

Chronic hepatitis B virus (HBV) infection continues to be a major global health concern, and patients who fail to mount an efficient immune response to clear the virus will develop a life-long chronic infection that can progress to chronic active hepatitis, cirrhosis, and primary hepatocellular carcinoma. There is no definite cure for chronic hepatitis B, and alpha interferon (IFN-α) is the only available immunomodulatory drug, to which only a minority of chronic patients are responsive, with hepatitis B e antigen (HBeAg)-negative patients responding better than HBeAg-positive patients. We herein report that the intracellular HBeAg, also known as precore or p22, inhibits the antiviral signaling of IFN-α, which sheds light on the enigmatic function of precore protein in shaping HBV chronicity and provides a perspective toward areas that need to be further studied to make the current therapy better until a cure is achieved.
KEYWORDS: hepatitis B virus, interferons
ABSTRACT
Antagonism of host immune defenses against hepatitis B virus (HBV) infection by the viral proteins is speculated to cause HBV persistence and the development of chronic hepatitis. The circulating hepatitis B e antigen (HBeAg, p17) is known to manipulate host immune responses to assist in the establishment of persistent viral infection, and HBeAg-positive (HBeAg+) patients respond less effectively to IFN-α therapy than do HBeAg-negative (HBeAg−) patients in clinical practice. However, the function(s) of the intracellular form of HBeAg, previously reported as the precore protein intermediate (p22) without the N-terminal signal peptide, remains elusive. Here, we report that the cytosolic p22 protein, but not the secreted HBeAg, significantly reduces interferon-stimulated response element (ISRE) activity and the expression of interferon-stimulated genes (ISGs) upon alpha interferon (IFN-α) stimulation in cell cultures. In line with this, HBeAg+ patients exhibit weaker induction of ISGs in their livers than do HBeAg− patients upon IFN-α therapy. Mechanistically, while p22 does not alter the total STAT1 or pSTAT1 levels in cells treated with IFN-α, it blocks the nuclear translocation of pSTAT1 by interacting with the nuclear transport factor karyopherin α1 through its C-terminal arginine-rich domain. In summary, our study suggests that HBV precore protein, specifically the p22 form, impedes JAK-STAT signaling to help the virus evade the host innate immune response and, thus, causes resistance to IFN therapy.
IMPORTANCE Chronic hepatitis B virus (HBV) infection continues to be a major global health concern, and patients who fail to mount an efficient immune response to clear the virus will develop a life-long chronic infection that can progress to chronic active hepatitis, cirrhosis, and primary hepatocellular carcinoma. There is no definite cure for chronic hepatitis B, and alpha interferon (IFN-α) is the only available immunomodulatory drug, to which only a minority of chronic patients are responsive, with hepatitis B e antigen (HBeAg)-negative patients responding better than HBeAg-positive patients. We herein report that the intracellular HBeAg, also known as precore or p22, inhibits the antiviral signaling of IFN-α, which sheds light on the enigmatic function of precore protein in shaping HBV chronicity and provides a perspective toward areas that need to be further studied to make the current therapy better until a cure is achieved.
INTRODUCTION
Hepatitis B is a potentially life-threatening liver infection caused by the hepatitis B virus (HBV) (1, 2). Approximately 250 million people worldwide are chronically infected with HBV, and more than 780,000 people die every year due to hepatitis B-associated complications, including cirrhosis and liver cancer (3). The longevity of the virus within the host is maintained by nuclear, episomal, nucleosome-decorated viral DNA called covalently closed circular DNA (cccDNA) (4, 5). Concurrently, inability of the host immune system to clear the virus contributes to the development of the chronic infection. Over the decades, varied theories have been proposed to explain the development of a persistent infection, including the presence of viral proteins that antagonize immune defenses, such as the circulating hepatitis B surface antigen (HBsAg) and hepatitis B e antigen (HBeAg) (6, 7).
Upon HBV infection, the virion containing the relaxed circular DNA (rcDNA) genome is transported into the cell nucleus and converted to an episomal cccDNA, which assembles into a minichromosome and serves as a transcription template for all the viral mRNAs (8, 9). Four groups of viral RNA are transcribed from cccDNA, namely, the 3.5-kb precore mRNA that encodes the precore protein, another 3.5-kb pregenomic RNA (pgRNA) that encodes the core and the polymerase, a 2.4-kb mRNA for the large (L) envelope protein, a 2.1-kb mRNA for the middle (M) and major surface (S) proteins, and a 0.7-kb mRNA for the X protein (1, 2). The pgRNA, upon being exported to the cytoplasm, gets packaged into the nucleocapsid together with viral polymerase. The viral polymerase then reverse transcribes pgRNA into viral minus-strand DNA, followed by asymmetric plus-strand DNA synthesis to yield the rcDNA genome within the capsid (10, 11). The mature nucleocapsid is either packaged by viral envelope proteins and secreted as a virion particle or redirects the rcDNA to the nucleus to replenish the cccDNA reservoir (1, 11). The viral envelope proteins can also be secreted alone as subviral particles (HBsAg). The precore protein is translated from precore mRNA and further processed in the endoplasmic reticulum (ER) and is released as HBeAg through the ER-Golgi apparatus secretory pathway (12, 13).
HBeAg is a 17-kDa secreted viral protein. As illustrated in Fig. 1A, HBeAg is derived from the precursor protein, precore (p25). The precore core open reading frame (ORF) contains two in-frame initiation codons that encode precore (p25) and core protein (p21), respectively. The translation of p25 on precore mRNA starts at the first ATG site, while translation of the core protein on pgRNA starts at the second ATG. In contrast to the core protein, which spontaneously oligomerizes into the viral capsid, the p25 protein is a hypothetical 25-kDa monomer with a 29-amino-acid (aa) N-terminal extension. The initially translated 19-aa signal peptide bound to the signal recognition particle (SRP) complex guides the precore mRNA to the ER membrane. The signal peptide sequence is then cotranslationally cleaved off by signal peptidase, resulting in p22. This 22-kDa protein is either translocated into the ER or released back to the cytoplasm (14). Due to the presence of a nuclear localization signal (NLS) on the arginine-rich C-terminal domain (CTD) of p22, this protein can also be transported into the nucleus (15–17). In the trans-Golgi network, the CTD is removed by furin endoprotease to release the 17-kDa mature HBeAg into the blood in a dimeric form (18, 19). p22 is a precursor of HBeAg and, hence, referred to as intracellular HBeAg. As an auxiliary protein of HBV, HBeAg is not a structural component of viral and subviral particles, and the precise function of precore/HBeAg in the HBV life cycle is currently unknown. It is, however, known that precore is not required for HBV DNA replication (20).
FIG 1.

HBV precore protein exists as a p22 form intracellularly. (A) Schematic illustration of the translation of HBV precore and core proteins. While HBV core protein is formed from the pregenomic RNA (pgRNA), the ORF of precore (p25) is translated from the precore mRNA and processed into p22 by removal of the N-terminal signal peptide (SP) by signal peptidase (SPase); p22 then gets further processed at the C-terminal domain (CTD) to form the secreted HBeAg (p17). The figure shows a comparison of the differences in the various domains among the above-mentioned proteins. The position of the HA tag insertion in the precore domain of HA-tagged precore is indicated. (B) HepG2 cells were seeded in 12-well plates and mock transfected or transfected with pHBV1.3, pHBV1.3ΔC, pcHBc, pcHBe, pcHBeΔSP, and pcHA-HBe, as indicated. Cells were harvested at day 5 posttransfection, and the proteins were analyzed by Western blotting using antibodies against CTD or HA tag. β-Actin served as the loading control.
In the clinic, HBeAg positivity is generally used as a surrogate marker for high levels of viral replication. Loss of detectable HBeAg in individuals with chronic viral hepatitis and the appearance of detectable levels of antibody to HBeAg is usually considered to be a beneficial milestone and/or evidence of reduced viral replication, suggesting a role of HBeAg in HBV persistence (21). In line with this, previous studies have demonstrated that the circulating HBeAg elicits T cell tolerance and depletes inflammatory HBeAg- and HBcAg-specific Th1 cells that are necessary for viral clearance (22, 23), and the HBV core-specific CTL responses are significantly weaker in HBeAg-positive (HBeAg+) than in HBeAg (HBeAg−) patients (24). During HBV vertical transmission, more than 90% of untreated infants born to HBeAg+ mothers become chronically infected due to T cell immune tolerance to HBV infection (20, 25, 26). A mechanistic study in HBV-transgenic mice revealed that such impairment of CD8+ T cell responses can be attributed to the maternal HBeAg-mediated upregulation of PD-L1 in fetal liver macrophages (27). Thus, HBeAg may represent a viral strategy to persist in the host through inducing immune tolerance and/or exhaustion. In terms of innate immunity, it has been reported that HBeAg downregulates NK cell-mediated gamma interferon (IFN-γ) production and interleukin-18 (IL-18) signaling (28) and precore protein suppresses the Toll-like receptor 2 (TLR2) and IL-1 signaling pathways intracellularly (29–31). However, another study demonstrated that HBeAg triggers an IL-1 response through binding to the IL-1 receptor accessory protein (32).
Although it remains debatable whether HBV is able to activate or block IFN production upon infection of hepatocytes (33–44), alpha interferon (IFN-α) is used clinically to treat chronic hepatitis B patients; however, only approximately one-third of the patients respond to IFN-α treatment (45). This limited antiviral activity of IFN-α against HBV could be due to blockade of the IFN-elicited JAK-STAT pathway by an HBV protein, an idea which is supported by a study showing that HBV prevents the nuclear translocation of STAT1 and inhibits interferon-stimulated gene (ISG) expression in humanized mice (46). However, which viral protein(s) inhibits IFN signaling needs further investigation. Interestingly, it was found that patients with low HBeAg levels prior to treatment are more likely to respond to IFN-α therapy, and HBeAg− patients with virus bearing basal core promoter mutations or the commonly occurring G1896A mutation (a mutation of G to A at position 1896) respond better to IFN-α therapy than HBeAg+ patients (47–51), suggesting that HBeAg or its intracellular precursor may blunt the IFN signaling.
In this study, we observed that HBeAg+ patients exhibit weaker induction of ISGs in their livers than HBeAg− patients upon IFN-α therapy. Furthermore, the cytosolic p22 protein, but not the secreted HBeAg, significantly reduces interferon-stimulated response element (ISRE) activity and the expression of interferon stimulated genes (ISGs) upon IFN-α stimulation in cell cultures. Mechanistically, while p22 does not alter the total STAT1 or pSTAT1 levels in cells treated with IFN-α, it blocks the nuclear translocation of pSTAT1 by interacting with karyopherin α1 (Kα1) through its CTD domain. In summary, our study suggests that HBV precore protein, specifically the p22 form, impedes JAK-STAT signaling to help the virus evade the host innate immune response and, thus, causes resistance to IFN therapy.
RESULTS
HBV precore protein exists in a p22 form in cell cultures.
