

Viruses have evolved to inhibit cellular DNA damage response pathways that possess antiviral activities and utilize DNA damage response pathways that possess proviral activities. Adenovirus has evolved, primarily, to inhibit DNA damage response pathways by engaging with the ubiquitin-proteasome system and promoting the degradation of key cellular proteins. Adenovirus differentially regulates ATR DNA damage response signaling pathways during infection. The cellular adenovirus E1B-55K binding protein E1B-AP5 participates in ATR signaling pathways activated during infection, while adenovirus 12 E4orf6 negates Chk1 activation by promoting the proteasome-dependent degradation of the ATR activator TOPBP1. The studies detailed here indicate that adenovirus utilizes ATR kinase and CDKs during infection to promote the degradation of SMARCAL1 to attenuate normal cellular DNA replication. These studies further our understanding of the relationship between adenovirus and DNA damage and cell cycle signaling pathways during infection and establish new roles for E1B-55K in the modulation of cellular DNA replication.
KEYWORDS: DNA damage response, adenoviruses
ABSTRACT
Here, we show that the cellular DNA replication protein and ATR substrate SMARCAL1 is recruited to viral replication centers early during adenovirus infection and is then targeted in an E1B-55K/E4orf6- and cullin RING ligase-dependent manner for proteasomal degradation. In this regard, we have determined that SMARCAL1 is phosphorylated at S123, S129, and S173 early during infection in an ATR- and CDK-dependent manner, and that pharmacological inhibition of ATR and CDK activities attenuates SMARCAL1 degradation. SMARCAL1 recruitment to viral replication centers was shown to be largely dependent upon SMARCAL1 association with the RPA complex, while Ad-induced SMARCAL1 phosphorylation also contributed to SMARCAL1 recruitment to viral replication centers, albeit to a limited extent. SMARCAL1 was found associated with E1B-55K in adenovirus E1-transformed cells. Consistent with its ability to target SMARCAL1, we determined that E1B-55K modulates cellular DNA replication. As such, E1B-55K expression initially enhances cellular DNA replication fork speed but ultimately leads to increased replication fork stalling and the attenuation of cellular DNA replication. Therefore, we propose that adenovirus targets SMARCAL1 for degradation during infection to inhibit cellular DNA replication and promote viral replication.
IMPORTANCE Viruses have evolved to inhibit cellular DNA damage response pathways that possess antiviral activities and utilize DNA damage response pathways that possess proviral activities. Adenovirus has evolved, primarily, to inhibit DNA damage response pathways by engaging with the ubiquitin-proteasome system and promoting the degradation of key cellular proteins. Adenovirus differentially regulates ATR DNA damage response signaling pathways during infection. The cellular adenovirus E1B-55K binding protein E1B-AP5 participates in ATR signaling pathways activated during infection, while adenovirus 12 E4orf6 negates Chk1 activation by promoting the proteasome-dependent degradation of the ATR activator TOPBP1. The studies detailed here indicate that adenovirus utilizes ATR kinase and CDKs during infection to promote the degradation of SMARCAL1 to attenuate normal cellular DNA replication. These studies further our understanding of the relationship between adenovirus and DNA damage and cell cycle signaling pathways during infection and establish new roles for E1B-55K in the modulation of cellular DNA replication.
INTRODUCTION
Cellular DNA damage response (DDR) signaling pathways coordinated by the phosphoinositide 3-kinase (PI3K)-like kinase proteins ataxia telangiectasia mutated (ATM), ATM-Rad3-related gene (ATR), and DNA-dependent protein kinase (DNA-PK) are often targeted by viruses during infection in order to facilitate viral replication (1, 2). As such, viruses often exploit the ubiquitin-proteasome system to inhibit DDR pathway components that possess antiviral activities and utilize DDR pathway components that possess proviral activities (1, 3). In this regard, adenovirus (Ad) types from all groups have evolved, almost exclusively, to inhibit DDR pathways during infection. Early work determined that Ad5 E1B 55-kDa protein (E1B-55K) and E4orf6 assemble an Ad ubiquitin (Ub) ligase complex, consisting of cullin RING ligase 5 (CRL5), elongin B, elongin C, and Rbx1, that was capable of promoting the specific degradation of the tumor suppressor gene product p53 during infection (4, 5). In this regard, BC box motifs within E4orf6 served to recruit CRL5 through association with elongins B and C, whereas E1B-55K served to recruit p53 to the Ad Ub ligase through interaction with E4orf6 (6). Later studies indicated that group A viruses, such as Ad12, utilized CRL2 to promote the degradation of p53 during infection (7, 8).
The Ad Ub ligase was subsequently shown to inhibit the ATM-coordinated response to viral infection by promoting the degradation of MRE11 and BLM to ensure that viral genome processing, resection, recombination, and concatenation are all negated (9, 10). Adenovirus was also shown to inhibit nonhomologous end-joining pathways coordinated by DNA-PK by targeting DNA ligase IV for Ad Ub ligase-mediated degradation, which also served to prevent viral genome concatenation (11). The Ad Ub ligase has also been shown to promote the degradation of cellular proteins not involved in DDR signaling but do, nevertheless, possess antiviral activities. As such, cellular proteins involved in cell signaling, cell adhesion, and cell contacts, such as integrin α3, ALCAM, EPHA2, and PTPRF, are all targeted for degradation during infection (12, 13). E1B-55K can also, in isolation, promote the proteasome-mediated degradation of Daxx, a component of PML nuclear bodies and transcriptional regulator that has antiviral activities (14), while Ad E4orf3, which possesses inherent SUMO ligase activity, can target cellular proteins such as TIF1γ and TFII-I for SUMO-targeted ubiquitin ligase (STUbL)-mediated degradation during infection (15–17).
The ATR kinase serves specifically to regulate pathways that control DNA replication in response to replication stress (18). ATR is an essential gene; hypomorphic mutations cause Seckel syndrome, a pleiotropic disease characterized primarily by growth retardation and microcephaly (18). ATR signaling pathways are targeted specifically during Ad infection. It has long been known that the single-stranded DNA (ssDNA)-binding protein complex RPA, which participates in ATR signaling pathways through its association with ssDNA during cellular DNA replication, and following resection at double-stranded DNA (dsDNA) breaks (DSBs), is recruited to viral replication centers (VRCs) during Ad infection and presumably associates with viral ssDNA replication intermediates during genome replication (19, 20). As such, RPA has often served as a surrogate marker for VRCs. More recently, a number of ATR signaling components required for ATR activation, such as ATR-interacting protein (ATRIP) components of the RAD9-HUS1-RAD1 (9-1-1) clamp complex and Rad17, have all been shown to be recruited to VRCs following both Ad5 and Ad12 infection (19, 20). It has also been suggested that Ad5, but not Ad12, inhibits the ATR-dependent activation of Chk1 by promoting the E4orf3-dependent immobilization of the MRE11-RAD50-NBS1 complex in nuclear tracks, while Ad12 E4orf6 alone associates with CRL2-Rbx1 to promote the degradation of the ATR activator TOPBP1 and ensures that Chk1 is not activated during Ad12 infection (7, 20). It has been determined that the ATR pathway is differentially regulated during Ad infection. ATR kinase has been shown to be activated during both Ad5 and Ad12 infection and that the cellular Ad E1B-55K-associated protein E1B-AP5 (hnRNPUL1) is required for ATR activation under these circumstances (20). Indeed, E1B-AP5 was shown to be required for the ATR-dependent phosphorylation of RPA32 during infection and also contributed to the Ad-induced phosphorylation of Smc1 and H2AX. However, it is not apparent why ATR kinase activity is not fully inactivated during Ad infection. This situation suggests that the virus promotes the selective ATR-dependent phosphorylation of specific substrates during infection to inhibit cellular replication and facilitate viral replication (20).
SMARCAL1 (SWI/SNF-related matrix-associated actin-dependent regulator of chromatin subfamily A-like protein 1) is a DNA-dependent ATPase and ATP-dependent annealing helicase that has the capacity to interact with both dsDNA and ssDNA through DNA-binding domains (DBDs) within its primary structure and its interaction with the RPA complex, respectively (21–25). Biallelic inactivation of SMARCAL1 causes Schimke immuno-osseous dysplasia (SIOD), which is characterized by renal failure, immune deficiencies, bone growth retardation, and predisposition to different types of cancer (26). SMARCAL1 has the capacity to remodel replication forks and serves to prevent replication fork collapse and promote replication restart (21–25). As such, SMARCAL1 is recruited to stalled forks through its interaction with RPA to promote fork regression and the restoration of fork structure. SMARCAL1 function is regulated by the ATR kinase; in response to replication stress, ATR phosphorylates SMARCAL1 on S652 and limits its fork regression and fork-processing activities (27). Indeed, when ATR is inhibited pharmacologically such that SMARCAL1 activity is not tightly regulated, uncoordinated SMARCAL1 activity promotes fork collapse (28). SMARCAL1 also participates directly in response to different types of DNA damage and is recruited in an RPA-dependent manner to DSBs that have been processed to generate ssDNA, and it serves to both stabilize replication forks and restore fork integrity (21–25).
As our understanding of the relationship between ATR signaling pathways and adenovirus is incomplete, this study sought to further our knowledge in this area. As such, we determined that the ATR substrate SMARCAL1 is phosphorylated in an ATR- and CDK-dependent manner and then targeted for degradation during adenovirus infection, presumably to disable its cellular activities during infection. Consistent with this notion, E1B-55K, which associates specifically with SMARCAL1, was shown to dysregulate cellular DNA replication fork speed and promote replication fork stalling. Therefore, we propose that adenovirus inhibits SMARCAL1 activity to effectively inactivate cellular DNA replication during infection.
RESULTS
SMARCAL1 localizes to ad replication centers during the early stages of infection.
