

Abstract
In most eukaryotes, telomerase counteracts chromosome erosion by adding repetitive sequence to terminal ends. Drosophila melanogaster instead relies on specialized retrotransposons that insert exclusively at telomeres. This exchange of goods between host and mobile element—wherein the mobile element provides an essential genome service and the host provides a hospitable niche for mobile element propagation—has been called a “genomic symbiosis.” However, these telomere-specialized, jockey family retrotransposons may actually evolve to “selfishly” overreplicate in the genomes that they ostensibly serve. Under this model, we expect rapid diversification of telomere-specialized retrotransposon lineages and, possibly, the breakdown of this ostensibly symbiotic relationship. Here we report data consistent with both predictions. Searching the raw reads of the 15-Myr-old melanogaster species group, we generated de novo jockey retrotransposon consensus sequences and used phylogenetic tree-building to delineate four distinct telomere-associated lineages. Recurrent gains, losses, and replacements account for this retrotransposon lineage diversity. In Drosophila biarmipes, telomere-specialized elements have disappeared completely. De novo assembly of long reads and cytogenetics confirmed this species-specific collapse of retrotransposon-dependent telomere elongation. Instead, telomere-restricted satellite DNA and DNA transposon fragments occupy its terminal ends. We infer that D. biarmipes relies instead on a recombination-based mechanism conserved from yeast to flies to humans. Telomeric retrotransposon diversification and disappearance suggest that persistently “selfish” machinery shapes telomere elongation across Drosophila rather than completely domesticated, symbiotic mobile elements.
Transposable elements (TEs) infest eukaryotic genomes, ever-evolving to increase in copy number over time (Feschotte and Pritham 2007; Beauregard et al. 2008). These so-called “selfish genetic elements” enhance their own transmission relative to other elements in the genome, imposing neutral or deleterious consequences on the host (Werren 2011). Deleterious consequences arise when TE insertions disrupt host genes (Hancks and Kazazian 2012), nucleate local epigenetic silencing (Slotkin and Martienssen 2007; Lee and Karpen 2017), and trigger catastrophic recombination between nonhomologous genomic regions (Langley et al. 1988; Beck et al. 2011). TEs also provide raw material for genome adaptation (Jangam et al. 2017): Across eukaryotes, host genomes repurpose TE-derived sequence for basic cellular and developmental processes, from immune response (van de Lagemaat et al. 2003) to placental development (Lynch et al. 2015) to programmed genome rearrangements (Cheng et al. 2010, 2016). These diverse “molecular domestication” events share a common feature—the degeneration or deletion of the TE's capacity to propagate. Consequently, the TE-derived sequence resides permanently at a single genome location, just like any other host gene sequence. Inability to increase copy number via transposition resolves prior conflict of interest between the host and the TE (Jangam et al. 2017). However, not all adaptive molecular domestication events necessitate TE immobilization; in rare cases, essential host functions rely on retention of the mobilization machinery that promotes recurrent TE insertions into host DNA. The noncanonical telomere elongation mechanism of Drosophila is exemplary (Pardue and DeBaryshe 2003; Casacuberta 2017).
In most eukaryotes beyond Drosophila, telomerase-added DNA repeats (Greider and Blackburn 1989; Zakian 1989, 1996; Blackburn 1991) counteract the “end-replication problem” that otherwise erodes unique DNA sequence at chromosome termini (Watson 1972). However, telomeres of select fungi (Starnes et al. 2012), algae (Higashiyama et al. 1997), moths (Osanai-Futahashi and Fujiwara 2011), crustaceans (Gladyshev and Arkhipova 2007), and DNA repair–deficient mammalian cells (Morrish et al. 2007) encode not only telomerase-added repeat elements but also TEs that insert preferentially at chromosome termini. These telomeric mobile elements are typically derived from a single class of TE—the non-long terminal repeat (“non-LTR”) retrotransposons (Beck et al. 2011), which mobilize via reverse transcription and insertion into new genomic locations. The most extreme example of this cooption is found in Drosophila melanogaster, whose telomeres harbor no telomerase-added repeats. In fact, the 220-Myr-old “true fly” insect Order, Diptera, completely lacks the genes encoding the telomerase holoenzyme (Pardue and DeBaryshe 2003; Casacuberta 2017). Instead, D. melanogaster harbors three telomere-specialized retrotransposons—HeT-A, TART, and TAHRE—that preserve distal, unique sequence (Pardue and DeBaryshe 2011). These three retrotransposons represent a monophyletic clade within the larger “jockey” element family (Villasante et al. 2007), whose members are more typically found along chromosome arms (Xie et al. 2013). The telomere-specialized jockey subclade, in contrast, rarely inserts outside the telomere (Pardue and DeBaryshe 2003, 2011; Berloco et al. 2005).
This molecular domestication of still-mobile retrotransposons into an essential genome function is often referred to as a “genomic symbiosis” (Pardue and DeBaryshe 2008). Evidence for such a mutualism is compelling. The elements that maintain telomere ends in D. melanogaster comprise a monophyletic clade ostensibly specialized to replicate only at chromosome ends (Pardue and DeBaryshe 2008). Elements from this jockey clade also appear at terminal ends in distant Drosophila species, consistent with a single domestication event >40 Myr ago followed by faithful vertical transmission (Casacuberta and Pardue 2003; Villasante et al. 2007). Moreover, mobile elements from other families rarely appear at terminal ends (Mason and Biessmann 1995; Biessmann et al. 2005; Mason et al. 2016). These data suggest that retrotransposon-mediated chromosome elongation represents a long-term, conserved relationship between a host genome and its domesticated, but still-mobile, retrotransposons.
This picture of cooperativity was complicated by the discovery that the telomere-associated subclade of jockey elements evolves rapidly across Drosophila. Leveraging 12 Drosophila genomes that span 40 Myr of evolution, Villasante, Abad, and colleagues detected at least one jockey-like, candidate telomeric retrotransposon in all 12 species (Villasante et al. 2007). These data implicated a single evolutionary event in a common ancestral sequence that conferred telomere specificity. However, these elements represent distinct phylogenetic lineages rather than species-specific versions of the HeT-A, TART, and TAHRE elements well-studied in D. melanogaster. For example, D. melanogaster and its close relatives encode HeT-A, TART, and TAHRE, whereas the 15-Myr-diverged Drosophila ananassae encodes a single, phylogenetically distinct candidate retrotransposon lineage, “TR2.” The 30-Myr-diverged Drosophila pseudoobscura species encodes yet other phylogenetically distinct lineages within this jockey subclade. This expansive evolutionary lens revealed a previously unappreciated, dynamic evolutionary history of these jockey subclade retrotransposons (Villasante et al. 2008). However, the large evolutionary distances between species left fine-scale dynamics unknown, and telomere-specific localization was not explored (Villasante et al. 2007). The evolutionary origin(s), ages, between-species differences, and genome locations of these candidate telomere elongators remain obscure. Elucidating the evolutionary history of these elements is essential to address the possibility that this molecular domestication event is less a stable, long-term genomic symbiosis and instead an ever-evolving relationship between the domesticator and the domesticated. Here we investigate the fine-scale evolutionary history of telomere-specialized elements to evaluate the possibility that these elements harbor signatures of diversification and disappearance typical of undomesticated mobile elements (Yang and Barbash 2008; de la Chaux and Wagner 2009; Dias et al. 2015).
Results
Candidate telomere-specialized retrotransposons identified in the melanogaster species group
D. melanogaster encodes telomere-specialized, non-LTR retrotransposons that increase in copy number by a copy-and-paste mechanism (Pardue and DeBaryshe 2011). Transcripts encoded by these elements are localized, reverse-transcribed, and integrated at the terminal nucleotides of chromosome ends, resulting in stereotypical head-to-tail arrays (Pardue and DeBaryshe 2003). Full-length, autonomous retrotransposons typically contain two open reading frames (ORFs) between the variable 5′ and 3′ UTRs. ORF1 encodes an RNA-binding domain (gag) and ORF2 encodes a reverse transcriptase (RT) domain and an endonuclease domain (EN) (Supplemental Fig. S1). The telomere-specialized HeT-A (ORF1 only), TART, and TAHRE elements form a monophyletic clade within the jockey family of non-LTR retrotransposons (Villasante et al. 2007), as stated above.