As schematically illustrated in Fig. 1A, the biogenesis of HBeAg requires a multiple-step intracellular process of the precore precursor protein, and a 22-kDa p22 intermediate, harboring the intact CTD domain but devoid of the N-terminal signal peptide, has been identified by a previous study (15). To further validate the presence of p22 during HBV replication and HBeAg production, the plasmid pHBV1.3, which expresses all HBV proteins, was transfected into HepG2 cells. The expression of intracellular core protein (p21) and p22 was detected by Western blotting using antibodies against the last 14 amino acids of precore/core CTD (Fig. 1B, lane 1) (52, 53). The expression of core protein (p21) and precore-derived p22 from pHBV1.3 was further confirmed by transfection of the core-null HBV plasmid pHBV1.3ΔC (Fig. 1B, lane 2) and by transfection of the core-expressing plasmid pHBc alone (Fig. 1B, lane 3) and the precore-expressing plasmid pcHBe alone (Fig. 1B, lane 4). The protein size of the p22 intermediate was further confirmed by the expression of the precore ORF without the 19-aa signal peptide coding sequence, which was predicted to express a 22-kDa protein (Fig. 1B, lanes 4 and 5). Transfection of pcHA-HBe produced an intracellular N-terminally hemagglutinin (HA)-tagged precore protein of 24 kDa (HA-p22) (Fig. 1B, lane 6). However, the secreted HA-HBeAg (HA-p17) was not detected intracellularly, which is consistent with previous studies demonstrating that HBeAg/p17 is rapidly secreted out of the cell from the Golgi apparatus after furin cleavage and is not retained in the Golgi apparatus (18, 52). Collectively, the results demonstrate that the intracellular HBV precore protein predominantly exists as the p22 form.
p22 does not form capsid or support DNA replication.
HBV core protein and p22 share the exact same amino acid sequence except for the 10-aa N-terminal extension in the p22 protein, as shown in Fig. 1A. While core protein can form dimers and these dimers in turn lead to the formation of a capsid, Schodel et al. had shown that, unlike core, the bacterially expressed HBeAg (p17) failed to form a capsid in Escherichia coli due to a putative disulfide bridge between cysteines (C) at residues −7 and 61, and mutation of −7C in the precore domain to a glutamine (C−7Q) restored particle formation (54). In the present study, we tested the ability of full-length precore/p22 to form a capsid in mammalian cells. As shown by the results in Fig. 2A, compared to the results for the replication-competent plasmid pHBV1.3, the core-null pHBV1.3ΔC failed to support HBV pgRNA encapsidation and DNA replication due to the defect of capsid formation, though p22 was expressed (Fig. 2A, lane 2 versus lane 1). The viral replication of pHBV1.3ΔC was rescued by transcomplementing core but not p22 or HA-tagged p22 (Fig. 2A, lanes 3 to 5). Furthermore, p22 with the C−7Q mutation remains defective in capsid formation (Fig. 2B). Thus, unlike core, p22 protein is incapable of forming capsid, suggesting that the N-terminal 10-aa precore domain of p22 negatively regulates capsid assembly and that p22 does not play an essential role in HBV DNA replication.
FIG 2.

Inability of precore/p22 to support HBV capsid formation and DNA replication. (A) HepG2 cells in 12-well plates were cotransfected with pHBV1.3 and control vector, pHBV1.3ΔC and control vector, or pcHBc, pcHBe, or pcHA-HBe, as indicated. Cells were harvested at day 5 posttransfection, and viral total RNA and encapsidated pgRNA were analyzed by Northern blotting, core DNA was detected by Southern blotting, and cytoplasmic capsid was analyzed by native capsid gel EIA. The expression of core and precore protein was detected by Western blotting using core antibody against the CTD domain, and the HA-tagged p22 was detected by Western blotting using anti-HA antibody. β-Actin served as the loading control. For RNA analysis, each lane was loaded with 10 μg of total RNA and probed with a genome-length, plus-strand-specific HBV riboprobe. The levels of ribosomal RNAs (28S and 18S) are presented as loading controls. The positions of HBV pgRNA (3.5 kb) and subgenomic surface RNAs (2.4 kb and 2.1 kb) are indicated. HBV core DNA was probed with genome-length, minus-strand-specific HBV riboprobe. The positions of relaxed circular (RC) and single-stranded (SS) DNAs are indicated. (B) HepG2 cells in 12-well plates were cotransfected with pHBV1.3 and control vector, pHBV1.3ΔC and control vector, or pcHBeC−7Q, as indicated, for 5 days. The analyses of viral RNA, DNA, and proteins were performed in the same way as described for panel A.
Subcellular distribution of p22.
Next, we examined the distribution of p22 among different cellular compartments, using the HepHBe4 cell line, which constitutively expresses HA-tagged p22 (HA-p22) and HBeAg (HA-HBeAg), and the tetracycline-inducible HepBHAe82 cell line, which expresses HA-p22 and HA-HBeAg in a cccDNA-dependent manner (52). Immunofluorescence microscopy demonstrated that while HA-p22 is predominantly found in the cytoplasm, it is also able to translocate to the nucleus (Fig. 3A). This was further validated with subcellular fractionation of HepG2 cells transfected with HA-p22-expressing plasmid, which showed the presence of both the cytoplasmic and nuclear forms of p22 (Fig. 3B). Since p22 is translated in the ER lumen and has been suggested to partly retrotranslocate to the cytosol through the ER-associated degradation pathway (14, 55), we did a microsomal fractionation of cells transfected with HA-p22 and found that p22 exists in both the microsomal and cytosolic compartments (Fig. 3C, lane 1). Furthermore, when cells were treated with furin inhibitor I to block furin-mediated cleavage of p22 and the subsequent release of HBeAg from the Golgi apparatus, p22 was seen to accumulate significantly in the microsomal fraction without further increase in the cytosolic compartment (Fig. 3C, lane 2 versus lane 1), indicating that, once p22 reaches the Golgi apparatus, it cannot be transported back to the cytosol. The mechanism underlying the sorting of p22 into the Golgi apparatus for the HBeAg process and ER-to-cytosol retrotranslocation awaits further investigation.
FIG 3.
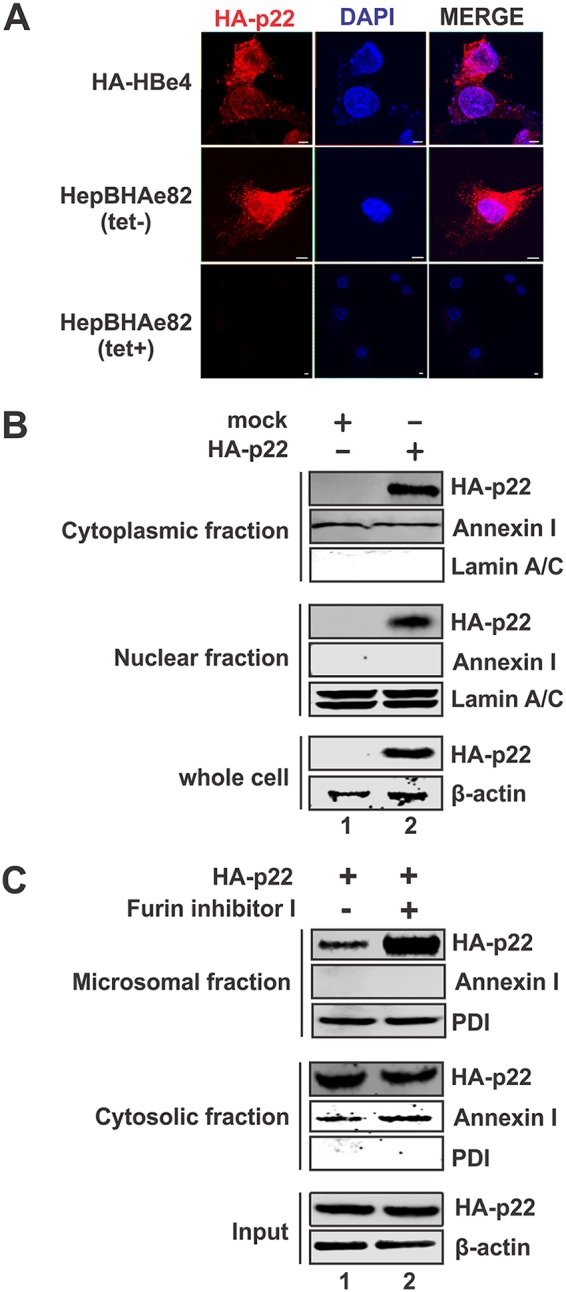
Subcellular localization of p22. (A) HepHA-HBe4 cells and noninduced (Tet+) and induced (Tet−; day 14) HepBHAe82 cells were subjected to immunofluorescence microscopy for HA-p22 (red) staining. The cell nuclei were stained by DAPI (blue). (B) HepG2 cells in 12-well plates were transfected with either control vector or pcHA-HBe (HA-p22). Cells were harvested at day 5 posttransfection, and subcellular fractionation was carried out to detect HA-p22 in cytoplasmic and nuclear lysates by Western blotting using anti-HA antibody. Annexin I and lamin A/C served as a marker and loading control for the cytoplasmic and the nuclear fractions, respectively, and β-actin served as a loading control for whole-cell lysate. (C) 293T cells in 12-well plates were transfected with HA-p22 and left untreated or treated with furin inhibitor I (1 μM). Cells were harvested at day 5 posttransfection and subjected to microsomal fractionation analysis. Membrane-associated HA-p22 and cytosolic HA-p22 were detected by Western blotting using anti-HA antibody. Annexin I and PDI served as a marker and loading control for the cytosolic and the membrane fractions, respectively, and β-actin served as a loading control for whole-cell lysate.
p22 inhibits ISRE promoter activity and ISG expression.
It has been reported that HBeAg− patients respond better to IFN therapy than HBeAg+ patients (47–51), but the underlying mechanisms remain elusive. In this study, consecutive liver biopsy specimens were collected from 11 HBeAg+ and 10 HBeAg− patients at baseline and after 24 weeks of IFN-α monotherapy and used for transcriptome sequencing (RNA-seq) analysis with special focus on the ISG induction profile (Table 1). As shown by the results in Fig. 4, across a group of 28 ISGs with more than 2-fold induction in each patient at the end of IFN therapy, HBeAg+ patients had an overall weaker ISG induction than HBeAg− patients, indicating that circulating HBeAg and/or intracellular p22 might have a negative effect on the IFN-signaling pathway, though the variable levels of viral DNA and/or other viral antigens might be additional contributing factors.
TABLE 1.
Baseline characteristics of enrolled patients
| Characteristica | Value [mean (range) or as indicated] for patients who were: |
P value | |
|---|---|---|---|
| HBeAg+ (n = 11) | HBeAg− (n = 10) | ||
| Male [no. (%)] | 9 (81.8) | 8 (80.0) | 1.000 |
| Age (yr) | 30 (16, 44) | 30.5 (25, 57) | 0.306 |
| HBsAg [log10 (IU/ml)] | 4.39 (3.50, 4.72) | 3.23 (1.50, 4.27) | 0.001 |
| HBeAg (s/co) | 831.42 (3.75, 1,565.70) | 0.37 (0.34, 0.84) | <0.001 |
| HBV DNA [log10 (IU/ml)] | 7.30 (2.70, 7.70) | 5.58 (2.70, 7.05) | 0.024 |
| ALT (IU/liter) | 178 (56, 325) | 100 (41, 474) | 0.067 |
| AST (IU/liter) | 59 (46, 147) | 58.5 (28, 250) | 0.290 |
| TB (μmol/liter) | 12.0 (7.0, 22.0) | 11.6 (9.0, 23.4) | 0.778 |
s/co, signal-to-cutoff ratio.
FIG 4.

Induction of intrahepatic ISGs in HBeAg+ and HBeAg− chronic hepatitis B patients under IFN-α treatment. Liver biopsies were performed for 11 HBeAg+ and 10 HBeAg− patients at baseline and after 24 weeks of IFN-α monotherapy. Total cellular mRNA samples were subjected to RNA sequencing, and the expression levels of genes were determined. The fold changes of the expression abundance for 28 ISGs (filter criteria, P ≤ 0.05 and fold change of ≥2) in HBeAg+ and HBeAg− groups were plotted. The histogram represents the ratio of FPKM for each gene between baseline and after therapy, whose values were then logarithmically transformed.
