

Herpes simplex virus 1 (HSV-1) establishes lifelong latent infections in neurons within trigeminal ganglia (TG); periodically, reactivation from latency occurs, leading to virus transmission and recurrent disease. Chronic or acute stress increases the frequency of reactivation from latency; how this occurs is not well understood. Here, we demonstrate that the synthetic corticosteroid dexamethasone stimulated explant-induced reactivation from latency. Conversely, a glucocorticoid receptor (GR) antagonist significantly impaired reactivation from latency, indicating that GR activation stimulated explant-induced reactivation. The viral regulatory protein VP16 was readily detected in TG neurons prior to infected-cell protein 0 (ICP0) and ICP4 during explant-induced reactivation. Dexamethasone induced expression of all three viral regulatory proteins following TG explant. Whereas the immunosuppressive properties of corticosteroids would facilitate viral spread during reactivation from latency, these studies indicate GR activation increases the number of TG neurons that express viral regulatory proteins during early stages of explant-induced reactivation.
KEYWORDS: HSV-1, reactivate, stress response
ABSTRACT
Herpes simplex virus 1 (HSV-1) establishes lifelong latent infections in neurons. Reactivation from latency can lead to serious recurrent disease, including stromal keratitis, corneal scarring, blindness, and encephalitis. Although numerous studies link stress to an increase in the incidence of reactivation from latency and recurrent disease, the mechanism of action is not well understood. We hypothesized that stress, via corticosteroid-mediated activation of the glucocorticoid receptor (GR), stimulates viral gene expression and productive infection during reactivation from latency. Consequently, we tested whether GR activation by the synthetic corticosteroid dexamethasone influenced virus shedding during reactivation from latency using trigeminal ganglion (TG) explants from Swiss Webster mice latently infected with HSV-1, strain McKrae. TG explants from the latently infected mice shed significantly higher levels of virus when treated with dexamethasone. Conversely, virus shedding from TG explants was significantly impaired when they were incubated with medium containing a GR-specific antagonist (CORT-108297) or stripped fetal bovine serum, which lacks nuclear hormones and other growth factors. TG explants from latently infected, but not uninfected, TG contained significantly more GR-positive neurons following explant when treated with dexamethasone. Strikingly, VP16 protein expression was detected in TG neurons at 8 hours after explant whereas infected-cell protein 0 (ICP0) and ICP4 protein expression was not readily detected until 16 hours after explant. Expression of all three viral regulatory proteins was stimulated by dexamethasone. These studies indicated corticosteroid-mediated GR activation increased the number of TG neurons expressing viral regulatory proteins, which enhanced virus shedding during explant-induced reactivation from latency.
IMPORTANCE Herpes simplex virus 1 (HSV-1) establishes lifelong latent infections in neurons within trigeminal ganglia (TG); periodically, reactivation from latency occurs, leading to virus transmission and recurrent disease. Chronic or acute stress increases the frequency of reactivation from latency; how this occurs is not well understood. Here, we demonstrate that the synthetic corticosteroid dexamethasone stimulated explant-induced reactivation from latency. Conversely, a glucocorticoid receptor (GR) antagonist significantly impaired reactivation from latency, indicating that GR activation stimulated explant-induced reactivation. The viral regulatory protein VP16 was readily detected in TG neurons prior to infected-cell protein 0 (ICP0) and ICP4 during explant-induced reactivation. Dexamethasone induced expression of all three viral regulatory proteins following TG explant. Whereas the immunosuppressive properties of corticosteroids would facilitate viral spread during reactivation from latency, these studies indicate GR activation increases the number of TG neurons that express viral regulatory proteins during early stages of explant-induced reactivation.
INTRODUCTION
Infection of craniofacial mucosal membranes with herpes simplex virus 1 (HSV-1) leads to lifelong latent infections within sensory neurons of trigeminal ganglia (TG) (1), as well as neurons within the central nervous system (2, 3). In contrast to productive infection of cultured cells, abundant viral protein expression and infectious virus are not readily detected when latency is established. The only abundantly expressed viral transcript detected during latency is the latency-associated transcript (LAT), which expresses several transcripts, six microRNAs, and two small noncoding regions (4–6). LAT protects neurons from cell death, partly by inhibiting apoptosis and viral gene expression (7–12). Hence, LAT enhances reactivation in small-animal models of infection (1, 13–17).
Increased stress correlates with an increased frequency of HSV-1 reactivation from latency in humans (18–20). Furthermore, the synthetic corticosteroid dexamethasone (DEX) increases the incidence of reactivation from latency in TG neuronal cultures prepared from latently infected mice (21) and latently HSV-1-infected TG organ cultures (22). Corticosteroids that mimic the effects of stress also consistently stimulate reactivation from latency in canine herpesvirus 1 (23) and bovine herpesvirus 1 (BoHV-1) (24), indicating stress has effects on other Alphaherpesvirinae subfamily members.
Stressful stimuli generally increase levels of corticosteroids, which enter a cell and bind to the glucocorticoid receptor (GR) or mineralocorticoid receptor (MR), and then the hormone–nuclear-receptor complex enters the nucleus (25). A GR or MR homodimer specifically binds consensus glucocorticoid response elements (GREs) and stimulates transcription in the absence of de novo protein synthesis (26, 27). The consensus GRE is gGa/tACANNNTGTc/tCT: a capital letter denotes a conserved nucleotide, a lowercase letter denotes a nucleotide not conserved, a/t or c/t indicates that either of those nucleotides can be found in a GRE, and N means that any nucleotide can be in that position. A GR monomer can also bind certain ½ GR binding sites and activate transcription (28, 29). Two ½ GREs have been identified to date: GGACAN and GG/ANAC/gAT/G, where a capital letter denotes a well-conserved nucleotide, a lowercase letter denotes a not-well-conserved nucleotide, an N denotes a not-conserved nucleotide, and G/A or T/G means that either nucleotide can be present. In addition to stimulating gene expression, corticosteroids have potent anti-inflammatory and immunosuppressive effects (25, 30, 31), which would likely enhance virus shedding and spread during reactivation from latency.
Several published studies suggest that the viral protein infected-cell protein 0 (ICP0), independently of other viral gene products, promotes reactivation from latency (32–35) and enhances the ability of transfected viral DNA to initiate productive infection in cell culture (36). It has also been proposed that a viral tegument protein, VP16, is crucial for inducing viral gene expression in quiescently HSV-1-infected embryonic-rat neuronal cultures (37, 38), as well as initiating HSV-1 reactivation in vivo from latently infected mice exposed to hyperthermic stress (38, 39). Given that VP16 selectively activates immediate-early (IE) gene expression (40–42), its expression during early stages of reactivation from latency would enhance the expression of lytic cycle viral genes. Regardless of whether IE genes or VP16 is initially induced, it is likely that cellular transcription factors are crucial for initiating viral transcription during reactivation from latency.
In this study, we provide evidence that HSV-1 explant-induced reactivation from latency results from cooperation between viral protein expression and host GR. Virus shedding from latently infected TG explants was significantly impaired when they were incubated with charcoal-stripped fetal bovine serum (FBS), which has most nuclear hormones and additional growth factors removed; however, addition of DEX to stripped-serum explants restored efficient reactivation from latency. When explants were incubated with the GR-specific antagonist CORT-108297, explant-induced reactivation from latency was significantly impaired. Moreover, DEX-treated TG explants showed higher levels of GR expression, as well as detectable expression of the viral proteins ICP0, ICP4, and VP16, than latent TG neurons. Conversely, a LAT-null mutant exhibited reduced reactivation efficiency following DEX treatment, and CORT-108297 had little effect on explant-induced reactivation.
RESULTS
Dexamethasone stimulates explant-induced reactivation from latency.