To elucidate the fine-scale evolutionary history of telomere-specialized elements in Drosophila, we searched for jockey-like, telomere-specialized retrotransposons across lineages that span 3 to 15 Myr of evolution captured by the “melanogaster species group” (Drosophila 12 Genomes Consortium 2007; Chen et al. 2014). The group includes D. melanogaster and its close relatives, which share the well-studied retrotransposon lineages HeT-A, TART, and TAHRE (Danilevskaya et al. 1998; Casacuberta and Pardue 2002; Berloco et al. 2005; Villasante et al. 2007), and the 15-Myr-diverged D. ananassae, which encodes only a single, distinct lineage (called TR2) yet to be validated cytogenetically as telomere specialized (Villasante et al. 2007). Spanning these two clades are our five focal species, Drosophila rhopaloa, D. biarmipes, Drosophila takahashii, Drosophila elegans, and Drosophila ficusphila (Chen et al. 2014). The well-characterized telomeres of D. melanogaster and its close relatives (e.g., Drosophila yakuba) served as positive controls for our de novo identification of retrotransposons phylogenetically related to the jockey subclade specialized at telomeres. D. ananassae served as an outgroup.
We developed a custom pipeline to discover jockey family elements ancestrally related to previously defined lineages that maintain chromosome ends (Supplemental Fig. S2). We conducted TBLASTN searches against raw reads from each of the 10 species using a query that included both fully and minimally validated telomere-specialized retrotransposons (gag and RT domains, specifically) from across Drosophila (Supplemental Table S1). We also included the reference nontelomeric jockey element (from D. melanogaster) defined in Repbase (Supplemental Table S1; www.girinst.org/repbase/). A detailed description of our pipeline of iterative BLAST searches to raw reads, de novo consensus building, and phylogenetic tree-assisted sorting can be found in the Methods. This pipeline (Supplemental Fig. S2) generated a refined list of consensus sequences that included previously described telomere-specialized elements from D. melanogaster and its close relatives (our positive controls), as well as jockey family elements outside the specialized telomeric subclade (Supplemental Table S2). These latter elements indicated that our search was exhaustive—we effectively overshot the jockey subclade associated with telomeres for all species. We built Bayesian phylogenetic trees based on the gag and RT domains of all final consensus sequences (Fig. 1; Supplemental Fig. S3 for genus-wide). Our gag-based trees revealed that the 15 consensus sequences form a well-supported, monophyletic subclade within the jockey family but are distinct from 18 generalist jockey element consensus sequences that also form a distinct, well-supported monophyletic clade (Fig 1A, gray). The candidate telomeric gag consensus sequences form four distinct lineages: TAHRE, TART, TR2, and a previously undefined lineage that we named “TARTAHRE” for its labile phylogenetic position between TART and TAHRE. Moreover, our trees support previous inferences that HeT-A gags are phylogenetically indistinguishable from TAHRE gags (Supplemental Fig. S3; Villasante et al. 2007)–HeT-A encodes its own 5′ UTR, gag, and 3′ UTR (Pardue and DeBaryshe 2003), yet its phylogenetic position reveals that this single-domain element is effectively a TAHRE element missing an RT domain. Henceforth, we refer to the HeT-A gag as HeT-A/TAHRE gag. The RT domain–based tree (Fig. 1B) revealed a similar topology. Our pipeline detected only a partial TAHRE gag in D. takahashii (Supplemental Table S2) and no evidence at all of the telomere-associated, jockey subclade elements in its close relative, D. biarmipes (see below).
Figure 1.
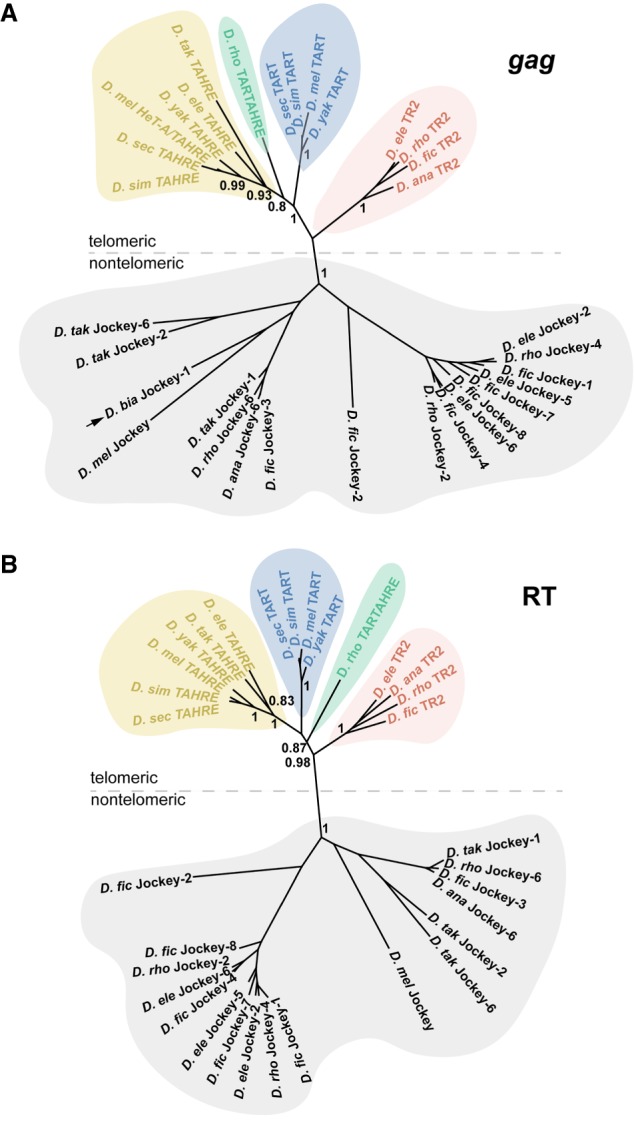
Phylogenetic relationships among previously and newly defined jockey subclade elements. Unrooted phylogenetic trees built from gag domain (A) and RT domain (B) consensus sequences. Node support values are posterior probabilities generated by MrBayes. Gray designates jockey elements that passed the final pipeline filter but are distantly related to the telomere-specialized subclade. Various colors delineate candidate telomere-specialized elements along with previously characterized elements that form monophyletic clades. Only the D. rhopaloa–restricted element, TARTAHRE (green), occupies different positions across the two trees and may represent long branch attraction or, instead, a chimera of the two lineages. The black arrow corresponds to the closest D. biarmipes jockey family element to the telomere-specialized subclade.
PCR and cytogenetic validation of computationally predicted telomeric retrotransposons
Despite our search being inherently conservative—our consensus-building approach may average out sublineages within major named retrotransposon lineages—we uncovered rapid diversification across only 15 Myr of Drosophila evolution. By virtue of phylogenetic relatedness, we predict that the jockey subclade retrotransposons uncovered by our pipeline specialize at chromosome ends in their respective host species. Testing this prediction first requires molecular biology to confirm that (1) these in silico–generated consensus sequences represent actual elements in the targeted genomes and (2) the elements are arrayed in the characteristic head-to-tail orientation that arises from exclusive end-integration of the poly-adenylated 3′ end (Pardue and DeBaryshe 2008, 2011). By using Sanger sequencing of PCR products amplified from genomic DNA, we discovered that the in silico sequences represent true DNA elements in their host genomes (Fig. 2A; Supplemental Table S3). The PCR-amplified/Sanger-sequenced domains typically share ∼97.5% sequence identity to a given consensus (Supplemental Table S3). For D. takahashii and D. ananassae, we successfully amplified the predicted partial TAHRE and TR2 RT domains, respectively, as well as D. ananassae’s partial TR2 gag domain. We also confirmed head-to-tail orientation using primers that annealed to the 3′ “tail” of one copy and the 5′ “head” of another copy predicted to reside at the same telomere (Fig. 2A; Supplemental Table S3). PCR-based and Sanger sequencing–based validation suggests that virtually all consensus sequences represented in Figure 1 correspond to actual jockey subclade elements found in their respective genomes and are in an orientation stereotypical of telomere-specialized retrotransposons. Restriction to telomere ends, however, has only been shown previously for the HeT-A/TAHRE and TART lineages.