In order to investigate the potential effects of p22 and HBeAg on interferon-sensitive response element (ISRE) activity, we cotransfected HepG2 cells with an ISG56 promoter-controlling luciferase reporter plasmid, ISG56-Luc, along with control vector or different HBV protein-expressing plasmids, followed by IFN-α treatment. As shown by the results in Fig. 5A, while the control group exhibited a high ISG56 promoter activity upon IFN-α stimulation, the expression of HBV core or p22 (wild type or HA tagged) reduced this activity significantly, confirming an inhibitory effect of HBV p22 or its extracellular product, HBeAg, on IFN signaling. No such effect was seen when other HBV proteins, such as HBx, HBV surface proteins, or HBV polymerase, were coexpressed, suggesting that the inhibitory effect on ISG56 promoter activity is specific to p22 and core proteins. In order to further identify whether the inhibition was mediated by intracellular p22 or extracellular HBeAg, ISG56-Luc-transfected cells were treated with recombinant HBeAg or furin inhibitor I. The cells treated with recombinant HBeAg did not show any inhibition of IFN-α activity, and the cells treated with furin inhibitor I, which prevents the secretion of HBeAg in p22-transfected cells, did not show attenuation of the p22-mediated reduction of ISG56 promoter activity, suggesting that intracellular p22 but not secreted HBeAg blocks IFN-α signaling. It is worth noting that blocking HBeAg secretion with the furin inhibitor did not further inhibit ISG56-Luc activity, which is consistent with the fact that the furin inhibitor-arrested intracellular p22 does not retrotranslocate to the cytosol (Fig. 3C), where the IFN signaling takes place.
FIG 5.

Suppression of IFN-α signaling by intracellular p22. (A) HepG2 cells were seeded in 96-well plates and cotransfected with ISG56-Luc and control vector or HBV protein-expressing plasmids, as indicated, together with pRL-CMV. Five days later, the transfected cells were left untreated or treated with human IFN-α (1,000 IU/ml) for 18 h. Where indicated, exogenous treatment with recombinant HBeAg (rHBeAg; 50 ng/ml) or treatment with furin inhibitor I (1 μM) was carried out. Firefly and Renilla luciferase activities were measured where the latter served as the internal control to normalize transfection efficiency. The relative luciferase activities were plotted as fold changes versus the results for the control group without IFN-α treatment (mean ± SD, n = 4). (B) HepG2 cells in 12-well plates were transfected with control vector, pcHBc (core/p21), or pcHBe (p22) as indicated. At day 5 posttransfection, the transfected cells were treated with human IFN-α (1,000 IU/ml) for 30 min and total cellular RNA was extracted and subjected to MxA and ISG56 mRNA qPCR analysis. The relative levels of individual gene induction were plotted as fold changes versus the results for the control group (mean ± SD, n = 4).
Furthermore, the effect of HBV core and p22 on ISG mRNA levels upon IFN-α treatment was examined. Quantitative PCR analysis of the relative expression levels of ISG56 and MxA demonstrated that HBV p22 and core protein markedly reduced the ISG transcripts induced by IFN-α (Fig. 5B), further confirming an inhibitory effect of these HBV proteins on the IFN-elicited ISG production.
p22 inhibits ISG production in HBV infection system in vitro.
In order to validate the above-described effects of p22 in a more physiological setting, we resorted to the infection model using HepG2-NTCP12 cells. We made two types of p22-null viruses, one containing an A1814T start codon mutation of the precore ORF in the viral genome and the other harboring the precore G1896A stop codon mutation. The wild-type and p22-null virus were used to infect the HepG2-NTCP cells with the same inoculum size for 7 days, and 3TC (lamivudine) was included during the infection to prevent de novo HBV DNA replication and intracellular cccDNA amplification. Core immunofluorescence demonstrated that both wild-type and p22-null viruses successfully established the infection, though a slightly reduced core signal was seen in p22-null-virus-infected cells (Fig. 6A and D). The true precore-null nature of the mutant viruses was confirmed by HBeAg and HA-HBeAg chemiluminescence immunoassay (CLIA) (Fig. 6B and E). The infected cells were treated with IFN-α, and the expression levels of several ISGs were analyzed by quantitative PCR (qPCR). A stronger induction of analyzed ISGs was seen in cells infected with p22-null HBV than in those infected with wild-type virus (Fig. 6C and F). The above-described results from the HBV infection system further validated the inhibitory effect of p22 on IFN-α signaling and ruled out the possibility of the observed effects being the result of overexpression of the protein.
FIG 6.

Precore downregulates IFN-α-mediated ISG induction in HBV infection system in vitro. HepG2-NTCP12 cells in 96-well plates were left uninfected or were infected with the wild-type HBV and G1896A and A1814T precore-null viruses, respectively, at 500 VGE/cell for 24 h. The infected cells were then cultured in the presence of 10 μM 3TC for 7 days. (A and D) HBV infectivity was assessed by HBcAg immunofluorescence. (B and E) The precore-null nature of the mutant viruses was confirmed with supernatant HBeAg and HA-HBeAg CLIA. (C and F) For another set of cells uninfected or infected with wild-type or mutant viruses in the same way as described above, the cells were treated with IFN-α (1,000 IU/ml) for 30 min prior to cell harvest, and the induced expression of MxA, ISG56, OAS2, IRF9, and ISG20 was measured by qPCR analysis. The relative levels of individual gene induction by IFN-α were plotted as fold changes versus the results for the uninfected control group (mean ± SD, n = 3).
p22 inhibits the nuclear translocation of pSTAT1/2 through competing with their interaction with karyopherin.
It is well known that IFN-α induces ISG production through the canonical JAK-STAT pathway, leading to the nuclear translocation of phosphorylated STAT1/2 heterodimer in the ISGF3 complex and binding with ISG promoters containing ISRE elements (Fig. 7A) (56). In order to elucidate the mechanism of p22-mediated inhibition of IFN signaling, we assessed the effect of p22 on STAT phosphorylation upon IFN-α stimulation. As shown by the results in Fig. 7B, the levels of pSTAT1 and pSTAT2 in whole-cell lysates of HepG2 cells activated by IFN-α were unchanged in the presence of p22 compared to the levels in the control group, suggesting a proper binding of IFN to its receptors and phosphorylation of STAT1/2. However, subcellular fractionation analysis showed that p22 led to an accumulation of pSTAT1 and pSTAT2 in the cytoplasm that corresponded to reduced levels of pSTAT1/2 in the nucleus, suggesting that p22 blocks the nuclear translocation of the pSTAT1/2 complex (Fig. 7C). Interestingly, HBV core protein did not prevent the nuclear importation of pSTAT1 or pSTAT2 (Fig. 7D), indicating a different mode of inhibition of IFN signaling by the core protein, which awaits further investigation.
FIG 7.

p22 blocks nuclear translocation of STAT1/2. (A) Schematic illustration of IFN-α-elicited JAK-STAT signaling and the activation of ISG expression. IFN-α binds to its cognate receptor IFNAR1 and IFNAR2 on the cell membrane and activates the JAK-STAT pathway. The phosphorylated STAT1 and STAT2 form a heterodimer, followed by interaction with IRF9 to form interferon-stimulated gene factor 3 (ISGF3). Karyopherin α1 (Kα1), a nuclear transporter protein binding ISGF3, is essential to mediate the nuclear import of ISGF3. The ISGF3 binds to interferon-stimulated response element (ISRE) in DNA to activate transcription of ISGs. (B) HepG2 cells in 12-well plates were transfected with either control vector or pcHBe (p22). At day 5 posttransfection, the transfected cells were left untreated or treated with IFN-α (1,000 IU/ml) for 30 min. Total STAT1, pSTAT1, and pSTAT2 were detected by Western blotting, with β-actin serving as a loading control. (C) HepG2 cells in 6-well plates were transfected with control vector or p22 for 5 days, followed by mock treatment or IFN-α treatment (1,000 IU/ml) for 30 min. The harvested cells were subjected to cell fractionation, and the levels of the pSTATs were detected in cytoplasmic (top) and nuclear (bottom) fractions. Annexin I and lamin A/C served as a marker and loading control for the cytoplasmic and nuclear fraction, respectively. (D) HepG2 cells in 6-well-plate were transfected with control vector or core/p21. At day 5 posttransfection, the cells were left untreated or treated with IFN-α (1,000 IU/ml) for 30 min. Cell fractionation and pSTAT1/2 detection in cytoplasmic and nuclear fractions were conducted as described for panel C.
In order to further study the mechanism underlying p22-mediated blockage of nuclear translocation of the ISGF3 complex, we looked at the protein-protein interaction between p22 and the nuclear transporter karyopherin alpha 1 (Kα1). The ISGF3 complex requires the interaction with Kα1 for its nuclear entry and activation of ISG expression. Hence, we performed a coimmunoprecipitation experiment with HA-tagged p22 and FLAG-tagged Kα1. Immunoprecipitation with anti-HA antibody and detection of Kα1 through anti-FLAG antibody confirmed p22’s ability to interact with Kα1 (Fig. 8A). The HA-tagged Ebola virus VP24 has been shown to interact with Kα1 and block the nuclear translocation of pSTAT1 and, thus, served as a positive control (57). Since both p22 and the ISGF3 complex bind to Kα1 to enter the nucleus, we hypothesized that p22 might be competing with the ISGF3 complex to bind to Kα1 and preventing the entry of the ISGF3 complex into the nucleus. Hence, we performed a competition assay where we treated cells with IFN-α in the absence or presence of p22. Immunoprecipitation with anti-pSTAT1 antibody followed by detection of FLAG-Kα1 demonstrated a reduced amount of Kα1 binding to pSTAT1 in the presence of p22 (Fig. 8B). Thus, we conclude that p22 competes with pSTAT1 to bind to the nuclear transporter Kα1, preventing the entry of pSTAT1 into the nucleus.
FIG 8.
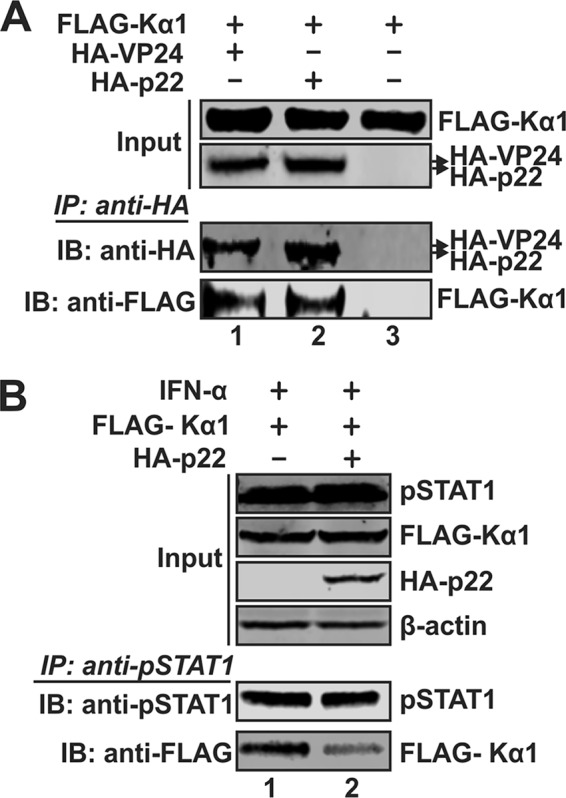
p22 competes with the binding of Kα1 with pSTAT1. (A) 293T cells in 6-well plates were cotransfected with FLAG-Kα1 and HA-VP24, HA-p22, or control vector as indicated. Five days later, the transfected cells were lysed and subjected to coimmunoprecipitation with beads coated by anti-HA antibody, and the presence of coprecipitated proteins was analyzed by Western blotting using anti-HA and anti-FLAG antibodies. (B) 293T cells in 6-well plates were cotransfected with FLAG-Kα1 plus control vector or HA-p22 for 5 days, followed by mock treatment or IFN-α treatment (1,000 IU/ml) for 30 min. The cells were then subjected to coimmunoprecipitation with anti-pSTAT1 antibody-conjugated beads, and the presence of pulled-down FLAG-Kα1 was analyzed by Western blotting using anti-FLAG antibody.
p22 blocks IFN-α signaling through its CTD domain.