As we and others have shown that the RPA complex and other components of ATR signaling pathways are recruited to VRCs during infection, we decided initially to determine whether SMARCAL1, a known ATR substrate and RPA-binding protein, was also recruited to VRCs following infection of human A549 cells with wild-type (wt) Ad5 or wt Ad12. Confocal microscopy revealed that like the RPA complex component RPA2, SMARCAL1 was distributed predominantly throughout the nucleus in mock-infected, interphase A549 cells, although there also appeared to be a proportion of cytoplasmic SMARCAL1 (Fig. 1i to iii). Following infection with either wt Ad5 or wt Ad12, and consistent with previous studies, RPA2 relocalized to VRCs (Fig. 1iv to vi, Ad5; vii to ix, Ad12). Importantly, SMARCAL1 was also recruited to VRCs, and colocalized with RPA2, following either wt Ad5 or wt Ad12 infection (Fig. 1iv to vi, Ad5; vii to ix, Ad12). Interestingly, the levels of SMARCAL1 in the Ad12-infected cells appeared to be reduced relative to those of mock-infected cells (Fig. 1i and ii). Taken together, these data indicate that SMARCAL1 is recruited to VRCs during Ad infection.
FIG 1.
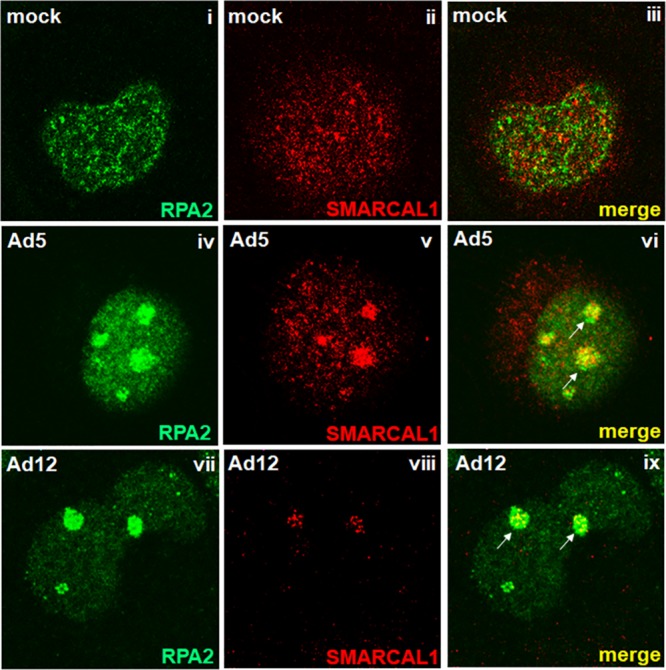
SMARCAL1 is reorganized to viral replication centers during the early stages of Ad infection. A549 cells were either mock infected (i to iii) or infected with 10 PFU/cell of wt Ad5 (iv to vi) or wt Ad12 (vii to ix). At 18 h postinfection, cells were fixed, permeabilized, and costained for SMARCAL1 and RPA2. Arrows indicate regions of RPA2/SMARCAL1 colocalization. In all instances, images were recorded using a Zeiss LSM510-Meta confocal microscope.
SMARCAL1 protein levels are reduced following Ad5 and Ad12 infection.
Given that the immunofluorescence studies suggested that SMARCAL1 levels were reduced following Ad12 infection (Fig. 1), we next sought to determine whether absolute SMARCAL1 protein levels are affected by viral infection. To do this, we infected A549 cells with either wt Ad5 or wt Ad12 and analyzed SMARCAL1 protein levels at various stages postinfection. Western blot (WB) analyses revealed that akin to p53, SMARCAL1 protein levels were reduced substantially following wt Ad5 infection (Fig. 2A). WB analyses revealed that SMARCAL1 protein levels were similarly reduced following wt Ad12 infection (Fig. 2B). Interestingly, WB analyses revealed that SMARCAL1 appeared to undergo posttranslational modification at early time points postinfection, as judged by an apparent increase in its molecular weight, following infection with either wt Ad5 or wt Ad12 (Fig. 2A and B). These data suggest that SMARCAL1 is targeted for degradation during Ad infection.
FIG 2.
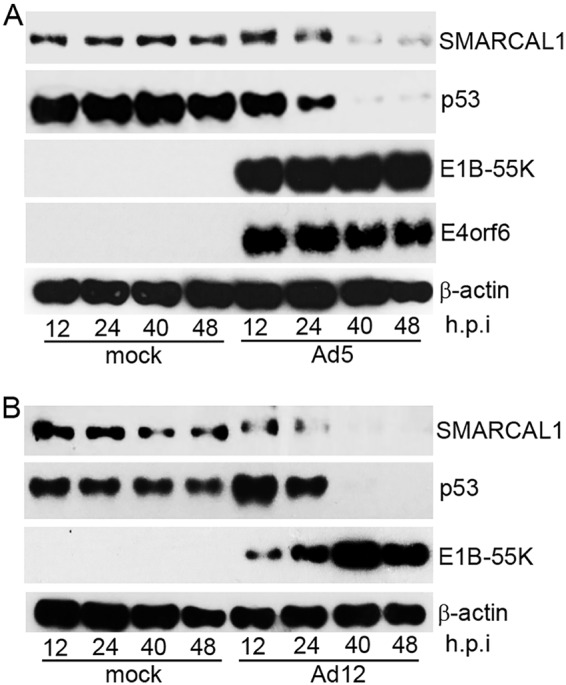
SMARCAL1 is targeted for degradation during Ad infection. A549 cells were either mock infected or infected with 10 PFU/cell of wt Ad5 or wt Ad12 and harvested at the appropriate times postinfection. (A) Ad5 cell lysates were then subjected to WB for SMARCAL1, p53, E1B-55K, E4orf6, and β-actin. (B) Ad12 cell lysates were subjected to WB for SMARCAL1, p53, E1B-55K, and β-actin. h.p.i, hours postinfection. Data are representative of more than three independent experiments.
SMARCAL1 is degraded during ad infection in an E1B-55K/E4orf6- and CRL-dependent manner.
As E1B-55K/E4orf6 complexes and E1B-55K, E4orf3, and E4orf6 alone have all been implicated in the targeting of cellular proteins for degradation, we next investigated which early region viral proteins were required to induce SMARCAL1 degradation during infection. To do this, we infected A549 cells with wt Ad5, the E1B-55K deletion mutant, Ad5 dl1520, the E4orf3 deletion mutant, pm4150, and the Ad5 E4orf6 deletion mutant, pm4154, and then analyzed SMARCAL1 protein levels at 24 h and 48 h postinfection (Fig. 3A). In line with previous studies, WB analyses revealed that p53 degradation was dependent on the expression of both E1B-55K and E4orf6 (Fig. 3A). Consistent with the notion that the Ad Ub ligase was also required to promote the degradation of SMARCAL1 during infection, WB analyses also revealed that SMARCAL1 degradation was dependent upon the expression of both E1B-55K and E4orf6 (Fig. 3A). Consistent with a role for E1B-55K in the degradation of SMARCAL1 in Ad12-infected cells, the Ad12 E1B-55K deletion mutant Ad12 dl620 was not as efficient as wt Ad12 in promoting the degradation of SMARCAL1 (Fig. 3B).
FIG 3.

SMARCAL1 is degraded during Ad infection in an E1B-55K/E4orf6- and CRL-dependent manner. (A) A549 cells were either mock infected, infected with wt Ad5, or infected with E1B-55K (dl1520), E4orf3 (H5pm4150), or E4orf6 (H5pm4154) deletion virus. At 24 h and 48 h postinfection, cells were harvested and subjected to WB for SMARCAL1, p53, E1B-55K, E4orf3, E4orf6, and β-actin. (B) A549 cells were either mock infected, infected with wt Ad12, or infected with the E1B-55K (dl620) deletion virus. At 24 h and 48 h postinfection, cells were harvested and Western blotted for SMARCAL1, p53, E1B-55K, and β-actin. (C and D) A549 cells were either mock infected or infected with wt Ad5 or wt Ad12 in the absence or presence of 100 nM or 500 nM MLN4924. At 24 h and 48 h postinfection, cells were harvested and subjected to WB for SMARCAL1, p53, E1B-55K, and β-actin. h.p.i, hours postinfection. Data are representative of three independent experiments.
To investigate the role for cellular CRLs in the E1B-55K/E4orf6-dependent degradation of SMARCAL1, we utilized the NEDD8-activating enzyme (NAE) inhibitor MLN4924, which inhibits cullin neddylation and activation (29). As MLN4924 has been shown to be effective in the low- to high-nanomolar range and has been shown to activate p53 at high-nanomolar concentrations (29, 30), we used two different doses to assess its efficacy as a CRL inhibitor during Ad infection. We therefore infected A549 cells with wt Ad5 or wt Ad12 and then incubated infected cells in the absence or presence of MLN4924 and analyzed SMARCAL1 protein levels at 24 h and 48 h postinfection (Fig. 3C and D). WB analyses revealed that 500 nM MLN4924 reduced markedly the ability of wt Ad5 and wt Ad12 to promote SMARCAL1 degradation (Fig. 3C and D, compare the third and fourth lanes to the eleventh and twelfth lanes). As noted in other studies, MLN4924 treatment in the absence of infection promoted p53 stabilization and, consistent with other reports, limited p53 degradation following Ad infection (30) (Fig. 3C and D, compare the first and second lanes to the fifth, sixth, ninth, and tenth lanes). Pertinently, however, MLN4924 treatment did not affect the levels of SMARCAL1 in mock-infected cells (Fig. 3C and D, compare the first and second lanes to the fifth, sixth, ninth, and tenth lanes). Taken together, these data suggest that E1B-55K/E4orf6 recruit cellular CRLs to promote the degradation of SMARCAL1 during Ad infection.
SMARCAL1 is phosphorylated in the early stages of Ad5 and Ad12 infection.