Figure 2.

PCR- and cytology-based validation of in silico–predicted, telomere-specialized elements. (A) Cartoon representation of our PCR-based validation of in silico–predicted gag and RT domains and head-to-tail orientation of candidate telomeric retrotransposons (and the previously validated HeT-A/TAHRE and TART). Primer orientation is represented above as cartoons in white and black. gag (arrowhead) and RT (rectangle) domains are represented by lighter or darker shades, respectively. PCR-validated partial gag and partial RT domains are represented as truncated symbols in D. takahashii and D. ananassae. (B) DNA-FISH or oligopainting with probes/paints cognate to HeT-A/TAHRE gag, TART, TARTAHRE, and TR2 on polytene chromosomes from representative species. HeT-A/TAHRE and TART from D. melanogaster serve as positive controls. All insets show telomere hybridization exclusively except TARTAHRE, which hybridized to both telomeric and nontelomeric locations (insets designated with an asterisk).
To investigate chromosome localization of the newly defined elements, TR2 and TARTAHRE, we conducted either DNA FISH or oligopainting (Beliveau et al. 2012) on polytene chromosomes in representative species. HeT-A/TAHRE and TART probes in D. melanogaster served as positive controls. Like the telomere restriction of HeT-A/TAHRE and TART in D. melanogaster, the TR2 probe hybridized exclusively to telomere ends (Fig. 2B). However, TARTAHRE probe hybridization revealed both telomeric localization and nontelomeric localization, especially around chromocenters rich in heterochromatin (Fig. 2B). The promiscuous localization of TARTAHRE implicates either incomplete domestication or “escape” from domestication. Its nested phylogenetic position within a clade of telomere-specialized elements favors an innovation event possibly leading to escape from the telomere (see Discussion). Consistent with this possibility, only the TARTAHRE ORF2 encodes nontelomeric jockey-like residues in the EN domain responsible for DNA recognition and internal nicking (Supplemental Fig. S4).
Recurrent turnover of telomere-specialized retrotransposon lineages in the melanogaster species group
To infer telomeric retrotransposon lineage turnover across the melanogaster species group, we summarized the presence/absence of these validated elements across the species tree (Fig. 3A; Chen et al. 2014). The TART lineage, well-known from D. melanogaster, is relatively young, emerging in the ancestor of the “melanogaster subgroup” between 10 and 15 Myr ago. The TAHRE lineage is more ancient, emerging after the split from D. ananassae or instead, before the common ancestor of the melanogaster species group and then subsequently being lost along the D. ananassae lineage. TAHRE has been lost at least three times, along lineages leading to D. rhopaloa, D. ficusphila, and D. biarmipes. D. biarmipes’ closest relative on the tree, D. takahashii, encodes only a truncated TAHRE. These two species together suggest that TAHRE loss began in their common ancestor, although only in D. biarmipes is the loss event complete. The unusual TARTAHRE lineage appears in D. rhopaloa only, making it the sole jockey subclade element restricted to a single species on the densely sampled, melanogaster species group tree. TR2 absence from D. melanogaster and its close relatives suggests the possibility that this lineage was functionally replaced by TAHRE and/or TART. Finally, we infer that TR2 was lost at least once along the lineage leading to D. takahashii/D. biarmipes and melanogaster subgroup clades.
Figure 3.

Telomeric retrotransposon identity, copy number, and history across the melanogaster species group. (A) Presence/absence of telomere-localized elements across the melanogaster species group. Each column represents a phylogenetically distinct lineage defined and validated in Figures 1 and 2, respectively. Hatched lines delineate elements for which only a degraded version was recovered. gag and RT domains are represented by lighter and darker shaded boxes, respectively. (B) Estimated gag (light) and RT (dark) copy number per species calculated from the average read depth of a consensus sequence relative to genome-wide estimates. (C) Repeat landscapes of telomere-specialized retrotransposons captured by Kimura two-parameter distance between genomic reads with significant BLAST hits (>90% identity). Copy number (consensus read no. / genome-wide average read no.) appears on the y-axis, and binned divergence classes appears on the x-axis. The bins closest to zero putatively represent the youngest classes. We estimated divergence for the gag (lighter shade) and RT (darker shade) separately.
Although some lineages appear to rely on only one element, others harbor multiple retrotransposon lineages. The observation that D. elegans, for example, encodes both TAHRE and TR2, D. rhopaloa both TARTAHRE and TR2, and D. melanogaster both TART and TAHRE rejects the null expectation that the retrotransposon lineage tree recapitulates the species tree. Instead, retrotransposon lineages are alternately retained and lost across the species phylogeny; that is, not all species are represented in each element subclade. Overall, these data are consistent with diversification via gain and loss, both with and without replacement, of major retrotransposon lineages across 3 to 15 Myr of evolution. Across the melanogaster species group, we observe a pervasive lineage-specific presence/absence of TR2, TARTAHRE, and TAHRE, and even extreme cases of wholesale loss of jockey subclade, telomere-specialized elements.
Expansions and contractions of DNA content derived from telomere-specialized elements
Retrotransposon expansions and contractions over time result in contemporary species restriction of specific retrotransposon lineages. To evaluate such bulk sequence changes across species, we mapped raw reads to a given domain consensus sequence and quantified read depth (Fig. 3B; Supplemental Table S4). This copy number estimate serves also as a proxy for relative telomere length for all elements except TARTAHRE, which localizes to both telomeric and nontelomeric sites. We note that the highly variable telomeric retrotransposon abundance within D. melanogaster (Wei et al. 2017) suggests that our single-genome estimates offer only a partial picture of divergence in bulk content.
We observe broad between-species differences in copy number, even for species that share common retrotransposon lineages. The degree and direction of these between-species differences were robust to multiple percentage similarity thresholds to the consensus, minimizing the likelihood that we inadvertently underestimated copy number owing to highly diverged variants (Supplemental Table S4). qPCR on genomic DNA in cases of extreme copy number differences across species validated these computationally generated estimates (Supplemental Fig. S5). Across melanogaster subgroup species, telomeric retrotransposon content is twofold larger in D. yakuba compared with D. melanogaster, at least in the sequenced strains. Moreover, D. melanogaster and D. simulans telomeres are predominantly composed of the HeT-A/TAHRE gag, whereas D. yakuba encodes relatively more TART RTs (Fig. 3B). The abundance of the gag-only HeT-A element in D. melanogaster and D. simulans relative to other telomeric elements in these genomes has been attributed to its dependency on the RT encoded by TART (Rashkova et al. 2003). These data suggest that this ostensible parasitism may not be the rule: Most other species harbor elements with similar gag and RT copy numbers. TARTAHRE in D. rhopaloa is especially abundant, consistent with its uniquely broad localization (Fig. 2B). The telomere-restricted element, TR2, is also highly abundant in D. rhopaloa but depauperate in D. ananassae. D. takahashii also harbors low copy numbers of its partial TAHRE gag and RT domains. These copy number–poor elements are depauperate of highly similar reads (Supplemental Fig. S6), consistent with mutation accumulation at inactive copies. These data highlight the divergent jockey subclade content even across species that share a common retrotransposon lineage and suggest low functional constraint on telomere length.
To further refine our snapshot of retrotransposon invasion and degeneration history, we generated frequency distributions of pairwise read divergence for each element in each species. If transposition events were recent, we expect the highest frequency classes to show the lowest divergence. Alternatively, an element that has undergone only an ancient episode of expansion will show an excess of high frequency reads with similar but elevated divergence from the consensus. Because fully functional Drosophila telomeres must constantly renew their telomere-specialized retrotransposon template, we expect actively transposing telomeric elements to have high similarity between consensus-mapping reads. Profiles of sequence divergence (estimated by Kimura two-parameter distance) support this mechanism; virtually all distributions reveal an enrichment of reads diverging minimally or not at all (Fig. 3C). Indeed, most distributions are typical of recent transposition bursts characterized by a preponderance of highly similar sequences. We cannot formally rule out the homogenizing force of gene conversion to the excess of highly similar reads; however, the empirically verified rates of transposition in D. melanogaster (Kahn et al. 2000) suggest that gene conversion may contribute only moderately to the observed patterns. The distributions of D. ananassae TR2, D. simulans TART, and D. elegans TAHRE instead appear more uniform, consistent with ancient activity followed by mutation accumulation.
