

Disturbances of the lipid metabolism in chickens infected with MDV contribute to the pathogenesis of disease. However, the role of lipid metabolism in MDV replication remained unknown. Here, we demonstrate that MDV infection activates FAS and induces LD formation. Moreover, our results demonstrate that MDV replication is highly dependent on the FAS pathway and the downstream metabolites. Finally, our results reveal that MDV also activates the COX-2/PGE2 pathway, which supports MDV replication by activating PGE2/EP2 and PGE2/EP4 signaling pathways.
KEYWORDS: fatty acid synthesis, Marek’s disease virus, PGE2, avian viruses, virus replication
ABSTRACT
Marek’s disease virus (MDV) causes deadly lymphoma and induces an imbalance of the lipid metabolism in infected chickens. Here, we discovered that MDV activates the fatty acid synthesis (FAS) pathway in primary chicken embryo fibroblasts (CEFs). In addition, MDV-infected cells contained high levels of fatty acids and showed increased numbers of lipid droplets (LDs). Chemical inhibitors of the FAS pathway (TOFA and C75) reduced MDV titers by approximately 30-fold. Addition of the downstream metabolites, including malonyl-coenzyme A and palmitic acid, completely restored the inhibitory effects of the FAS inhibitors. Furthermore, we could demonstrate that MDV infection activates the COX-2/prostaglandin E2 (PGE2) pathway, as evident by increased levels of arachidonic acid, COX-2 expression, and PGE2 synthesis. Inhibition of the COX-2/PGE2 pathway by chemical inhibitors or knockdown of COX2 using short hairpin RNA reduced MDV titers, suggesting that COX-2 promotes virus replication. Exogenous PGE2 completely restored the inhibition of the COX-2/PGE2 pathway in MDV replication. Unexpectedly, exogenous PGE2 also partially rescued the inhibitory effects of FAS inhibitors on MDV replication, suggesting that there is a link between these two pathways in MDV infection. Taken together, our data demonstrate that the FAS and COX-2/PGE2 pathways play an important role in the replication of this deadly pathogen.
IMPORTANCE Disturbances of the lipid metabolism in chickens infected with MDV contribute to the pathogenesis of disease. However, the role of lipid metabolism in MDV replication remained unknown. Here, we demonstrate that MDV infection activates FAS and induces LD formation. Moreover, our results demonstrate that MDV replication is highly dependent on the FAS pathway and the downstream metabolites. Finally, our results reveal that MDV also activates the COX-2/PGE2 pathway, which supports MDV replication by activating PGE2/EP2 and PGE2/EP4 signaling pathways.
INTRODUCTION
Marek’s disease virus (MDV) is a highly oncogenic herpesvirus that infects chickens and causes deadly lymphoma. The virus is transmitted to naive chickens via the respiratory tract. Macrophages or dendritic cells subsequently transfer the virus to the major lymphoid organs, where it infects B and T cells (1). The infection of T cell subsets plays a crucial role in MDV pathogenesis, while B cells are dispensable for this process (2). CD4+ T cells are the predominant cells for the establishment of MDV latency and can be transformed, resulting in T cell lymphomas. T cells also transport the virus to the feather follicle epithelia that produce infectious virus and shed it into the environment (reviewed in reference 3). MDV has been shown to disturb the lipid metabolism of the infected chickens, as it causes atherosclerotic plaque formation (4). Vaccination prevents the development of these plaques (5). Lipid analysis of the arterial smooth muscles from MDV-infected birds revealed a significant increase in nonesterified fatty acids, cholesterol, cholesterol esters, squalene, phospholipids, and triacylglycerol. Furthermore, excess lipid biosynthesis triggers cellular deposition of lipid droplets in MDV-infected cells (4, 5). Despite these intriguing observations, the role of lipid metabolism in MDV-infected cells remained unknown.
In fatty acid synthesis (FAS), acetyl-coenzyme A (CoA) is converted to malonyl-CoA and subsequently to fatty acids. The first step toward FAS is the conversion of citric acid into acetyl-CoA by direct phosphorylation of ATP-citrate lyase (ACLY). The subsequent committed step involves the conversion of acetyl-CoA into malonyl-CoA by acetyl-CoA carboxylase (ACC). The final step involves committed elongation by utilizing both acetyl-CoA and malonyl-CoA coupled to the multifunctional fatty acid synthase (FASN) to generate fatty acids (6). Dengue virus (DENV) (7), West Nile virus (WNV) (8) and hepatitis C virus (HCV) (9) have been shown to preferentially enhance FASN activity and fatty acid synthesis. Fatty acid can contribute to several key biological functions, such as fatty acid oxidation (FAO), β-oxidation, posttranslational modification of proteins, and generation of very-long-chain fatty acids. Prostaglandin E2 (PGE2), a potent lipid modulator, is derived from enzymatic activity of inducible COX-2 on arachidonic acid (AA). A direct association between induction of COX-2 activity and enhancement of human cytomegalovirus (HCMV) replication has been reported (10, 11).
Here, we demonstrate that MDV infection activates both FAS and COX-2/PGE2 pathways, which are crucial for MDV replication. Interestingly, exogenous malonyl-CoA and palmitic acid completely restore the inhibitory effects of FAS inhibitors on MDV replication, suggesting that the synthetized fatty acids are crucial for MDV replication. Intriguingly, addition of PGE2 partially restored the inhibitory effects of FAS inhibitors on MDV replication, indicating that the two pathways are connected. Taken together, our results demonstrate that FAS and PGE2 synthesis contribute to MDV replication.
(This article was submitted to an online preprint archive [12].)
RESULTS
Increased levels of lipid metabolites in MDV-infected cells.
Relative production of a panel of metabolites was determined in mock- and MDV-infected primary chicken embryo fibroblasts (CEFs) at 48 and 72 h postinfection (hpi) using two-dimensional gas chromatography with mass spectrometry (GC×GC-MS). The 16 lipid metabolites were detected and analyzed. Eight lipid metabolites, including palmitic acid, were increased at 72 hpi (Fig. 1A), 4 lipid metabolites showed no changes at any time point postinfection (Fig. 1B), and 4 lipid metabolites were decreased (Fig. 1C) at either 48 or 72 hpi. Enhanced levels of fatty acids in the MDV-infected cells can be attributed to increase in FAS and/or breakdown of lipids.
FIG 1.

Alteration of fatty acid and arachidonic acid in MDV-infected CEFs. Metabolomics analysis of relative ratio (ratio of area to internal standard) of lipid metabolites from mock (control; Con)- and MDV (RB1B; Inf)-infected CEFs are shown at 48 hpi and 72 hpi. Box and whisker plots show minimum and maximum relative levels of named lipid metabolites that are increased (A), show no change (B), or are decreased (C) in MDV-infected CEFs. Nonparametric Wilcoxon tests (Mann-Whitney) were used to assess normal distribution and test significance, with the results shown as means ± SD. * (P = 0.01), ** (P = 0.001), and *** (P = 0.0005) indicate a statistically significant difference compared to the control. NS, no significant difference. The experiments were performed in biological triplicates with six technical replicates per biological replicate.
MDV replication depends on fatty acid synthesis.