As ATR kinase is known to be activated following Ad infection and SMARCAL1 migration on SDS-PAGE was retarded following infection, we next investigated whether SMARCAL1 was phosphorylated in response to Ad infection. To do this, we first infected A549 cells with either wt Ad5 or wt Ad12 and then immunoprecipitated (IP) SMARCAL1 from mock-infected or Ad-infected cells with an anti-SMARCAL1 antibody. Immunoprecipitates were then either left untreated or treated with λ-phosphatase prior to investigating the migratory properties of SMARCAL1 on SDS-PAGE. Consistent with the notion that SMARCAL1 is phosphorylated following Ad infection, WB analyses revealed that when anti-SMARCAL1 immunoprecipitates from Ad-infected cells were treated with λ-phosphatase, the migration of SMARCAL1 was increased relative to that of untreated samples and comparable to that of the migration of SMARCAL1 from mock-infected cells (Fig. 4A, compare the sixth and eighth lanes with the first lane). Treatment with the NAE inhibitor promoted limited phosphorylation of SMARCAL1 (Fig. 4A, third and fourth lanes). To determine which SMARCAL1 residues were phosphorylated following Ad infection, we immunoprecipitated SMARCAL1 from mock-, Ad5-, and Ad12-infected A549 cells. Following SDS-PAGE and gel slice processing, we subjected isolated tryptic peptides to tandem array mass spectrometry (MS/MS). MS analyses revealed that SMARCAL1 was phosphorylated at three major sites following both Ad5 and Ad12 infection: S123, S129, and S173 (Fig. 4B). S123 and S129 formed part of a minimal CDK consensus phosphorylation motif, SP, while S173 formed part of an ATR consensus phosphorylation motif, SQE. Sequence homology searches revealed that these residues were conserved among primates but less well conserved for lower mammals (Fig. 4C).
FIG 4.

SMARCAL1 is phosphorylated during the early stages of Ad infection. (A) A549 cells were either mock infected, treated with MLN4924, or infected with 10 PFU/cell of wt Ad5 or wt Ad12 and harvested at 18 h postinfection. Cells were harvested in IP buffer and subjected to immunoprecipitation for SMARCAL1. Anti-SMARCAL1 immunoprecipitates collected on protein G-Sepharose were treated in the absence or presence of λ-phosphatase and then subjected to SDS-PAGE and WB for SMARCAL1. (B) SMARCAL1 was immunoprecipitated from mock-infected and wt Ad5- or wt Ad12-infected A549 cells 18 h postinfection and separated by SDS-PAGE. Protein bands excised from the gel were subjected to trypsinization and mass spectrometric analysis. Identified SMARCAL1 phosphorylated peptides from Ad-infected cells are presented. (C) S123, S129, and S173 are conserved between primates but less well conserved in lower mammals. SMARCAL1 primary sequences from a number of species were aligned using CLUSTAL Omega. Shaded areas indicate conserved residues.
Pharmacological inhibition of ATR kinase and CDK activities limits SMARCAL1 degradation following Ad5 and Ad12 infection.
Given that SMARCAL1 phosphorylation precedes its degradation following Ad infection, we next investigated whether the ATR- and CDK-dependent phosphorylation of SMARCAL1 during Ad infection was an essential prerequisite for the Ad-induced degradation of SMARCAL1. To do this, we studied the effects of the selective ATR kinase inhibitor AZD6738 and the CDK inhibitor RO-3306 on the ability of both wt Ad5 and wt Ad12 to induce the degradation of SMARCAL1. Initially, therefore, A549 cells were either mock infected or infected with wt Ad5 or wt Ad12 and then incubated in the absence or presence of AZD6738 for specific times postinfection. WB analyses revealed that treatment of A549 cells with AZD6738 reduced modestly the ability of wt Ad5 to promote the degradation of SMARCAL1 (Fig. 5A, compare the seventh and eighth lanes with the fifth and sixth lanes). Interestingly, however, the effect of AZD6738 treatment on the ability of wt Ad12 to promote SMARCAL1 degradation was much more dramatic; the ATR kinase inhibitor reduced appreciably the ability of wt Ad12 to stimulate SMARCAL1 degradation during infection, with no observable degradation at 24 h postinfection (Fig. 5B, compare the seventh and eighth lanes with the fifth and sixth lanes). To establish whether CDKs cooperate with ATR to promote SMARCAL1 degradation following Ad infection, we infected A549 cells with either wt Ad5 or wt Ad12 and then incubated infected cells in the absence or presence of AZD6738 and RO-3306 for specific times postinfection. WB analyses revealed that the use of both inhibitors reduced substantially the ability of wt Ad5 to promote the degradation of SMARCAL1, particularly at 48 h postinfection (Fig. 5C, compare the fifth and sixth lanes with the seventh and eighth lanes). Similarly, the combined effects of AZD6738 and RO-3306 almost entirely abated the ability of wt Ad12 to induce the degradation of SMARCAL1 (Fig. 5D, compare the fifth and sixth lanes with the seventh and eighth lanes). Taken together, these data suggest strongly that the combined ATR kinase- and CDK-dependent phosphorylation of SMARCAL1 facilitates the E1B-55K/E4orf6-dependent degradation of SMARCAL1 during Ad infection. As such, these studies are important in establishing that Ad can activate, and then utilize, cellular kinases during infection to promote viral replication.
FIG 5.
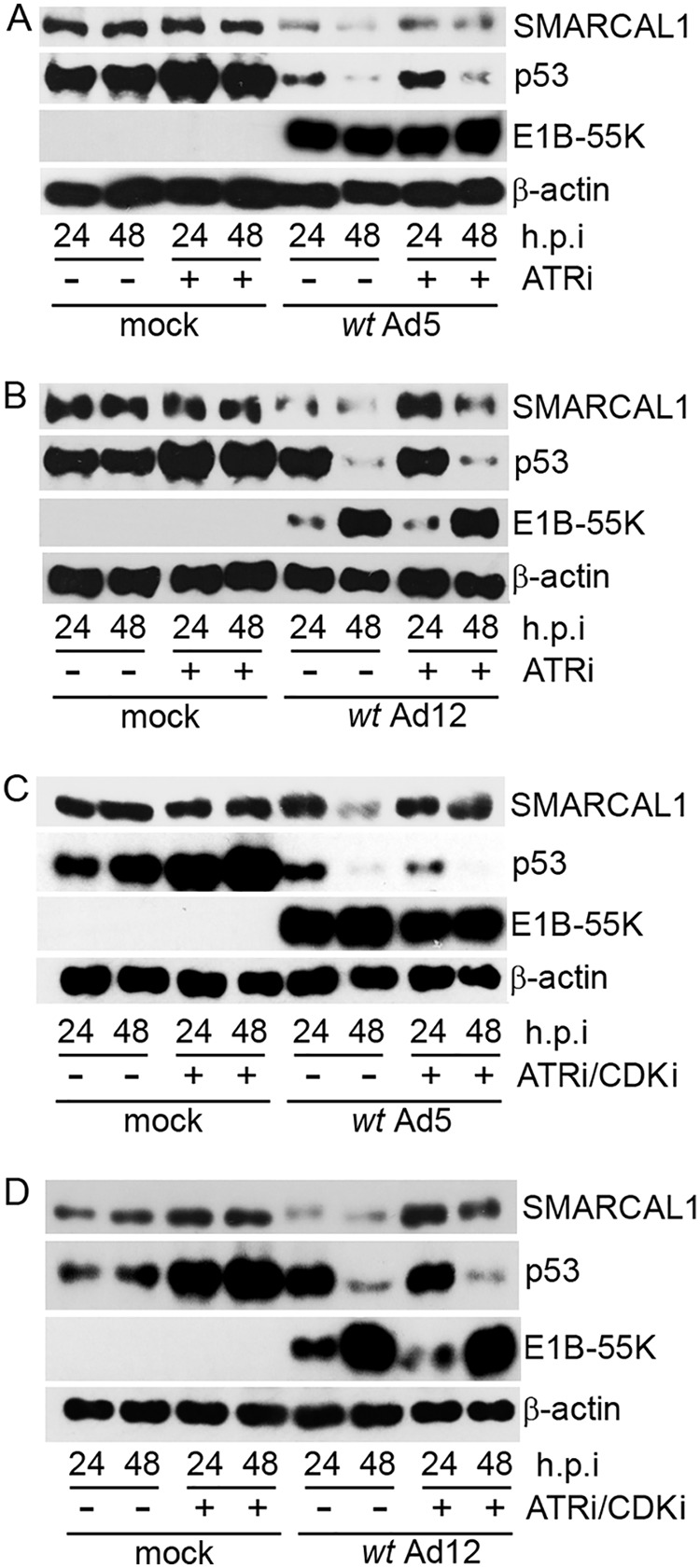
ATR kinase and CDKs promote SMARCAL1 degradation following Ad5 and Ad12 infection. A549 cells were either mock infected or infected with 10 PFU/cell of wt Ad5 (A and C) or wt Ad12 (B and D). Cells were then incubated in the absence or presence of ATR inhibitor (AZD6738 [ATRi], 1 μM; A and B) or ATR and CDK inhibitors (AZD6738, 1 μM and RO-3306 [CDKi], 9 μM; C and D) and harvested at the appropriate times postinfection. Cell lysates were then separated by SDS-PAGE and subjected to WB for SMARCAL1, p53, E1B-55K, and β-actin. h.p.i, hours postinfection. Data are representative of three independent experiments.
SMARCAL1 recruitment to VRCs is largely dependent upon its association with the RPA complex but is also regulated by ATR- and CDK-dependent phosphorylation.