HBV core protein possesses a bipartite nuclear localization signal in its arginine-rich C-terminal domain (CTD) (16). Since p22 shares the same C terminus with core protein, we hypothesized that the CTD might signal p22 for its nuclear translocation and, hence, might be responsible for competing with the ISGF3 complex to bind to Kα1. We created a CTD deletion mutant of HA-p22 (HA-p22ΔCTD), and this mutant showed its inability to translocate to the nucleus from the cytoplasm (Fig. 9A). We also examined the mutant’s ability to interact with Kα1 through coimmunoprecipitation. While HA-p22 was seen to interact with Kα1, the HA-p22ΔCTD mutant did not coprecipitate with Kα1, suggesting the role of p22’s CTD in its interaction with the nuclear transporter and in the nuclear translocation of p22 (Fig. 9B). Moreover, an ISG56-Luc reporter assay demonstrated that the HA-p22ΔCTD mutant failed to inhibit IFN-α-induced ISG56 promoter activity (Fig. 9C). Thus, the CTD domain of p22 is responsible for its inhibition of IFN-induced JAK-STAT signaling.
FIG 9.

The CTD domain is required for the p22-mediated inhibition of IFN signaling. (A) HepG2 cells in 6-well plates were transfected with plasmid expressing HA-p22 or HA-p22ΔCTD for 5 days and subjected to cell fractionation assay. The presence of full-length and CTD-truncated HA-p22 in the whole-cell and subcellular fractions was assessed by Western blotting using anti-HA antibody. β-Actin served as the loading control for whole-cell lysate. Annexin I and lamin A/C served as a marker and loading control for cytoplasmic and nuclear lysate, respectively. (B) 293T cells in 6-well plates were cotransfected with FLAG-Kα1-expressing plasmid plus HA-p22- or HA-p22ΔCTD-expressing plasmid or control vector as indicated for 5 days. The transfected cells were then lysed and subjected to coimmunoprecipitation with anti-HA antibody-coated beads, and the coimmunoprecipitated FLAG-Kα1 was detected by Western Blotting using anti-FLAG antibody. (C) HepG2 cells in 96-well plates were cotransfected with ISG56-Luc and control vector or p22, HA-p22, or HA-p22ΔCTD as indicated, plus pRL-CMV. Five days later, the transfected cells were left untreated or treated with IFN-α (1,000 IU/ml) for 18 h. The cells were then harvested for dual-luciferase assay. Renilla luciferase activity served as the internal control to normalize transfection efficiency. The relative firefly luciferase activities in each sample were plotted as fold changes versus the results for the control group without IFN-α treatment (mean ± SD, n = 3).
DISCUSSION
HBeAg is a serological marker of hepatitis B and a known nonstructural protein of HBV. Circulating HBeAg has been reported to regulate various host immune responses and is often associated with high viremia in chronic hepatitis B patients, suggesting a proviral activity of HBeAg (7, 20, 26). However, the function of the intracellular precore protein p22, the precursor for HBeAg, remains largely elusive. In the present study, we sought to characterize p22’s ability to support replication and to identify the function of its cytosolic form. Our results reconfirmed the presence of intracellular p22, as well as its cytosolic and nuclear distribution (Fig. 1 and 3) (14, 15, 55). Although the cytosolic p22 shares the exact same amino acid sequence as the core protein except for a 10-aa N-terminal extension, it fails to form a capsid-like structure and support HBV DNA replication (Fig. 2A), which is consistent with previous studies showing that precore is not required for HBV infection in vivo or replication in vitro (58, 59), though it can form heterocapsid with core protein (60, 61). It has been shown that the presence of a cysteine at the −7 position in the N-terminal precore domain of HBeAg causes a preference for the formation of an intramolecular disulfide bond between the −7 and 61 cysteines over an intermolecular disulfide bond, as seen in the case of the core protein, resulting in HBeAg dimerization in an inverted orientation relative to the core dimer and, thus, precluding capsid formation from HBeAg. In line with this, bacterially expressed HBeAg/p17 with the −7 cysteine-to-glutamine (C−7Q) mutation or under reducing conditions restores particle formation (19, 54). However, we found that p22 with the C−7Q mutation remains nonparticulate in HepG2 cells and does not support HBV DNA replication (Fig. 2B), indicating that the N-terminally extended precore domain blocks the ability of p22 to assemble into capsid-like structures in cell cultures. However, whether the intracellular p22 exists as a monomeric or dimeric form remains unknown.
A previous study with hepatitis B patient serum samples suggested that a precore-derived aberrant 22-kDa protein devoid of the CTD domain but harboring the N-terminal signal peptide, termed p22cr, could self-assemble into an HBV genome-free capsid and acquire envelope, to be secreted as an empty virion (62). In our study, we did not detect capsid formation in cells from precore expressed alone or in the context of core-null HBV transfection (Fig. 2). It is unclear whether the p22cr species is present in our experimental system or not, but nonetheless, we were always able to detect the canonical CTD-containing p22.
Despite the dispensable role of precore in the HBV replication cycle, the observed high viremia and low immune system activation in HBeAg+ chronic hepatitis B patients directs attention to the role of HBeAg and/or p22 in relation to the regulation of host defense and virus persistence (7, 9, 20, 26). Furthermore, it has been reported that HBeAg− patients respond better to standard IFN-α therapy than do HBeAg+ patients (47–51), suggesting that either HBeAg or its precursor, p22, might have an inhibitory effect that prevents the IFN therapy from working to its full potential in HBeAg-positive patients. Following this direction, we, for the first time, performed a comparative RNA-seq analysis of ISG transcriptome profiles in consecutive liver biopsy specimens from HBeAg+ and HBeAg− patients at baseline and the endpoint of 24 weeks of IFN-α therapy. We found a statistically stronger induction of ISGs in HBeAg− patients than in HBeAg+ patients (Fig. 4), indicating a potential inhibitory effect of HBeAg and/or p22 on IFN signaling. Furthermore, a cell-based ISG56 promoter reporter assay revealed that the intracellular p22 but not the exogenously treated HBeAg is able to suppress IFN-α activity (Fig. 5A), and the mRNA levels of MxA and ISG56 were found to be reduced in the presence of p22 (Fig. 5B), further confirming that p22 inhibits IFN-α-induced JAK-STAT signaling and ISG expression. In addition, infection of HepG2-NTCP12 cells with the precore-null virus followed by IFN-α treatment led to similar levels of ISG expression (MxA, ISG56, OAS2, IRF9, and ISG20) as in uninfected control cells, while the ISG induction levels were significantly reduced in the wild-type-HBV-infected cells, which validated the role of p22 in antagonizing IFN signaling in a more physiologically relevant setting (Fig. 6).
In order to elucidate the mechanism of p22-mediated inhibition of IFN signaling, we examined the effect of p22 on cellular STAT1/2 phosphorylation and nuclear translocation upon IFN-α treatment. The results demonstrated that p22 does not affect the IFN-α-activated tyrosine phosphorylation of STAT1/2 but significantly downregulates the nuclear translocation of pSTAT1/2 (Fig. 7). We first performed a coimmunoprecipitation assay of p22 and pSTAT1/2 but did not detect obvious interaction between p22 and pSTAT1/2 (data not shown). Considering that pSTAT1 engages a subset of nuclear localization signal (NLS) receptors, including Kα1, for nuclear entry to bind to the ISRE for activation of ISG expression, we next examined the interaction between p22 and Kα1. The results demonstrate that p22 efficiently binds to Kα1 and, thus, competes with pSTAT1 to interact with the same receptor and prevents the nuclear translocation of pSTAT1 (Fig. 8). As expected, the NLS-containing CTD domain of p22 is responsible for Kα1 binding and the deletion of the CTD abrogates the nuclear transportation of p22 and the inhibitory effect of p22 on IFN-α signaling (Fig. 9). Thus, we conclude that HBV p22 inhibits the IFN-α-elicited JAK-STAT pathway by blocking pSTAT nuclear translocation through competing in Kα1 binding (Fig. 10).
FIG 10.

Model of action of p22-mediated inhibition of IFN-α signaling. HBV virion infects hepatocyte via sodium-taurocholate cotransporting polypeptide (NTCP)-mediated entry, followed by nuclear import of nucleocapsid and the establishment of cccDNA episome. cccDNA serves as the transcription template to produce viral mRNAs. During the translation of precore protein from the 3.5-kb precore mRNA, the initially translated N-terminal signal peptide is recognized by the cellular signal recognition particle (SRP) receptor and directed to the rough ER. After the cleavage of signal peptide by ER-associated signal peptidase (SPase), the translation resumes and p22 is synthesized and enters the ER lumen. Subsequently, p22 is sorted into the Golgi apparatus, where the CTD is cleaved off by Golgi apparatus-resident endopeptidase furin to generate the secreted HBeAg/p17. In the meantime, a portion of p22 is retrotranslocated into the cytosol from the ER and can be further imported into the nuclear compartment due to the presence of the NLS motif on the CTD domain, which is recognized by cellular nuclear transportation receptor karyopherin α1 (Kα1). When the cell receives IFN-α treatment, the cytoplasmic p22 can blunt the IFN-α-elicited JAK-STAT pathway through competing with the binding of Kα1 with activated pSTAT, leading to downregulated IFN signaling and ISG expression. This mode of action represents a viral strategy to evade the innate antiviral response. This scheme was illustrated by using the Biology Bundle of Motifolio Drawing Toolkits.
Prevention of nuclear import of activated STAT1 is employed by many other viruses to antagonize IFN-mediated antiviral innate defense (63). For instance, Ebola virus protein VP24 has been reported to inhibit STAT1 nuclear translocation by interacting with karyopherin α1, α5, and α6 (57, 64, 65). However, except for Kα1, we did not detect the binding of p22 with other karyopherin α proteins using coimmunoprecipitation (data not shown). The preferred interaction of p22 with Kα1 over other karyopherin α proteins may be attributed to the fact that the NLS of p22 possesses optimal affinity to Kα1’s binding sequence or structure and may partly explain the observed incomplete inhibition of pSTAT1 nuclear translocation (Fig. 7C). On the other hand, it may also ensure a selective inhibition of STAT nuclear translocation without altering the transport of other nuclear proteins essential for virus replication and cell viability.
