 and a fold change of >1.5-fold were considered significant.
Statistical analysis.
One-way analysis of variance (ANOVA) was performed using SigmaPlot, version 12.5, by comparing the individual normalized expression values for each gene region during latency between the wt virus and the ΔCTRL2 recombinant. The P values are indicated on the graphs where a significant change was determined by one-way ANOVA.
Data availability.
The ChIP-seq data has been deposited in the NCBI Gene Expression Omnibus (GEO) database under accession number GSE128258.
ACKNOWLEDGMENTS
This work was supported in part by grants R01AI134807 (D.M.N.) and R01AI048633 (D.C.B.) from the NIH, NIAID, and an unrestricted grant from Research to Prevent Blindness (Department of Ophthalmology and Visual Sciences, University of Wisconsin).
REFERENCES
- 1.Austin A, Lietman T, Rose-Nussbaumer J. 2017. Update on the management of infectious keratitis. Ophthalmology 124:1678–1689. doi: 10.1016/j.ophtha.2017.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bloom DC. 2016. Alphaherpesvirus latency: a dynamic state of transcription and reactivation. Adv Virus Res 94:53–80. doi: 10.1016/bs.aivir.2015.10.001. [DOI] [PubMed] [Google Scholar]
- 3.Stevens JG, Wagner EK, Vi-Rao GB, Cook ML, Feldman LT. 1987. RNA complementary to a herpesvirus alpha gene mRNA is prominent in latently infected neurons. Science 235:1056–1059. doi: 10.1126/science.2434993. [DOI] [PubMed] [Google Scholar]
- 4.Cliffe AR, Coen DM, Knipe DM. 2013. Kinetics of facultative heterochromatin and polycomb group protein association with the herpes simplex viral genome during establishment of latent infection. mBio 4:e00590-12. doi: 10.1128/mBio.00590-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Knipe DM, Cliffe A. 2008. Chromatin control of herpes simplex virus lytic and latent infection. Nat Rev Microbiol 6:211–221. doi: 10.1038/nrmicro1794. [DOI] [PubMed] [Google Scholar]
- 6.Cliffe AR, Knipe DM. 2008. Herpes simplex virus ICP0 promotes both histone removal and acetylation on viral DNA during lytic infection. J Virol 82:12030–12038. doi: 10.1128/JVI.01575-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cliffe AR, Garber DA, Knipe DM. 2009. Transcription of the herpes simplex virus latency-associated transcript promotes the formation of facultative heterochromatin on lytic promoters. J Virol 83:8182–8190. doi: 10.1128/JVI.00712-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kristie TM. 2016. Chromatin modulation of herpesvirus lytic gene expression: managing nucleosome density and heterochromatic histone modifications. mBio 7:e00098-16. doi: 10.1128/mBio.00098-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kristie TM, Liang Y, Vogel JL. 2010. Control of alpha-herpesvirus IE gene expression by HCF-1 coupled chromatin modification activities. Biochim Biophys Acta 1799:257–265. doi: 10.1016/j.bbagrm.2009.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kubat NJ, Amelio AL, Giordani NV, Bloom DC. 2004. The herpes simplex virus type 1 latency-associated transcript (LAT) enhancer/rcr is hyperacetylated during latency independently of LAT transcription. J Virol 78:12508–12518. doi: 10.1128/JVI.78.22.12508-12518.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kubat NJ, Tran RK, McAnany P, Bloom DC. 2004. Specific histone tail modification and not DNA methylation is a determinant of herpes simplex virus type 1 latent gene expression. J Virol 78:1139–1149. doi: 10.1128/JVI.78.3.1139-1149.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Neumann DM, Bhattacharjee PS, Giordani NV, Bloom DC, Hill JM. 2007. In vivo changes in the patterns of chromatin structure associated with the latent herpes simplex virus type 1 genome in mouse trigeminal ganglia can be detected at early times after butyrate treatment. J Virol 81:13248–13253. doi: 10.1128/JVI.01569-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.West AG, Gaszner M, Felsenfeld G. 2002. Insulators: many functions, many mechanisms. Genes Dev 16:271–288. doi: 10.1101/gad.954702. [DOI] [PubMed] [Google Scholar]