To test whether the synthetic corticosteroid DEX influenced explant-induced reactivation, TG explants from latently infected mice were incubated with minimal essential medium (MEM) plus 2% charcoal-stripped FBS in the presence or absence of DEX. FBS passed through a column containing activated charcoal removes hormones, lipid-based molecules, certain growth factors, and cytokines, yielding stripped FBS: however, the process does not remove salts, glucose, and most amino acids. FBS contains bioactive corticosteroids, because nearly all Neuro-2A cells incubated with MEM plus 10% FBS (43) or 2% FBS (data not shown) contain GR in the nucleus. In contrast, nearly all Neuro-2A cells contain GR in the cytoplasm when incubated with 2% stripped FBS (43). Corticosterone levels in 10% FBS were 3,517.4 pg/ml (703.5 pg/ml in 2% FBS) but only 192.7 pg/ml in 2% stripped FBS when we measured corticosterone with a specific enzyme-linked immunosorbent assay (ELISA) (Enzo Life Sciences). In general, cortisol and corticosterone have similar levels under different physiological or stressful stimuli (44), which is why corticosterone was measured. dLAT2903R was used for these studies because it is a marker rescued virus generated from a LAT-null mutant (dLAT2903) that has growth properties identical to those of wild-type (WT) McKrae, and thus, it was designated WT virus (13, 14). Virus shedding from supernatants of TG explant cultures was assessed for efficiency of reactivation. When DEX was added to TG explants, WT-virus shedding was significantly higher at 5 and 9 days after explant than with MEM plus 2% stripped FBS lacking DEX (Fig. 1A and C, respectively). Although WT viral titers in the presence of DEX 7 days after explant were consistently higher than those of dLAT2903 when incubated with MEM plus 2% stripped FBS plus DEX, the differences were not significant (Fig. 1B).
FIG 1.

Explant-induced reactivation is regulated by GR. Female Swiss Webster mice (6 to 8 weeks old) were infected in the ocular cavity with ∼2 × 105 PFU of WT HSV-1 strain McKrae (dLAT2903R) or LAT-null virus (dLAT2903) as described in Materials and Methods. Thirty days after infection, TG were harvested in MEM containing 2% stripped FBS, 100 IU/ml penicillin, and 100 μg/ml streptomycin. The TG were minced and incubated at 37°C, 5% CO2, and where indicated, 10 μM water-soluble DEX was added to the cultures. (A to C) At 5 (A), 7 (B), or 9 (C) days postexplant, 100 μl supernatant from each sample was removed and virus shedding was measured by plaque assay. (D to F) Thirty days after infection, TG were harvested, minced, and incubated in MEM containing 10% FBS and 10 μM CORT-108297 or vehicle (DMSO). At 5 (D), 8 (E), or 10 (F) days postexplant, 100 μl supernatant was removed and used for plaque assays to measure virus shedding. The data are shown as means and standard deviations (SD) of the results from duplicate experiments. *, P < 0.05; ***, P < 0.0005; Student's t test.
Because the LAT-null mutant (dLAT2903) reactivates with reduced efficiency relative to the LAT+ McKrae strain in rabbits and mice (13, 14), the strain was compared to dLAT2903R. Virus shedding from TG explants prepared from mice latently infected with dLAT2903 was detected at very low levels 5 days after explant relative to WT virus (Fig. 1A), suggesting FBS contains a factor (not DEX, as DEX was added) lacking in stripped FBS that is important for accelerated shedding of dLAT2903 after explant. At 7 (Fig. 1B) and 9 (Fig. 1C) days after explant, more dLAT2903 was released when DEX was added: however, virus shedding was consistently less than that of WT McKrae (dLAT2903R). By 9 days after explant, WT virus shedding declined, presumably because stripped FBS lacked factors, not replaced by DEX alone, that were essential for tissue integrity and virus production 7 days after explant.
GR knockout mice were developed, but most pups died at birth due to respiratory failure (45). A conditional GR mutant that did not express GR in the nervous system (GrNesCre) was also developed (46, 47). However, GrNesCre mice are smaller than parental mice and have altered fat deposition because they express significantly more corticosteroids in serum than WT mice due to disruption of the hypothalamic/pituitary axis. Since corticosteroids have anti-inflammatory and immunosuppressive properties (30, 31), GrNesCre mice may be very susceptible to virulent HSV-1 strains, including McKrae. Consequently, we used CORT-108297 to further evaluate the effect GR functions have on explant-induced reactivation. CORT-108297 is a GR-specific antagonist that has no effect on the progesterone receptor or mineralocorticoid receptor and has no other known off-target effects (48–51). TG from latently infected mice were explanted in MEM containing FBS and dimethyl sulfoxide (DMSO) or 10 μM CORT-108297. Relative to controls, incubating TG explants with MEM plus FBS plus CORT-108297 significantly reduced virus replication, as judged by plaque assays, at 5, 8, and 10 days after infection (Fig. 1D to F). Interestingly, virus shedding from TG explants latently infected with the LAT-null mutant dLAT2903 was only slightly reduced by CORT-108297 at 8 and 10 days after explant. CORT-108297 had similar effects on TG explants incubated with 2% stripped FBS plus DEX (data not shown). In summary, these studies indicated GR was important for explant-induced reactivation with WT McKrae (dLAT2903R).
DEX treatment of TG from latently infected mice increased the number of GR+ neurons.
A subset of TG neurons express GR in rats (52) and bovines (53). Following TG explant from latently infected mice, when TG were incubated with MEM plus 2% stripped FBS plus DEX, a significant increase in GR+ TG neurons was detected at 4 and 8 h after explant relative to TG not treated with DEX (Fig. 2; see Fig. 4). In the absence of primary antibody, TG neuron staining from latently infected mice was not evident at 8 h with DEX, indicating GR staining was specific (Fig. 2, No 1° Ab). TG from uninfected mice that were not explanted (Fig. 3, U, and Fig. 4) contained GR+ TG neurons, which was consistent with the finding that 30 to 50% of rat TG neurons express GR (52). In contrast to latently infected mice, TG explants from uninfected mice did not contain significantly more GR+ TG neurons at 4 and 8 h after explant when cultures were incubated with MEM plus 2% stripped FBS plus DEX (Fig. 3 and 4).
FIG 2.

GR protein expression in TG explants from latently infected mice. TG explants from mice latently infected with HSV-1 (Latency), 4 h or 8 h, or 4 h or 8 h in the presence of DEX were subsequently formalin fixed and paraffin embedded. TG thin sections were stained with a GR antibody (Santa-Cruz; Sc-393232; diluted 1:250). The arrows denote GR-positive neurons. TG explants from mice latently infected for 8 h were incubated in MEM plus 2% stripped FBS plus DEX, and IHC was performed but the primary antibody (GR) was not added (No 1° antibody). Magnification, ×400.
FIG 4.
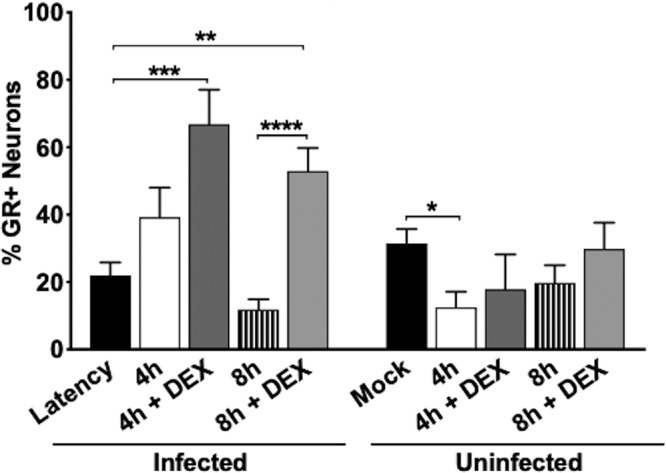
Comparison of GR+ TG neurons after TG explant. The number of GR+ neurons was estimated from approximately 500 total neurons for each treatment shown in Fig. 2 and 3; the results show a comparison of samples from latently infected TG and a comparison of samples from uninfected TG. Sections were obtained from 5 mice/treatment. The asterisks indicate significant differences as determined by Student's t test. *, P < 0.05; **, P < 0.005; ***, P < 0.0005; ****, P < 0.0001.