It is worth noting that HBV core protein also exhibits inhibitory activity against IFN-α-induced activation of the ISG56 promoter and ISG production (Fig. 5), and it may also influence the responses to IFN-α therapy in hepatitis B patients. However, core is unable to inhibit the nuclear translocation of pSTAT1/2 (Fig. 7D), suggesting that core and p22 inhibit IFN-α signaling via distinct mechanisms. Although core shares the entire amino acid sequence with p22, including the CTD domain, core predominantly exists in a capsid structure where the CTD domains are localized on the interior side of the capsid shell and are inaccessible to karyopherins (53, 66). We thus reason that core protein may exert its inhibition of ISG expression in the nucleus. It has been shown that HBV core can shuttle between the cytoplasm and nucleus via its NLS and nuclear export signal (NES) (16, 67, 68), and the nuclear core plays a role in regulating host gene expression and viral episomal integrity (69, 70). Interestingly, a substantial portion of p22 is also found in the nucleus (Fig. 3) (15). In future studies, it would be of interest to investigate the potential inhibitory effect of nuclear p22 and/or core on ISG expression at the late stages of the JAK-STAT cascade, such as competitive binding to the conserved ISRE motif or specific promoters of ISGs, leading to decreased transcription of the latter or affecting the epigenetic profiles of the ISG promoters negatively. In addition, considering that the CTD of core harbors an array of serine-threonine kinase phosphoacceptor sites where the dynamic phosphorylation and dephosphorylation events differentially regulate core functions, including nuclear import (68, 71–73), the potential phosphorylation of the CTD of p22 and its effects on p22 nuclear translocation and p22-mediated blockage of the JAK-STAT pathway await further investigations.
Taken together, our study gives a further inkling into the biological functions of the enigmatic intracellular HBV precore protein in viral pathogenesis. Though precore or HBeAg is not absolutely required for HBV infection, a growing body of evidence suggests that it serves as an immunomodulator to maintain persistent HBV infection and the intracellular precore (p22) mainly antagonizes the innate immune responses (7, 20, 26). Therefore, precore and HBeAg may hold promise for being considered as additional antiviral targets in developing novel hepatitis B therapeutics or, at least, in improving the effectiveness of IFN-based immunomodulatory therapies.
MATERIALS AND METHODS
Plasmids.
HBV replication-competent plasmid pHBV1.3, which contains a 1.3-mer replicon of the HBV genome, and pCMVHBV, which transcribes HBV pgRNA under the control of a human cytomegalovirus immediate early (CMV-IE) promoter, have been described previously (74–77). The core-null HBV plasmid pHBV1.3ΔC is a 1.3-mer HBV plasmid with mutation of the start codon (ATG to ATA) of the core protein open reading frame (ORF) (provided by Robert Lanford) (78). The precore/HBeAg-null HBV plasmid pCMVHBVΔe-A1814T was made by replacing the sequence between the RsrII and AflII restriction sites on pCMVHBV with a synthetic sequence containing the precore start codon mutation (AUG to TTG [A1814T]). The plasmid pTREHBV-HAe supports the replication of the HBV genome under an inducible CMV promoter; it has been previously used together with plasmid pTet-off (Clontech) to generate the HepBHAe82 stable cell line, which produces HA-tagged HBeAg (HA-HBeAg) in a cccDNA-dependent manner (52). Plasmid pTREHBVΔHAe-G1896A was made by replacing the sequence between the SacI and BspEI restriction sites on pTREHBV-HAe with a synthetic sequence containing the precore stop codon mutation G1896A. HBV protein expression plasmids, including pcHBc, expressing HBV core protein, pcHBe (also known as p22), encoding HBV precore and HBeAg, and pcHA-HBe (also known as HA-p22), expressing HA-tagged HBV precore and HBeAg, have been described in our previous publication (52). Plasmid pcHBeΔSP was constructed by deleting the N-terminal signal peptide (SP; aa 2 to 19)-coding sequence from pcHBe. Plasmid pcHBeC−7Q, expressing precore with the cysteine (C) of aa −7 in the precore domain being changed to glutamine (Q), was made through site-directed mutagenesis of pcHBe. Plasmid HA-p22ΔCTD was constructed by deleting the HBV precore/core C-terminal domain (CTD; aa 149 to 183)-coding sequence from plasmid HA-p22. Plasmid pLMS, expressing the HBV large (L), middle (M), and small (S) envelope proteins, was provided by Youhua Xie (79). Plasmid pCMV-FLAG-Pol, expressing 3×FLAG-tagged HBV Pol, was provided by Wang-Shick Ryu (80). Plasmid FL1-154HBx, expressing FLAG-tagged HBx, was provided by Michael Bouchard (81). Firefly luciferase reporter plasmid ISG56-Luc was provided by Kui Li (82). Plasmid expressing N-terminal FLAG-tagged karyopherin-α1 to -6 (FLAG-Kα1 − 6) and plasmid expressing HA-tagged Ebola virus VP24 (HA-VP24) were provided by Christopher Basler through BEI Resources (57, 65).
Cell lines.
HepG2 and 293T cells were purchased from ATCC and cultured in Dulbecco's modified Eagle medium (DMEM)–F-12 medium (Gibco) supplemented with 10% fetal bovine serum, 100 U/ml penicillin, and 100 μg/ml streptomycin. The tetracycline-inducible HBV stable cell line HepBHAe82 was established previously (52) and maintained in the same way as HepG2 cells, except for the addition of 1 μg/ml tetracycline (Tet) and 400 μg/ml G418. When required, the culture medium was switched to Tet-free to initiate HBV replication and cccDNA-dependent HA-HBeAg expression in HepBHAe82 cells. The HepHA-HBe4 stable cell line, which constitutively expresses HA-HBeAg (52), and the HepG2-NTCP12 cells, which support HBV infection (83), were described previously.
Transfection.
Cells were seeded in the collagen-coated plate and were transfected with the plasmid(s) indicated in the figures by using Lipofectamine 3000 (Life Technologies) according to the manufacturer’s directions.
HBV RNA and DNA analyses.
HBV total RNA, cytoplasmic encapsidated pgRNA, and core DNA were isolated and detected by Northern and Southern blotting as previously described (77, 84, 85). Quantitative PCR (qPCR) analysis of HBV core DNA was conducted according to a method in the literature (83).
Preparation of virus inoculum.
HepG2 cells were transfected with wild-type pCMVHBV or precore-null pCMVHBVΔe-A1814T or with pTREHBV-HAe or precore-null pTREHBVΔHAe-G1896A plasmid, plus pTet-off and pLMS, in a 2:1:2 ratio, and the cells were maintained in tetracycline-free medium posttransfection. Supernatant from the transfected cells was collected every other day over an 8-day period. The pooled supernatant was mixed with polyethylene glycol 8000 (PEG 8000) powder (final concentration of 10%) and gently rotated at 4°C overnight. HBV particles were then precipitated by centrifugation at 1,000 × g for 30 min at 4°C and redissolved in serum-free DMEM–F-12 medium with 1% volume of the original supernatant samples. The concentrated virus stocks of wild-type or two different kinds of precore-null viruses were aliquoted and stored at −80°C. HBV virus genome equivalents (VGE) were determined using HBV core DNA qPCR and a standard curve using titrated HBV plasmid (83).
HBV infection.
HepG2-NTCP12 cells were seeded into collagen-coated 96-well plates at a density of 6 × 105 cells/well and cultured in regular DMEM–F-12 medium overnight, and then the culture medium was switched to Cellartis hepatocyte maintenance medium (TaKaRa). After 24 h, the cells were incubated with HBV inoculum at 500 VGE per cell in primary hepatocyte maintenance medium (PMM) (described in detail below) containing 4% PEG 8000. The cells with HBV inoculation were transferred to a CO2 incubator at 37°C. Twenty-four hours later, the HBV inocula were removed and the infected cells were maintained in regular PMM, specifically, Williams E medium supplemented with 10% fetal bovine serum (FBS), 5 μg/ml transferrin, 10 ng/ml human epidermal growth factor (hEGF), 3 μg/ml insulin, 2 mM l-glutamine, 18 μg/ml hydrocortisone, 40 ng/ml dexamethasone, 5 ng/ml sodium selenite, 2% dimethyl sulfoxide (DMSO; cell culture grade), 100 U/ml penicillin, and 100 μg/ml streptomycin for 7 days before harvest. Where indicated, the infected cells were treated with lamivudine (3TC; 10 μM) to block de novo synthesis of HBV DNA replication.
Promoter reporter assay.
HepG2 cells were plated in a 96-well plate and transfected with reporter plasmid ISG56-Luc plus control vector or vectors expressing the indicated gene of interest (see the figures) individually. Renilla luciferase reporter plasmid pRL-CMV was cotransfected to normalize the transfection efficiency across different transfected wells. Each transfection received the same amount of total DNA (200 ng/well), with ISG56-Luc, the gene of interest, and pRL-CMV in a 2:2:1 ratio. Recombinant IFN-α2a was purchased from PBL Biomedical Laboratories, and the treatment was carried out at 1,000 IU/ml for 18 h. Recombinant HBeAg was procured commercially (product number 8915; Virostat). Furin inhibitor I was purchased from Calbiochem. Firefly luciferase and Renilla luciferase activities were measured by using the dual-luciferase assay kit (Promega) and the BioTek Synergy 2 multimode plate reader.
Cellular mRNA qPCR.
DNase I-treated total cellular RNA was used to generate cDNA by using SuperScript III reverse transcriptase (Life Technologies). Real-time PCR was performed with SYBR green master mix (Roche) and the LightCycler 96 system (Roche) to detect MxA, ISG56, OAS2, IRF9, and ISG20 mRNA by using gene-specific primers (Table 2). The relative expression levels of HBV RNA were normalized to the level of β-actin from the same samples. The qPCR setting was 0.8 μM primers, with annealing and extension at 64°C for 45 cycles.
TABLE 2.
Primers for cellular mRNA qPCR
| Gene | Primer |
|
|---|---|---|
| Forward (5′→3′) | Reverse (5′→3′) | |
| MxA | TGATCCAGCTGCTGCATCCC | GGCGCACCTTCTCCTCATAC |
| ISG56 | TCTCAGAGGAGCCTGGCTAAG | CCACACTGTATTTGGTGTCTAGG |
| OAS2 | AACTGCTTCCGACAATCAAC | CCTCCTTCTCCCTCCAAAA |
| IRF9 | TTCAGGATGGCCTCAGGCAAAGTA | GAACAAGTCTATTTCCATGGAGACG |
| ISG20 | TGAGGGAGAGATCACCGATT | TAGCCGCTCATGTCCTCTTT |
| β-Actin | CCTGGCACCCAGCACAATGA | ACTAAGTCATAGTCCGCCTAGA |
Subcellular fractionation.
Cytoplasmic and nuclear fractionations were prepared by using the Qiagen cell compartment kit (catalog number 37502; Qiagen); microsomal fractionations were performed by using the BioVision microsome isolation kit (catalog number K249-50; BioVision) according to the manufacturer’s directions.
Immunoblotting.
Cells were lysed in 1× Laemmli buffer and denatured at 95°C for 10 min. Cell lysate was resolved in a 12% SDS-PAGE gel, and proteins were transferred onto an Immobilon-FL polyvinylidene difluoride (PVDF) membrane (Millipore). The membranes were blocked with Western Breeze blocker and probed with antibodies against aa 170 to 183 of precore/core (HBc170) (53), HA tag (catalog number H3663, Sigma-Aldrich, or catalog number 3724, Cell Signaling Technology), FLAG tag (catalog number F3165, Sigma-Aldrich), annexin I (catalog number sc-65872, Santa Cruz Biotechnology), lamin A/C (catalog number sc-20681, Santa Cruz Biotechnology), protein disulfide isomerase (PDI) (catalog number 2446, Cell Signaling Technology), β-actin (catalog number sc-4778, Santa Cruz Biotechnology, or catalog number MAB1501, Millipore), STAT1 (catalog number 9175, Cell Signaling Technology), pSTAT1 (Tyr701) (catalog number 7649, Cell Signaling Technology), and pSTAT2 (catalog number 4441, Cell Signaling Technology). Bound antibodies were revealed by IRDye secondary antibodies and visualized using the LI-COR Odyssey system. Band intensity was measured using LI-COR Image Studio software.