To explore in more detail the factors that modulate the recruitment of SMARCAL1 to VRCs during Ad infection, we generated a phosphorylation-defective GFP-SMARCAL1-ΔP (S123A, S129A, and S173A) mutant in order to ablate the ATR- and CDK-dependent phosphorylation of SMARCAL1 in response to Ad infection and utilized a GFP-SMARCAL1-ΔRPA mutant that is unable to bind the RPA complex (21). We then generated clonal RPE-1 cell lines that expressed constitutively either GFP alone, wt GFP-SMARCAL1, GFP-SMARCAL1-ΔP, or GFP-SMARCAL1-ΔRPA. To investigate the role SMARCAL1 phosphorylation and the RPA complex play in SMARCAL1 recruitment to VRCs, we infected these cell lines with either wt Ad5 or wt Ad12 and analyzed GFP-SMARCAL1 cellular distribution throughout the infection process. Pertinently, Ad infection of RPE-1 cells expressing green fluorescent protein (GFP) alone had no effect upon the pancellular distribution of GFP (data not shown). In mock-infected RPE-1 cells, wt GFP-SMARCAL1, GFP-SMARCAL1-ΔP, and GFP-SMARCAL1-ΔRPA were distributed evenly throughout the nucleus (Fig. 6Ai to iii). Following infection of RPE-1 cells with either wt Ad5 or wt Ad12, wt GFP-SMARCAL1 was redistributed to VRCs (Fig. 6Aiv and vii). Interestingly, the ability of both wt Ad5 and wt Ad12 to promote the recruitment of the GFP-SMARCAL1-ΔP mutant to VRCs relative to that of wt GFP-SMARCAL1 was reduced significantly, but only by one-third (Fig. 6Av and viii and B). Moreover, the ability of both wt Ad5 and wt Ad12 to promote the recruitment of GFP-SMARCAL1-ΔRPA relative to that of wt GFP-SMARCAL1 was also reduced significantly by approximately two-thirds (Fig. 6Avi and ix and B). Taken together, these data suggest that the RPA complex plays a major role in the recruitment of SMARCAL1 to VRCs during Ad infection, while the ATR- and CDK-dependent phosphorylation of SMARCAL1, although not essential, also contributes to SMARCAL1 recruitment to VRCs following Ad infection.
FIG 6.
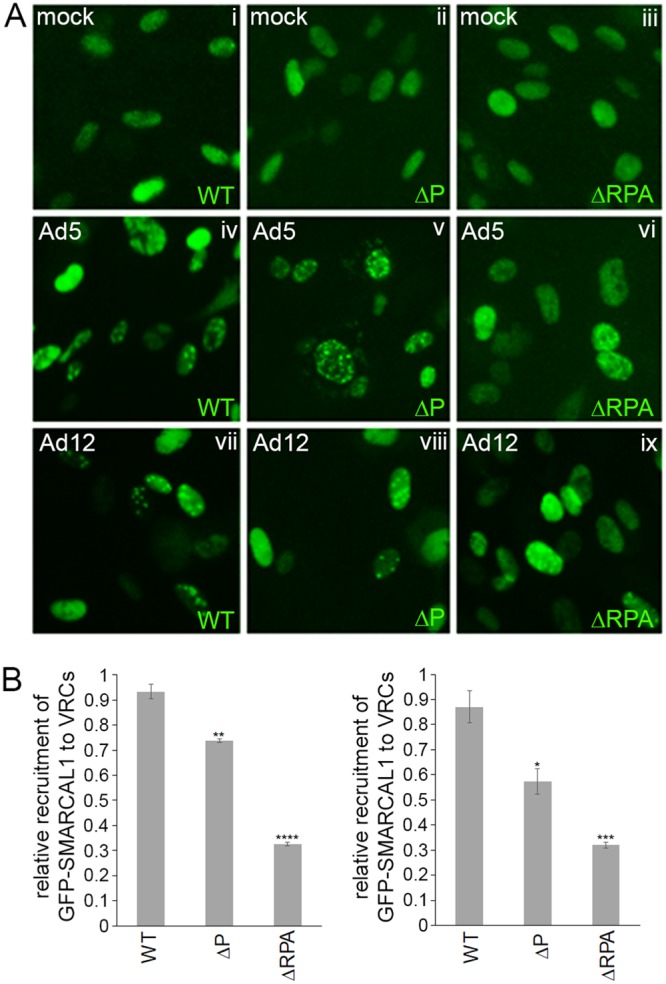
SMARCAL1 is recruited to VRCs in an RPA-dependent and ATR- and CDK-dependent manner. (A) Microscopic images depicting the cellular localization of wt GFP-SMARCAL1, GFP-SMARCAL1-ΔP, and GFP-SMARCAL1-ΔRPA in mock-infected (i to iii), wt Ad5-infected (iv to vi), or wt Ad12-infected cells (vii to ix) 18 h postinfection. (B) Bar graph (± SEM) showing the percentage of GFP-labeled cells that are recruited to VRCs following Ad5 or Ad12 infection. n = 3 (300 cells per experiment, 900 cells in total). Only those cells that exhibited clear GFP-SMARCAL1 structures in Ad-infected cells, comparable to the known architecture of VRCs at different stages of infection, were counted as VRC positive. Data presented were subjected to analysis of variance with a two-tailed t test. For significance testing for difference in recruitment of GFP-SMARCAL1-ΔP to VRCs relative to that of the wt GFP-SMARCAL1 following Ad5 infection, P = 0.0065 (**); for difference in recruitment of GFP-SMARCAL1-ΔRPA to VRCs relative to that of wt GFP-SMARCAL1 following Ad5 infection, P = 8.8E−05 (****); for difference in recruitment of GFP-SMARCAL1-ΔP to VRCs relative to that of wt GFP-SMARCAL1 following Ad12 infection, P = 0.04 (*); for difference in recruitment of GFP-SMARCAL1-ΔRPA to VRCs relative to that of wt GFP-SMARCAL1 following Ad5 infection, P = 0.002 (***).
Given that ATR and CDK inhibitors restricted the ability of both wt Ad5 and wt Ad12 to promote SMARCAL1 degradation during infection, we also wished to use this experimental system to explore the specific roles of S123, S129, and S173 phosphorylation in the Ad-mediated degradation of SMARCAL1. Unfortunately, Ad infection of RPE-1 cells that constitutively expressed GFP-SMARCAL1 species resulted in the enhanced expression of GFP-SMARCAL1 species, probably as a result of E1A transactivation of the cytomegalovirus (CMV) promoter driving the expression of GFP-SMARCAL1 species (data not shown). As such, we were not able to determine the individual contributions of specific SMARCAL1 phosphorylation sites in the Ad-induced degradation process.
Ad5 and Ad12 E1B-55K associate with SMARCAL1 in Ad-transformed cells.
As E1B-55K has previously been shown to function as a substrate adaptor in the recruitment of cellular proteins, such as p53 and MRE11, for CRL-dependent degradation during infection, we next investigated whether E1B-55K also served as an adaptor for SMARCAL1 and could be found associated with SMARCAL1 in Ad-transformed cells. To investigate whether Ad5 and Ad12 E1B-55K were found associated with SMARCAL1 in Ad-transformed cells, we performed reciprocal coimmunoprecipitation studies using Ad5 HEK293 cells and Ad12 HER2 cells. Consistent with the notion that E1B-55K and SMARCAL1 associate in vivo, anti-E1B-55K antibodies coimmunoprecipitated SMARCAL1 and anti-SMARCAL1 antibodies coimmunoprecipitated E1B-55K from both Ad5 HEK293 cells and Ad12 HER2 cells (Fig. 7A and B, respectively). Given that p53 is a known E1B-55K-interacting protein, we performed reciprocal p53 and E1B-55K coimmunoprecipitation studies to validate the approach taken (Fig. 7A and B, respectively).
FIG 7.
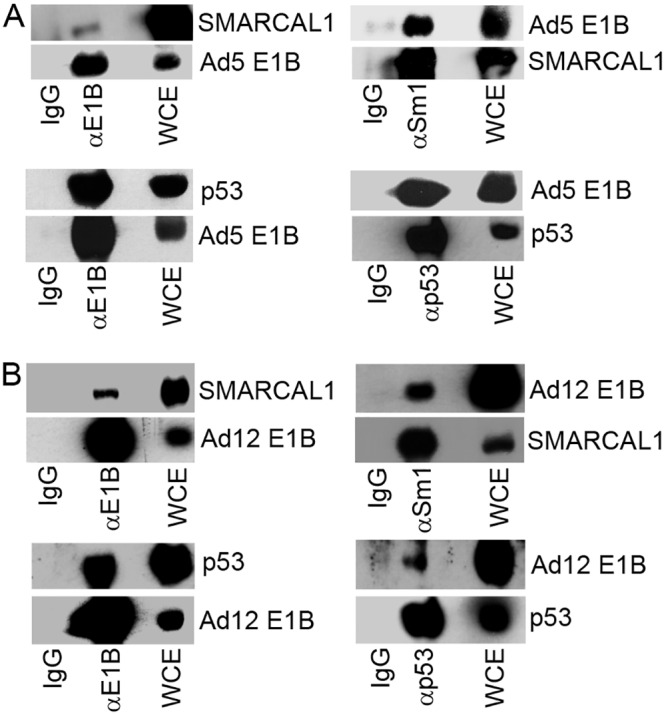
Ad E1B-55K associates with SMARCAL1 in Ad-transformed cells. (A) Ad E1B-55K and SMARCAL1 were immunoprecipitated from Ad5 HEK 293 cells (A) and Ad12 HER2 cells (B) and subjected to WB for E1B-55K and SMARCAL1. IgG, immunoglobulin control IP.
Generation of Ad5 and Ad12 E1B-55K FlpIn T-REX U2OS clonal cell lines.
As we have shown that Ad E1B-55K can associate with SMARCAL1 in Ad-transformed cells (Fig. 7), we wished to investigate the specific effects of E1B-55K expression in isolation upon SMARCAL1 function. To begin to do this, we first generated clonal TET-inducible Ad5 and Ad12 E1B-55K FlpIn U2OS cells that, upon induction with the tetracycline analog doxycycline, expressed Ad5 and Ad12 E1B-55K (Fig. 8). Consistent with the role for Ad E1B-55K in the stabilization of the p53 tumor suppressor, p53 protein levels were also increased following both Ad5 and Ad12 E1B-55K expression (Fig. 8). Unlike p53, the protein levels of SMARCAL1 and another E1B-55K binding partner, MRE11, were not altered appreciably following E1B-55K expression (Fig. 8). Taken together, these data demonstrate that we have generated TET-inducible Ad5 and Ad12 E1B-55K FlpIn U2OS cells that express functional E1B-55K following treatment with doxycycline.