Telomere-specialized retrotransposons are absent in D. biarmipes
Our data suggest that jockey subclade, telomere-specialized retrotransposons frequently occupy the melanogaster species group's telomeres. However, these elements encode a complete gag or complete RT domain in only a subset of our focal species. To evaluate the possibility that active insertions by telomere-specialized retrotransposons can be lost completely, we exploited the most extreme case of telomeric retrotransposon degeneration uncovered by our pipeline. We detected no D. biarmipes telomere-specialized elements branching inside the monophyletic jockey subclade (Fig. 1). From these short-read data, we recovered instead a jockey family gag (Fig. 1A, arrow). Hybridizing a FISH probe cognate to “jockey_1” confirmed our phylogeny-based inference. Specifically, this computationally predicted jockey element localized along D. biarmipes chromosome arms (Supplemental Fig. S7), a typical jockey family chromosomal distribution (Kaminker et al. 2002; Xie et al. 2013). To test the hypothesis that this exceptional species has lost jockey family retrotransposons at its chromosome ends, we performed long-read sequencing using the Pacific Biosciences (PacBio) SMRT sequencing platform. We predicted that upon assembling whole telomeres from D. biarmipes, we would discover a newly domesticated, telomere-specialized mobile element lineage unrelated to the jockey family or instead, the wholesale loss of telomere-specialized mobile elements.
Our long read–based assembly of the D. biarmipes genome revealed a highly diverged chromosome-end composition from the well-studied telomeres of D. melanogaster (Fig. 4A). Using the packages PBcR (Berlin et al. 2015) or DBG2OLC (Ye et al. 2016), we assembled D. biarmipes telomeres corresponding to D. melanogaster’s 2L, 2R, 3L, and 3R Chromosomes. To determine composition in both species, we delineated D. melanogaster and D. biarmipes telomeric DNA as distal to the most terminally annotated, orthologous genes from D. melanogaster (the two species share a common karyotype) (Deng et al. 2007). Consistent with previous literature (Pardue and DeBaryshe 2011; Mason et al. 2016), D. melanogaster telomeres are dominated by telomere-specialized elements (HeT-A/TAHRE and TART) and satellite repeats (primarily “HETRP_DM”) (Fig. 4A). In sharp contrast, the 50- to 200-kb D. biarmipes telomeres harbor no jockey subclade retrotransposons, consistent with our short read–based pipeline results above. We uncovered instead DNA transposons called Helitrons and, to a lesser extent, gypsy family LTR elements (Fig. 4A). We validated these D. biarmipes telomere assemblies in two ways. First, we observed 99% correspondence and no structural discrepancies between our assembled D. biarmipes telomeres and those of an independent, recently published hybrid assembly based on Illumina and Oxford Nanopore reads (Supplemental Fig. S8; Miller et al. 2018). Second, hybridization of FISH probes cognate to the telomeric Helitron and telomeric satellite sequence confirmed that we successfully assembled the termini of D. biarmipes chromosomes (Fig. 4B, insets).
Figure 4.

Collapse of the retrotransposon-based telomere elongation mechanism in D. biarmipes. (A) Composition of D. melanogaster and D. biarmipes chromosomes between the most distal, protein-coding gene and the terminal nucleotide. Fractions estimated from the distal sequence (assembled from PacBio-generated long reads for both species) for Muller elements corresponding to 2L, 2R, 3L, and 3R. Purple corresponds to telomere-specialized, jockey family retrotransposons found in D. melanogaster (but not in D. biarmipes). (B) Fluorescent in situ hybridization of SAR2 and Helitron probes to polytene chromosomes from D. biarmipes. Insets I and II show telomere localization of SAR2 and Helitrons, respectively, on D. biarmipes polytene chromosomes. (C) Schematic representation of the long-read–based assembly of D. melanogaster 2R and 3L telomeric DNA. Telomere-specialized, jockey-like elements (purple) are distal to a block of simple and complex satellites (black), consistent with previous reports. Triangles represent full-length elements, and rectangles represent partially degenerated elements. (D) Schematic representation of the long read–based assembly of 2R and 3L telomeres from D. biarmipes in which simple and complex satellite DNA (black) is juxtaposed with primarily Helitron DNA transposons (green).
The rough equivalency of D. melanogaster and D. biarmipes telomeric DNA attributed to mobile elements and to satellite sequence (Fig. 4A) suggests functional replacement of jockey subclade, telomere-specialized retrotransposons with other mobile element families. However, the integrity of these elements and their physical distribution challenge this inference. Starting at the most distally annotated host gene, D. melanogaster encodes long, uniform tracts of satellite DNA (Karpen and Spradling 1992) followed by equivalently uniform tracts of the end-integrating TART and HeT-A/TAHRE (Fig. 4C; Supplemental Table S5). Many of these elements are full length and so are presumably still active. Pervasive degradation instead dominates the Helitrons of D. biarmipes (Supplemental Fig. S9): We observed no full-length Helitrons at the telomere. Moreover, these fragments are interspersed amid highly abundant satellite sequence (“SAR” and “SAR2”) (Fig. 4D) and oriented randomly (Supplemental Table S6), unlike the head-to-tail arrays of jockey subclade retrotransposons in D. melanogaster. All assembled D. biarmipes telomeres show this pattern, despite varying in length from 50 to 200 kb and varying in enrichment for TE family (Fig. 4D; Supplemental Fig. S10; Supplemental Table S6). Finally, the abundant Helitron signal at the chromocenter (Fig. 4B) and the patterns of divergence between copies (Supplemental Fig. S11) are consistent with “generalist” mobile elements occupying a niche once restricted to telomere-specialized elements.
Patterns of divergence between Helitron insertions and repeat structure of satellite sequence further implicate an alternative lengthening mechanism in D. biarmipes. Using the uniquely long Chromosome 2R as model, we observed that Helitron divergence is uniformly elevated between both nearby and distant insertions, whereas HeT-A/TAHRE gag divergence is uniformly low (Supplemental Fig. S12). We attribute this pattern of divergence to an ancient Helitron invasion followed by mutation accumulation in D. biarmipes versus constant “refreshment” of end-inserting HeT-A/TAHRE gag elements in D. melanogaster. The absence of full-length Helitrons anywhere in our long-read D. biarmipes assembly (Supplemental Fig. S9) suggests that these D. biarmipes telomeric Helitron fragments do not jump via Helitron mobilization machinery in trans. The D. biarmipes telomere tracts of repeats are instead reminiscent of Drosophila pericentromeric heterochromatin, which encodes long tracts of simple and complex satellites interspersed with dead TEs (Hoskins et al. 2007). D. biarmipes telomeres reveal the wholesale loss of >40-Myr-old telomere elongation mechanism.
Discussion
Most eukaryotes rely on end-targeting and reverse transcription by telomerase to add DNA repeats to chromosome termini (Greider and Blackburn 1989; Zakian 1989, 1996; Blackburn 1991). The chromosome ends of D. melanogaster instead rely on telomere-specialized retrotransposons (Pardue and DeBaryshe 2011). These “domesticated” mobile elements also end-target and reverse-transcribe RNA into repetitive DNA at the most terminal base pairs, preserving unique genes several thousand nucleotides away. These alternative mechanisms differ not only in the molecular players that preserve chromosome ends but also in the rates of molecular evolution of telomeric DNA sequence. The telomerase-associated, nucleotide repeat unit composition changes slowly across eukaryotes (Meyne et al. 1989; Mason et al. 2016; Podlevsky and Chen 2016). Our investigation of the melanogaster species group reveals instead rapid diversification of the repeats charged with telomere length maintenance in Drosophila. Building on previous discoveries of phylogenetically distinct, candidate telomeric jockey elements (Villasante et al. 2007), we document recurrent turnover of major retrotransposon lineages as well as rapid expansions and contractions across only a few million years of evolution.