FIG 3.

GR expression in TG explants from uninfected mice (U). TG explant was performed as described in the legend to Fig. 2 using uninfected Swiss Webster mice that were 8 weeks old.
Expression of viral proteins during explant-induced reactivation from latency.
Several studies examined viral RNA expression during explant-induced reactivation (54–57): however, viral protein expression was not examined. We also performed immunohistochemistry (IHC) because reverse transcription (RT)-PCR does not necessarily mean the RNA is translated or the viral protein is expressed in neurons. Expression of three viral regulatory proteins (ICP0, ICP4, and VP16) was examined during explant-induced reactivation, as well as the effect DEX had on expression, using IHC protocols we established to identify proteins expressed in bovine TG (53, 58–62). VP16 (Fig. 5), but not ICP0 (Fig. 6) or ICP4 (Fig. 7), was readily detected in TG neurons 8 h after explant in the presence of DEX. All three viral proteins were detected in a subset of TG neurons at 16 h after explant, particularly when DEX was added. Significantly more TG neurons were stained by the VP16 antibody at 8 and 16 h when DEX was added to TG explants than with ICP0 and ICP4 (Fig. 8). If TG explants were not treated with DEX at 8 or 16 h, significantly more VP16+ TG neurons than ICP0+ or ICP4+ TG neurons were also detected. The percentage of TG neurons that contained VP16, ICP0, and ICP4 in the nucleus was estimated, because it was clear certain VP16+ (Fig. 5, blue arrows) and ICP0+ (Fig. 6) TG neurons contained the protein primarily in the cytoplasm. Only neurons in which the nucleus was clearly detected were counted, as certain sections contained stained neurons but the nucleus was not visible, and thus, it was not clear whether the neuron contained the viral protein in the nucleus. For VP16, 45 out of 54 VP16+ TG neurons were present in the nucleus at 8 h after explant (DEX treated). At 16 h after explant (DEX treated), 62 out of 72 neurons contained VP16 in the nucleus. Only 13 out of 58 ICP0+ TG neurons and 29 out of 48 ICP4+ TG neurons were detected in the nucleus. Since these three proteins are generally regarded as being localized to the nucleus, the results for ICP0 and ICP4 were surprising.
FIG 5.

Analysis of VP16 protein expression during explant-induced reactivation. Thin sections were cut from formalin-fixed, paraffin-embedded TG sections from uninfected mice (U), latently infected mice (Latency), and TG explants incubated in MEM plus 2% stripped FBS (with or without DEX) for 8 or 16 h. IHC was performed using an anti-VP16 antibody (Abcam; ab110226) that was diluted 1:100 and then biotinylated goat anti-mouse IgG (Vector Laboratories) as described in Materials and Methods. The arrows indicate TG neurons recognized by the VP16 antibody. The blue arrows indicate TG neurons with VP16 primarily in the cytoplasm. Magnification, approximately ×400.
FIG 6.

Analysis of ICP0 protein expression during explant-induced reactivation. Thin sections were cut from formalin-fixed, paraffin-embedded TG sections from latently infected mice and TG explants incubated in MEM plus 2% stripped FBS (with or without DEX) for 8 or 16 h. IHC was performed using an anti-ICP0 antibody (Santa Cruz Biotechnology; sc-53070) that was diluted 1:50 and then biotinylated goat anti-mouse IgG (Vector Laboratories). The arrows indicate TG neurons recognized by the ICP0 antibody. The blue arrows indicate TG neurons with ICP0 primarily in the cytoplasm. Magnification, ×400 for all sections except 16 h plus DEX, which was ×600.
FIG 7.

Analysis of ICP4 protein expression during explant-induced reactivation. Thin sections were cut from formalin-fixed, paraffin-embedded TG sections from latently infected mice and TG explants incubated in MEM plus 2% stripped FBS (with or without DEX) for 8 or 16 h. IHC was performed using an anti-ICP4 antibody (Abcam; ab6514) that was diluted 1:250, and then biotinylated goat anti-mouse IgG (Vector Laboratories) was added. The arrows indicate TG neurons recognized by the ICP4 antibody. Magnification, approximately ×400.
FIG 8.
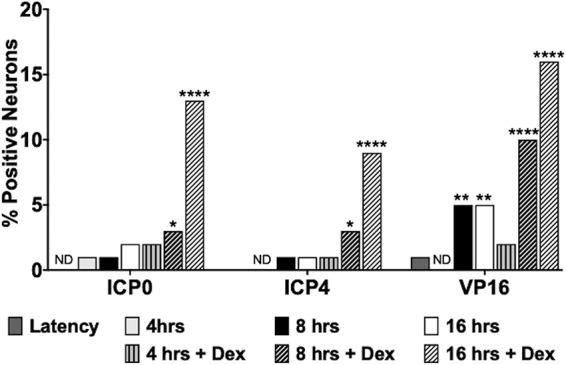
Quantification of viral protein expression during explant-induced reactivation. The numbers of viral-protein-positive TG neurons were estimated from approximately 250 total neurons for each viral protein from each time after explant (plus DEX and no DEX treatment). The data are shown as percent positive out of the total neurons counted. The asterisks indicate significant differences compared to latency as determined by the “n − 1” chi-square test (102): *, P < 0.05; **, P < 0.005; ****, P < 0.0001; ND, none detected.
Analysis of apoptosis in TG explants.
Cleaved caspase 3 was examined in TG sections because caspase 3 is activated following cleavage, and caspase 3 activation is considered to be a “point of no return” during induction of apoptosis (63). In latently infected mice or uninfected mice (Fig. 9 and 10), we occasionally detected TG neurons that were stained weakly by the cleaved caspase 3 antibody. At 8 and 16 h after TG explant from uninfected TG (Fig. 9) or latently infected mice (Fig. 10), cleaved caspase 3 TG neurons were readily detected regardless of DEX treatment. Euthanization of mice and then dissection of TG resulted in low levels of caspase 3 cleavage in approximately 15% of TG neurons from uninfected or latently infected mice (Fig. 11). Following TG explant, significant differences in cleaved caspase 3 were detected at 4 and 16 h when DEX was added to uninfected samples, but not infected samples, suggesting apoptosis induction following explant did not directly correlate with reactivation. This conclusion is consistent with the finding that neuronal apoptosis is not required for reactivation from latency if neurons derived from superior cervical ganglia are used as a model of in vitro latency (64). These studies also clearly demonstrated that TG explant led to caspase cleavage regardless of whether the TG were obtained from infected or uninfected mice.
FIG 9.

Analysis of caspase 3 cleavage from uninfected mice following TG explant or during latency. Thin sections were cut from formalin-fixed, paraffin-embedded TG sections from uninfected 8-week-old Swiss Webster mice or TG from mice latently infected with WT virus (dLAT2903R). TG explants were incubated in MEM plus 2% stripped FBS (with or without DEX) for 4, 8, or 16 h. The sections were examined for cleaved caspase 3 (Cell Signaling Technology; 9661; 1:150) by IHC as previously described (103). Biotinylated goat anti-rabbit IgG (Vector Laboratories) was used as a secondary antibody to detected cleaved-caspase 3+ cells. The arrows indicate TG neurons recognized by the cleaved-caspase 3 antibody. Magnification, approximately ×600.
FIG 10.

Analysis of caspase 3 cleavage of TG explants from latently infected mice. TG explants from mice latently infected with WT virus (dLAT2903R) were incubated in MEM plus 2% stripped FBS (with or without DEX) for 8 or 16 h. IHC was performed as described in the legend to Fig. 9.
FIG 11.