Here, we examined whether MDV infection increases FAS. For a better understanding of FAS, a schematic illustration of the FAS pathway is shown in Fig. 2A. Gene expression of the cellular enzymes involved in the FAS pathway was analyzed in mock- and MDV-infected CEF cells using quantitative PCR (qPCR). The results demonstrate that acetyl-CoA carboxylase (ACC) and fatty acid synthase (FASN) genes were highly upregulated at 72 hpi (Fig. 2B). Analysis of ACC protein levels in mock- and MDV-infected cells was performed using Western blotting. The results demonstrate that the expression of ACC subunit 1 (62 kDa) (Fig. 2C) and FASN proteins (Fig. 2D) is increased in MDV-infected cells. Specific chemical inhibitors for ACC (TOFA) and FASN (C75) were utilized to determine the role of the FAS pathway in MDV replication. Nontoxic concentrations of TOFA, C75, or TOFA and C75 (T/C) were determined based on viability and confluence of the treated CEFs (data not shown). At 72 hpi, viral titers were determined in the presence of the pharmacological inhibitors with addition of the exogenous downstream metabolites (malonyl-CoA and palmitic acid). The inhibitors of ACC (TOFA; 1.54 μM) and FASN (C75; 5.9 μM) significantly reduced MDV titer by 27 (Fig. 2E)- and 28 (Fig. 2F)-fold, respectively. MDV titer was further reduced when the cells were treated with nontoxic concentrations (data not shown) of TOFA (1.54 μM) and C75 (5.9 μM) combined (T/C) compared to those of C75 or TOFA alone (Fig. 2G). Therefore, we used T/C in some experiments to observe maximum inhibitory effects. Addition of malonyl-CoA to the culture in the presence of TOFA restored MDV replication (Fig. 2H). Similarly, palmitic acid restored virus replication in the presence of C75 (Fig. 2I). Treatment of the cells with C75 or TOFA did not reduce plaque sizes (data not shown). To confirm the role of the FAS pathway in MDV replication, we also analyzed MDV genome copy numbers at 72 hpi in the cells treated with TOFA or C75 using qPCR. The results demonstrated that TOFA and C75 also reduce virus copy numbers (Fig. 2J), suggesting that reduction in virus titer by TOFA or C75 is associated with virus copy numbers. Taken together, the results demonstrate that MDV activates the FAS pathway and blocking ACC and FASN decreases MDV titer, which can be rescued by the downstream metabolites.
FIG 2.

Fatty acid synthesis is involved in MDV replication in CEF cells. (A) Schematic FAS pathways highlighting the relevant pharmacological inhibitors (red) and the respective enzymes (yellow box) as well as metabolites (green box). (B) Fold changes in gene expression in mock- or MDV-infected CEF cells are shown at 24, 48, and 72 hpi. (C and D) Protein levels of ACC (C) and FASN (D) from mock- and MDV-infected CEFs at 72 hpi. (E to G) Alteration in MDV viral titer (PFU/ml) in MDV-infected CEFs treated with TOFA (0.03, 0.077, 0.154, 0.31, 0.77, and 1.54 μM) (E), C75 (0.393, 1.97, 3.93, and 5.9 μM) (F), and TOFA (1.54 μM) (G) in combination with C75 (5.9 μM) at 72 hpi are shown. (H and I) Analysis of MDV viral titer (PFU/ml) in infected CEFs in the presence of malonyl-CoA (30, 25, 15, 10 and 5 μM) with TOFA (1.54 μM) (H) or palmitic acid with C75 (5.9 μM) (I). (J) MDV (RB1B) genome copy numbers per 104 cells (Meq gene with reference ovotransferrin gene) in the presence TOFA (1.54 μM) or C75 (5.9 μM) at 72 hpi. ** (P < 0.01), *** (P < 0.001), and **** (P < 0.0001) indicate a statistically significant difference compared to the control. NS, no significant difference. All viral titer experiments were performed in 6 replicates for plaque assays and 3 replicates for real-time PCR and Western blot assays. The data are representative of 3 independent experiments.
MDV infection increases the formation of neutral LDs.
Lipid droplets (LDs) are endoplasmic reticulum (ER)-derived organelles that consist of a neutral lipid core with a phospholipid bilayer, and an increase in the number of LDs is an indication of lipogenesis. Here, the role of FAS on lipid metabolism was evaluated in MDV-infected cells based on LD formation. The presence of lipid droplets was examined in mock- and MDV-infected CEFs using oil red O staining at 72 hpi (Fig. 3A). An increased accumulation of LDs was observed in the MDV-infected cells (Fig. 3Ai) compared to that of the mock-infected cells (Fig. 3Aii). To quantify the number of LDs, pRB1B UL35-GFP-infected CEFs (488 nm) were stained with Red LipidTOX (568 nm), and LD formation was analyzed using confocal microscopy. Larger numbers of LDs per cell were observed in MDV-infected cells than in the mock-infected cells, as determined using the IMARIS software (P = 0.0001) (Fig. 3B and C). Treatment of CEFs with TOFA or C75 decreases (P = 0.0001) the LD numbers per cell in MDV-infected cells (Fig. 3D). Altogether, these data showed that MDV infection induced the accumulation of LDs and FAS inhibitors reduced LD formation, indicating that there was a direct link between FAS and LD formation in MDV infection.
FIG 3.

MDV infection increases the formation of neutral lipid droplets. (A) Visualization of cytoplasmic lipids in neutral lipid droplet organelles of CEF cells in MDV-infected (i) and mock-infected (ii) cells (magnification, ×10) at 72 hpi. Black arrows indicate lipid droplets. (B) Confocal microscopy imaging with maximum projection of z-stacks for each channel demonstrating nuclear and cytoplasmic distribution of pRB1B UL35-GFP virus (green) and lipid droplets (red). Mock- and pRB1B UL35-GFP-infected cells were fixed at 72 hpi and stained with DAPI (nuclear stain) and the neutral lipid stain LipidTOX-568nm. Images 5 and 10 are three-dimensional representative images analyzed using IMARIS software. z-stacks were analyzed using the IMARIS spot function analysis tool to quantify the relative amount of lipid droplets per cell in MDV-infected and mock-infected CEFs (50 to 90 cells). (C) The numbers of lipid droplets per cell in MDV-infected and mock-infected CEFs at 72 hpi. (D) The numbers of lipid droplets per cell in MDV-infected CEFs treated with TOFA (1.54 μM) or C75 (5.9 μM) are shown. ****, statistically significant difference compared to the control (P < 0.0001). All experiments were performed in duplicates, and data are representative of 3 independent experiments.
MDV activates the COX-2/PGE2 pathway.