- 14.Ghirlando R, Felsenfeld G. 2016. CTCF: making the right connections. Genes Dev 30:881–891. doi: 10.1101/gad.277863.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Paris C, Pentland I, Groves I, Roberts DC, Powis SJ, Coleman N, Roberts S, Parish JL. 2015. CCCTC-binding factor recruitment to the early region of the human papillomavirus 18 genome regulates viral oncogene expression. J Virol 89:4770–4785. doi: 10.1128/JVI.00097-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Martinez FP, Cruz R, Lu F, Plasschaert R, Deng Z, Rivera-Molina YA, Bartolomei MS, Lieberman PM, Tang Q. 2014. CTCF binding to the first intron of the major immediate early (MIE) gene of human cytomegalovirus (HCMV) negatively regulates MIE gene expression and HCMV replication. J Virol 88:7389–7401. doi: 10.1128/JVI.00845-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kang H, Lieberman PM. 2009. Cell cycle control of Kaposi's sarcoma-associated herpesvirus latency transcription by CTCF-cohesin interactions. J Virol 83:6199–6210. doi: 10.1128/JVI.00052-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tempera I, Klichinsky M, Lieberman PM. 2011. EBV latency types adopt alternative chromatin conformations. PLoS Pathog 7:e1002180. doi: 10.1371/journal.ppat.1002180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Amelio AL, McAnany PK, Bloom DC. 2006. A chromatin insulator-like element in the herpes simplex virus type 1 latency-associated transcript region binds CCCTC-binding factor and displays enhancer-blocking and silencing activities. J Virol 80:2358–2368. doi: 10.1128/JVI.80.5.2358-2368.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Washington SD, Musarrat F, Ertel MK, Backes GL, Neumann DM. 2018. CTCF binding sites in the herpes simplex virus 1 genome display site-specific CTCF occupation, protein recruitment, and insulator function. J Virol 92:e00156-18. doi: 10.1128/JVI.00156-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Washington SD, Edenfield SI, Lieux C, Watson ZL, Taasan SM, Dhummakupt A, Bloom DC, Neumann DM. 2018. Depletion of the insulator protein CTCF results in HSV-1 reactivation in vivo. J Virol 92:e00173-18. doi: 10.1128/jvi.00173-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ertel MK, Cammarata AL, Hron RJ, Neumann DM. 2012. CTCF occupation of the herpes simplex virus 1 genome is disrupted at early times postreactivation in a transcription-dependent manner. J Virol 86:12741–12759. doi: 10.1128/JVI.01655-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lee JS, Raja P, Pan D, Pesola JM, Coen DM, Knipe DM. 2018. CCCTC-binding factor acts as a heterochromatin barrier on herpes simplex viral latent chromatin and contributes to poised latent infection. mBio 9:e02372-17. doi: 10.1128/mBio.02372-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kwiatkowski DL, Thompson HW, Bloom DC. 2009. The polycomb group protein Bmi1 binds to the herpes simplex virus 1 latent genome and maintains repressive histone marks during latency. J Virol 83:8173–8181. doi: 10.1128/JVI.00686-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vogel JL, Kristie TM. 2013. The dynamics of HCF-1 modulation of herpes simplex virus chromatin during initiation of infection. Viruses 5:1272–1291. doi: 10.3390/v5051272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Simon JA, Kingston RE. 2009. Mechanisms of polycomb gene silencing: knowns and unknowns. Nat Rev Mol Cell Biol 10:697–708. doi: 10.1038/nrm2763. [DOI] [PubMed] [Google Scholar]