FIG 8.
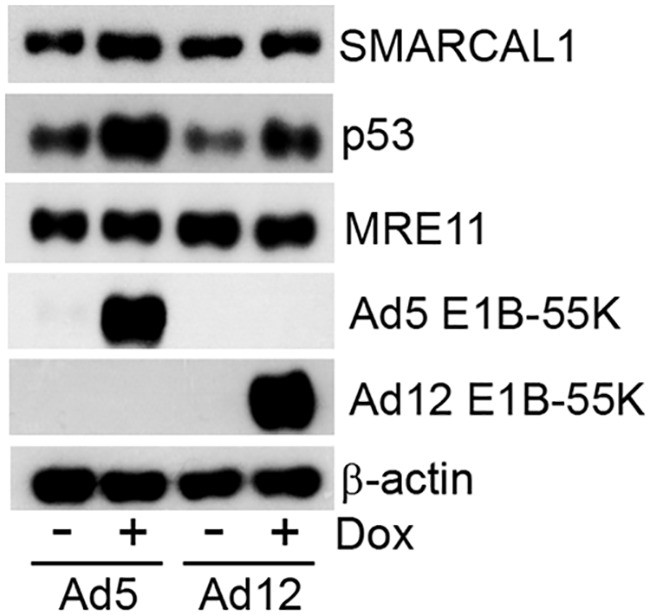
Generation and characterization of tetracycline-inducible Ad5 and Ad12 E1B-55K FlpIn U2OS cells. FlpIn U2OS cells were transfected with Ad5 E1B-55K and Ad12 E1B-55K pcDNA5/FRT/TO plasmids and the recombination plasmid pOG44. Cells were incubated in selection medium containing hygromycin (200 μg/ml). Individual colonies were isolated, expanded, and treated with 0.1 μg/ml doxycycline. Twenty-four h postinduction, cell lysates were harvested, separated by SDS-PAGE, and subjected to WB analysis for Ad5 and Ad12 E1B-55K. WB analyses were also performed to gauge the levels of SMARCAL1, p53, MRE11, and β-actin for Ad5 E1B-55K and Ad12 E1B-55K FlpIn U2OS cells. Data are representative of more than three independent experiments.
Ad5 and Ad12 E1B-55K dysregulate DNA fork speed during cellular DNA replication and promote replication fork collapse.
It is well established that in addition to its role as a substrate adaptor in the CRL-dependent degradation of p53 during Ad infection, E1B-55K also can, in isolation, inhibit the transactivation properties of p53 (31). As SMARCAL1 possesses the inherent ability to prevent replication fork collapse in unperturbed S phase and, in response to agents that promote replication stress, promote replication fork restart after fork collapse, we wished to establish whether Ad E1B-55K could also modulate the cellular functions of SMARCAL1. To measure the effects of Ad E1B-55K expression upon replication fork speed during unperturbed S phase, we utilized the DNA fiber assay. To do this, we pulse-labeled FlPIn U2OS cells (with or without Ad5 or Ad12 E1B-55K expression) successively with the thymidine analogs CldU and IdU for 20 min each to label DNA at replication forks. DNA fiber analyses revealed that in the presence of Ad5 E1B-55K or Ad12 E1B-55K, CldU-labeled tracks of newly synthesized DNA were significantly longer than those of mock controls, suggesting that both Ad5 and Ad12 E1B-55K expression led specifically to accelerated speeds of replication fork progression (Fig. 9A and B). Interestingly, however, this accelerated fork speed at ongoing DNA replication forks, in the presence of Ad E1B-55K, was not maintained when cells were subsequently labeled with IdU, such that IdU track length was comparable to that of cells that did not express Ad E1B-55K (Fig. 9A and B). As an increased CldU/IdU ratio can be indicative of fork stalling or collapse (32), we next quantified the effects of Ad E1B-55K expression on replication fork collapse. Consistent with the notion that the Ad E1B-55K-dependent acceleration in fork speed results in replication fork collapse, cells that expressed either Ad5 or Ad12 E1B-55K had a significantly increased number of stalled replication forks (CldU-only labeled DNA fibers) relative to that of cells that do not express Ad E1B-55K (Fig. 9C). Taken together, these data indicate that Ad E1B-55K can, in isolation, modulate cellular DNA replication. In consideration of the known functions of SMARCAL1, this finding is supportive of the notion that Ad E1B-55K interaction with SMARCAL1 contributes to dysregulated cellular DNA replication.
FIG 9.
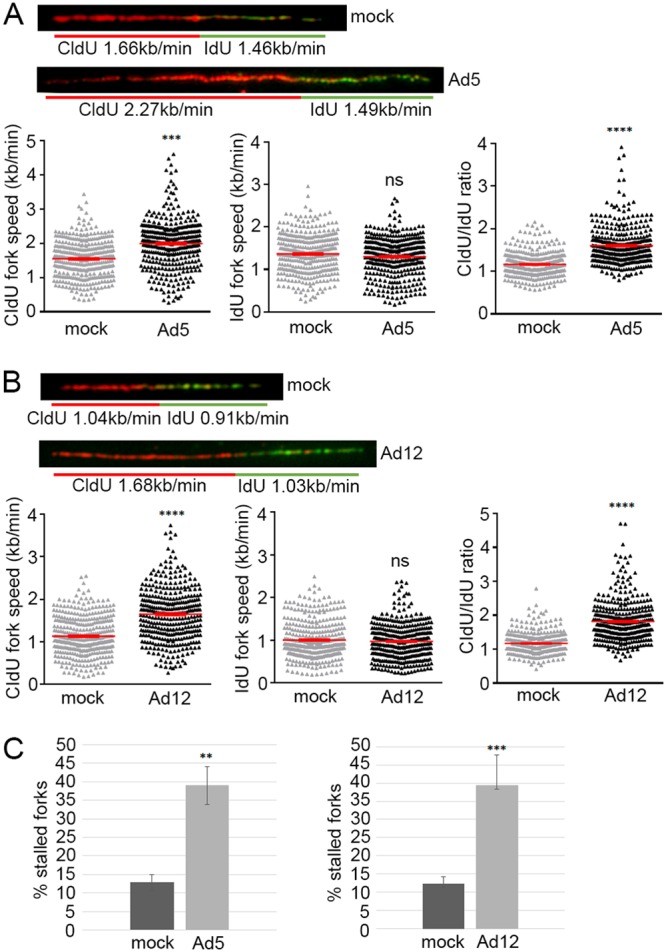
Ad5 and Ad12 E1B-55K modulate cellular DNA replication rates and promote replication fork stalling. Uninduced and doxycycline-induced Ad5 and Ad12 E1B-55K FlpIn U2OS cells were labeled with 25 μM CldU and 250 μM IdU for 20 min each. DNA fiber spreads were then prepared and denatured with 2.5 M HCl. DNA fibers were labeled with the appropriate primary and secondary antibodies and visualized using a Nikon E600 microscope. (A and B) Representative DNA spreads (with or without Ad5 or Ad12 E1B-55K) are shown indicating the mean fork speeds; CldU and IdU fork lengths were quantified and presented as dot plots (± standard deviations [SD]), with the mean fork speed shown as a red bar. n = 3 (total fibers analyzed: Ad5 mock infected, 347; Ad5 E1B-55K, 368; Ad12 mock infected, 370; Ad12 E1B-55K, 364). (C) Percent stalled forks (CldU-only labeled forks) were quantified and presented as a bar chart (±SD). In all instances data presented were subjected to analysis of variance with two-tailed t test: Ad5 E1B-55K CldU tract length relative to the mock CldU tract length, P = 4.8E−20 (***); Ad5 E1B-55K CldU/IdU ratio relative to the mock CldU tract length, P = 9.44E−45 (****); Ad12 E1B-55K CldU tract length relative to the mock CldU tract length, P = 1.29E−32 (****); Ad12 E1B-55K CldU/IdU ratio relative to the mock CldU tract length, P = 6.32E−61 (****); ns, not significant. For significance for stalled forks, Ad5 E1B-55K relative to mock infection, P = 0.009 (**); Ad12 E1B-55K relative mock infection, P = 0.002 (**).
DISCUSSION
It is now well established that Ad engages with cellular CRLs to stimulate the ubiquitin-mediated degradation of a small number of cellular DDR proteins in order to promote viral replication (1, 2). Typically, E4orf6 serves to recruit CRLs to protein substrates through direct interaction with CRL components elongin B and elongin C, while E1B-55K, through direct interaction with both E4orf6 and protein substrates, recruits cellular proteins to CRLs for polyubiquitylation and proteasome-mediated degradation (1, 2). Using well-established Ad5 and Ad12 mutant viruses, we show that Ad likely utilizes this canonical pathway to promote the degradation of the cellular replication protein SMARCAL1 during infection (Fig. 2 and 3). Indeed, treatment with the NAE inhibitor reduced the extent of degradation of SMARCAL1 during infection, suggesting that CRLs contribute to this degradation process.
It was evident during our studies that, prior to its degradation, a higher-molecular-weight form of SMARCAL1 was observed upon SDS-PAGE (Fig. 2). In this regard, we used mass spectrometry to establish that SMARCAL1 was phosphorylated on residues S123, S129, and S173 early during both Ad5 and Ad12 infection (Fig. 4). S123 and S129 form part of minimal CDK consensus SP motifs, and S173 forms part of a consensus ATM/ATR SQE motif. Although all of these residues have been shown previously to be phosphorylated in vivo, the biological significance of these phosphorylation events has yet to be determined (28). Given that S123 and S129 are likely to be phosphorylated by a CDK and S173 is likely phosphorylated by ATR, we investigated whether small-molecule inhibitors of ATR kinase and CDKs could affect the ability of Ad to promote SMARCAL1 degradation. Significantly, studies with the ATR inhibitor AZD6738 and CDK inhibitor RO-3306 determined that ATR and CDKs cooperate to promote the Ad-targeted degradation of SMARCAL1 during infection (Fig. 5), suggesting that S123, S129, and S173 all contribute to SMARCAL1 stability in vivo. Although RO-3306 has greater selectivity for CDK1 than CDK2 and CDK4 (33), Ad infection is known to stimulate the activity of all three kinases (34), such that we cannot, at present, state which CDK(s) is responsible for phosphorylating SMARCAL1 during Ad infection. We wished to investigate further the role of phosphorylation of these specific residues in the Ad-mediated degradation of SMARCAL1. To this end, we made GFP-SMARCAL1 RPE-1 cell lines where S123, S129, and S173 residues were all mutated to A to ablate phosphorylation at these sites. Although we were able to generate clonal cell lines that expressed these mutations, we were unable to undertake these studies, as Ad infection results in the transactivation of the CMV promoter that regulates GFP-SMARCAL1 expression (data not shown).