LDs are reservoirs of COX-2 and the site of PGE2 synthesis (13, 14) by which arachidonic acid (AA) can be converted into eicosanoids, including PGE2, thromboxane A2 (TXA2), prostaglandin D2 (PGD2), prostaglandin I2 (PGI2), and prostaglandin F2 alpha (PGF2α) (Fig. 4A). AA is converted from elongation/desaturation of 18:3 ω-6 fatty acid, an essential fatty acid which can be freed by lipases. In our experiments, we demonstrate that AA levels were elevated in the MDV-infected CEFs (P = 0.003) at 48 and 72 hpi (Fig. 1A). No significant difference in COX-1 mRNA transcript expression levels was observed between the mock- and MDV-infected cells at 72 hpi using reverse transcription-PCR (RT-PCR). In contrast, COX-2 mRNA transcripts (Fig. 4B) and protein levels (Fig. 4C) were increased in the MDV-infected cells at 72 hpi. The expression levels of COX-2 in five independent experiments are shown (Fig. 4D). Higher levels of PGE2 were detected in the supernatant of MDV-infected cells than in mock-infected cells at 72 hpi using a specific enzyme-linked immunosorbent assay (ELISA) (Fig. 4E). Treatment of the cells with a COX-2 inhibitor, SC-236, reduced MDV titer in a dose-dependent manner (Fig. 4F). To confirm the role of COX-2 in MDV replication, we also demonstrated that short hairpin RNA (shRNA) targeting COX-2 (Fig. 4 G) reduces MDV titers (Fig. 4H). Exogenous PGE2, TXA2, PGD2, PGI2, or PGF2α was added to the cells treated with SC-236, and the results demonstrated that PGE2 was the only prostanoid that rescued the inhibitory effects of the COX-2 inhibitor on MDV titer (Fig. 4I). Exogenous PGE2 (0.1 to 5 μg/ml) rescued the inhibitory effects of SC-236 on MDV titer (Fig. 4J), while it did not alter MDV titer in the absence of the COX-2 inhibitor (Fig. 4K). Strikingly, exogenous PGE2 also restored the inhibitory effects of TOFA (Fig. 4L) or C75 (Fig. 4M) on virus titer, suggesting that the inhibitory effect of FAS pathway inhibitors was at least partially dependent on inhibition of PGE2 synthesis. Exogenous PGE2 recovered an MDV titer in the MDV-infected cells treated with low (0.31 μM), intermediate (0.77 μM), or high (1.54 μM) concentrations of TOFA (Fig. 4L). Similarly, exogenous PGE2 rescued the inhibitory effects of low (1.97 μM) and high (5.9 μM) concentrations of C75 on MDV titer (Fig. 4M). To demonstrate that PGE2 does not block the function of TOFA and C75 on lipid metabolism, the cells were treated with T/C in the presence or absence of PGE2 (5 μg/ml), and the fold reduction in the numbers of lipid droplets per cell were analyzed using confocal microscopy. The results demonstrated that the presence of PGE2 did not affect the ability of T/C to reduce lipid droplet numbers (Fig. 4N).
FIG 4.

MDV activates COX-2/PGE2 pathway. (A) Schematic pathway outlines the relevant pharmacological inhibitor (red) and the respective enzyme (yellow box) as well as the metabolite (green box) studied within the eicosanoid biosynthesis pathway. (B) Fold change in expression of upregulation of COX-2, but not COX-1, gene expression in MDV-infected CEFs at 72 hpi. (C) Western blot analysis showing relative COX-2 protein expression in mock- and MDV-infected CEF cells at 72 hpi. (D) Relative fluorescence representing the expression of COX-2 protein in mock- and MDV-infected cells from five independent experiments at 72 hpi. Levels of β-actin expression were used for normalization. (E) PGE2 (pg/ml) concentrations in supernatant of mock- and MDV-infected CEF cells at 72 hpi. (F) MDV titer (PFU/ml) in the MDV-infected CEFs in the presence of a COX-2 inhibitor, SC-236 (0.005, 0.025, 0.05, 0.1, and 0.25 μg/ml). (G) Gel visualization of COX-2 gene silencing by shRNA compared to that of scramble RNA in MDV-infected CEFs. β-Actin was used as an internal control for each reaction. (H) CEFs transfected with shRNA targeting COX-2 silencing or control shRNA (scramble) compared to SC-236 (0.25 μg/ml) at 72 hpi. (I) MDV titer (PFU/ml) in the MDV-infected CEFs treated with SC-236 (0.25 μg/ml) or in combination with PGE2 (5.0 μg/ml), PGI2 (0.5 μg/ml), PGD2 (0.5 μg/ml), PGF2α (0.5 μg/ml), and TXA2 (0.5 μg/ml) at 72 hpi. (J and K) MDV titer in the presence of SC-236 (0.25 μg/ml) and different concentrations of PGE2 (0.25, 0.5, 1.0, 2.5, and 5.0 μg/ml) (J) or PGE2 (0.25, 0.5, 1.0, 2.5, and 5.0 μg/ml) without SC-236 (K) at 72 hpi. (L and M) MDV titer in the presence of different concentrations of TOFA (0.31, 0.77, and 1.54 μM) (L) or C75 (1.97, 3.93, and 5.9 μM) (M) with and without exogenous PGE2 (5.0 μg/ml) in the presence of SC-236, T/C, or vehicle at 72 hpi. (N) Fold reduction in numbers of lipid droplets per cell in response to TOFA (1.54 μM) and C75 (5.9 μM) in the presence or absence of exogenous PGE2 (5.0 μg/ml) at 72 hpi. * (P < 0.01), ** (P = 0.0022), *** (P = 0.0007), and **** (P = 0.0001) indicate statistically significant differences. NS, no statistical difference. Experiments were performed in 6 replicates for plaque assays and 3 replicates for real-time PCR, shRNA silencing, Western blotting, and ELISAs. The data are representative of 3 independent experiments.
PGE2 supports MDV replication through EP2 and EP4 receptors.
In mammals, PGE2 exerts its biological activities through prostaglandin receptors EP1 to EP4 (Fig. 5A). Chicken EP2, EP3, and EP4 receptors have been cloned and characterized (15, 16), while EP1 receptor has not yet been identified in chickens. Analysis of gene expression in the mock- and MDV-infected CEFs demonstrated that the EP2 and EP4, but not EP3, receptors were upregulated upon MDV infection (Fig. 5B), suggesting that PGE2 supported MDV replication through an EP2- and/or EP4-mediated mechanism. Receptor antagonists for EP1 (SC-51322), EP2 (TG 4-155), and EP4 (ER-819762) were utilized to examine the role of different EP receptors in MDV replication in vitro. As expected, the EP1 receptor antagonist (SC-51322; 0.1, 1, 5, and 10 μM) had no effect on MDV replication (Fig. 5C). In contrast, TG 4-155 (2 and 4 μM) (Fig. 5D) and ER-819762 (0.1, 0.5 and 1 μM (Fig. 5E) reduced MDV titer (P = 0.0001), suggesting that PGE2 supports MDV replication through the EP2 and EP4 receptors. To confirm these data, we also used an shRNA system to downregulate the EP2 and EP4 receptors in CEFs (Fig. 4F). The results demonstrate that RNA silencing of EP2 and EP4 by shRNAs and chemical inhibitors (TG 4-155, 4 μM; ER-819762, 1.0 μM) significantly reduced total numbers of MDV plaques (Fig. 5G). Taken together, our data indicate that virus-induced PGE2 synthesis supports MDV replication through the EP2 and EP4 receptors.
FIG 5.

PGE2 supports MDV replication through EP2 and EP4 receptors. (A) Schematic pathway outlines the respective metabolites (green box) and the relevant pharmacological inhibitor (red) that were used in this study to block prostaglandin receptors. (B) Fold change in gene expression of PGE2 receptors EP2, EP3, and EP4 in MDV-infected CEFs at 72 hpi. (C to E) MDV titer in MDV-infected CEFs treated with different concentrations of SC-51322 (EP1 antagonist; 0.1, 1, 5, and 10 μM) (C), TG 4-155 (EP2 antagonist; 1, 2, and 4 μM) (D), and ER-819762 (EP4 antagonist; 0.1, 0.5, and 1.0 μM) (E) at 72 hpi. (F) Gel visualization of EP2 and EP4 gene silencing by shRNA compared to scramble RNA in MDV-infected CEFs. β-Actin was used as an internal control for each reaction. (G) MDV titer (PFU/ml) in the MDV-infected CEFs transfected with shRNA targeting EP2 and EP4 silencing or control shRNA (scramble) compared to TG 4-155 (4 μM) and ER-819762 (1 μM) at 72 hpi. ****, statistically significant differences (P = 0.0001). NS, no statistical difference. Experiments were performed in 6 replicates for plaque assays and 3 replicates for real-time PCR and gene silencing using shRNA. The data are representative of 3 independent experiments.