- 27.Owen DJ, Crump CM, Graham SC. 2015. Tegument assembly and secondary envelopment of alphaherpesviruses. Viruses 7:5084–5114. doi: 10.3390/v7092861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Buch A, Muller O, Ivanova L, Dohner K, Bialy D, Bosse JB, Pohlmann A, Binz A, Hegemann M, Nagel CH, Koltzenburg M, Viejo-Borbolla A, Rosenhahn B, Bauerfeind R, Sodeik B. 2017. Inner tegument proteins of Herpes Simplex Virus are sufficient for intracellular capsid motility in neurons but not for axonal targeting. PLoS Pathog 13:e1006813. doi: 10.1371/journal.ppat.1006813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ohlsson R, Renkawitz R, Lobanenkov V. 2001. CTCF is a uniquely versatile transcription regulator linked to epigenetics and disease. Trends Genet 17:520–527. doi: 10.1016/S0168-9525(01)02366-6. [DOI] [PubMed] [Google Scholar]
- 30.Ohlsson R, Bartkuhn M, Renkawitz R. 2010. CTCF shapes chromatin by multiple mechanisms: the impact of 20 years of CTCF research on understanding the workings of chromatin. Chromosoma 119:351–360. doi: 10.1007/s00412-010-0262-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kim YW, Kim A. 2013. Histone acetylation contributes to chromatin looping between the locus control region and globin gene by influencing hypersensitive site formation. Biochim Biophys Acta 1829:963–969. doi: 10.1016/j.bbagrm.2013.04.006. [DOI] [PubMed] [Google Scholar]
- 32.Chen HS, Martin KA, Lu F, Lupey LN, Mueller JM, Lieberman PM, Tempera I. 2014. Epigenetic deregulation of the LMP1/LMP2 locus of Epstein-Barr virus by mutation of a single CTCF-cohesin binding site. J Virol 88:1703–1713. doi: 10.1128/JVI.02209-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tempera I, Lieberman PM. 2010. Chromatin organization of gammaherpesvirus latent genomes. Biochim Biophys Acta 1799:236–245. doi: 10.1016/j.bbagrm.2009.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Stedman W, Kang H, Lin S, Kissil JL, Bartolomei MS, Lieberman PM. 2008. Cohesins localize with CTCF at the KSHV latency control region and at cellular c-myc and H19/Igf2 insulators. EMBO J 27:654–666. doi: 10.1038/emboj.2008.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tempera I, Wiedmer A, Dheekollu J, Lieberman PM. 2010. CTCF prevents the epigenetic drift of EBV latency promoter Qp. PLoS Pathog 6:e1001048. doi: 10.1371/journal.ppat.1001048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Morse AM, Calabro KR, Fear JM, Bloom DC, McIntyre LM. 2017. Reliable detection of herpes simplex virus sequence variation by high-throughput resequencing. Viruses 9:E226. doi: 10.3390/v9080226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Amelio AL, Giordani NV, Kubat NJ, O'Neil JE, Bloom DC. 2006. Deacetylation of the herpes simplex virus type 1 latency-associated transcript (LAT) enhancer and a decrease in LAT abundance precede an increase in ICP0 transcriptional permissiveness at early times postexplant. J Virol 80:2063–2068. doi: 10.1128/JVI.80.4.2063-2068.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Andrews S. 2010. FastQC: a quality control tool for high throughput sequence data. http://www.bioinformatics.babraham.ac.uk/projects/fastqc.
- 39.Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, Liu XS. 2008. Model-based analysis of ChIP-Seq (MACS). Genome Biol 9:R137. doi: 10.1186/gb-2008-9-9-r137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Boyd J. 2018. seqsetyis: set-based visualizations for next-gen sequencing data. https://rdrr.io/bioc/seqsetvis/.
- 41.Stark R, Brown G. 2011. DiffBind: differential binding analysis of ChIP-seq peak data. http://bioconductor.org/packages/release/bioc/vignettes/DiffBind/inst/doc/DiffBind.pdf.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The ChIP-seq data has been deposited in the NCBI Gene Expression Omnibus (GEO) database under accession number GSE128258.