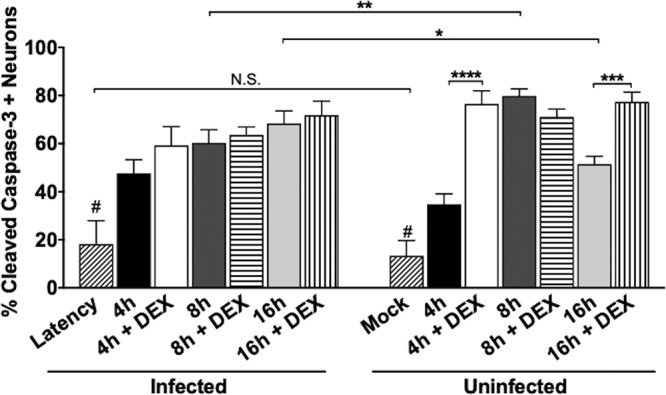
Quantification of cleaved-caspase 3-positive TG neurons during explant-induced reactivation. The percentage of cleaved-caspase 3+ TG neurons was determined from approximately 250 total neurons per sample shown in Fig. 8 and 9. The asterisks indicate significant difference compared to mock-infected neurons, and the statistical difference was determined using Student's t test. *, P < 0.05; **, P < 0.005; ***, P < 0.0005; ****, P < 0.0001; N.S., not significant. # indicates that mock infection and latency data were significantly different than those of every other column for which the P value was <0.005.
DISCUSSION
The results of this study demonstrated that explant-induced reactivation from latency was stimulated by the synthetic corticosteroid DEX but was significantly reduced by a GR-specific antagonist (CORT-108297). CORT-108297 also significantly reduced viral replication in cultured cells (65), consistent with TG explant findings. Previous studies reported DEX accelerated explant-induced reactivation (21, 22); however, these studies may have underestimated the effects of adding DEX to TG explants, because FBS was used in the studies.
With the exception of LAT, viral genes are not abundantly expressed during latency (66–71). Following a reactivation stimulus, a switch from silent chromatin to active viral chromatin occurs, which is coincident with activation of viral transcription and successful reactivation from latency (35, 64, 68, 72–74). Although many details related to initiating viral gene expression during reactivation are lacking, we suggest activated GR has certain properties that make it well suited to remodel a quiescent viral genome into an actively transcribing genome. For example, GR has rapid effects on gene expression because de novo protein synthesis is not required for transcriptional activation (25). Secondly, activated GR can specifically bind transcriptionally silent chromatin (75–77) and form a nuclease-hypersensitive site, culminating in transcriptional activation (78, 79). Thirdly, certain GREs in chromatin can stimulate transcription when located more than 5 kb from a transcriptional start site (80). These properties are consistent with GR being called a “pioneer transcription factor” (81, 82). The HSV-1 genome contains many consensus GREs distributed across the viral genome (data not shown): hence, GR activation can potentially stimulate many viral genes following a stressful stimulus. Interestingly, oriL, but not oriS, contains a functional GRE that stimulates replication of a plasmid in neuron-like cells (83). Introducing point mutations into oriL reduces viral pathogenesis during acute infection and impairs reactivation from latency in mouse models of infection (84).
Strikingly, these studies provided evidence that VP16, but not ICP0 and ICP4, was readily detected in TG neurons at 8 h after explant, even when TG explants were not treated with DEX. These three antibodies strongly and specifically recognize their respective targets in Western blots (data not shown). However, in formalin-fixed TG sections, it is possible the VP16 antibody specifically recognized lower levels of the VP16 protein relative to the ability of the ICP0 or ICP4 antibody to recognize its respective target. These findings were consistent with published reports that concluded VP16 is critical for reactivation from latency (37–39) and further suggest the VP16 promoter is active during early stages of explant-induced reactivation. The VP16 promoter contains three motifs, one in the TATA box and 2 downstream of the transcriptional start site, that are important for expression in TG neurons following heat stress (85). Conversely, the same virus replicates like the WT in cultured cells and in the ocular cavity in acutely infected mice (86, 87). A VP16 coactivator, host cell factor 1 (HCF-1), also localizes to the nucleus during explant-induced reactivation from latency (88), suggesting VP16 can transactivate IE gene expression during early stages of reactivation. Although the proximal VP16, ICP0, and ICP4 promoters do not contain consensus “whole” GREs, the ICP0 promoter is cooperatively transactivated by GR, DEX, and Krüppel-like transcription factor 15 (65). A ½ GRE in ICP0 promoter sequences was important for DEX-induced transcription mediated by GR and KLF15, suggesting multiple stress-induced factors are important for activating viral gene expression during early stages of reactivation.
Heat shock and UV light also induce HSV-1 reactivation from latency (1, 89, 90). On the surface, these reactivation stimuli are not similar to stress-induced reactivation from latency via GR activation. However, UV light induces ligand-independent GR phosphorylation, which correlates with GR transcriptional activation (91, 92) and increased expression of certain enzymes regulated by GR (93). Moreover, inhibitors of cortisol production impair heat shock-induced HSV-1 reactivation (94). Interestingly, UV light and stress both activate a serine/threonine protein kinase, JNK (c-Jun N-terminal kinase) (92), that is crucial for HSV-1 chromatin remodeling during reactivation from latency in an in vitro neuronal model (64). Thus, dissimilar reactivation stressors can activate common signaling proteins that regulate crucial steps during reactivation from latency, for example, JNK and GR.
In addition to stimulating transcription, corticosteroids interfere with immune and inflammatory responses (reviewed in references 28, 95, and 96). For example, activated GR interacts with two cellular transcription factors, activating protein 1 (AP-1) and NF-κB (nuclear factor kappa light chain enhancer of activated B cells), that are crucial for an inflammatory response (reviewed in references 25, 30, and 31). AP-1 and NF-kB activate the beta-interferon enhanceosome following virus infection (97), which is significant, because beta-interferon has antiviral activity (98). Furthermore, infiltrating lymphocytes in TG (98–101) that persist in TG during latency impair reactivation from latency, and increased corticosteroids can lead to apoptosis of these lymphocytes. In summary, we predict that GR activation directly stimulates viral gene expression in a subset of latently infected neurons during early stages of reactivation from latency. Immune-inhibitory functions of corticosteroids are predicted to promote virus shedding and spread during later stages of reactivation from latency.
MATERIALS AND METHODS
Virus, cells, and mouse infections.
HSV-1 strains McKrae dLAT2903R (WT; LAT positive) and dLAT2903 (LAT−/−) (13, 14) were grown in Vero cell monolayers in MEM supplemented with 10% FBS, 2 mM l-glutamine, 100 IU/ml penicillin, 100 μg/ml streptomycin. Viral aliquots were obtained through serial freeze-thawing of cells and subsequent centrifugation to remove cellular debris. Virus was titered on Vero monolayers to determine the number of PFU per milliliter for each stock prior to animal infections. Female 8-week-old Swiss Webster mice were purchased from Charles River Laboratories and housed and handled in agreement with Oklahoma State University Institutional Animal Care and Use Committee guidelines. The mice were acclimated for 1 week prior to ocular infection using ∼2 × 105 PFU/eye in 2 μl MEM without scarification, as described previously (8).
Explant-induced reactivation.
TG from ∼5 animals/treatment were harvested approximately 30 days postinfection in MEM containing 2% stripped FBS and antibiotics. Tissue was explanted into 6-well dishes, minced into 3 or 4 pieces each, and incubated at 37°C, 5% CO2, with or without 10 μM water-soluble DEX (Sigma; catalog no. D2915). Where indicated, 10 μM CORT-108297 (Sigma; catalog no. ADV509447843) reconstituted in DMSO was added to the TG explants. Each day, 100 μl medium was removed and used to perform plaque assays on Vero cell monolayers as previously described (65).
Immunohistochemistry.