DISCUSSION
MDV is an Alphaherpesvirus that infects chickens and causes a deadly lymphoproliferative disease. In addition to transformation of CD4+ T cells, MDV causes atherosclerosis by disturbing the lipid metabolism in infected chickens (17), which can be inhibited by vaccine-induced immunity (4, 18). MDV is a cell-associated virus, and cell-free virus can only be produced by feather follicle epithelial cells in vivo. MDV is transmitted to other chickens via inhalation of virus. No cell-free virus can be generated in vitro, and surprisingly, little is known about the mechanism of MDV entry, replication, and egress in vitro or in vivo. After infection of chickens, MDV can be detected in both immune and nonimmune cells; however, the virus can only be detected in less than 2% of CD4+ T cells in the spleen. As there is no specific marker for identification of MDV-transformed T cells (3) and the majority of these cells undergo apoptosis even following in vitro T cell activation, which can potentially modulate lipid metabolism, analysis of MDV-induced lipid metabolism in primary lymphoma cells is highly challenging. Similarly, MDV can only be detected in less than 2% of MDV-transformed CD4+ T cell lines (3), and lipid metabolism is highly activated in lymphoma cells. Therefore, CEF cells represent more reproducible and reliable cells for studying the role of lipid metabolism in MDV infection in an in vitro model. MDV infection can be readily detected in the majority of primary CEF cells, which are the standard cells to investigate the MDV replication cycle in vitro. Recently, an in vitro infection system for primary B and T cells has been developed that we will also include in future studies (19).
Our analysis of MDV-infected CEFs demonstrates that MDV infection enhances the expression of genes involved in FAS and increases the levels of AA, the prostaglandin precursor. Our data suggest that MDV hijacks host metabolic pathways to provide essential macromolecular synthesis to support infection and replication. De novo FAS generates the metabolic intermediate acetyl-CoA, malonyl-CoA, and finally palmitate. Targeted inhibition studies against the enzymes involved in FAS during infection have yielded an alternative understanding of alteration of lipid metabolism in infections with HCMV (20), Epstein-Barr virus (21), and HCV (22, 23). The first step toward FAS is the conversion of citric acid into acetyl-CoA by direct phosphorylation of ACLY. The subsequent step involves the conversion of acetyl-CoA into malonyl-CoA by acetyl-coA carboxylase (ACC) and finally elongation by utilizing both acetyl-CoA and malonyl-CoA coupled to the multifunctional fatty acid synthase (FASN) to make fatty acids. Our results demonstrated that blocking ACC and FASN activity significantly reduces MDV replication, suggesting that MDV preferentially modulates the FAS pathway to generate a variety of lipids that contribute to several key cellular processes. Our data confirm that the inhibitory effects of FAS inhibitors on MDV replication could be overcome by the addition of palmitic acid, a metabolite downstream of FASN in the FAS pathway. This indicates that inhibition of MDV by FAS inhibitors is not simply detrimental to the cell but is essential for the production of infectious virus. There is very limited information on the mechanism involved in alteration of cellular metabolism by viruses. There are only two examples where the viral gene products necessary for changes in each specific metabolic pathway have been identified (reviewed in reference 24). Similarly, there is no information on the mechanism involved in activation of FAS by MDV. Interestingly, MDV encodes a secreted glycoprotein, vLIP, which has homology with lipase but does not have any enzymatic activity (25). Further research is required to determine the role of MDV genes, including vLIP, in activation of lipid metabolism.
It is known that fatty acids are essential components for the initiation of key cellular processes, such as membrane lipid synthesis, generation of LDs, and eicosanoid synthesis (26). LDs are classically defined as organelles with stored neutral lipids, and some reports suggest that some viruses promote LD formation, which is involved in virus replication and assembly (27–29). Our data demonstrate that MDV infection also increases LD formation; however, the exact role of LDs in MDV infection is still unknown. LD formation in MDV-infected cells is dependent on the FAS pathway, as FAS inhibitors reduced the numbers of LDs. This organelle is a significant source of triglyceride-derived arachidonic acid (AA), reservoir of COX-2 and site of PGE2 synthesis (13, 14, 30). Our results demonstrate that MDV infection increases the levels of FAS, AA, COX-2, and PGE2 synthesis; however, further studies are required to establish whether PGE2 synthesis occurs in LDs of MDV-infected cells. Furthermore, our results confirm previous observations on the role of FAS in LD formation, replication of pathogens, and PGE2 synthesis (13). In MDV-infected cells, both metabolic and nonmetabolic factors could be involved in COX-2 activation. Upregulation of COX-2 in the MDV-infected cells could be induced by several mechanisms, including transforming growth factor beta (TGF-β) (31, 32), cyclic AMP (33), and activation of NF-κB (34). Further studies are required to support the role of these factors in the activation of COX-2 during MDV infection. COX-2-inducing factors such as TGF-β (32), which also activates fatty acid synthesis (31, 35), might be involved in activation of COX-2 in MDV-infected cells. In fact, we have recently shown that MDV infection increases the induction of TGF-β in vivo (36); however, further studies are required to examine the role of MDV-induced TGF-β in the induction of COX-2 during MDV infection. It has been shown that COX-2 in LDs, but not in cytoplasm, is involved in PGE2 synthesis following infection or activation of the cells (14). Further experiments are required to establish the role of FAS-induced LDs in the activation of the COX-2/PGE2 pathway by MDV. Finally, we demonstrated that PGE2 promotes MDV replication via EP2 and EP4 receptors, which are upregulated in MDV-infected cells. PGE2 can contribute to virus replication by inhibition of nitric oxide production (37), type I interferon production (38), and modulation of immune cells, including antigen-presenting cells and T cells (39). We have recently reported that soluble factors released from MDV-transformed T cells inhibit the function of chicken T cells in a COX-2-dependent manner (36). The proposed model for the role of FAS and COX-2/PGE2 pathways in MDV infection is summarized in Fig. 6. This study has assisted us in the identification of potential pathways involved in MDV pathogenesis and may eventually lead to the development of control measures. However, we do not anticipate the use of FAS inhibitors as an antiviral agent in poultry because of their systemic side effects and potential residue in meat and eggs. Taken together, our results demonstrate that MDV activates FAS and COX-2/PGE2 pathways, and replication of MDV is dependent on PGE2 synthesis, which supports MDV infectivity through EP2 and EP4 receptors.
FIG 6.