We were, however, able to use the wt GFP-SMARCAL1 and GFP-SMARCAL1 phosphomutant RPE-1 cell lines to address the role of SMARCAL1 phosphorylation in the recruitment of SMARCAL1 to VRCs. As such, we determined that ATR and CDKs, although not essential, contributed to some extent to the recruitment of SMARCAL1 to VRCs during infection (Fig. 6). Moreover, using a GFP-SMARCAL1 species lacking its N-terminal RPA interaction motif, we were also able to establish that SMARCAL1 association with RPA is a major determinant in SMARCAL1 recruitment to VRCs (Fig. 6). SMARCAL1 was initially characterized as an RPA-interacting protein, and its recruitment to replication forks and sites of DNA damage was shown to be dependent upon its interaction with RPA (21–25). More recent studies have determined that RPA, in addition to its ability to control SMARCAL1 localization, also confers substrate specificity and regulates SMARCAL1 fork-remodeling reactions through the orientation of its high-affinity DNA-binding domains (35). RPA is a single-stranded DNA binding protein complex that has long been known to promote large T-antigen-dependent simian virus 40 (SV40) DNA replication (36). Although RPA has been shown to be recruited to Ad VRCs during infection, its precise role in Ad replication is not known (19, 20). Given that SMARCAL1 is an RPA-binding protein and that most of its activities are controlled by RPA, it is interesting to speculate that any proviral RPA functions during Ad infection are not coordinated through the activation of SMARCAL1-dependent remodeling activities. Indeed, as SMARCAL1 is degraded during infection (Fig. 2), it is highly likely that SMARCAL1 possesses antiviral activities. As the mechanism of SV40 DNA replication is well established, it would be interesting to determine the requirement for SMARCAL1 in RPA-dependent SV40 DNA replication.
Given the role of SMARCAL1 in cellular DNA replication, we investigated the effects of Ad E1B-55K expression on cellular DNA replication. We observed that E1B-55K expression enhanced nascent cellular DNA replication fork speed, but ultimately, E1B-55K expression resulted in increased replication fork stalling (Fig. 9). It has been determined previously that loss of SMARCAL1 prevents replication restart after replication stress, resulting in stalled replication, while knockdown of p53 and MRE11 also promoted stalled cellular DNA replication (28, 37, 38). More generally, it has been determined that oncogene product expression can enhance replication stress to either increase or decrease DNA replication initiation, elongation, fork speed, fork stalling, and fork restart through the modulation of origin firing, replication-transcription collisions, reactive oxygen species, and defective nucleotide metabolism (39). Therefore, it is plausible that the E1B-55K oncoprotein promotes replication stress in Ad-infected cells through interaction with p53, MRE11, SMARCAL1, and potentially other cellular targets, ultimately resulting in cellular DNA replication inhibition. Given the known role of E1B-55K in the promotion of late viral mRNA accumulation and the inhibition of cellular mRNA transport and translation in the mediation of host protein shutoff, as well as the proposed role for Ad-mediated protein degradation in mRNA export (40, 41), we postulate that E1B-55K similarly inhibits cellular DNA replication and promotes viral replication through the specific targeting of cellular E1B-55K-interacting proteins for degradation during infection.
MATERIALS AND METHODS
Cells.
A549 human lung carcinoma cells, TERT-immortalized RPE-1 (retinal pigment epithelial) cells, FlpIn T-REX U2OS cells, and GP2-293 cells were grown in HEPES-modified Dulbecco’s modified Eagle’s medium (DMEM; Sigma-Aldrich) supplemented with 8% (vol/vol) fetal calf serum (FCS; Sigma-Aldrich) and 2 mM l-glutamine (Sigma-Aldrich). Ad5 and Ad12 E1B-55K FlpIn T-Rex U2OS cells were maintained in HEPES-modified DMEM in the presence of 200 μg/ml hygromycin (Life Technologies), while clonal RPE-1 cells that express wild-type (wt) GFP-SMARCAL1 or GFP-SMARCAL1 mutants were also maintained in HEPES-modified DMEM in the presence of 500 μg/ml G418 (Gibco). All cells were maintained at 37°C in a humidified 5% CO2 atmosphere (Nuaire Autoflow).
Viruses.
wt Ad5 and wt Ad12 Huie viruses were from the ATCC. Ad5 dl1520, Ad5 pm4150, Ad5 pm4154, Ad5 pm4155, and Ad12 dl620 viruses have all been described previously (15). Ad5 and Ad12 viruses were propagated on permissive human embryonic kidney 293 (HEK293) cells and human embryonic retinoblastoma 3 (HER3) cells, respectively, and titers were determined by plaque assay on HER911 and HER3 cells. Viruses were diluted in DMEM without FCS, and cells were typically infected at a multiplicity of infection (MOI) of 10. Infected cells were incubated at 37°C with agitation every 10 min. After 2 h of infection, virus-containing medium was removed and replaced with fresh culture medium supplemented with 8% (vol/vol) FCS.
Plasmids.
wt SMARCAL1 and ΔN-SMARCAL1 (lacking the N-terminal RPA interaction domain; ΔRPA) constructs cloned into the retroviral vector pLEGFP-C1 (Clontech) were provided by David Cortez. pLEGFP-C1 S123A, S129A, and S173A SMARCAL1 phosphomutants were generated using the QuikChange II XL site-directed mutagenesis kit (Agilent) and validated by Sanger sequencing. Using wt Ad5 E1B-55K and Ad12 E1B-55K cDNA templates, both Ad5 and Ad12 E1B-55K were amplified by PCR, digested with BamHI and XhoI, and subcloned into the pcDNA5/FRT/TO plasmid for the generation of tetracycline-inducible cell lines. Ad5 E1B-55K was amplified using the primers Ad5 E1B55K BamHI forward, AGGTTGGATCCATGGAGCGAAGAAACCCATCTGAG, and Ad5 E1B55K XhoI reverse, AGGTTCTCGAGTCAATCTGTATCTTCATCGCTAGA. Ad12 E1B-55K was amplified using the primers Ad12 E1B55K BamHI forward, TTGCAGGATCCATGGAGCGAGAAATCCCACCTGAG, and Ad12 E1B55K XhoI reverse, TTGCACTCGAGTCAGTTGTCGTCTTCATCACTTGA. Clones were validated by Sanger sequencing using the primers pcDNA5 forward, CGCAAATGGGCGGTAGGCGTG, pcDNA5 reverse, TAGAAGGCACAGTCGAGG, Ad5 E1B-55K seq1, GGCTACAGAGGAGGCTAGGAATCTA, Ad5 E1B-55K seq2, CCTGGCCAATACCAACCTTATCCT, Ad5 E1B-55K seq3, TGCTGACCTGCTCGGACGGCAACT, Ad12 E1B-55K seq1, AACTGTATATTGGCAGGAGTTGCAG, Ad12 E1B-55K seq2, AATACCTGTCTTGTCTTGCATGGT, and Ad12 E1B-55K seq3, ATAACATGTTTATGCGCTGTACCAT.
Generation of clonal cell lines.
FlpIn T-REX U2OS cells were grown to 90% confluence prior to transfection. The Ad5 E1B-55K and Ad12 E1B-55K pcDNA5/FRT/TO plasmids were mixed with the recombination plasmid pOG44 at a 1:9 ratio in Opti-MEM (Life Technologies) and transfected according to the manufacturer’s instructions into FlpIn T-REX U2OS cells with the use of Lipofectamine 2000 (Life Technologies). Cells were then incubated in a CO2-humidified incubator at 37°C for 6 h. Following transfection, cells were incubated in fresh HEPES-modified DMEM supplemented with 8% (vol/vol) FCS and 2 mM glutamine. Twenty-four h posttransfection, cells from one plate were passaged onto four plates, and 48 h posttransfection cells were incubated with growth medium containing 200 μg/ml hygromycin (Life Technologies) for clonal selection. Cells were then fed every 3 days; individual colonies were ultimately selected, expanded, and assessed for Ad E1B-55K expression following incubation with 0.1 μg/ml doxycycline for 24 h. To generate GFP-SMARCAL1 cell lines, pLEGFP-C1 SMARCAL1 constructs were transfected in a 1:1 ratio with the pVSV envelope plasmid in the GP2-293 retrovirus packaging cell line (Clontech) using Lipofectamine 2000. Seventy-two h posttransfection, the virus-containing supernatants were collected and filtered through a 0.45-μm filter (Sartorius). Retroviral transduction of RPE-1 cells, at 20% density, was then performed. Seventy-two h postransduction, clonal cells were selected using G418 (500 μg/ml). Individual colonies were ultimately expanded and assessed for GFP-SMARCAL1 expression.
Antibodies and inhibitors.
The anti-Ad5 E1B-55K monoclonal antibody (MAb) 2A6, anti-Ad12 E1B-55K MAb XPH9, and the anti-p53 MAb DO-1 were all obtained as supernatant fluid from cultures of the appropriate hybridoma cell lines. The anti-SMARCAL1 (A-2) MAb was from Santa Cruz (sc-376377). Horseradish peroxidase (HRP)-conjugated secondary anti-mouse and anti-rabbit antibodies used for Western blotting were from Agilent. Secondary anti-mouse and anti-rabbit Alexa 488/594 antibodies used for immunofluorescence were from Thermo Fisher. The ATR inhibitor AZD6738 and the CRL inhibitor MLN4924 were purchased from Cayman chemicals, while the CDK inhibitor RO-3306 was purchased from Merck Millipore.