TG from either mock- or latently infected mice were harvested and either directly placed in formalin or explanted for the indicated times prior to formalin fixation. Slide preparation and IHC were performed as previously described (61, 62). Briefly, 4- to 5-μm sections of paraffin-embedded TG were cut and mounted onto glass slides. The slides were incubated at 65°C for ∼20 min before deparaffinization in xylene and serial rehydration using decreasing concentrations of ethanol. Endogenous peroxidases were blocked by incubating the slides in 0.045% H2O2 for 20 min. Antigen retrieval was performed using proteinase K (Agilent Dako, Santa Clara, CA), and the slides were blocked using animal-free blocking solution (AFBS) (Cell Signaling Technologies, Danvers, MA) for 45 min at room temperature. Avidin and biotin were blocked using an avidin/biotin-blocking kit (Vector Laboratories, Burlingame, CA) according to the manufacturer’s instructions. The slides were incubated overnight at 4°C with the indicated antibody in AFBS. The following day, a Vectastain ABC HRP kit (Vector Laboratories) was used according to the manufacturer’s instructions. Biotinylated secondary antibody was prepared in AFBS, and the slides were incubated for 30 min at room temperature. The slides were then developed with NovaRED substrate (Vector Laboratories) and lightly counterstained with Mayer’s hematoxylin (Sigma-Aldrich, St. Louis, MO). The slides were imaged using an Olympus BX43 microscope. The TG neurons that were stained by the respective antibodies were counted in a blinded fashion.
Statistical analysis.
All graphs were created and comparisons performed using GraphPad Prism software (v7.0d). P values of less than 0.05 were considered significant for all calculations.
ACKNOWLEDGMENTS
This research was supported by grants from the National Institute of Neurological Disorders and Stroke of the National Institutes of Health under award number R21NS102290, the USDA-NIFA Competitive Grants Program (16-09370), and the Sitlington Endowment and with support from the Oklahoma Center for Respiratory and Infectious Diseases (National Institutes of Health Centers for Biomedical Research Excellence grant no. P20GM103648). L.Z. was partially supported by Chinese National Science Foundation grants (no. 31472172 and no. 31772743) and the National Key Research Program (no. 2016YFD0500704).
REFERENCES
- 1.Perng G-C, Jones C. 2010. Towards an understanding of the herpes simplex virus type 1 latency-reactivation cycle. Interdiscip Perspect Infect Dis 2010:1–18. doi: 10.1155/2010/262415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lewandowski G, Zimmerman MN, Denk LL, Porter DD, Prince GA. 2002. Herpes simplex type 1 infects and establishes latency in the brain and trigeminal ganglia during primary infection of the lip in cotton rats and mice. Arch Virol 147:167–179. doi: 10.1007/s705-002-8309-9. [DOI] [PubMed] [Google Scholar]
- 3.Fraser N, Lawrence WC, Wroblewska Z, Gilde DH, Koprowski H. 1981. Herpes simplex virus type 1 DNA in human brain tissue. Proc Natl Acad Sci U S A 78:6461–6465. doi: 10.1073/pnas.78.10.6461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Umbach JL, Kramer MF, Jurak I, Karnowski HW, Coen DM, Cullen BR. 2008. MicroRNAs expressed by herpes simplex virus 1 during latent infection regulate viral mRNAs. Nature 454:780–785. doi: 10.1038/nature07103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Umbach JL, Kramer MF, Jural I, Karnowski HW, Coen DM, Cullen BR. 2009. Analysis of human alphaherpesvirus microRNA expression in latently infected human trigeminal ganglia. J Virol 83:10677–10683. doi: 10.1128/JVI.01185-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Peng W, Vitvitskaia O, Carpenter D, Wechsler SL, Jones C. 2008. Identification of two small RNAs within the first 1.5-kb of the herpes simplex virus type 1 (HSV-1) encoded latency-associated transcript (LAT). J Neurovirol 14:41–52. doi: 10.1080/13550280701793957. [DOI] [PubMed] [Google Scholar]
- 7.Thompson RL, Sawtell NM. 2001. Herpes simplex virus type 1 latency-associated transcript gene promotes neuronal survival. J Virol 75:6660–6675. doi: 10.1128/JVI.75.14.6660-6675.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Inman M, Perng G-C, Henderson G, Ghiasi H, Nesburn AB, Wechsler SL, Jones C. 2001. Region of herpes simplex virus type 1 latency-associated transcript sufficient for wild-type spontaneous reactivation promotes cell survival in tissue culture. J Virol 75:3636–3646. doi: 10.1128/JVI.75.8.3636-3646.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Perng G-C, Jones C, Ciacci-Zanella J, Stone M, Henderson G, Yukht A, Slanina SM, Hoffman FM, Ghiasi H, Nesburn AB, Wechsler SL. 2000. Virus-induced neuronal apoptosis blocked by the herpes simplex virus latency-associated transcript (LAT). Science 287:1500–1503. doi: 10.1126/science.287.5457.1500. [DOI] [PubMed] [Google Scholar]
- 10.Ahmed M, Lock M, Miller CG, Fraser NW. 2002. Regions of the herpes simplex virus type 1 latency-associated transcript that protect cells from apoptosis in vitro and protect neuronal cells in vivo. J Virol 76:717–729. doi: 10.1128/JVI.76.2.717-729.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Branco FJ, Fraser NW. 2005. Herpes simplex virus type 1 latency-associated transcript expression protects trigeminal ganglion neurons from apoptosis. J Virol 79:9019–9025. doi: 10.1128/JVI.79.14.9019-9025.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hamza MA, Higgins DM, Feldman LT, Ruyechan WT. 2007. The latency-associated transcript of herpes simplex virus type 1 promotes survival and stimulates axonal regeneration in sympathetic and trigeminal ganglia. J Neurovirol 13:56–66. doi: 10.1080/13550280601156297. [DOI] [PubMed] [Google Scholar]
- 13.Perng G-C, Dunkel EC, Geary PA, Slanina SM, Ghiasi H, Kaiwar R, Nesburn AB, Wechsler SL. 1994. The latency-associated transcript gene of herpes simplex virus type 1 (HSV-1) is required for efficient in vivo spontaneous reactivation of HSV-1 from latency. J Virol 68:8045–8055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Perng G-C, Ghiasi H, Slanina SM, Nesburn AB, Wechsler SL. 1996. The spontaneous reactivation function of the herpes simplex virus type 1 LAT gene resides completely within the first 1.5 kilobases of the 8.3-kilobase primary transcript. J Virol 70:976–984. doi: 10.1006/viro.1998.9492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Thompson RL, Sawtell NM. 1997. The herpes simplex virus type 1 latency-associated transcript gene regulates the establishment of latency. J Virol 71:5432–5440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Thompson RL, Sawtell NM. 2011. The herpes simplex virus type 1 latency associated transcript locus is required for the maintenance of reactivation competent latent infections. J Neurovirol 17:552–558. doi: 10.1007/s13365-011-0071-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sawtell NM, Thompson RL. 1992. Herpes simplex virus type 1 latency-associated transcription unit promotes anatomical site-dependent establishment and reactivation from latency. J Virol 66:2157–2169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Freeman ML, Sheridan BS, Bonneau RH, Hendricks RL. 2007. Psychological stress compromises CD8+ T cell control of latent herpes simplex virus type 1 infection. J Immunol 179:322–328. doi: 10.4049/jimmunol.179.1.322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Padgett DA, Sherida JF, Dorne J, Berntson GG, Candelora J, Glaser R. 1998. Social stress and the reactivation of latent herpes simplex virus type 1. Proc Natl Acad Sci U S A 95:7231–7235. doi: 10.1073/pnas.95.12.7231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Uchakin PN, Parish DC, Dane FC, Uchakina ON, Scheetz AP, Agarwal NK, Smith BE. 2011. Fatigue in medical residents leads to reactivation of herpes virus latency. Interdiscip Perspect Infect Dis 2011:1. doi: 10.1155/2011/571340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Halford WP, Gebhardt BM, Carr DJ. 1996. Persistent cytokine expression in trigeminal ganglion latently infected with herpes simplex virus type 1. J Immunol 157:3542–3549. [PubMed] [Google Scholar]