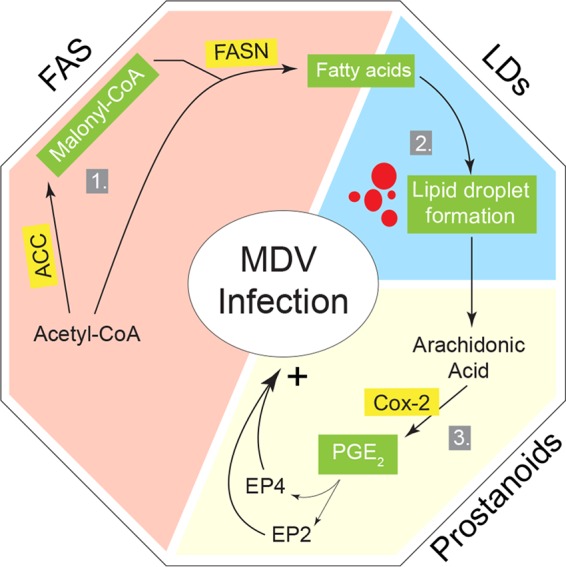
Schematic representation of the model. MDV systematically modulates cellular lipid metabolic pathways to support its replication through FAS and COX-2/PGE2 pathways. (1) Infection of CEFs with MDV results in an increase of de novo FAS pathway generation, which is required for MDV replication. (2) Fatty acid can be stored in lipid droplet organelles as a site for eicosanoid biosynthesis. (3) Subsequently, PGE2 is biosynthesized enzymatically from arachidonic acid by inducible COX-2. Signaling of PGE2 is mediated through its EP2 and EP4 receptors in MDV-infected cells. Specific enzymes studied along the pathway and their relative importance for MDV infection are highlighted in yellow. Major metabolites which were identified as essential lipids are highlighted in green.
MATERIALS AND METHODS
Ethics statement.
Ten-day-old specific-pathogen-free embryonated chicken eggs (Valo Biomedia GmbH) were used to generate primary chicken embryonic fibroblast cells (CEFs). All embryonated chicken eggs were handled in accordance with the guidance and regulations of Europe and the United Kingdom Home Office under project license number 30/3169. As part of this process, the work has undergone scrutiny and approval by the ethics committee at The Pirbright Institute.
Chemicals and antibodies.
Chemicals used include SB 204990 (Thermo Fisher Scientific, Paisley, UK), TOFA, C75, clofibrate, palmitic acid, and SC-236 (Sigma-Aldrich, Dorset, UK), PGD2, PGI2, and PGE2 (Cambridge Bioscience, Cambridge, UK), and SC-51322, TG-4-155, and ER-819762 (Bio-Techne Ltd., Abingdon, UK), and all were reconstituted in dimethyl sulfoxide. TXA2 (Cambridge Bioscience, Cambridge, UK) was reconstitute in ethanol. PGF2α (Cambridge Bioscience, Cambridge, UK) and etomoxir and malonyl-CoA (Sigma-Aldrich, Dorset, UK) were reconstituted in E199 medium. Anti-β-actin mouse monoclonal antibody (MAb), anti-FASN goat polyclonal antibody (pAb), anti-COX2 goat pAb, and anti-ACC rabbit MAb (Abcam, Cambridge, UK) were used for Western blot analysis. Donkey pAb to goat IgG, goat pAb to mouse IgG, and goat pAb to rabbit IgG were also purchased from Abcam, Cambridge, UK.
CEF culture and plaque assay.
CEFs were seeded at a rate of 1.5 × 105 cells/ml in 24-well plates in E199 medium supplemented with 5% fetal calf serum (FCS), and virus titer was determined 72 h postinfection with cell-associated MDV in the presence or absence of the inhibitors. To identify nontoxic concentrations of the chemicals, mock- and MDV-infected CEFs were exposed to the chemicals or vehicles, cell morphology and adherence/confluence were monitored under light microscopy, and then cells were stained with 7-AAD (BD Bioscience, Oxford, UK) and acquired using MACSQuant flow cytometry and FlowJo software for analysis of the data. Nontoxic concentrations of the inhibitors and chemicals were selected based on flow cytometry data and confluence. MDV-infected cells were titrated onto fresh CEFs, and 72 h postinfection, the cells were fixed and incubated with mouse anti-gB MAb (clone HB-3), as previously described (40), followed by horseradish peroxidase-conjugated rabbit anti-mouse Ig. After development of the plaques using AEC substrate, the cells were washed with super Q water and viral plaques were counted using a light microscope.
Metabolomics.
CEFs were either mock infected or infected with the RB1B strain of MDV (100 PFU per 1.5 × 105 cells) in triplicates and harvested at 48 and 72 h postinfection (hpi). The levels of metabolites were determined using GC×GC-MS (Target Discovery Institute, University of Oxford), and the data were analyzed as described previously (41). Those metabolites for which we had high confidence in their identifications are shown. The lipid profiles of mock- and MDV-infected cells were analyzed in biological triplicates with up to six technical replicates per biological replicate. The data were adjusted and normalized based on protein content. Virus infection did not change the size of the cells, as determined by microscopy.
Knockdown of EP2, EP4, and COX-2 gene expression.
Small short hairpin RNAs (shRNAs) and control scrambled RNAs were designed using short interfering RNA (siRNA) Wizard software, and these were chemically synthesized (InvivoGene, Toulouse, France) (Table 1). Construction of the shRNA harboring plasmid was performed per the manufacturer’s instructions. For silencing purpose, CEFs were transfected with the silencing and scrambled plasmid backbone containing green fluorescent protein using Lipofectamine stem reagent (Life Technologies, Warrington, UK). The transfection efficiency was observed at 24 h posttransfection using fluorescence microscopy, and cells showing more than 70% transfection efficiency were used for MDV infection.
TABLE 1.
siRNA sequences against COX-2, EP2, and EP4
| Gene and target | Oligonucleotide sequencea |
|---|---|
| COX-2(PTGER2) | |
| siRNA 1 | 5′-ACCTCGATTGACAGCCCACCAACATATCAAGAGTATGTTGGTGGGCTGTCAATCTT-3′ |
| 5′-CAAAAAGATTGACAGCCCACCAACATACTCTTGATATGTTGGTGGGCTGTCAATCG-3′ | |
| Scrambled 1 | 5′-ACCTCGTGCCCAACACGCAATTAACATCAAGAGTGTTAATTGCGTGTTGGGCACTT-3′ |
| 5′-CAAAAAGTGCCCAACACGCAATTAACACTCTTGATGTTAATTGCGTGTTGGGCACG-3′ | |
| siRNA 2 | 5′-ACCTCGTGGGATGATGAGCAGCTATTTCAAGAGAATAGCTGCTCATCATCCCACTT-3′ |
| 5′-CAAAAAGTGGGATGATGAGCAGCTATTCTCTTGAAATAGCTGCTCATCATCCCACG-3′ | |
| Scrambled 2 | 5′-ACCTCGCTGTGAGTGCGAAGAATTGTTCAAGAGACAATTCTTCGCACTCACAGCTT-3′ |
| 5′-CAAAAAGCTGTGAGTGCGAAGAATTGTCTCTTGAACAATTCTTCGCACTCACAGCG-3′ | |
| EP2 (PTGER2) | |
| siRNA 1 | 5′-ACCTCGCGCCTACGTCAACAAGTTCATCAAGAGTGAACTTGTTGACGTAGGCGCTT-3′ |
| 5′-CAAAAAGCGCCTACGTCAACAAGTTCACTCTTGATGAACTTGTTGACGTAGGCGCG-3′ | |
| Scrambled 1 | 5′-ACCTCGGCCGAACTAAGCCCACTTTATCAAGAGTAAAGTGGGCTTAGTTCGGCCTT-3′ |
| 5′-CAAAAAGGCCGAACTAAGCCCACTTTACTCTTGATAAAGTGGGCTTAGTTCGGCCG-3′ | |
| EP4 (PTGER4) | |
| siRNA 1 | 5′-ACCTCGCTCCTTCATCCTCCTCTTCTTCAAGAGAGAAGAGGAGGATGAAGGAGCTT-3′ |
| 5′-CAAAAAGCTCCTTCATCCTCCTCTTCTCTCTTGAAGAAGAGGAGGATGAAGGAGCG-3′ | |
| Scrambled 1 | 5′-ACCTCGCTCCTCTCCCCTTCTCTATTTCAAGAGAATAGAGAAGGGGAGAGGAGCTT-3′ |
| 5′-CAAAAAGCTCCTCTCCCCTTCTCTATTCTCTTGAAATAGAGAAGGGGAGAGGAGCG-3′ |
The boldface at 5' ends represents sequences for duplex stability, which provide resistance to RNase degradation. Boldface in the middle of a segment represents the site for hairpin loop folding. Boldface at the 3' region represent oligonucleotide overhangs, which are resistant to nuclease degradation.