Most focal species described here encode one or two full-length retrotransposon lineages restricted to chromosome ends. In contrast, D. takahashii encodes only degenerated elements. The absence of full-length elements suggest that active transposition may not always be the primary mechanism of length regulation genus-wide (Casacuberta and Pardue 2003; Villasante et al. 2007). Active retrotransposons indeed populate the well-studied telomeres of D. melanogaster; the long read–based assembly of D. melanogaster’s telomeres (Fig. 4C) supports decades of work defining this system (Biessmann et al. 1990; Mason and Biessmann 1995; Pardue and DeBaryshe 2003, 2011; Mason et al. 2016). Indeed, all Drosophila species investigated over the past several decades harbor telomere-specialized jockey subclade elements (Casacuberta and Pardue 2003; Berloco et al. 2005; Villasante et al. 2007). Our discovery of species that lack active telomere-specialized elements raises the possibility that chromosome length maintenance depends on an alternative class of telomere-specialized mobile elements or instead, a mobile element–independent mechanism.
If newly domesticated mobile elements readily replace ancestral, telomere-specialized lineages, we would expect the species lacking active jockey subclade elements—D. biarmipes—to encode such new recruits. The extreme terminal ends of D. biarmipes instead harbor AT-rich satellite “SAR” DNA (Mirkovitch et al. 1984; Käs and Laemmli 1992) at all telomeres, as well as Helitron fragments (Kapitonov and Jurka 2007) and gypsy-derived LTR fragments (Nefedova and Kim 2017) at variable frequencies across different telomeres. These inactive elements are found both inside and outside the D. biarmipes telomeres (Fig. 4D) and found more typically outside the telomere in other Drosophila species (Käs and Laemmli 1992; Kapitonov and Jurka 2007; Nefedova and Kim 2017). Moreover, we found not one full-length Helitron in the D. biarmipes genome, rejecting the possibility of contemporary propagation using mobilization proteins in trans (Supplemental Fig. S9). These data suggest that active transposition is not responsible for telomere length regulation, at least recently, in D. biarmipes. Dias et al. (2015) previously reported lineage-restricted bursts of a Helitron tandem repeat, DINE-TRI, along the lineage leading to D. biarmipes and along the lineage leading to Drosophila virilis/Drosophila americana compared with 25 other Drosophila genomes (Dias et al. 2015). Hybridization of DINE-TRI probes to D. virilis polytene chromosomes revealed localization to multiple telomeres, in addition to pericentromeric heterochromatin, reminiscent of the localization we detected in D. biarmipes. Like D. biarmipes telomeres, telomeric Helitrons in D. virilis represent only partial fragments rather than full-length, actively transposing copies (Dias et al. 2015). However, unlike D. biarmipes, D. virilis telomeres also encode jockey subclade retrotransposons predicted to maintain its chromosome length (Casacuberta and Pardue 2003).
In the absence of active telomeric transposition, how might D. biarmipes (and possibly D. takahashii) telomeres be maintained? D. melanogaster telomeres elongate not only by transposition but also by a recombination-based mechanism called “terminal gene conversion” (Kahn et al. 2000; Melnikova and Georgiev 2002). This alternative lengthening pathway, used across eukaryotes alongside or instead of telomerase (McEachern and Haber 2006; Sakofsky and Malkova 2017; Sobinoff and Pickett 2017), relies on the shorter telomere resecting and invading a longer telomere or even invading itself. Second-strand synthesis extends the chromosome end. This newly synthesized strand serves as the template for synthesis of its sister.
We predict that D. biarmipes depends on this ancient mechanism of terminal gene conversion. All studied non-Drosophila species from the insect order Diptera lack both telomerase-added repeats and telomere-specialized TEs and so are presumed to depend on terminal gene conversion (Mason et al. 2016). Specifically, dipterans such as Anopheles gambiae (Biessmann et al. 1998), Chironomous sp. (López et al. 1996; Rosen and Edstrom 2000), and Rhynchosciara americana (López et al. 1996) encode only simple and/or complex satellites at chromosome ends. We predict that Drosophila species lacking full-length, telomere-specialized retrotransposons rely exclusively on this alternative elongation mechanism. Consistent with recombination shaping D. biarmipes telomere sequence evolution, we observe a higher-order repeat unit structure of telomeric SAR (Supplemental Fig. S13). The concomitant proliferation of nonautonomous Helitrons at D. biarmipes telomeres may serve to replenish the repetitive sequence that mediates resected end homology searches and to serve as a template for repair. D. biarmipes’ close relative, D. takahashii, likely represents a transitional state (as does D. biarmipes’ even closer relative, D. suzukii) ( Supplemental Fig. S14). In D. takahashii, we detected no evidence of a full-length TAHRE. Instead, we recovered at least one chimeric TAHRE–Helitron instance (Supplemental Table S3). The variable retention of a retrotransposon-based telomere elongation mechanism, together with the inferred ancestral dipteran telomere state, helps us to contextualize what seemed initially like an aberration. Specifically, D. biarmipes’ jockey element-poor, Helitron-rich telomeres may, in fact, represent a reversion to an ancestral, dipteran-like state rather than a previously unseen innovation (Fig. 5).
Figure 5.

Model of telomere elongation mechanism evolution pre- and postbirth of the genus Drosophila. The dipteran ancestor of Drosophila encodes neither telomerase nor telomere-specialized mobile elements. Instead, a recombination-based mechanism, “terminal gene conversion,” likely lengthens the repetitive DNA. Exclusive chromosome-end insertions by a jockey family element becomes the primary, Drosophila-wide telomere elongation mechanism. Major jockey family lineages turn over across Drosophila species that retain this lengthening mechanism (bottom left). In species like D. biarmipes, the loss of telomere-specialized elements, and the presence of “generalist” mobile elements, illustrates how some Drosophila species may revert to the ancestral, predominantly recombination-based telomere lengthening mechanism (bottom right).
The discovery of undomesticated Helitrons at D. takahashii and D. biarmipes telomeres raises the possibility that some telomeric mobile elements act akin to ecological “facilitators” rather than “mutualists.” Like a canopy tree inadvertently cultivating a favorable environment for shade-tolerant herbs, these immobile elements, by virtue of where they inserted during an ancient transposition burst, serve inadvertently the genome's recombination-based telomere-lengthening mechanism. Moreover, we cannot rule out the possibility that reverse transcription of satellite or Helitron RNA may also extend these telomeres (and other species’) by using a nontelomeric retrotransposon's RT in trans (Gorab 2003). We speculate that these back-up mechanisms reduce constraint on telomere maintenance by active transposition across Drosophila, setting the stage for “selfish” overreplication by ostensibly domesticated telomere-specialized retrotransposons. Evolutionary pressure to police these telomeric retrotransposons may shape the adaptive evolution detected at telomere proteins encoded by the host (Lee et al. 2017). Future work will determine whether intra-genomic conflict lies beneath this dynamic relationship between the domesticator and the domesticated.
Methods
Drosophila genome sequence data used to define telomeric retrotransposons
Reference genomes assembled from short-read sequences rarely include the repetitive DNA elements that accumulate at chromosome ends. We searched instead the publicly available raw sequence reads derived from 10 species (Supplemental Table S7) sequenced with either Sanger (ABI 3700) and/or 454 (Genome Sequencer FLX). Although sequencing depth varies across these 10 species, we discovered jockey subclade, candidate telomere-specialized elements in even the lowest-coverage genome. The only exception was D. biarmipes, a species to which we ultimately subjected long-read sequencing (see below). For all other genomes, we detected no biased enrichment of jockey family lineages in genomes sequenced using one platform or another (Supplemental Fig. S15).