- 22.Du T, Zhou G, Roizman B. 2012. Induction of apoptosis accelerates reactivation from latent HSV-1 in ganglionic organ cultures and replication in cell cultures. Proc Natl Acad Sci U S A 109:14616–14621. doi: 10.1073/pnas.1212661109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ledbetter EC, Kim SG, Dubovi EJ, Bicalho RC. 2009. Experimental reactivation of latent canine herpesvirus-1 and induction of recurrent ocular disease in adult dogs. Vet Microbiol 138:98–105. doi: 10.1016/j.vetmic.2009.03.013. [DOI] [PubMed] [Google Scholar]
- 24.Jones C. 2014. Reactivation from latency by alpha-herpesvirinae subfamily members: a stressful situation. Curr Top Virol 12:99–118. [Google Scholar]
- 25.Oakley RH, Cidlowski JA. 2013. The biology of the glucocorticoid receptor: new signaling mechanisms in health and disease. J Allergy Clin Immunol 132:1033–1044. doi: 10.1016/j.jaci.2013.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang J-C, Derynck MK, Nonaka DF, Khodabakhsh DB, Haqq C, Yamamoto KR. 2004. Chromatin immunoprecipitation (ChIP) scanning identifies primary glucocorticoid receptor target genes. Proc Natl Acad Sci U S A 101:15603–15608. doi: 10.1073/pnas.0407008101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Giguere V, Hollenberg SM, Rosenfeld MG, Evans RM. 1986. Functional domains of the human glucocorticoid receptor. Cell 46:645–652. doi: 10.1016/0092-8674(86)90339-9. [DOI] [PubMed] [Google Scholar]
- 28.Schoneveld OJLM, Gaemers IC, Lamers WH. 2004. Mechanisms of glucocorticoid signalling. Biochim Biophys Acta 1680:114–128. doi: 10.1016/j.bbaexp.2004.09.004. [DOI] [PubMed] [Google Scholar]
- 29.Taniguchi-Yanai K, Koike Y, Hasegawa T, Furuta Y, Serizawa M, Ohshima N, Kato N, Yanai K. 2010. Identification and characterization of glucocorticoid receptor-binding sites in the human gneome. J Recept Signal Transduct Res 30:88–105. doi: 10.3109/10799891003614816. [DOI] [PubMed] [Google Scholar]
- 30.Rhen T, Cidlowski JA. 2005. Antiinflammatory action of glucocorticoids—new mechanisms of old drugs. N Engl J Med 353:1711–1723. doi: 10.1056/NEJMra050541. [DOI] [PubMed] [Google Scholar]
- 31.Smoak KL, Cidlowski JA. 2004. Mechanisms of glucocorticoid receptor signaling during inflammation. Mech Aging Dev 125:697–706. doi: 10.1016/j.mad.2004.06.010. [DOI] [PubMed] [Google Scholar]
- 32.Halford WP, Kemp CD, Isler JA, Davido DJ, Schaffer PA. 2001. ICP0, ICP4, or VP16 expressed from adenovirus vectors induces reactivation of latent herpes simplex virus type 1 in primary cultures of latently infected trigeminal ganglion cells. J Virol 75:6143–6153. doi: 10.1128/JVI.75.13.6143-6153.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Halford WP, Schaffer PA. 2001. ICP0 is required for efficient reactivation of herpes simplex virus type 1 from neuronal latency. J Virol 75:3240–3249. doi: 10.1128/JVI.75.7.3240-3249.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.McMahan L, Schaffer PA. 1990. The repressing and enhancing functions of the herpes simplex virus regulatory protein ICP27 map to C-terminal regions and are required to modulate viral gene expression very early in infection. J Virol 64:3471–3485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Whitlow ZW, Kristie TM. 2009. Recruitment of the transcriptional coactivator HCF-1 to viral immediate-early promoters during initiation of reactivation from latency of herpes simplex virus type 1. J Virol 83:9591–9595. doi: 10.1128/JVI.01115-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cai WZ, Schaffer PA. 1989. Herpes simplex virus type 1 ICP0 plays a critical role in the de novo synthesis of infectious virus following transfection of viral DNA. J Virol 63:4579–4589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Camarena V, Kobayashi M, Kim JK, Roehm P, Perez R, Gardner J, Wilson AC, Mohr I, Chao MV. 2010. Nature and duration of growth factor signaling through receptor tyrosine kinases regulates HSV-1 latency in neurons. Cell Host Microbe 8:320–330. doi: 10.1016/j.chom.2010.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kim JY, Mandarino A, Chao MV, Mohr I, Wilson AC. 2012. Transient reversal of episome silencing precedes VP16-dependent transcription during reactivation of HSV-1 in neurons. PLoS Pathog 8:e1002540. doi: 10.1371/journal.ppat.1002540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Thompson RL, Preston CM, Sawtell NM. 2009. De novo synthesis of VP16 coordinates the exit from HSV latency in vivo. PLoS Pathog 5:e1000352. doi: 10.1371/journal.ppat.1000352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.O'Hare P. 1993. The virion transactivator of herpes simplex virus. Semin Virol 4:145–155. doi: 10.1006/smvy.1993.1010. [DOI] [Google Scholar]
- 41.O'Hare P, Goding CR. 1988. Herpes simplex virus regulatory elements and the immunoglobulin octamer domain bind a common factor and are both targets for virion transactivation. Cell 52:435–445. doi: 10.1016/S0092-8674(88)80036-9. [DOI] [PubMed] [Google Scholar]
- 42.O'Hare P, Hayward GS. 1985. Three trans-acting regulatory proteins of herpes simplex virus modulate immediate-early gene expression in a pathway involving positive and negative feedback regulation. J Virol 56:723–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kook I, Henley C, Meyer F, Hoffmann FG, Jones C. 2015. Bovine herpesvirus 1 productive infection and the immediate early transcription unit 1 are stimulated by the synthetic corticosteroid dexamethasone. Virology 484:377–385. doi: 10.1016/j.virol.2015.06.010. [DOI] [PubMed] [Google Scholar]
- 44.Gong S, Miao Y-L, Jiao G-Z, Sun M-J, Li H, Lin J, Luo M-J, Tan J-H. 2015. Dynamics and correlation of serum cortisol and corticosterone under different physiological or stressful conditions in mice. PLoS One 10:e0117503. doi: 10.1371/journal.pone.0117503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cole TJ, Blendy JA, Monaghan AP, Krieglstein K, Schmid W, Aguzzi A, Fantuzzi G, Hummler E, Unsicker K, Schutz G. 1995. Targeted disruption of the glucocorticoid receptor gene blocks adrenergic chromaffin cell development and severely retards lung maturation. Genes Dev 9:1608–1621. doi: 10.1101/gad.9.13.1608. [DOI] [PubMed] [Google Scholar]
- 46.Oitz MS, de Kloet ER, Joels M, Schmid W, Cole TJ. 1997. Spatial learning deficits in mice with a targeted glucocorticoid receptor gene disruption. Eur J Neurosci 9:2284–2296. doi: 10.1111/j.1460-9568.1997.tb01646.x. [DOI] [PubMed] [Google Scholar]
- 47.Tronche F, Kellendonk C, Kretz O, Gass P, Anlag K, Orban P, Bock R, Klein R, Schutz G. 1999. Disruption of the glucocorticoid receptor gene in the nervous system results in reduced anxiety. Nat Genet 23:99–103. doi: 10.1038/12703. [DOI] [PubMed] [Google Scholar]