Coimmunoprecipitation assay.
HepG2 or 293T cells were cotransfected with HA-p22, HA-p22ΔCTD, or HA-VP24 and FLAG-Kα1 for 6 days. The harvested cells were lysed on ice with cell lysis buffer containing 1% NP-40, 10 mM Tris-HCl (pH 7.5), 1 mM EDTA, 50 mM NaCl, protease inhibitor cocktail, and Benzonase. After centrifugation to remove the cell debris, the clarified cell lysates were incubated with EZview red anti-HA affinity gel (Sigma-Aldrich) or protein A/G beads precoated with anti-pSTAT1 or anti-pSTAT2 antibody at 4°C overnight with gentle rotation. The beads were spun down the next day and resuspended gently with cold low-salt wash buffer (50 mM Tris-HCl [pH 7.4], 5 mM EDTA, 150 mM NaCl, and protease inhibitors) three times at 4°C, followed by one wash with high-salt buffer (50 mM Tris-HCl [pH 7.4], 5 mM EDTA, 500 mM NaCl, and protease inhibitors) and a desalting step with 20 mM Tris-HCl, pH 7.4. The washed beads were resuspended in Laemmli buffer without dithiothreitol (DTT) and spun down at high speed, followed by collection of the supernatant, addition of DTT, and immunoblotting following the technique described above.
Immunofluorescence assay.
Cells were fixed with 4% paraformaldehyde for 20 min and permeabilized with 0.5% Triton X-100 in phosphate-buffered saline (PBS) for 60 min at room temperature. Cells were subsequently blocked with blocking buffer (10% FBS plus 2% bovine serum albumin [BSA] in 1× PBS) for 60 min at room temperature and then incubated with anti-HA antibody (catalog number 3724, Cell Signaling Technology, or catalog number H3663, Sigma-Aldrich) or anti-HBcAg antibody (catalog number B0586, Dako) diluted in the above-mentioned blocking buffer overnight at 4°C. After being washed with PBS, the cells were stained with Alexa Fluor 594 dye-conjugated secondary antibody (Life Technologies), and the nuclei were counterstained with DAPI (4′,6-diamidino-2-phenylindole) for 60 min at room temperature. Finally, the cells were washed with PBS and subjected to Olympus FV1000 MPE confocal microscopy analysis with the 60× or 20× objective. Images were analyzed using FV10-ASW 3.0 Viewer software.
Enzyme-linked immunoassay.
The cytoplasmic HBV capsid particles were resolved in native agarose gel by electrophoresis and transferred onto nitrocellulose membrane, followed by capsid enzyme immunoassay assay (EIA) using antibodies against HBV core (catalog number B0586, Dako) as previously described (86, 87). Untagged HBeAg in the supernatant was detected by using a chemiluminescence immunoassay (CLIA) kit (catalog number CL0312-2, Autobio Diagnostics). HA-HBeAg in the supernatant was detected by an in-house CLIA as previously described (52).
Statistical analysis.
All data were analyzed by using GraphPad Prism 5 and expressed as mean values ± standard deviations (SD) unless otherwise specified. Student’s t test was used to determine the statistical significance for in vitro experiments. A P value of <0.001 was considered statistically significant.
Patients.
Between 2015 and 2016, patients with chronic HBV infection at Huashan Hospital of Fudan University (Shanghai, China) were evaluated. Individuals with concurrent hepatitis C, hepatitis D, HIV, autoimmune liver disease, or alcoholic liver disease were excluded. Twenty-one treatment-naive patients (11 HBeAg+ and 10 HBeAg−) with liver biopsies performed at baseline were enrolled. HBV sequence analysis revealed that all of the HBeAg− patients were infected with virus encoding the precore stop codon mutation (G1896A). Both HBeAg+ and HBeAg− patients received IFN-α monotherapy, and a second liver biopsy was performed at 24 weeks. In the present study, the upper limit of normal (ULN) for alanine transaminase (ALT) was set at 50 U liter−1. Patient characteristics are summarized in Table 1. All human subjects were recruited upon obtaining informed written consent. The study was approved by the Institutional Ethics Committee for human studies at Huashan Hospital (IRB numbers 2016-123 and 2018-131).
Library construction for RNA-seq, sequencing procedures, and data analysis.
Total RNA of liver biopsy specimens was extracted by using the RNeasy minikit (Qiagen). Strand-specific libraries were prepared using the TruSeq stranded total RNA sample preparation kit (Illumina), following the manufacturer’s instructions. Briefly, rRNA was removed from total RNA by Ribo-Zero rRNA removal beads. Following purification, the mRNA was fragmented into small pieces using divalent cations at under 94°C for 8 min. The RNA fragments were copied into the first-strand cDNA by using reverse transcriptase and random primers, followed by second-strand cDNA synthesis using DNA polymerase I and RNase H. These cDNA fragments then went through an end repair process, the addition of a single A base, and then ligation of the adapters. The products were then purified and enriched with PCR to create the final cDNA library. Purified libraries were quantified by using the Qubit 2.0 fluorometer (Life Technologies) and validated by using the Agilent 2100 bioanalyzer to confirm the insert size and calculate the mole concentration. Clusters were generated by cBot with the library diluted to 10 pM and then were sequenced on the Illumina HiSeq 2500. The library construction and sequencing were performed at Shanghai Biotechnology Corporation.
Raw reads for sequencing were preprocessed by filtering out rRNA reads, sequencing adapters, short-fragment reads, and other low-quality reads. TopHat version 2.1.0 was used to map the cleaned reads to the human h19 reference genome with two mismatches (88). After genome mapping, Cufflinks version 2.1.1 was run with a reference annotation to generate fragments per kilobase per million (FPKM) values for known gene models, and the differentially expressed genes (DEGs) were identified using Cuffdiff (89). The P value significance threshold in multiple tests was set by the false discovery rate (FDR) (90). The fold changes were also estimated according to the FPKM in each sample. The differentially expressed ISG genes were selected using the following filter criteria: P value of ≤0.05 and fold change of ≥2.
ACKNOWLEDGMENTS
We thank Christopher Basler, Michael Bouchard, Robert Lanford, Kui Li, Wang-Shick Ryu, and Youhua Xie for providing plasmids to this study.
This study is supported by the U.S. National Institutes of Health (grants number R01AI110762, R01AI123271, and R01AI134818 to H.G.), the National Natural Science Foundation of China (grants number 81672009, 81471933, 81670528, and 81400625 to J.Z.), and the Major Science and Technology Special Project of China (grants number 2017ZX10202202 and 2017ZX10202203-007 to J.Z.). H.G. is the Showalter Scholar of Indiana University School of Medicine. B.M. received the Cagiantas Scholarship of the Indiana University School of Medicine and the Merilyn Hester Scholarship of the Indiana University School of Medicine Simon Cancer Center.
REFERENCES
- 1.Seeger C, Mason WS. 2000. Hepatitis B virus biologies. Microbiol Mol Biol Rev 64:51–68. doi: 10.1128/MMBR.64.1.51-68.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Block TM, Guo H, Guo JT. 2007. Molecular virology of hepatitis B virus for clinicians. Clin Liver Dis 11:685–706. doi: 10.1016/j.cld.2007.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Alter H, Block T, Brown N, Brownstein A, Brosgart C, Chang KM, Chen PJ, Chisari FV, Cohen C, El-Serag H, Feld J, Gish R, Glenn J, Greten T, Guo H, Guo JT, Hoshida Y, Hu J, Kowdley KV, Li W, Liang J, Locarnini S, Lok AS, Mason W, McMahon B, Mehta A, Perrillo R, Revill P, Rice CM, Rinaudo J, Schinazi R, Seeger C, Shetty K, Tavis J, Zoulim F. 2018. A research agenda for curing chronic hepatitis B virus infection. Hepatology 67:1127–1131. doi: 10.1002/hep.29509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nassal M. 2015. HBV cccDNA: viral persistence reservoir and key obstacle for a cure of chronic hepatitis B. Gut 64:1972–1984. doi: 10.1136/gutjnl-2015-309809. [DOI] [PubMed] [Google Scholar]
- 5.Guo JT, Guo H. 2015. Metabolism and function of hepatitis B virus cccDNA: implications for the development of cccDNA-targeting antiviral therapeutics. Antiviral Res 122:91–100. doi: 10.1016/j.antiviral.2015.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chisari FV, Isogawa M, Wieland SF. 2010. Pathogenesis of hepatitis B virus infection. Pathol Biol (Paris) 58:258–266. doi: 10.1016/j.patbio.2009.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tsai KN, Kuo CF, Ou JJ. 2018. Mechanisms of hepatitis B virus persistence. Trends Microbiol 26:33–42. doi: 10.1016/j.tim.2017.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hong X, Kim ES, Guo H. 2017. Epigenetic regulation of hepatitis B virus covalently closed circular DNA: implications for epigenetic therapy against chronic hepatitis B. Hepatology 66:2066–2077. doi: 10.1002/hep.29479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mitra B, Thapa RJ, Guo H, Block TM. 2018. Host functions used by hepatitis B virus to complete its life cycle: Implications for developing host-targeting agents to treat chronic hepatitis B. Antiviral Res 158:185–198. doi: 10.1016/j.antiviral.2018.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nassal M. 2008. Hepatitis B viruses: reverse transcription a different way. Virus Res 134:235–249. doi: 10.1016/j.virusres.2007.12.024. [DOI] [PubMed] [Google Scholar]
- 11.Hu J, Seeger C. 2015. Hepadnavirus genome replication and persistence. Cold Spring Harb Perspect Med 5:a021386. doi: 10.1101/cshperspect.a021386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liu S, Zhou B, Valdes JD, Sun J, Guo H. 2019. Serum hepatitis B virus RNA: a new potential biomarker for chronic hepatitis B virus infection. Hepatology 69:1816–1827. doi: 10.1002/hep.30325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tong S, Revill P. 2016. Overview of hepatitis B viral replication and genetic variability. J Hepatol 64:S4–S16. doi: 10.1016/j.jhep.2016.01.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Garcia PD, Ou JH, Rutter WJ, Walter P. 1988. Targeting of the hepatitis B virus precore protein to the endoplasmic reticulum membrane: after signal peptide cleavage translocation can be aborted and the product released into the cytoplasm. J Cell Biol 106:1093–1104. doi: 10.1083/jcb.106.4.1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ou JH, Yeh CT, Yen TS. 1989. Transport of hepatitis B virus precore protein into the nucleus after cleavage of its signal peptide. J Virol 63:5238–5243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Eckhardt SG, Milich DR, McLachlan A. 1991. Hepatitis B virus core antigen has two nuclear localization sequences in the arginine-rich carboxyl terminus. J Virol 65:575–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yeh CT, Liaw YF, Ou JH. 1990. The arginine-rich domain of hepatitis B virus precore and core proteins contains a signal for nuclear transport. J Virol 64:6141–6147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ito K, Kim KH, Lok AS, Tong S. 2009. Characterization of genotype-specific carboxyl-terminal cleavage sites of hepatitis B virus e antigen precursor and identification of furin as the candidate enzyme. J Virol 83:3507–3517. doi: 10.1128/JVI.02348-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.DiMattia MA, Watts NR, Stahl SJ, Grimes JM, Steven AC, Stuart DI, Wingfield PT. 2013. Antigenic switching of hepatitis B virus by alternative dimerization of the capsid protein. Structure 21:133–142. doi: 10.1016/j.str.2012.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Milich D, Liang TJ. 2003. Exploring the biological basis of hepatitis B e antigen in hepatitis B virus infection. Hepatology 38:1075–1086. doi: 10.1053/jhep.2003.50453. [DOI] [PubMed] [Google Scholar]