qPCR to amplify MDV genes.
DNA samples were isolated from 5 × 106 cells using the DNeasy-96 kit (Qiagen, Manchester, UK) according to the manufacturer’s instructions. A master mix was prepared containing primers Meq-FP and Meq-RP (0.4 μM), Meq probes (0.2 μM), ovo forward and reverse primers (0.4 μM), and ovo probe (0.2 μM; 5′Yakima Yellow-3′TAMRA; Eurogentec) and ABsolute Blue qPCR, low Rox, master mix (Thermo Fisher Scientific, Paisley, UK). A standard curve generated for both Meq (10-fold serial dilutions prepared from plasmid construct with Meq target) and the ovo gene (10-fold serial dilutions prepared from plasmid construct with ovo target) were used to normalize DNA samples and to quantify MDV genomes per 104 cells. All reactions were performed in triplicates to detect both Meq and the chicken ovotransferrin (ovo) gene on an ABI7500 system (Applied Biosystems) using standard conditions. MDV genomes were normalized and are reported as viral genome per 104 cells.
Real-Time PCR.
Total RNA was extracted from mock- and MDV-infected CEFs using TRIzol (Thermo Fisher Scientific, Paisley, UK) according to the manufacturer’s protocol and treated with DNA-free DNase. Subsequently, 1 μg of purified RNA was reverse transcribed to cDNA using a Superscript III first-strand synthesis kit (Thermo Fisher Scientific, Paisley, UK) and oligo(dT) primers according to the manufacturer’s recommended protocol. The resulting cDNA was diluted 1 : 10 in diethyl pyrocarbonate-treated water. Quantitative real-time PCR using SYBR green was performed on diluted cDNA using a LightCycler 480 II (Roche Diagnostics GmbH, Mannheim, GER) as described previously (36). Data represent means from 6 biological replicates. The fold decreases in the mRNA transcript for EP2, EP4, and COX-2 genes were detected by one-step RT-PCR performed with a Luna Universal one-step RT-qPCR kit (NEB) on an Applied Biosystems 7500 qPCR system using primers outlined in Table 2.
TABLE 2.
List of primers used for RT-PCR
| Gene name | Accession no. | Primer and sequence | Tma (oC) | Product size (bp) |
|---|---|---|---|---|
| Acetyl-CoA carboxylase (ACC) | J03541.1 | Fwd, ACGTTCGAAGGGCGTACATT | 60 | 161 |
| Rev, TACGTGGACCATCCCGTAGT | ||||
| Fatty acid synthase (FASN) | J03860.1 | Fwd, CTTTGGTGGTTCGAGGTGGT | 60 | 170 |
| Rev, CTGTGGGAACCTTGCTTGGA | ||||
| Rev, AATATCCGCTCCAATGCCTCC | ||||
| Prostaglandin receptor 2 (EP2) | NM_001083365.1 | Fwd, CCTTCACGATCTGCGCCTAC | 60 | 92 |
| Rev, GGGGTTGATGGAGAGGAAGC | ||||
| Prostaglandin receptor 3 (EP3) | NM_001040468.1 | Fwd, GCTGCTGGTAACGATGCTGA | 60 | 177 |
| Rev, CGGAGCAGCAGATAAACCCA | ||||
| Prostaglandin receptor 4 (EP4) | NM_001081503.1 | Fwd, ATGTTCCAGGGTACAGGTTTTGT | 60 | 175 |
| Rev, GCCTAGCCTGCACGGTGTT | ||||
| Clyclooxygenase 1 (COX-1) | XM_425326 | Fwd, TCAGGTGGTTCTGGGACATCA | 60 | 123 |
| Rev, TGTAGCCGTACTGGGAGTTGAA | ||||
| Clyclooxygenase 2 (COX-2) | NM_001167718 | Fwd, CTGCTCCCTCCCATGTCAGA | 60 | 123 |
| Rev, CACGTGAAGAATTCCGGTGTT | ||||
| Cytoplasmic beta actin | X00182 | Fwd, TGCTGTGTTCCCATCTATCG | 60 | 150 |
| Rev, TTGGTGACAATACCGTGTTCA |
Tm, melting temperature.
Western blotting.
Mock- and MDV-infected CEFs were lysed in radioimmunoprecipitation assay buffer and quantified by spectrophotometry. The lysate was suspended in the Laemmli’s sample loading buffer (Sigma-Aldrich, Dorset, UK) and loaded in 10% SDS-PAGE. After semidry transfer of SDS-PAGE on a nitrocellulose membrane, the membrane was blocked in 5% skim milk powder for 2 h. The membranes were incubated with primary antibody for 12 h at 4°C, washed, and incubated with secondary antibodies. Finally, the blots were probed with the Odyssey CLx imaging system (Li-Cor, USA), and bands were quantified with Image Studio Lite software.
Prostaglandin E2 ELISA.
PGE2 was quantified in supernatant of mock- and MDV-infected cells using a colorimetric assay (R&D Systems, Abingdon, UK) based on competition between unlabeled PGE2 in the sample and a fixed amount of conjugated PGE2. The assay was performed according to the recommendations of the assay kit manufacturer. In brief, CEFs were either mock infected or infected with RB1B, and the levels of PGE2 in the supernatant were determined at 72 hpi or the assay results were measured using optical density at 450 nm. The concentrations of PGE2 were determined against a standard curve.
Oil red O staining.
For analysis of lipid droplets, cell monolayer was washed with phosphate-buffered saline (PBS) and fixed with 4% formaldehyde for 30 min. Cells were subsequently stained with oil red O solution for 30 min, followed by a wash in PBS. Cells were counterstained with hematoxylin for 3 min, followed by a wash with super Q water. Plates were visualized and imaged using a light microscope, and the pictures were processed using Adobe Photoshop software.
Fluorescence confocal microscopy.