- 48.Solomon MB, Wulsin AC, Rice T, Wick D, Myers B, McKlveen J, Flak JN, Ulrich-Lai Y, Herman JP. 2014. The selective glucocorticoid receptor antagonist CORT 108297 decreases neuroendocrine stress responses and immobility in the forced swim test. Horm Behav 65:363–371. doi: 10.1016/j.yhbeh.2014.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Belanoff JK, Blasey CM, Clark RD, Roe RL. 2010. Selective glucocorticoid receptor (type II) antagonist prevents and reverses olanzapine induced weight gain. Diabetes Obes Metab 12:545–547. doi: 10.1111/j.1463-1326.2009.01185.x. [DOI] [PubMed] [Google Scholar]
- 50.Clark R, Ray NC, Williams K, Blaney P, Ward S, Crackett PH, Hurley C, Dyke HJ, Clark DE, Lockey P, Devos R, Wong M, Porres SS, Bright CP, Jenkins RE, Belanoff J. 2008. 1H-pyrazolo[3,4-g]hexahydro-isoquinolines as selective glucocorticoid receptor antagonists with high functional activity. Bioorg Med Chem Lett 18:1312–1317. doi: 10.1016/j.bmcl.2008.01.027. [DOI] [PubMed] [Google Scholar]
- 51.Morgan BP, Swick AG, Hargrove DM, LaFlamme JA, Moynihan MS, Carroll RS, Martin KA, Lee E, Decosta D, Bordner J. 2002. Discovery of potent, nonsteroidal, and highly selective glucocorticoid receptor antagonists. J Med Chem 45:2417–2424. doi: 10.1021/jm0105530. [DOI] [PubMed] [Google Scholar]
- 52.DeLeon M, Covenas R, Chadi G, Narvaez JA, Fuxe K, Cintra A. 1994. Subpopulations of primary sensory neurons show coexistence of neuropeptides and glucocorticoid receptors in the rat spinal and trigeminal ganglia. Brain Res 14:338–342. doi: 10.1016/0006-8993(94)91034-0. [DOI] [PubMed] [Google Scholar]
- 53.Frizzo da Silva L, Kook I, Doster A, Jones C. 2013. Bovine herpesvirus 1 regulatory proteins, bICP0 and VP16, are readily detected in trigeminal ganglionic neurons expressing the glucocorticoid receptor during the early stages of reactivation from latency. J Virol 87:11214–11222. doi: 10.1128/JVI.01737-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Du T, Zhou G, Roizman B. 2011. HSV-1 gene expression from reactivated ganglia is disordered and concurrent with suppression of latency-associated transcript and miRNAs. Proc Natl Acad Sci U S A 108:18820–18824. doi: 10.1073/pnas.1117203108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Du T, Zhou G, Roizman B. 2013. Modulation of reactivation of latent herpes simplex virus 1 in ganglionic organ cultures by p300/CBP and STAT3. Proc Natl Acad Sci U S A 110:E2621–E2628. doi: 10.1073/pnas.1309906110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Amelio AL, Giordani NV, Kubat NJ, O'Neil JE, Bloom DC. 2006. Deacetylation of the herpes simplex virus type 1 latency-associated transcript (LAT) enhancer and a decrease in LAT abundance precede an increase in ICP0 transcriptional permissiveness at early times postexplant. J Virol 80:2063–2068. doi: 10.1128/JVI.80.4.2063-2068.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Devi-Rao GB, Bloom DC, Stevens JG, Wagner EK. 1994. Herpes simplex virus type 1 DNA replication and gene expression during explant-induced reactivation of latently infected murine sensory ganglia. J Virol 68:1271–1282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kook I, Doster A, Jones C. 2015. Bovine herpesvirus 1 regulatory proteins are detected in trigeminal ganglionic neurons during the early stages of stress-induced escape from latency. J Neurovirol 21:585–591. doi: 10.1007/s13365-015-0339-x. [DOI] [PubMed] [Google Scholar]
- 59.Liu Y, Hancock M, Workman A, Doster A, Jones C. 2016. Beta-catenin, a transcription factor activated by canonical Wnt signaling, is expressed in sensory neurons of calves latently infected with bovine herpesvirus 1. J Virol 90:3148–3159. doi: 10.1128/JVI.02971-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Workman A, Eudy J, Smith L, Frizzo da Silva L, Sinani D, Bricker H, Cook E, Doster A, Jones C. 2012. Cellular transcription factors induced in trigeminal ganglia during dexamethasone-induced reactivation from latency stimulate bovine herpesvirus 1 productive infection and certain viral promoters. J Virol 86:2459–2473. doi: 10.1128/JVI.06143-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Workman A, Zhu L, Keel BN, Smith TPL, Jones C. 2018. The Wnt signaling pathway is differentially expressed during the bovine herpesvirus 1 latency-reactivation cycle: evidence that two protein kinases associated with neuronal survival, Akt3 and BMPR2, are expressed at higher levels during latency. J Virol 92:e01937-17. doi: 10.1128/JVI.01937-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhu L, Workman A, Jones C. 2017. A potential role for a beta-catenin coactivator (high mobility group AT-hook 1 protein) during the latency-reactivation cycle of bovine herpesvirus 1. J Virol 91:e02132-36. doi: 10.1128/JVI.02132-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Nicholson DW, Thornberry NA. 1997. Caspases: killer proteases. Trends Biochem Sci 22:299–306. doi: 10.1016/S0968-0004(97)01085-2. [DOI] [PubMed] [Google Scholar]
- 64.Cliffe A, Arbuckle JH, Vogel JL, Geden MJ, Rothbart SB, Cusack CL, Strahl BD, Kristie TM, Deshmukh M. 2015. Neuronal stress pathway mediating a histone methyl/phospho switch is required for herpes simplex virus reactivation. Cell Host Microbe 18:649–658. doi: 10.1016/j.chom.2015.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ostler J, Harrison KS, Schroeder K, Thunuguntla P, Jones C. 2019. The glucocorticoid receptor (GR) stimulates herpes simplex virus 1 productive infection, in part because the infected cell protein 0 (ICP0) promoter is cooperatively transactivated by the GR and Krüppel-like transcription factor 15. J Virol 93:e02063-18. doi: 10.1128/JVI.02063-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Bloom DC, Giordani NV, Kwiatkowski DL. 2010. Epigenetic regulation of latent HSV-1 gene expression. Biochim Biophys Acta 1799:246–256. doi: 10.1016/j.bbagrm.2009.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Knipe DM, Cliffe A. 2008. Chromatin control of herpes simplex virus lytic and latent infection. Nat Rev Microbiol 6:211–221. doi: 10.1038/nrmicro1794. [DOI] [PubMed] [Google Scholar]
- 68.Cliffe AR, Garber DA, Knipe DM. 2009. Transcription of the herpes simplex virus latency-associated transcript promotes the formation of facultative heterochromatin on lytic promoters. J Virol 83:8182–8190. doi: 10.1128/JVI.00712-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kwiatkowski D, Thompson HW, Bloom DC. 2009. The polycomb group protein Bim1 binds to the herpes simplex virus 1 latent genome and maintains repressive histone marks during latency. J Virol 83:8173–8181. doi: 10.1128/JVI.00686-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wang Q-Y, Zhou C, Johnson KE, Colgrove RC, Coen DM, Knipe DM. 2005. Herpesviral latency-associated transcript gene promotes assembly of heterochromatin on viral lytic-gene promoters in latent infection. Proc Natl Acad Sci U S A 102:16055–16059. doi: 10.1073/pnas.0505850102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Giordani N, Neumann DM, Kwiatkowski DL, Bhattacharjee PS, McAnany PK, Hill JM, Bloom DC. 2008. During herpes simplex virus type 1 infection of rabbits, the ability to express the latency-associated transcript increases latent-phase transcription of lytic genes. J Virol 82:6056–6060. doi: 10.1128/JVI.02661-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Neumann DM, Bhattacharjee PS, Giordani NV, Bloom DC, Hill JM. 2007. In vivo changes in the patterns of chromatin structure associated with the latent herpes simplex virus type 1 genome in mouse trigeminal ganglia can be detected at early times after butyrate treatment. J Virol 81:13248–13253. doi: 10.1128/JVI.01569-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Liand Y, Vogel JL, Arbuckle JH, Rai G, Jadhav A, Simeonov A, Maloney DJ, Kristie TM. 2013. Targeting the JMJD2 histone demethylases to epigenetically control herpesvirus infection and reactivation from latency. Sci Transl Med 5:167ra165. doi: 10.1126/scitranslmed.3005145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Liang Y, Vogel J, Narayanan A, Peng H, Kristie TM. 2009. Inhibition of the histone demethylase LSD1 blocks alpha-herpesvirus lytic replication and reactivation from latency. Nat Med 15:1312–1318. doi: 10.1038/nm.2051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Perlman T. 1992. Glucocorticoid receptor DNA-binding specificity is increased by the organization of DNA in nucleosomes. Proc Natl Acad Sci U S A 89:3884–3888. doi: 10.1073/pnas.89.9.3884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Perlman T, Wrange O. 1988. Specific glucocorticoid receptor binding to DNA reconstituted in nucleosome. EMBO J 7:3073–3079. doi: 10.1002/j.1460-2075.1988.tb03172.