IP.
Cells were harvested by washing twice in ice-cold phosphate-buffered saline and solubilized in immunoprecipitation (IP) buffer containing 20 mM Tris-HCl (pH 8.0), 150 mM NaCl, 1 mM ethylenediaminetetraacetic acid (EDTA), 1% (vol/vol) Nonidet P-40, 25 mM NaF, and 25 mM β-glycerophosphate. Cell lysates were then homogenized twice with 10 strokes while being kept on ice and centrifuged at 40,000 rpm for 30 min at 4°C. Immunoprecipitating antibodies were added to clarified supernatants at 4°C overnight with rotation. After this time, protein G-Sepharose beads (Sigma-Aldrich) were added to all samples to capture and isolate immune complexes for 2 h at 4°C with rotation. The beads were then washed five times by centrifugation at 3,000 rpm in ice-cold IP buffer, eluted in 30 μl of SDS-containing sample buffer, and run on SDS-PAGE gels for Western blotting.
SDS-PAGE and Western blot analysis.
Whole-cell protein lysates were prepared in 9 M urea, 150 mM β-mercaptoethanol, 50 mM Tris-HCl (pH 7.4). Lysates were clarified by sonication and centrifugation, and protein concentrations were determined by Bradford assay (Bio-Rad). Proteins were separated by SDS-PAGE in the presence of 100 mM Tris, 100 mM bicine, and 0.1% (wt/vol) SDS. Following SDS-PAGE, proteins where electrophoretically transferred onto nitrocellulose membranes (PALL) in transfer buffer (50 mM Tris, 190 mM glycine, 20% [vol/vol] methanol). Membranes were then blocked in 5% (wt/vol) dried milk powder in TBST (Tris-buffered saline containing 0.1% [vol/vol] Tween 80) for 1 h at room temperature with agitation. Membranes were incubated overnight with antibodies at the appropriate dilution in TBST containing 5% (vol/vol) milk at 4°C with agitation. The following day, membranes were washed four times in TBST and incubated with the appropriate HRP-conjugated secondary antibody made up in TBST containing 5% (vol/vol) milk at room temperature for 2 h with agitation. Finally, membranes were washed four times in TBST and antigens were detected using enhanced chemiluminescence (ECL) reagents (Millipore) and autoradiography film (SLS).
Microscopy.
GFP-SMARCAL1 cells were visualized using an EVOS fluorescent digital inverted microscope. Cells for confocal microscopy were seeded on glass 12-well multispot microscope slides (Hendley-Essex). Following mock or Ad infection, slides were fixed in 4% (wt/vol) paraformaldehyde in phosphate-buffered saline (PBS) and then permeabilized in ice-cold acetone. Slides were then air dried and blocked in HINGS buffer (20% [vol/vol] heat-inactivated normal goat serum, 0.2% [wt/vol] bovine serum albumin [BSA] in PBS) prior to incubation with the appropriate primary and Alexa Fluor secondary antibodies (Life Technologies) in HINGS buffer. Slides were then mounted in Vectashield (Vector Laboratories) containing 4’,6-diamidino-2-phenylindole (DAPI) and visualized using an LSM 510 META confocal laser scanning microscope (Carl Zeiss).
Mass spectrometry.
Anti-SMARCAL1 immunoprecipitates were isolated on protein G Sepharose beads and separated upon precast Novex NuPage 4 to 12% Bis-Tris gels (Life Technologies). Protein bands were stained with colloidal Coomassie brilliant blue (Fisher). After washing gels in distilled water, protein bands were excised and washed twice, by agitation, with a solution containing 50 mM ammonium bicarbonate and 50% (vol/vol) acetonitrile for 45 min at 37°C. The excised proteins were then reduced by incubation for 1 h at 56°C in a solution containing 50 mM dithiothreitol and 50 mM ammonium bicarbonate in 10% (vol/vol) acetonitrile. Proteins were then incubated in an alkylating solution (200 mM iodoacetamide, 50 mM ammonium bicarbonate, and 10% [vol/vol] acetonitrile) for 30 min at room temperature in the dark. The protein bands were then washed three times for 15 min each at room temperature in 10% (vol/vol) acetonitrile–40 mM ammonium bicarbonate on a shaker and then dried in a DNA minivacuum centrifuge for 3 to 4 h. The dried samples were then resuspended and digested by rehydration in sequencing-grade modified trypsin (Promega). An equal volume of 10% (vol/vol) acetonitrile–40 mM ammonium bicarbonate was then added to the protein bands and left to incubate with agitation overnight at 37°C. The resultant peptides were then analyzed using a Q Exactive HF hybrid quadrupole-Orbitrap mass spectrometer (ThermoFisher Scientific).
DNA fiber analysis.
Cells were labeled with 25 μM CldU (Sigma-Aldrich) and 250 μM IdU (Sigma-Aldrich) for 20 min each, and DNA fiber spreads were prepared in 200 mM Tris-HCl, pH 7.4, 50 mM EDTA, 0.5% (wt/vol) SDS and fixed with a 3:1 mixture of methanol-acetic acid. DNA fiber spreads were then denatured with 2.5 M HCl for 80 min and incubated with blocking buffer (PBS plus 1% [wt/vol] BSA plus 0.1% [vol/vol] Tween 20) for 1 h prior to incubation with rat anti-bromodeoxyuridine (BU1/75; ab6326; 1:250; Abcam) and mouse anti-bromodeoxyuridine (B44; 347580, 1:500; Becton Dickinson) in blocking buffer for 1 h. Fibers were then fixed with 4% (wt/vol) paraformaldehyde and incubated further with anti-rat Alexa Fluor 555 and anti-mouse Alexa Fluor 488 for 1.5 h prior to mounting and analysis on a Nikon E600 microscope with a Nikon Plan Apo 60× (1.3-numeric-aperture) oil lens, a Hamamatsu digital camera (C4742-95), and the Volocity acquisition software (Perkin Elmer). Images were analyzed using ImageJ.
ACKNOWLEDGMENTS
We thank David Cortez (Vanderbilt University, Nashville, TN) for SMARCAL1 reagents and Thomas Dobner (Heinrich Pette Institute, Hamburg, Germany) for adenovirus 5 mutants.
We also thank the BBSRC MIBTP program for funding for M.T.T. and S.D. (BB/M01116X/1; BB/J014532/1) and Jazan and Taibah Universities, Saudi Arabia, for funding F.S.I.Q. and A.S.A., respectively.
REFERENCES
- 1.Turnell AS, Grand RJ. 2012. Viral regulation of DNA damage response pathways. J Gen Virol 93:2076–2097. doi: 10.1099/vir.0.044412-0. [DOI] [PubMed] [Google Scholar]
- 2.Weitzman MD, Fradet-Turcotte A. 2018. Virus DNA replication and the host DNA damage response. Annu Rev Virol 5:141–164. doi: 10.1146/annurev-virology-092917-043534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dybas JM, Herrmann C, Weitzman MD. 2018. Ubiquitination at the interface of tumor viruses and DNA damage responses. Curr Opin Virol 32:40–47. doi: 10.1016/j.coviro.2018.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Querido E, Blanchette P, Yan Q, Kamura T, Morrison M, Boivin D, Kaelin WG, Conaway RC, Conaway JW, Branton PE. 2001. Degradation of p53 by adenovirus E4orf6 and E1B55K proteins occurs via a novel mechanism involving a cullin-containing complex. Genes Dev 15:3104–3117. doi: 10.1101/gad.926401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Harada JN, Shevchenko A, Shevchenko A, Pallas DC, Berk AJ. 2002. Analysis of the adenovirus E1B-55K-anchored proteome reveals its link to ubiquitination machinery. J Virol 6:9194–9206. doi: 10.1128/JVI.76.18.9194-9206.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Blanchette P, Cheng CY, Yan Q, Ketner G, Ornelles DA, Dobner T, Conaway RC, Conaway JW, Branton PE. 2004. Both BC-box motifs of adenovirus protein E4orf6 are required to efficiently assemble an E3 ligase complex that degrades p53. Mol Cell Biol 24:9619–9629. doi: 10.1128/MCB.24.21.9619-9629.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Blackford AN, Patel RN, Forrester NA, Theil K, Groitl P, Stewart GS, Taylor AMR, Morgan IM, Dobner T, Grand RJA, Turnell AS. 2010. Adenovirus 12 E4orf6 inhibits ATR activation by promoting TOPBP1 degradation. Proc Natl Acad Sci U S A 107:12251–12256. doi: 10.1073/pnas.0914605107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cheng CY, Gilson T, Wimmer P, Schreiner S, Ketner G, Dobner T, Branton PE, Blanchette P. 2013. Role of E1B55K in E4orf6/E1B55K E3 ligase complexes formed by different human adenovirus serotypes. J Virol 87:6232–6245. doi: 10.1128/JVI.00384-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stracker TH, Carson CT, Weitzman MD. 2002. Adenovirus oncoproteins inactivate the MRE11-Rad50-NBS1 DNA repair complex. Nature 418:348–352. doi: 10.1038/nature00863. [DOI] [PubMed] [Google Scholar]