CEFs were seeded in 24-well plates that contained 12-mm-diameter round coverslips at a rate of 1.0 × 105 cells per well. At 72 h after mock infection or infection with the pRB1B UL35-GFP virus (the virus was kindly provided by V. K. Nair, The Pirbright Institute), the samples were prepared for imaging. In brief, mock- or MDV-infected CEFs were fixed with 4% formaldehyde for 30 min and incubated with HCS LipidTOX Red neutral lipid stain, and then nuclei were labeled with 4′,6-diamidino-2-phenylindole (DAPI). Cells were viewed using a Leica SP2 laser-scanning confocal microscope. The data are presented as maximum projections of z-stacks (23 to 25 sections; spacing, 0.3 mm), which were analyzed using IMARIS (Bitplane Scientific Software). Ninety MDV-infected cells and 40 mock-infected cells were analyzed, and their LipidTOX-labeled neutral lipid-containing organelles were detected with the spot function of IMARIS. Images were processed using Adobe Photoshop software.
Statistical analysis.
All data are presented as means ± standard deviations (SD) from at least three independent experiments. Quantification was performed using Graph Pad Prism 7 for Windows. The differences between groups, in each experiment, were analyzed by nonparametric Wilcoxon tests (Mann-Whitney) or by Kruskal-Wallis test (one-way analysis of variance, nonparametric). Results were considered statistically significant at a P value of <0.05 (*).
ACKNOWLEDGMENTS
This work was supported by funding from the Biotechnology and Biological Sciences Research Council (award no. BB/N002598/1, BBS/E/I/00007030, BBS/E/I/00007031, and BBS/E/I/00007032) to S.B. The funder had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
REFERENCES
- 1.Jarosinski KW, Tischer BK, Trapp S, Osterrieder N. 2006. Marek's disease virus: lytic replication, oncogenesis and control. Expert Rev Vaccines 5:761–772. doi: 10.1586/14760584.5.6.761. [DOI] [PubMed] [Google Scholar]
- 2.Bertzbach LD, Laparidou M, Hartle S, Etches RJ, Kaspers B, Schusser B, Kaufer BB. 2018. Unraveling the role of B cells in the pathogenesis of an oncogenic avian herpesvirus. Proc Natl Acad Sci U S A 115:11603–11607. doi: 10.1073/pnas.1813964115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Boodhoo N, Gurung A, Sharif S, Behboudi S. 2016. Marek's disease in chickens: a review with focus on immunology. Vet Res 47:119. doi: 10.1186/s13567-016-0404-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hajjar DP, Fabricant CG, Minick CR, Fabricant J. 1986. Virus-induced atherosclerosis. Herpesvirus infection alters aortic cholesterol metabolism and accumulation. Am J Pathol 122:62–70. [PMC free article] [PubMed] [Google Scholar]
- 5.Fabricant CG, Hajjar DP, Minick CR, Fabricant J. 1981. Herpesvirus infection enhances cholesterol and cholesteryl ester accumulation in cultured arterial smooth muscle cells. Am J Pathol 105:176–184. [PMC free article] [PubMed] [Google Scholar]
- 6.Berg JM, Tymoczko JL, Stryer L. 2002. Chapter 22. Fatty acid metabolism In Biochemistry, 5th ed W. H. Freeman, New York, NY. [Google Scholar]
- 7.Heaton NS, Perera R, Berger KL, Khadka S, Lacount DJ, Kuhn RJ, Randall G. 2010. Dengue virus nonstructural protein 3 redistributes fatty acid synthase to sites of viral replication and increases cellular fatty acid synthesis. Proc Natl Acad Sci U S A 107:17345–17350. doi: 10.1073/pnas.1010811107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Merino-Ramos T, Vázquez-Calvo Á, Casas J, Sobrino F, Saiz J-C, Martín-Acebes MA. 2016. Modification of the host cell lipid metabolism induced by hypolipidemic drugs targeting the acetyl coenzyme A carboxylase impairs West Nile virus replication. Antimicrob Agents Chemother 60:307–315. doi: 10.1128/AAC.01578-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Oem JK, Jackel-Cram C, Li YP, Zhou Y, Zhong J, Shimano H, Babiuk LA, Liu Q. 2008. Activation of sterol regulatory element-binding protein 1c and fatty acid synthase transcription by hepatitis C virus nonstructural protein 2. J Gen Virol 89:1225–1230. doi: 10.1099/vir.0.83491-0. [DOI] [PubMed] [Google Scholar]
- 10.Zhu H, Cong JP, Yu D, Bresnahan WA, Shenk TE. 2002. Inhibition of cyclooxygenase 2 blocks human cytomegalovirus replication. Proc Natl Acad Sci U S A 99:3932–3937. doi: 10.1073/pnas.052713799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sharma-Walia N, Raghu H, Sadagopan S, Sivakumar R, Veettil MV, Naranatt PP, Smith MM, Chandran B. 2006. Cyclooxygenase 2 induced by Kaposi's sarcoma-associated herpesvirus early during in vitro infection of target cells plays a role in the maintenance of latent viral gene expression. J Virol 80:6534–6552. doi: 10.1128/JVI.00231-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Boodhoo N, Kamble N, Kaufer BB, Behboudi S. 2019. Replication of Marek's disease virus is dependent on synthesis of de novo fatty acid and prostaglandin E2. bioRxiv doi: 10.1101/323840. [DOI] [PMC free article] [PubMed]
- 13.Accioly MT, Pacheco P, Maya-Monteiro CM, Carrossini N, Robbs BK, Oliveira SS, Kaufmann C, Morgado-Diaz JA, Bozza PT, Viola JP. 2008. Lipid bodies are reservoirs of cyclooxygenase-2 and sites of prostaglandin-E2 synthesis in colon cancer cells. Cancer Res 68:1732–1740. doi: 10.1158/0008-5472.CAN-07-1999. [DOI] [PubMed] [Google Scholar]
- 14.D'Avila H, Melo RC, Parreira GG, Werneck-Barroso E, Castro-Faria-Neto HC, Bozza PT. 2006. Mycobacterium bovis bacillus Calmette-Guerin induces TLR2-mediated formation of lipid bodies: intracellular domains for eicosanoid synthesis in vivo. J Immunol 176:3087–3097. doi: 10.4049/jimmunol.176.5.3087. [DOI] [PubMed] [Google Scholar]
- 15.Kwok AH, Wang Y, Leung FC. 2012. Molecular characterization of prostaglandin F receptor (FP) and E receptor subtype 3 (EP3) in chickens. Gen Comp Endocrinol 179:88–98. doi: 10.1016/j.ygcen.2012.07.019. [DOI] [PubMed] [Google Scholar]
- 16.Kwok AH, Wang Y, Wang CY, Leung FC. 2008. Molecular cloning and characterization of chicken prostaglandin E receptor subtypes 2 and 4 (EP2 and EP4). Gen Comp Endocrinol 157:99–106. doi: 10.1016/j.ygcen.2008.04.001. [DOI] [PubMed] [Google Scholar]
- 17.Benditt EP, Barrett T, McDougall JK. 1983. Viruses in the etiology of atherosclerosis. Proc Natl Acad Sci U S A 80:6386–6389. doi: 10.1073/pnas.80.20.6386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fabricant CG, Fabricant J. 1999. Atherosclerosis induced by infection with Marek's disease herpesvirus in chickens. Am Heart J 138:S465–S468. doi: 10.1016/S0002-8703(99)70276-0. [DOI] [PubMed] [Google Scholar]