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Lin Q, Wrange O. 1993. Translational positioning of a nucleosome: glucocorticoid response element modulates glucocorticoid receptor affinity. Genes Dev 7:2471–2482. doi: 10.1101/gad.7.12a.2471. [DOI] [PubMed] [Google Scholar]
- 78.Foy R, Horz W. 2003. Sequence-specific positioning of nucleosomes over the steroid-inducible MMTV promoter. EMBO J 6:2321–2328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Zaret KS, Yamamoto KR. 1984. Reversible and persistent changes in chromatin structure accompany activation of a glucocorticoid-dependent enhancer element. Cell 258:1780–1784. [DOI] [PubMed] [Google Scholar]
- 80.Polman JAE, Welten JE, Bosch DS, de Jonge RT, Balog J, van der Maarel SM, de Kloet E, Datson NA. 2012. A genome-wide signature of glucocorticoid receptor binding in neuronal PC12 cells. BMC Neurosci 13:118–125. doi: 10.1186/1471-2202-13-118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Zaret KS, Carrol JS. 2011. Pioneer transcription factors: establishing competence for gene expression. Genes Dev 25:2227–2241. doi: 10.1101/gad.176826.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Iwafuchi-Doi M, Zaret KS. 2014. Pioneer transcription factors in cell reprogramming. Genes Dev 28:2679–2692. doi: 10.1101/gad.253443.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Hardwicke MA, Schaffer PA. 1997. Differential effects of nerve growth factor and dexamethasone on herpes simplex virus type 1 oriL- and oriS-dependent DNA replication in PC12 cells. J Virol 71:3580–3587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Balliet JW, Schaffer PA. 2006. Point mutations in herpes simplex virus type 1 oriL, but not in oriS, reduce pathogenesis during acute infection of mice and impair reactivation from latency. J Virol 80:440–450. doi: 10.1128/JVI.80.1.440-450.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Sawtell N, Thompson RL. 2016. De novo herpes simplex virus VP16 expression gates a dynamic programmatic transition and sets the latent/lytic balance during acute infection in trigeminal ganglia. PLoS Pathog 12:e1005877. doi: 10.1371/journal.ppat.1005877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Lieu P, Pande NT, Rice MK, Wagner EK. 2000. The exchange of cognate TATA boxes results in a corresponding change in the strength of two HSV-1 early promoters. Virus Genes 20:5–10. doi: 10.1023/A:1008108121028. [DOI] [PubMed] [Google Scholar]
- 87.Lieu P, Wagner EK. 2000. Two leaky-late HSV-1 promoters differ significantly in structural architecture. Virology 272:191–203. doi: 10.1006/viro.2000.0365. [DOI] [PubMed] [Google Scholar]
- 88.Kristie TM, Vogel JL, Sears AE. 1999. Nuclear localization of the C1 factor (host cell factor) in sensory neurons correlates with reactivation of herpes simplex virus from latency. Proc Natl Acad Sci U S A 96:1229–1233. doi: 10.1073/pnas.96.4.1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Jones C. 1998. Alphaherpesvirus latency: its role in disease and survival of the virus in nature. Adv Virus Res 51:81–133. doi: 10.1016/S0065-3527(08)60784-8. [DOI] [PubMed] [Google Scholar]
- 90.Jones C. 2003. Herpes simplex virus type 1 and bovine herpesvirus 1 latency. Clin Microbiol Rev 16:79–95. doi: 10.1128/CMR.16.1.79-95.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Galliher-Beckley AJ, Williams JG, Cidlowski JA. 2011. Ligand-independent phosphorylation of the glucocorticoid receptor integrates cellular stress pathways with nuclear receptor signaling. Mol Cell Biol 31:4663–4675. doi: 10.1128/MCB.05866-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Davies L, Karthikeyan N, Lynch JT, Sial E-A, Gkourtsa A, Demonacos C, Krstic-Demonacos M. 2008. Cross talk of signaling pathways in the regulation of the glucocorticoid receptor function. Mol Endocrinol 22:1331–1344. doi: 10.1210/me.2007-0360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Skobowiat C, Sayre RM, Dowdy JC, Slominski AT. 2013. Ultraviolet radiation regulates cortisol activity in a waveband dependent manner in human skin ex-vivo. Br J Dermatol 168:595–601. doi: 10.1111/bjd.12096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Noisakran S, Halford WP, Veress L, Djj C. 1998. Role of the hypothalmic pituitary adrenal axis and IL-6 in stress-induced reactivation of latent herpes simplex virus type 1. J Immunol 160:5441–5447. [PubMed] [Google Scholar]
- 95.Barnes PJ. 1998. Anti-inflammatory actions of glucocorticoids: molecular mechanisms. Clin Sci 94:557–572. doi: 10.1042/cs0940557. [DOI] [PubMed] [Google Scholar]
- 96.Funder JW. 1997. Glucocorticoids and mineralocorticoid receptors: biology and clinical relevance. Annu Rev Med 48:231–240. doi: 10.1146/annurev.med.48.1.231. [DOI] [PubMed] [Google Scholar]
- 97.Goodbourn S, Zinn K, Maniatis T. 1985. Human beta-interferon gene expression is regulated by an inducible enhancer element. Cell 41:509–520. doi: 10.1016/S0092-8674(85)80024-6. [DOI] [PubMed] [Google Scholar]
- 98.Decman V, Freeman ML, Kinchington PR, Hendricks RL. 2005. Immune control of HSV-1 latency. Viral Immunol 18:466–473. doi: 10.1089/vim.2005.18.466. [DOI] [PubMed] [Google Scholar]
- 99.Knickelbein JE, Khanna KM, Yee MB, Baty CJ, Kinchington PR, Hendricks RL. 2008. Noncytotoxic lytic granule-mediated CD8+ T cell inhibition of HSV-1 reactivation from neuronal latency. Science 322:268–272. doi: 10.1126/science.1164164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Matundan H, Mott KR, Ghiasi H. 2014. Role of CD8+ T cells and lymphoid dendritic cells in protection from ocular herpes simplex virus 1 challenge in immunized mice. J Virol 88:8016–8027. doi: 10.1128/JVI.00913-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Mott KR, Allen SJ, Zandian M, Ghiasi H. 2014. Coregulatory interactions among CD8alpha dendritic cells, the latency-associated transcript, and programmed death 1 contribute to higher levels of herpes simplex virus 1 latency. J Virol 88:6599–6610. doi: 10.1128/JVI.00590-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Campbell I. 2007. Chi-squared and Fisher-Irwin tests of two-by-two tables with small sample recommendations. Stat Med 26:3661–3675. doi: 10.1002/sim.2832. [DOI] [PubMed] [Google Scholar]
- 103.Lovato L, Inman M, Henderson G, Doster A, Jones C. 2003. Infection of cattle with a bovine herpesvirus 1 (BHV-1) strain that contains a mutation in the latency related gene leads to increased apoptosis in trigeminal ganglia during the transition from acute infection to latency. J Virol 77:4848–4857. doi: 10.1128/JVI.77.8.4848-4857.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]