Identification of candidate telomeric retrotransposons from raw reads
We developed a custom pipeline (Supplemental Fig. S2; Supplemental Code) to search raw reads from 10 Drosophila species for the non-LTR retrotransposons related to the jockey subclade of telomere-specialized elements. We designed this two-step method to identify phylogenetically distinct elements that maintain chromosome ends—both active and degraded copies. Our initial query included all previously characterized, telomeric-specialized retrotransposons, as well as those uncharacterized elements only predicted to maintain telomere ends in more distant species (Supplemental Table S1). We also included the reference jockey element from D. melanogaster (Repbase, www.girinst.org/repbase/). These input sequences served as the query for a TBLASTN search of a given species’ raw read database (Supplemental Table S7). We retained any read that shared 80% sequence identity with any one of the query elements. We then de novo assembled this subset of raw reads into consensus sequences (i.e., two or more reads assembled by the Geneious assembler: medium sensitivity/fast parameters). We aligned each consensus sequence to the query sequences plus the reference jockey element from D. melanogaster using MAFFT (Katoh and Standley 2013) (k = 2, Gap penalty = 1.53, Offset = 0.123). For each alignment of either the gag or RT domain, we built a phylogenetic tree using FastTree (GTR + CAT) (Price et al. 2009) and determined if the focal consensus sequence branched as an ingroup or an outgroup (outside the jockey family). We retained only ingroup consensus sequences for subsequent analysis (482 out of 4583 consensus sequences). We repeated this pipeline by inputting the 482 consensus sequences from round one (length mean = 4143, standard deviation = 1656) as a new query in a BLASTN search of the raw reads from each species. This second iteration generated 3112 consensus sequences (length mean = 1531, standard deviation = 732) (Supplemental Table S8).
We next generated full-length gag and RT domains from our consensus sequences. We first translated the nucleotide sequences using “six-pack” (Rice et al. 2000) and aligned using MAFFT to known telomere-specialized retrotransposon domains (parameters: -k = 2, Gap penalty = 1.53, Offset = 0.123) (Supplemental Table S1; Katoh and Standley 2013). We then classified the gag and RT consensus sequences based on their branch position (i.e., TAHRE-like, TART-like, TR2-like, jockey-like) from the previous FastTree sorting step. Guided by the alignments, we removed frameshifts and unalignable sequence. We retained entire consensus sequences harboring <80% identity over 80% of its alignment length to any other consensus within the subset (Wicker et al. 2007). For each subset of consensus sequences per species, we generated final consensus sequences using majority rule. We also used Repbase (www.girinst.org/repbase/) to infer consensus identity for all consensus sequences diverged beyond the threshold described above. These sequences represented mostly jockey elements (Supplemental Table S2) and also several highly degraded, telomere-associated retrotransposons juxtaposed with DNA unrelated to the jockey family. We conducted a final refinement step for each complete gag and RT domain by mapping raw reads to our set of telomeric retrotransposon candidates. These raw reads came from Illumina-based short-read sequence databases generated independently of the Sanger and 454 reads used to build the consensus sequences (Supplemental Table S7). This step successfully called many sites previously designated ambiguous based on majority rule. In addition, we elongated the domain boundaries using reads that spanned the consensus sequence and sequence beyond the domain. Again, we used majority rule to infer the final sequence. We repeated cycles of mapping and extension up to 10 times until we detected a full-length ORF and, in most cases, the sequence spanning gag and RT for a given element. Details of our phylogenetic tree building using MrBayes 3.2.6 (Ronquist et al. 2012) can be found in the Supplemental Methods.
Validation of computationally defined candidate telomeric retrotransposons
We used PCR on genomic DNA prepared from 10 females to validate (1) domain consensus sequences and (2) head-to-tail tandem array orientations stereotypical of telomere-lengthening retrotransposons. Sequence traces are reported in Supplemental Table S3 and Drosophila stocks are reported in Supplemental Table S9.
PCR-validated elements may be phylogenetically related to telomere-specialized elements but may not necessarily be telomere-restricted. We used fluorescent in situ hybridization (FISH) probes and/or “oligopaints” (Beliveau et al. 2012) to determine whether the computationally predicted, PCR-validated retrotransposons localize to chromosome ends. Specifically, we hybridized to polytene chromosomes FISH probes cognate to HeT-A, TART (both D. melanogaster controls), and TARTAHRE (D. rhopaloa), as well as oligopaints cognate to TR2 (D. elegans) (Supplemental Table S10). We also evaluated whether a jockey family element from D. biarmipes localized to telomeres using FISH (previously referred to HTR0) (Villasante et al. 2007). Finally, we hybridized Helitron and SAR2 sequence-derived FISH probes (Supplemental Table S10) to D. biarmipes polytene chromosomes to validate our long read–based assembly of its chromosome ends. Experimental details of FISH and oligopaint experiments can be found in the Supplemental Methods.
Relative telomeric retrotransposon copy number and estimates of within-genome diversity
To estimate the relative abundance of our validated telomeric retrotransposons in a given species genome, we mapped raw reads (Sanger or 454) to both our consensus sequences and the most recent genome assembly (Supplemental Table S7) using SMALT (http://sourceforge.net/projects/smalt/) with varying parameters (k: length of the hashed word index, s: the sampling distance between successive words, and y: the percentage identity allowing a word to match). We selected the parameters that maximized coverage for both genome and TE consensus (index: -k 20, -s 13; map: -y 0.9). To infer retrotransposon copy number, we divided a given retrotransposon average coverage by the genome-wide average coverage. Previous reports suggested that a nontrivial fraction of telomere-specialized retrotransposons is partially degenerated (Mason and Biessmann 1995; George et al. 2006; Villasante et al. 2007). We indeed detected widespread degradation in all sampled genomes. To account for retrotransposon sequence represented by these reads encoding partially degraded sequence, we also conducted a BLAST-guided approach to capture all reads harboring >90% identity to the consensus sequence. We trimmed the nontelomeric TE sequence from these reads and recalculated normalized coverage with these trimmed reads. We report the latter analysis. Previously reported nontelomeric, Y-linked HeT-A and TART (Agudo et al. 1999) do not confound our copy number estimates: Virtually all internal, Y-linked sequences cognate to these elements map to 5′ UTR and 3′ UTR sequence, not to the gag and RT domains to which we mapped reads for copy number estimates (Supplemental Fig. S16; Chang and Larracuente 2019). Nevertheless, we experimentally validated our in silico estimates by conducting qPCR on genomic DNA prepared from 10 females per species. We designed primers to amplify the region of highest mean coverage (estimated from the BLAST analysis above) and used the ΔΔCq method to quantitate abundance relative to a single copy gene (rp49). To infer retrotransposon invasion history, we aligned in PHYLIP a given consensus sequence and all significant, >90% identity reads. After visual inspection, we estimated the rates of transitions and transversions from alignment files and calculated the Kimura two-parameter distances (Kimura 1980).
Single-molecule–based sequencing, assembly, and validation of D. biarmipes
Assembling D. biarmipes telomeres with long reads
Genomic DNA preparation for the PacBio SMRT platform can be found in the Supplemental Methods. We built both PacBio-only assemblies and PacBio/Illumina hybrid assemblies. We used the PBcR pipeline (Celera 8.3) to assemble PacBio reads only. First, we executed a self-correction step (“PBcR-MHAP”, k = 14, merSize = 16, sketches = 1024, coverage = 25) that generated a preassembly (N50 = 480 kb; longest contig = 4.3 Mb). We passed the Celera assembler (CA 8.3) the longest 25× coverage-corrected contigs, using parameters optimized for genomes >100 Mb (Berlin et al. 2015). The N50 and longest contig both increased to a final 718 kb and 9.1 Mb, respectively. We generated a hybrid assembly of our PacBio reads and the publicly available Illumina short reads from D. biarmipes (Supplemental Table S7) using the software package DGB2OLC (Ye et al. 2016), following parameters described previously (Chakraborty et al. 2018). This hybrid assembly increased the N50 and longest contig to 2.5 and 9.2 Mb, respectively.