- 10.Orazio NI, Naeger CM, Karlseder J, Weitzman MD. 2011. The adenovirus E1b55K/E4orf6 complex induces degradation of the Bloom helicase during infection. J Virol 85:1887–1892. doi: 10.1128/JVI.02134-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Baker A, Rohleder KJ, Hanakahi LA, Ketner G. 2007. Adenovirus E4 34k and E1b 55k oncoproteins target host DNA ligase IV for proteasomal degradation. J Virol 81:7034–7040. doi: 10.1128/JVI.00029-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dallaire F, Blanchette P, Groitl P, Dobner T, Branton PE. 2009. Identification of integrin alpha3 as a new substrate of the adenovirus E4orf6/E1B 55-kilodalton E3 ubiquitin ligase complex. J Virol 83:5329–5338. doi: 10.1128/JVI.00089-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fu YR, Turnell AS, Davis S, Heesom KJ, Evans VC, Matthews DA. 2017. Comparison of protein expression during wild-type, and E1B-55k-deletion, adenovirus infection using quantitative time-course proteomics. J Gen Virol 98:1377–1388. doi: 10.1099/jgv.0.000781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schreiner S, Wimmer P, Groitl P, Chen SY, Blanchette P, Branton PE, Dobner T. 2011. Adenovirus type 5 early region 1B 55K oncoprotein-dependent degradation of cellular factor Daxx is required for efficient transformation of primary rodent cells. J Virol 85:8752–8765. doi: 10.1128/JVI.00440-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Forrester NA, Patel RN, Speiseder T, Groitl P, Sedgwick GG, Shimwell NJ, Seed RI, Catnaigh PÓ, McCabe CJ, Stewart GS, Dobner T, Grand RJ, Martin A, Turnell AS. 2012. Adenovirus E4orf3 targets transcriptional intermediary factor 1γ for proteasome-dependent degradation during infection. J Virol 86:3167–3179. doi: 10.1128/JVI.06583-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sohn SY, Hearing P. 2016. The adenovirus E4-ORF3 protein functions as a SUMO E3 ligase for TIF-1γ sumoylation and poly-SUMO chain elongation. Proc Natl Acad Sci U S A 113:6725–6730. doi: 10.1073/pnas.1603872113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bridges RG, Sohn SY, Wright J, Leppard KN, Hearing P. 2016. The adenovirus E4-ORF3 protein stimulates SUMOylation of general transcription factor TFII-I to direct proteasomal degradation. mBio 7:e02184. doi: 10.1128/mBio.02184-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Blackford AN, Jackson SP. 2017. ATM, ATR, and DNA-PK: the trinity at the heart of the DNA damage response. Mol Cell 66:801–817. doi: 10.1016/j.molcel.2017.05.015. [DOI] [PubMed] [Google Scholar]
- 19.Carson CT, Orazio NI, Lee DV, Suh J, Bekker-Jensen S, Araujo FD, Lakdawala SS, Lilley CE, Bartek J, Lukas J, Weitzman MD. 2009. Mislocalization of the MRN complex prevents ATR signaling during adenovirus infection. EMBO J 28:652–662. doi: 10.1038/emboj.2009.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Blackford AN, Bruton RK, Dirlik O, Stewart GS, Taylor AM, Dobner T, Grand RJ, Turnell AS. 2008. A role for E1B-AP5 in ATR signaling pathways during adenovirus infection. J Virol 82:7640–7652. doi: 10.1128/JVI.00170-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bansbach CE, Bétous R, Lovejoy CA, Glick GG, Cortez D. 2009. The annealing helicase SMARCAL1 maintains genome integrity at stalled replication forks. Genes Dev 23:2405–2414. doi: 10.1101/gad.1839909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ciccia A, Bredemeyer AL, Sowa ME, Terret ME, Jallepalli PV, Harper JW, Elledge SJ. 2009. The SIOD disorder protein SMARCAL1 is an RPA-interacting protein involved in replication fork restart. Genes Dev 23:2415–2425. doi: 10.1101/gad.1832309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Postow L, Woo EM, Chait BT, Funabiki H. 2009. Identification of SMARCAL1 as a component of the DNA damage response. J Biol Chem 284:35951–35961. doi: 10.1074/jbc.M109.048330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yusufzai T, Kong X, Yokomori K, Kadonaga JT. 2009. The annealing helicase HARP is recruited to DNA repair sites via an interaction with RPA. Genes Dev 23:2400–2404. doi: 10.1101/gad.1831509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yuan J, Ghosal G, Chen J. 2009. The annealing helicase HARP protects stalled replication forks. Genes Dev 23:2394–2399. doi: 10.1101/gad.1836409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Boerkoel CF, Takashima H, John J, Yan J, Stankiewicz P, Rosenbarker L, André JL, Bogdanovic R, Burguet A, Cockfield S, Cordeiro I, Fründ S, Illies F, Joseph M, Kaitila I, Lama G, Loirat C, McLeod DR, Milford DV, Petty EM, Rodrigo F, Saraiva JM, Schmidt B, Smith GC, Spranger J, Stein A, Thiele H, Tizard J, Weksberg R, Lupski JR, Stockton DW. 2002. Mutant chromatin remodeling protein SMARCAL1 causes Schimke immuno-osseous dysplasia. Nat Genet 30:215–220. doi: 10.1038/ng821. [DOI] [PubMed] [Google Scholar]
- 27.Carroll C, Bansbach CE, Zhao R, Jung SY, Qin J, Cortez D. 2014. Phosphorylation of a C-terminal auto-inhibitory domain increases SMARCAL1 activity. Nucleic Acids Res 42:918–925. doi: 10.1093/nar/gkt929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Couch FB, Bansbach CE, Driscoll R, Luzwick JW, Glick GG, Bétous R, Carroll CM, Jung SY, Qin J, Cimprich KA, Cortez D. 2013. ATR phosphorylates SMARCAL1 to prevent replication fork collapse. Genes Dev 27:1610–1623. doi: 10.1101/gad.214080.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Soucy TA, Smith PG, Milhollen MA, Berger AJ, Gavin JM, Adhikari S, Brownell JE, Burke KE, Cardin DP, Critchley S, Cullis CA, Doucette A, Garnsey JJ, Gaulin JL, Gershman RE, Lublinsky AR, McDonald A, Mizutani H, Narayanan U, Olhava EJ, Peluso S, Rezaei M, Sintchak MD, Talreja T, Thomas MP, Traore T, Vyskocil S, Weatherhead GS, Yu J, Zhang J, Dick LR, Claiborne CF, Rolfe M, Bolen JB, Langston SP. 2009. An inhibitor of NEDD8-activating enzyme as a new approach to treat cancer. Nature 458:732–736. doi: 10.1038/nature07884. [DOI] [PubMed] [Google Scholar]
- 30.Bailly A, Perrin A, Bou Malhab LJ, Pion E, Larance M, Nagala M, Smith P, O'Donohue MF, Gleizes PE, Zomerdijk J, Lamond AI, Xirodimas DP. 2016. The NEDD8 inhibitor MLN4924 increases the size of the nucleolus and activates p53 through the ribosomal-Mdm2 pathway. Oncogene 35:415–426. doi: 10.1038/onc.2015.104. [DOI] [PubMed] [Google Scholar]
- 31.Yew PR, Berk AJ. 1992. Inhibition of p53 transactivation required for transformation by adenovirus early 1B protein. Nature 357:82–85. doi: 10.1038/357082a0. [DOI] [PubMed] [Google Scholar]
- 32.Petermann E, Maya-Mendoza A, Zachos G, Gillespie DAF, Jackson DA, Caldecott KW. 2006. Chk1 requirement for high global rates of replication fork progression during normal vertebrate S phase. Mol Cell Biol 26:3319–3326. doi: 10.1128/MCB.26.8.3319-3326.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Vassilev LT, Tovar C, Chen S, Knezevic D, Zhao X, Sun H, Heimbrook DC, Chen L. 2006. Selective small-molecule inhibitor reveals critical mitotic functions of human CDK1. Proc Natl Acad Sci U S A 103:10660–10665. doi: 10.1073/pnas.0600447103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Grand RJ, Ibrahim AP, Taylor AMR, Milner AE, Gregory CD, Gallimore PH, Turnell AS. 1998. Human cells arrest in S phase in response to adenovirus 12 E1A. Virology 244:330–342. doi: 10.1006/viro.1998.9102. [DOI] [PubMed] [Google Scholar]
- 35.Bhat KP, Bétous R, Cortez D. 2015. High-affinity DNA-binding domains of replication protein A (RPA) direct SMARCAL1-dependent replication fork remodeling. J Biol Chem 290:4110–4117. doi: 10.1074/jbc.M114.627083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wold MS, Kelly T. 1988. Purification and characterization of replication protein A, a cellular protein required for in vitro replication of simian virus 40 DNA. Proc Natl Acad Sci U S A 85:2523–2527. doi: 10.1073/pnas.85.8.2523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Klusmann I, Rodewald S, Müller L, Friedrich M, Wienken M, Li Y, Schulz-Heddergott R, Dobbelstein M. 2016. p53 activity results in DNA replication fork processivity. Cell Rep 17:1845–1857. doi: 10.1016/j.celrep.2016.10.036. [DOI] [PubMed] [Google Scholar]
- 38.Bryant HE, Petermann E, Schultz N, Jemth AS, Loseva O, Issaeva N, Johansson F, Fernandez S, McGlynn P, Helleday T. 2009. PARP is activated at stalled forks to mediate MRE11-dependent replication restart and recombination. EMBO J 28:2601–2615. doi: 10.1038/emboj.2009.206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kotsantis P, Petermann E, Boulton SJ. 2018. Mechanisms of oncogene-induced replication stress: jigsaw falling into place. Cancer Discov 8:537–555. doi: 10.1158/2159-8290.CD-17-1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Babiss LE, Ginsberg HS, Darnell JE. 1985. Adenovirus E1B proteins are required for accumulation of late viral mRNA and for effects on cellular mRNA translation and transport. Mol Cell Biol 5:2552–2558. doi: 10.1128/MCB.5.10.2552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Blanchette P, Kindsmuller K, Groitl P, Dallaire F, Speiseder T, Branton PE, Dobner T. 2008. Control of mRNA export by adenovirus E4orf6 and E1B55K proteins during productive infection requires E4orf6 ubiquitin ligase activity. J Virol 82:2642–2651. doi: 10.1128/JVI.02309-07. [DOI] [PMC free article] [PubMed] [Google Scholar]