- 21.Ou JH. 1997. Molecular biology of hepatitis B virus e antigen. J Gastroenterol Hepatol 12:S178–S187. doi: 10.1111/j.1440-1746.1997.tb00499.x. [DOI] [PubMed] [Google Scholar]
- 22.Milich DR, Chen MK, Hughes JL, Jones JE. 1998. The secreted hepatitis B precore antigen can modulate the immune response to the nucleocapsid: a mechanism for persistence. J Immunol 160:2013–2021. [PubMed] [Google Scholar]
- 23.Chen MT, Billaud JN, Sallberg M, Guidotti LG, Chisari FV, Jones J, Hughes J, Milich DR. 2004. A function of the hepatitis B virus precore protein is to regulate the immune response to the core antigen. Proc Natl Acad Sci U S A 101:14913–14918. doi: 10.1073/pnas.0406282101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Park J-J, Wong DK, Wahed AS, Lee WM, Feld JJ, Terrault N, Khalili M, Sterling RK, Kowdley KV, Bzowej N, Lau DT, Kim WR, Smith C, Carithers RL, Torrey KW, Keith JW, Levine DL, Traum D, Ho S, Valiga ME, Johnson GS, Doo E, Lok AS, Chang K-M, Hepatitis B Research Network. 2016. Hepatitis B virus-specific and global T-cell dysfunction in chronic hepatitis B. Gastroenterology 150:684–695.e5. doi: 10.1053/j.gastro.2015.11.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Milich DR, Jones JE, Hughes JL, Price J, Raney AK, McLachlan A. 1990. Is a function of the secreted hepatitis B e antigen to induce immunologic tolerance in utero? Proc Natl Acad Sci U S A 87:6599–6603. doi: 10.1073/pnas.87.17.6599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Walsh R, Locarnini S. 2012. Hepatitis B precore protein: pathogenic potential and therapeutic promise. Yonsei Med J 53:875–885. doi: 10.3349/ymj.2012.53.5.875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tian Y, Kuo CF, Akbari O, Ou JH. 2016. Maternal-derived hepatitis B virus e antigen alters macrophage function in offspring to drive viral persistence after vertical transmission. Immunity 44:1204–1214. doi: 10.1016/j.immuni.2016.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jegaskanda S, Ahn SH, Skinner N, Thompson AJ, Ngyuen T, Holmes J, De Rose R, Navis M, Winnall WR, Kramski M, Bernardi G, Bayliss J, Colledge D, Sozzi V, Visvanathan K, Locarnini SA, Kent SJ, Revill PA. 2014. Downregulation of interleukin-18-mediated cell signaling and interferon gamma expression by the hepatitis B virus e antigen. J Virol 88:10412–10420. doi: 10.1128/JVI.00111-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Visvanathan K, Skinner NA, Thompson AJ, Riordan SM, Sozzi V, Edwards R, Rodgers S, Kurtovic J, Chang J, Lewin S, Desmond P, Locarnini S. 2007. Regulation of Toll-like receptor-2 expression in chronic hepatitis B by the precore protein. Hepatology 45:102–110. doi: 10.1002/hep.21482. [DOI] [PubMed] [Google Scholar]
- 30.Lang T, Lo C, Skinner N, Locarnini S, Visvanathan K, Mansell A. 2011. The hepatitis B e antigen (HBeAg) targets and suppresses activation of the Toll-like receptor signaling pathway. J Hepatol 55:762–769. doi: 10.1016/j.jhep.2010.12.042. [DOI] [PubMed] [Google Scholar]
- 31.Wilson R, Warner N, Ryan K, Selleck L, Colledge D, Rodgers S, Li K, Revill P, Locarnini S. 2011. The hepatitis B e antigen suppresses IL-1β-mediated NF-κB activation in hepatocytes. J Viral Hepat 18:e499–e507. doi: 10.1111/j.1365-2893.2011.01484.x. [DOI] [PubMed] [Google Scholar]
- 32.Yang CY, Kuo TH, Ting LP. 2006. Human hepatitis B viral e antigen interacts with cellular interleukin-1 receptor accessory protein and triggers interleukin-1 response. J Biol Chem 281:34525–34536. doi: 10.1074/jbc.M510981200. [DOI] [PubMed] [Google Scholar]
- 33.Sato S, Li K, Kameyama T, Hayashi T, Ishida Y, Murakami S, Watanabe T, Iijima S, Sakurai Y, Watashi K, Tsutsumi S, Sato Y, Akita H, Wakita T, Rice CM, Harashima H, Kohara M, Tanaka Y, Takaoka A. 2015. The RNA sensor RIG-I dually functions as an innate sensor and direct antiviral factor for hepatitis B virus. Immunity 42:123–132. doi: 10.1016/j.immuni.2014.12.016. [DOI] [PubMed] [Google Scholar]
- 34.Lucifora J, Durantel D, Testoni B, Hantz O, Levrero M, Zoulim F. 2010. Control of hepatitis B virus replication by innate response of HepaRG cells. Hepatology 51:63–72. doi: 10.1002/hep.23230. [DOI] [PubMed] [Google Scholar]
- 35.Luangsay S, Gruffaz M, Isorce N, Testoni B, Michelet M, Faure-Dupuy S, Maadadi S, Ait-Goughoulte M, Parent R, Rivoire M, Javanbakht H, Lucifora J, Durantel D, Zoulim F. 2015. Early inhibition of hepatocyte innate responses by hepatitis B virus. J Hepatol 63:1314–1322. doi: 10.1016/j.jhep.2015.07.014. [DOI] [PubMed] [Google Scholar]
- 36.Liu Y, Li J, Chen J, Li Y, Wang W, Du X, Song W, Zhang W, Lin L, Yuan Z. 2015. Hepatitis B virus polymerase disrupts K63-linked ubiquitination of STING to block innate cytosolic DNA-sensing pathways. J Virol 89:2287–2300. doi: 10.1128/JVI.02760-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang H, Ryu WS. 2010. Hepatitis B virus polymerase blocks pattern recognition receptor signaling via interaction with DDX3: implications for immune evasion. PLoS Pathog 6:e1000986. doi: 10.1371/journal.ppat.1000986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kumar M, Jung SY, Hodgson AJ, Madden CR, Qin J, Slagle BL. 2011. Hepatitis B virus regulatory HBx protein binds to adaptor protein IPS-1 and inhibits the activation of beta interferon. J Virol 85:987–995. doi: 10.1128/JVI.01825-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang X, Li Y, Mao A, Li C, Li Y, Tien P. 2010. Hepatitis B virus X protein suppresses virus-triggered IRF3 activation and IFN-beta induction by disrupting the VISA-associated complex. Cell Mol Immunol 7:341–348. doi: 10.1038/cmi.2010.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wei C, Ni C, Song T, Liu Y, Yang X, Zheng Z, Jia Y, Yuan Y, Guan K, Xu Y, Cheng X, Zhang Y, Yang X, Wang Y, Wen C, Wu Q, Shi W, Zhong H. 2010. The hepatitis B virus X protein disrupts innate immunity by downregulating mitochondrial antiviral signaling protein. J Immunol 185:1158–1168. doi: 10.4049/jimmunol.0903874. [DOI] [PubMed] [Google Scholar]
- 41.Jiang J, Tang H. 2010. Mechanism of inhibiting type I interferon induction by hepatitis B virus X protein. Protein Cell 1:1106–1117. doi: 10.1007/s13238-010-0141-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yu S, Chen J, Wu M, Chen H, Kato N, Yuan Z. 2010. Hepatitis B virus polymerase inhibits RIG-I- and Toll-like receptor 3-mediated beta interferon induction in human hepatocytes through interference with interferon regulatory factor 3 activation and dampening of the interaction between TBK1/IKKepsilon and DDX3. J Gen Virol 91:2080–2090. doi: 10.1099/vir.0.020552-0. [DOI] [PubMed] [Google Scholar]
- 43.Cheng X, Xia Y, Serti E, Block PD, Chung M, Chayama K, Rehermann B, Liang TJ. 2017. Hepatitis B virus evades innate immunity of hepatocytes but activates cytokine production by macrophages. Hepatology 66:1779–1793. doi: 10.1002/hep.29348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wieland SF, Chisari FV. 2005. Stealth and cunning: hepatitis B and hepatitis C viruses. J Virol 79:9369–9380. doi: 10.1128/JVI.79.15.9369-9380.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Janssen HL, van Zonneveld M, Senturk H, Zeuzem S, Akarca US, Cakaloglu Y, Simon C, So TM, Gerken G, de Man RA, Niesters HG, Zondervan P, Hansen B, Schalm SW, HBV 99-01 Study Group, Rotterdam Foundation for Liver Research. 2005. Pegylated interferon alfa-2b alone or in combination with lamivudine for HBeAg-positive chronic hepatitis B: a randomised trial. Lancet 365:123–129. doi: 10.1016/S0140-6736(05)17701-0. [DOI] [PubMed] [Google Scholar]
- 46.Lutgehetmann M, Bornscheuer T, Volz T, Allweiss L, Bockmann JH, Pollok JM, Lohse AW, Petersen J, Dandri M. 2011. Hepatitis B virus limits response of human hepatocytes to interferon-alpha in chimeric mice. Gastroenterology 140:2074–2083.e2. doi: 10.1053/j.gastro.2011.02.057. [DOI] [PubMed] [Google Scholar]
- 47.Heijtink RA, Janssen HL, Hop WC, Osterhaus AD, Schalm SW. 2001. Interferon-alpha therapy for chronic hepatitis B: early response related to pre-treatment changes in viral replication. J Med Virol 63:217–219. doi: 10.1002/1096-9071(200103)63:3<217::AID-JMV1003>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 48.Aikawa T, Kanai K, Kako M, Kawasaki T, Hino K, Iwabuchi S, Tsubouchi H, Takehira Y, Tsuda F, Okamoto H. 1995. Interferon-alpha 2a for chronic hepatitis B with e antigen or antibody: comparable antiviral effects on wild-type virus and precore mutant. J Viral Hepat 2:243–250. doi: 10.1111/j.1365-2893.1995.tb00036.x. [DOI] [PubMed] [Google Scholar]
- 49.Zhang X, Han Y, Lu Z, Gao J, Luo Z, Zhang D. 2001. Effect of multiple mutations in the core promoter and pre-core/core region of hepatitis B virus genome on the response to interferon in e antigen-positive chronic hepatitis B. J Gastroenterol Hepatol 16:393–398. doi: 10.1046/j.1440-1746.2001.02451.x. [DOI] [PubMed] [Google Scholar]
- 50.Kanai K, Kako M, Aikawa T, Hino K, Tsubouchi H, Takehira Y, Iwabuchi S, Kawasaki T, Tsuda F, Okamoto H, Miyakawa Y, Mayumi M. 1996. Core promoter mutations of hepatitis B virus for the response to interferon in e antigen-positive chronic hepatitis B. Am J Gastroenterol 91:2150–2156. [PubMed] [Google Scholar]
- 51.Kako M, Kanai K, Aikawa T, Iwabuchi S, Takehira Y, Kawasaki T, Tsubouchi H, Hino K, Tsuda F, Okamoto H, Miyakawa Y, Mayumi M. 1997. Response to interferon-alpha 2a in patients with e antigen-negative chronic hepatitis B. J Clin Gastroenterol 25:440–445. doi: 10.1097/00004836-199709000-00009. [DOI] [PubMed] [Google Scholar]