- 19.Schermuly J, Greco A, Hartle S, Osterrieder N, Kaufer BB, Kaspers B. 2015. In vitro model for lytic replication, latency, and transformation of an oncogenic alphaherpesvirus. Proc Natl Acad Sci U S A 112:7279–7284. doi: 10.1073/pnas.1424420112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Munger J, Bennett BD, Parikh A, Feng XJ, McArdle J, Rabitz HA, Shenk T, Rabinowitz JD. 2008. Systems-level metabolic flux profiling identifies fatty acid synthesis as a target for antiviral therapy. Nat Biotechnol 26:1179–1186. doi: 10.1038/nbt.1500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li Y, Webster-Cyriaque J, Tomlinson CC, Yohe M, Kenney S. 2004. Fatty acid synthase expression is induced by the Epstein-Barr virus immediate-early protein BRLF1 and is required for lytic viral gene expression. J Virol 78:4197–4206. doi: 10.1128/JVI.78.8.4197-4206.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yang W, Hood BL, Chadwick SL, Liu S, Watkins SC, Luo G, Conrads TP, Wang T. 2008. Fatty acid synthase is up-regulated during hepatitis C virus infection and regulates hepatitis C virus entry and production. Hepatology 48:1396–1403. doi: 10.1002/hep.22508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fujino T, Nakamuta M, Yada R, Aoyagi Y, Yasutake K, Kohjima M, Fukuizumi K, Yoshimoto T, Harada N, Yada M, Kato M, Kotoh K, Taketomi A, Maehara Y, Nakashima M, Enjoji M. 2010. Expression profile of lipid metabolism-associated genes in hepatitis C virus-infected human liver. Hepatol Res 40:923–929. doi: 10.1111/j.1872-034X.2010.00700.x. [DOI] [PubMed] [Google Scholar]
- 24.Sanchez EL, Lagunoff M. 2015. Viral activation of cellular metabolism. Virology 479-480:609–618. doi: 10.1016/j.virol.2015.02.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kamil JP, Tischer BK, Trapp S, Nair VK, Osterrieder N, Kung HJ. 2005. vLIP, a viral lipase homolog, is a virulence factor of Marek's disease virus. J Virol 79:6984–6996. doi: 10.1128/JVI.79.11.6984-6996.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rohrig F, Schulze A. 2016. The multifaceted roles of fatty acid synthesis in cancer. Nat Rev Cancer 16:732–749. doi: 10.1038/nrc.2016.89. [DOI] [PubMed] [Google Scholar]
- 27.Miyanari Y, Atsuzawa K, Usuda N, Watashi K, Hishiki T, Zayas M, Bartenschlager R, Wakita T, Hijikata M, Shimotohno K. 2007. The lipid droplet is an important organelle for hepatitis C virus production. Nat Cell Biol 9:1089–1097. doi: 10.1038/ncb1631. [DOI] [PubMed] [Google Scholar]
- 28.Samsa MM, Mondotte JA, Iglesias NG, Assuncao-Miranda I, Barbosa-Lima G, Da Poian AT, Bozza PT, Gamarnik AV. 2009. Dengue virus capsid protein usurps lipid droplets for viral particle formation. PLoS Pathog 5:e1000632. doi: 10.1371/journal.ppat.1000632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cheung W, Gill M, Esposito A, Kaminski CF, Courousse N, Chwetzoff S, Trugnan G, Keshavan N, Lever A, Desselberger U. 2010. Rotaviruses associate with cellular lipid droplet components to replicate in viroplasms, and compounds disrupting or blocking lipid droplets inhibit viroplasm formation and viral replication. J Virol 84:6782–6798. doi: 10.1128/JVI.01757-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Toledo DA, D'Avila H, Melo RC. 2016. Host lipid bodies as platforms for intracellular survival of protozoan parasites. Front Immunol 7:174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fang L, Chang HM, Cheng JC, Leung PC, Sun YP. 2014. TGF-beta1 induces COX-2 expression and PGE2 production in human granulosa cells through Smad signaling pathways. J Clin Endocrinol Metab 99:E1217–E1226. doi: 10.1210/jc.2013-4100. [DOI] [PubMed] [Google Scholar]
- 32.Rodríguez-Barbero A, Dorado F, Velasco S, Pandiella A, Banas B, López-Novoa JM. 2006. TGF-beta1 induces COX-2 expression and PGE2 synthesis through MAPK and PI3K pathways in human mesangial cells. Kidney Int 70:901–909. doi: 10.1038/sj.ki.5001626. [DOI] [PubMed] [Google Scholar]
- 33.Klein T, Shephard P, Kleinert H, Komhoff M. 2007. Regulation of cyclooxygenase-2 expression by cyclic AMP. Biochim Biophys Acta 1773:1605–1618. doi: 10.1016/j.bbamcr.2007.09.001. [DOI] [PubMed] [Google Scholar]
- 34.Ulivi V, Giannoni P, Gentili C, Cancedda R, Descalzi F. 2008. p38/NF-kB-dependent expression of COX-2 during differentiation and inflammatory response of chondrocytes. J Cell Biochem 104:1393–1406. doi: 10.1002/jcb.21717. [DOI] [PubMed] [Google Scholar]
- 35.Jung MY, Kang JH, Hernandez DM, Yin X, Andrianifahanana M, Wang Y, Gonzalez-Guerrico A, Limper AH, Lupu R, Leof EB. 2018. Fatty acid synthase is required for profibrotic TGF-beta signaling. FASEB J 32:3803–3815. doi: 10.1096/fj.201701187R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gurung A, Kamble N, Kaufer BB, Pathan A, Behboudi S. 2017. Association of Marek's disease induced immunosuppression with activation of a novel regulatory T cells in chickens. PLoS Pathog 13:e1006745. doi: 10.1371/journal.ppat.1006745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Alfajaro MM, Choi JS, Kim DS, Seo JY, Kim JY, Park JG, Soliman M, Baek YB, Cho EH, Kwon J, Kwon HJ, Park SJ, Lee WS, Kang MI, Hosmillo M, Goodfellow I, Cho KO. 2017. Activation of COX-2/PGE2 promotes sapovirus replication via the inhibition of nitric oxide production. J Virol 91:e01656-16. doi: 10.1128/JVI.01656-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Coulombe F, Jaworska J, Verway M, Tzelepis F, Massoud A, Gillard J, Wong G, Kobinger G, Xing Z, Couture C, Joubert P, Fritz JH, Powell WS, Divangahi M. 2014. Targeted prostaglandin E2 inhibition enhances antiviral immunity through induction of type I interferon and apoptosis in macrophages. Immunity 40:554–568. doi: 10.1016/j.immuni.2014.02.013. [DOI] [PubMed] [Google Scholar]
- 39.Kunkel SL, Campbell DA Jr, Chensue SW, Higashi GI. 1986. Species-dependent regulation of monocyte/macrophage Ia antigen expression and antigen presentation by prostaglandin E. Cell Immunol 97:140–145. doi: 10.1016/0008-8749(86)90383-7. [DOI] [PubMed] [Google Scholar]
- 40.Baigent SJ, Smith LP, Currie RJ, Nair VK. 2005. Replication kinetics of Marek's disease vaccine virus in feathers and lymphoid tissues using PCR and virus isolation. J Gen Virol 86:2989–2998. doi: 10.1099/vir.0.81299-0. [DOI] [PubMed] [Google Scholar]
- 41.Yu Z, Huang H, Reim A, Charles PD, Northage A, Jackson D, Parry I, Kessler BM. 2017. Optimizing 2D gas chromatography mass spectrometry for robust tissue, serum and urine metabolite profiling. Talanta 165:685–691. doi: 10.1016/j.talanta.2017.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]