Defining telomeric DNA from D. biarmipes
To extract telomeric DNA sequence from the D. biarmipes PBcR assembly, we relied on the well-annotated, full-length chromosome arms of D. melanogaster (release r6.17). Starting with a query of terminal D. melanogaster genes (about 20 genes per chromosome arm), we used BLASTN to identify D. biarmipes contigs encoding terminal sequence (D. biarmipes and D. melanogaster share a common karyotype) (Deng et al. 2007). We identified exactly four D. biarmipes contigs with homology to D. melanogaster 2L, 2R, 3L, and 3R Chromosome ends. To further validate our assembly of four D. biarmipes telomeres, we compared them to a second hybrid assembly generated independently from Oxford Nanopore and Illumina reads (Miller et al. 2018). In no case did we observe in Miller et al. (2018) contigs that extend beyond the most distal sequence from our assembly (see Supplemental Fig. S8).
Analysis of telomeric DNA sequence in D. biarmipes and D. melanogaster
For both our D. biarmipes telomeric contigs and those defined using the same methods for D. melanogaster (Kim et al. 2014), we annotated the sequence distal to the most terminal gene using RepeatMasker (Smit et al. 2015). From this annotation, we plotted TE family abundance (Supplemental Fig. S10) and then focused our analyses on the two highest repeat classes, the Helitron 2N DNA transposon and the SAR2 complex satellite repeat. We extracted the repeats from annotated contigs and aligned them to their respective consensus. From these PHYLIP alignments, we calculated the Kimura two-parameter distance. We summarized distance estimates on heatmaps according to their position along telomere terminal sequences (Supplemental Fig. S12). We also annotated higher-order repeat structures of individual SAR2 variants from PHYLIP alignments by ordering SAR2 repeats according to their position along telomeres (Supplemental Fig. S13). Finally, we compared telomeric Helitrons to pericentric (those sharing a contig with most proximal genes) and euchromatic Helitrons by parsing copies from our PacBio assembly using the Helitron 2N consensus as a BLASTN query (>80% identity threshold). By using the methods described above, we calculated the Kimura two-parameter distance of copies derived from telomeric, pericentromeric, and euchromatic regions as above (Supplemental Fig. S11).
Data access
All Sanger sequenced PCR products from this study have been submitted to the NCBI GenBank database (https://www.ncbi.nlm.nih.gov/genbank/) under accession numbers MK645871–MK645883. The PacBio long reads for D. biarmipes and the draft, long read–only assembly generated in this study have been submitted to the NCBI BioProject database (https://www.ncbi.nlm.nih.gov/bioproject) under accession number PRJNA495839. All other processed data can be found in the Supplemental Material. Pipeline scripts are available as Supplemental Code and on GitHub (https://github.com/LevineLabUPenn/Diversification-Collapse-Telomeric-TEs).
Supplementary Material
Acknowledgments
We thank C. Leek for assistance with PCR-based validation experiments, J. Renfro for assistance with Amazon Web Services, and G. Lee, S. Zanders, M. Hahn, and three reviewers for their profoundly helpful comments on earlier versions of the manuscript. This work was supported by a National Institutes of Health (NIH) NIGMS grant R00GM107351 and an NIH NIGMS grant R35GM124684 to M.T.L.
Author contributions: M.T.L. and B.S.-L. designed the experiments. B.S.-L. performed the bioinformatics, the experiments, and the analyses. S.C.N. designed the oligopaints. M.T.L. and B.S.-L. wrote the manuscript.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.245001.118.
References
- Agudo M, Losada A, Abad JP, Pimpinelli S, Ripoll P, Villasante A. 1999. Centromeres from telomeres? The centromeric region of the Y chromosome of Drosophila melanogaster contains a tandem array of telomeric HeT-A- and TART-related sequences. Nucleic Acids Res 27: 3318–3324. 10.1093/nar/27.16.3318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beauregard A, Curcio MJ, Belfort M. 2008. The take and give between retrotransposable elements and their hosts. Annu Rev Genet 42: 587–617. 10.1146/annurev.genet.42.110807.091549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck CR, Garcia-Perez JL, Badge RM, Moran JV. 2011. LINE-1 elements in structural variation and disease. Annu Rev Genomics Hum Genet 12: 187–215. 10.1146/annurev-genom-082509-141802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beliveau BJ, Joyce EF, Apostolopoulos N, Yilmaz F, Fonseka CY, McCole RB, Chang Y, Li JB, Senaratne TN, Williams BR, et al. 2012. Versatile design and synthesis platform for visualizing genomes with Oligopaint FISH probes. Proc Natl Acad Sci 109: 21301–21306. 10.1073/pnas.1213818110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berlin K, Koren S, Chin CS, Drake JP, Landolin JM, Phillippy AM. 2015. Assembling large genomes with single-molecule sequencing and locality-sensitive hashing. Nat Biotechnol 33: 623–630. 10.1038/nbt.3238 [DOI] [PubMed] [Google Scholar]
- Berloco M, Fanti L, Sheen F, Levis RW, Pimpinelli S. 2005. Heterochromatic distribution of HeT-A- and TART-like sequences in several Drosophila species. Cytogenet Genome Res 110: 124–133. 10.1159/000084944 [DOI] [PubMed] [Google Scholar]
- Biessmann H, Mason JM, Ferry K, d'Hulst M, Valgeirsdottir K, Traverse KL, Pardue ML. 1990. Addition of telomere-associated HeT DNA sequences “heals” broken chromosome ends in Drosophila. Cell 61: 663–673. 10.1016/0092-8674(90)90478-W [DOI] [PubMed] [Google Scholar]
- Biessmann H, Kobeski F, Walter MF, Kasravi A, Roth CW. 1998. DNA organization and length polymorphism at the 2L telomeric region of Anopheles gambiae. Insect Mol Biol 7: 83–93. 10.1046/j.1365-2583.1998.71054.x [DOI] [PubMed] [Google Scholar]
- Biessmann H, Prasad S, Semeshin VF, Andreyeva EN, Nguyen Q, Walter MF, Mason JM. 2005. Two distinct domains in Drosophila melanogaster telomeres. Genetics 171: 1767–1777. 10.1534/genetics.105.048827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackburn EH. 1991. Structure and function of telomeres. Nature 350: 569–573. 10.1038/350569a0 [DOI] [PubMed] [Google Scholar]
- Casacuberta E. 2017. Drosophila: retrotransposons making up telomeres. Viruses 9: E192 10.3390/v9070192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casacuberta E, Pardue ML. 2002. Coevolution of the telomeric retrotransposons across Drosophila species. Genetics 161: 1113–1124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casacuberta E, Pardue ML. 2003. Transposon telomeres are widely distributed in the Drosophila genus: TART elements in the virilis group. Proc Natl Acad Sci 100: 3363–3368. 10.1073/pnas.0230353100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakraborty M, VanKuren NW, Zhao R, Zhang X, Kalsow S, Emerson JJ. 2018. Hidden genetic variation shapes the structure of functional elements in Drosophila. Nat Genet 50: 20–25. 10.1038/s41588-017-0010-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang CH, Larracuente AM. 2019. Heterochromatin-enriched assemblies reveal the sequence and organization of the Drosophila melanogaster Y chromosome. Genetics 211: 333–348. 10.1534/genetics.118.301765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen ZX, Sturgill D, Qu J, Jiang H, Park S, Boley N, Suzuki AM, Fletcher AR, Plachetzki DC, FitzGerald PC, et al. 2014. Comparative validation of the D. melanogaster modENCODE transcriptome annotation. Genome Res 24: 1209–1223. 10.1101/gr.159384.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng CY, Vogt A, Mochizuki K, Yao MC. 2010. A domesticated piggyBac transposase plays key roles in heterochromatin dynamics and DNA cleavage during programmed DNA deletion in Tetrahymena thermophila. Mol Biol Cell 21: 1753–1762. 10.1091/mbc.e09-12-1079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng CY, Young JM, Lin CG, Chao JL, Malik HS, Yao MC. 2016. The piggyBac transposon-derived genes TPB1 and TPB6 mediate essential transposon-like excision during the developmental rearrangement of key genes in Tetrahymena thermophila. Genes Dev 30: 2724–2736. 10.1101/gad.290460.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danilevskaya ON, Tan C, Wong J, Alibhai M, Pardue ML. 1998. Unusual features of the Drosophila melanogaster telomere transposable element HeT-A are conserved in Drosophila yakuba telomere elements. Proc Natl Acad Sci 95: 3770–3775. 10.1073/pnas.95.7.3770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de la Chaux N, Wagner A. 2009. Evolutionary dynamics of the LTR retrotransposons roo and rooA inferred from 12 complete Drosophila genomes. BMC Evol Biol 9: 205 10.1186/1471-2148-9-205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng Q, Zeng Q, Qian Y, Li C, Yang Y. 2007. Research on the karyotype and evolution of Drosophila melanogaster species group. J Genet Genomics 34: 196–213. 10.1016/S1673-8527(07)60021-6 [DOI] [PubMed] [Google Scholar]