- 52.Cai D, Wang X, Yan R, Mao R, Liu Y, Ji C, Cuconati A, Guo H. 2016. Establishment of an inducible HBV stable cell line that expresses cccDNA-dependent epitope-tagged HBeAg for screening of cccDNA modulators. Antiviral Res 132:26–37. doi: 10.1016/j.antiviral.2016.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Guo H, Mao R, Block TM, Guo JT. 2010. Production and function of the cytoplasmic deproteinized relaxed circular DNA of hepadnaviruses. J Virol 84:387–396. doi: 10.1128/JVI.01921-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Schodel F, Peterson D, Zheng J, Jones JE, Hughes JL, Milich DR. 1993. Structure of hepatitis B virus core and e-antigen. A single precore amino acid prevents nucleocapsid assembly. J Biol Chem 268:1332–1337. [PubMed] [Google Scholar]
- 55.Duriez M, Rossignol JM, Sitterlin D. 2008. The hepatitis B virus precore protein is retrotransported from endoplasmic reticulum (ER) to cytosol through the ER-associated degradation pathway. J Biol Chem 283:32352–32360. doi: 10.1074/jbc.M807178200. [DOI] [PubMed] [Google Scholar]
- 56.Sadler AJ, Williams BR. 2008. Interferon-inducible antiviral effectors. Nat Rev Immunol 8:559–568. doi: 10.1038/nri2314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Reid SP, Leung LW, Hartman AL, Martinez O, Shaw ML, Carbonnelle C, Volchkov VE, Nichol ST, Basler CF. 2006. Ebola virus VP24 binds karyopherin alpha1 and blocks STAT1 nuclear accumulation. J Virol 80:5156–5167. doi: 10.1128/JVI.02349-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tong SP, Diot C, Gripon P, Li J, Vitvitski L, Trepo C, Guguen-Guillouzo C. 1991. In vitro replication competence of a cloned hepatitis B virus variant with a nonsense mutation in the distal pre-C region. Virology 181:733–737. doi: 10.1016/0042-6822(91)90908-T. [DOI] [PubMed] [Google Scholar]
- 59.Ulrich PP, Bhat RA, Kelly I, Brunetto MR, Bonino F, Vyas GN. 1990. A precore-defective mutant of hepatitis B virus associated with e antigen-negative chronic liver disease. J Med Virol 32:109–118. doi: 10.1002/jmv.1890320208. [DOI] [PubMed] [Google Scholar]
- 60.Duriez M, Thouard A, Bressanelli S, Rossignol JM, Sitterlin D. 2014. Conserved aromatic residues of the hepatitis B virus Precore propeptide are involved in a switch between distinct dimeric conformations and essential in the formation of heterocapsids. Virology 462-463:273–282. doi: 10.1016/j.virol.2014.06.013. [DOI] [PubMed] [Google Scholar]
- 61.Yan Z, Wu D, Hu H, Zeng J, Yu X, Xu Z, Zhou Z, Zhou X, Yang G, Young JAT, Gao L. 21 January 2019. Direct inhibition of hepatitis B virus e antigen by core protein allosteric modulator. Hepatology doi: 10.1002/hep.30514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kimura T, Ohno N, Terada N, Rokuhara A, Matsumoto A, Yagi S, Tanaka E, Kiyosawa K, Ohno S, Maki N. 2005. Hepatitis B virus DNA-negative dane particles lack core protein but contain a 22-kDa precore protein without C-terminal arginine-rich domain. J Biol Chem 280:21713–21719. doi: 10.1074/jbc.M501564200. [DOI] [PubMed] [Google Scholar]
- 63.Fleming SB. 2016. Viral inhibition of the IFN-induced JAK/STAT signalling pathway: development of live attenuated vaccines by mutation of viral-encoded IFN-antagonists. Vaccines (Basel) 4:E23. doi: 10.3390/vaccines4030023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Xu W, Edwards MR, Borek DM, Feagins AR, Mittal A, Alinger JB, Berry KN, Yen B, Hamilton J, Brett TJ, Pappu RV, Leung DW, Basler CF, Amarasinghe GK. 2014. Ebola virus VP24 targets a unique NLS binding site on karyopherin alpha 5 to selectively compete with nuclear import of phosphorylated STAT1. Cell Host Microbe 16:187–200. doi: 10.1016/j.chom.2014.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Reid SP, Valmas C, Martinez O, Sanchez FM, Basler CF. 2007. Ebola virus VP24 proteins inhibit the interaction of NPI-1 subfamily karyopherin alpha proteins with activated STAT1. J Virol 81:13469–13477. doi: 10.1128/JVI.01097-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zlotnick A, Cheng N, Stahl SJ, Conway JF, Steven AC, Wingfield PT. 1997. Localization of the C terminus of the assembly domain of hepatitis B virus capsid protein: implications for morphogenesis and organization of encapsidated RNA. Proc Natl Acad Sci U S A 94:9556–9561. doi: 10.1073/pnas.94.18.9556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Rabe B, Delaleau M, Bischof A, Foss M, Sominskaya I, Pumpens P, Cazenave C, Castroviejo M, Kann M. 2009. Nuclear entry of hepatitis B virus capsids involves disintegration to protein dimers followed by nuclear reassociation to capsids. PLoS Pathog 5:e1000563. doi: 10.1371/journal.ppat.1000563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Li HC, Huang EY, Su PY, Wu SY, Yang CC, Lin YS, Chang WC, Shih C. 2010. Nuclear export and import of human hepatitis B virus capsid protein and particles. PLoS Pathog 6:e1001162. doi: 10.1371/journal.ppat.1001162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Guo Y, Kang W, Lei X, Li Y, Xiang A, Liu Y, Zhao J, Zhang J, Yan Z. 2012. Hepatitis B viral core protein disrupts human host gene expression by binding to promoter regions. BMC Genomics 13:563. doi: 10.1186/1471-2164-13-563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lucifora J, Xia Y, Reisinger F, Zhang K, Stadler D, Cheng X, Sprinzl MF, Koppensteiner H, Makowska Z, Volz T, Remouchamps C, Chou WM, Thasler WE, Huser N, Durantel D, Liang TJ, Munk C, Heim MH, Browning JL, Dejardin E, Dandri M, Schindler M, Heikenwalder M, Protzer U. 2014. Specific and nonhepatotoxic degradation of nuclear hepatitis B virus cccDNA. Science 343:1221–1228. doi: 10.1126/science.1243462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ning X, Basagoudanavar SH, Liu K, Luckenbaugh L, Wei D, Wang C, Wei B, Zhao Y, Yan T, Delaney W, Hu J. 2017. Capsid phosphorylation state and hepadnavirus virion secretion. J Virol 91:e00092-17. doi: 10.1128/JVI.00092-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kann M, Sodeik B, Vlachou A, Gerlich WH, Helenius A. 1999. Phosphorylation-dependent binding of hepatitis B virus core particles to the nuclear pore complex. J Cell Biol 145:45–55. doi: 10.1083/jcb.145.1.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Liao W, Ou JH. 1995. Phosphorylation and nuclear localization of the hepatitis B virus core protein: significance of serine in the three repeated SPRRR motifs. J Virol 69:1025–1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Mao R, Zhang J, Jiang D, Cai D, Levy JM, Cuconati A, Block TM, Guo JT, Guo H. 2011. Indoleamine 2,3-dioxygenase mediates the antiviral effect of gamma interferon against hepatitis B virus in human hepatocyte-derived cells. J Virol 85:1048–1057. doi: 10.1128/JVI.01998-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Guo H, Jiang D, Ma D, Chang J, Dougherty AM, Cuconati A, Block TM, Guo JT. 2009. Activation of pattern recognition receptor-mediated innate immunity inhibits the replication of hepatitis B virus in human hepatocyte-derived cells. J Virol 83:847–858. doi: 10.1128/JVI.02008-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Guo H, Zhou T, Jiang D, Cuconati A, Xiao GH, Block TM, Guo JT. 2007. Regulation of hepatitis B virus replication by the phosphatidylinositol 3-kinase-Akt signal transduction pathway. J Virol 81:10072–10080. doi: 10.1128/JVI.00541-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Mao R, Nie H, Cai D, Zhang J, Liu H, Yan R, Cuconati A, Block TM, Guo JT, Guo H. 2013. Inhibition of hepatitis B virus replication by the host zinc finger antiviral protein. PLoS Pathog 9:e1003494. doi: 10.1371/journal.ppat.1003494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Beames B, Lanford RE. 1993. Carboxy-terminal truncations of the HBV core protein affect capsid formation and the apparent size of encapsidated HBV RNA. Virology 194:597–607. doi: 10.1006/viro.1993.1299. [DOI] [PubMed] [Google Scholar]
- 79.Hong R, Bai W, Zhai J, Liu W, Li X, Zhang J, Cui X, Zhao X, Ye X, Deng Q, Tiollais P, Wen Y, Liu J, Xie Y. 2013. Novel recombinant hepatitis B virus vectors efficiently deliver protein and RNA encoding genes into primary hepatocytes. J Virol 87:6615–6624. doi: 10.1128/JVI.03328-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kim S, Lee J, Ryu WS. 2009. Four conserved cysteine residues of the hepatitis B virus polymerase are critical for RNA pregenome encapsidation. J Virol 83:8032–8040. doi: 10.1128/JVI.00332-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Clippinger AJ, Bouchard MJ. 2008. Hepatitis B virus HBx protein localizes to mitochondria in primary rat hepatocytes and modulates mitochondrial membrane potential. J Virol 82:6798–6811. doi: 10.1128/JVI.00154-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Foy E, Li K, Wang C, Sumpter R Jr, Ikeda M, Lemon SM, Gale M Jr.. 2003. Regulation of interferon regulatory factor-3 by the hepatitis C virus serine protease. Science 300:1145–1148. doi: 10.1126/science.1082604. [DOI] [PubMed] [Google Scholar]
- 83.Yan R, Zhang Y, Cai D, Liu Y, Cuconati A, Guo H. 2015. Spinoculation enhances HBV infection in NTCP-reconstituted hepatocytes. PLoS One 10:e0129889. doi: 10.1371/journal.pone.0129889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Liu Y, Nie H, Mao R, Mitra B, Cai D, Yan R, Guo J-T, Block TM, Mechti N, Guo H. 2017. Interferon-inducible ribonuclease ISG20 inhibits hepatitis B virus replication through directly binding to the epsilon stem-loop structure of viral RNA. PLoS Pathog 13:e1006296. doi: 10.1371/journal.ppat.1006296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Long Q, Yan R, Hu J, Cai D, Mitra B, Kim ES, Marchetti A, Zhang H, Wang S, Liu Y, Huang A, Guo H. 2017. The role of host DNA ligases in hepadnavirus covalently closed circular DNA formation. PLoS Pathog 13:e1006784. doi: 10.1371/journal.ppat.1006784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Yan R, Cai D, Liu Y, Guo H. 2017. Detection of hepatitis B virus particles released from cultured cells by particle gel assay. Methods Mol Biol 1540:193–202. doi: 10.1007/978-1-4939-6700-1_15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Yan R, Zhao X, Cai D, Liu Y, Block TM, Guo JT, Guo H. 2015. The interferon-inducible protein tetherin inhibits hepatitis B virus virion secretion. J Virol 89:9200–9212. doi: 10.1128/JVI.00933-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Trapnell C, Pachter L, Salzberg SL. 2009. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics 25:1105–1111. doi: 10.1093/bioinformatics/btp120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, Salzberg SL, Wold BJ, Pachter L. 2010. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol 28:511–515. doi: 10.1038/nbt.1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Benjamini Y, Yekutieli D. 2001. The control of the false discovery rate in multiple testing under dependency. Ann Statist 29:1165–1188. doi: 10.1214/aos/1013699998. [DOI] [Google Scholar]