- Dias GB, Heringer P, Svartman M, Kuhn GC. 2015. Helitrons shaping the genomic architecture of Drosophila: enrichment of DINE-TR1 in α- and β-heterochromatin, satellite DNA emergence, and piRNA expression. Chromosome Res 23: 597–613. 10.1007/s10577-015-9480-x [DOI] [PubMed] [Google Scholar]
- Drosophila 12 Genomes Consortium. 2007. Evolution of genes and genomes on the Drosophila phylogeny. Nature 450: 203–218. 10.1038/nature06341 [DOI] [PubMed] [Google Scholar]
- Feschotte C, Pritham EJ. 2007. DNA transposons and the evolution of eukaryotic genomes. Annu Rev Genet 41: 331–368. 10.1146/annurev.genet.40.110405.090448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- George JA, DeBaryshe PG, Traverse KL, Celniker SE, Pardue ML. 2006. Genomic organization of the Drosophila telomere retrotransposable elements. Genome Res 16: 1231–1240. 10.1101/gr.5348806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gladyshev EA, Arkhipova IR. 2007. Telomere-associated endonuclease-deficient Penelope-like retroelements in diverse eukaryotes. Proc Natl Acad Sci 104: 9352–9357. 10.1073/pnas.0702741104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorab E. 2003. Reverse transcriptase-related proteins in telomeres and in certain chromosomal loci of Rhynchosciara (Diptera: Sciaridae). Chromosoma 111: 445–454. 10.1007/s00412-003-0229-5 [DOI] [PubMed] [Google Scholar]
- Greider CW, Blackburn EH. 1989. A telomeric sequence in the RNA of Tetrahymena telomerase required for telomere repeat synthesis. Nature 337: 331–337. 10.1038/337331a0 [DOI] [PubMed] [Google Scholar]
- Hancks DC, Kazazian HH Jr. 2012. Active human retrotransposons: variation and disease. Curr Opin Genet Dev 22: 191–203. 10.1016/j.gde.2012.02.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higashiyama T, Noutoshi Y, Fujie M, Yamada T. 1997. Zepp, a LINE-like retrotransposon accumulated in the Chlorella telomeric region. EMBO J 16: 3715–3723. 10.1093/emboj/16.12.3715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoskins RA, Carlson JW, Kennedy C, Acevedo D, Evans-Holm M, Frise E, Wan KH, Park S, Mendez-Lago M, Rossi F, et al. 2007. Sequence finishing and mapping of Drosophila melanogaster heterochromatin. Science 316: 1625–1628. 10.1126/science.1139816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jangam D, Feschotte C, Betrán E. 2017. Transposable element domestication as an adaptation to evolutionary conflicts. Trends Genet 33: 817–831. 10.1016/j.tig.2017.07.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahn T, Savitsky M, Georgiev P. 2000. Attachment of HeT-A sequences to chromosomal termini in Drosophila melanogaster may occur by different mechanisms. Mol Cell Biol 20: 7634–7642. 10.1128/MCB.20.20.7634-7642.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaminker JS, Bergman CM, Kronmiller B, Carlson J, Svirskas R, Patel S, Frise E, Wheeler DA, Lewis SE, Rubin GM, et al. 2002. The transposable elements of the Drosophila melanogaster euchromatin: a genomics perspective. Genome Biol 3: RESEARCH0084 10.1186/gb-2002-3-12-research0084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapitonov VV, Jurka J. 2007. Helitrons on a roll: eukaryotic rolling-circle transposons. Trends Genet 23: 521–529. 10.1016/j.tig.2007.08.004 [DOI] [PubMed] [Google Scholar]
- Karpen GH, Spradling AC. 1992. Analysis of subtelomeric heterochromatin in the Drosophila minichromosome Dp1187 by single P element insertional mutagenesis. Genetics 132: 737–753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Käs E, Laemmli UK. 1992. In vivo topoisomerase II cleavage of the Drosophila histone and satellite III repeats: DNA sequence and structural characteristics. EMBO J 11: 705–716. 10.1002/j.1460-2075.1992.tb05103.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh K, Standley DM. 2013. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol 30: 772–780. 10.1093/molbev/mst010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim KE, Peluso P, Babayan P, Yeadon PJ, Yu C, Fisher WW, Chin CS, Rapicavoli NA, Rank DR, Li J, et al. 2014. Long-read, whole-genome shotgun sequence data for five model organisms. Sci Data 1: 140045 10.1038/sdata.2014.45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura M. 1980. A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. J Mol Evol 16: 111–120. 10.1007/BF01731581 [DOI] [PubMed] [Google Scholar]
- Langley CH, Montgomery E, Hudson R, Kaplan N, Charlesworth B. 1988. On the role of unequal exchange in the containment of transposable element copy number. Genet Res 52: 223–235. 10.1017/S0016672300027695 [DOI] [PubMed] [Google Scholar]
- Lee YCG, Karpen GH. 2017. Pervasive epigenetic effects of Drosophila euchromatic transposable elements impact their evolution. eLife 6: e25762 10.7554/eLife.25762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee YC, Leek C, Levine MT. 2017. Recurrent innovation at genes required for telomere integrity in Drosophila. Mol Biol Evol 34: 467–482. 10.1093/molbev/msw248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- López CC, Nielsen L, Edström JE. 1996. Terminal long tandem repeats in chromosomes form Chironomus pallidivittatus. Mol Cell Biol 16: 3285–3290. 10.1128/MCB.16.7.3285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch VJ, Nnamani MC, Kapusta A, Brayer K, Plaza SL, Mazur EC, Emera D, Sheikh SZ, Grutzner F, Bauersachs S, et al. 2015. Ancient transposable elements transformed the uterine regulatory landscape and transcriptome during the evolution of mammalian pregnancy. Cell Rep 10: 551–561. 10.1016/j.celrep.2014.12.052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mason JM, Biessmann H. 1995. The unusual telomeres of Drosophila. Trends Genet 11: 58–62. 10.1016/S0168-9525(00)88998-2 [DOI] [PubMed] [Google Scholar]
- Mason JM, Randall TA, Capkova Frydrychova R. 2016. Telomerase lost? Chromosoma 125: 65–73. 10.1007/s00412-015-0528-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McEachern MJ, Haber JE. 2006. Break-induced replication and recombinational telomere elongation in yeast. Annu Rev Biochem 75: 111–135. 10.1146/annurev.biochem.74.082803.133234 [DOI] [PubMed] [Google Scholar]
- Melnikova L, Georgiev P. 2002. Enhancer of terminal gene conversion, a new mutation in Drosophila melanogaster that induces telomere elongation by gene conversion. Genetics 162: 1301–1312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyne J, Ratliff RL, Moyzis RK. 1989. Conservation of the human telomere sequence (TTAGGG)n among vertebrates. Proc Natl Acad Sci 86: 7049–7053. 10.1073/pnas.86.18.7049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller DE, Staber C, Zeitlinger J, Hawley RS. 2018. Highly contiguous genome assemblies of 15 Drosophila species generated using nanopore sequencing. G3 (Bethesda) 8: 3131–3141. 10.1534/g3.118.200160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirkovitch J, Mirault ME, Laemmli UK. 1984. Organization of the higher-order chromatin loop: specific DNA attachment sites on nuclear scaffold. Cell 39: 223–232. 10.1016/0092-8674(84)90208-3 [DOI] [PubMed] [Google Scholar]
- Morrish TA, Garcia-Perez JL, Stamato TD, Taccioli GE, Sekiguchi J, Moran JV. 2007. Endonuclease-independent LINE-1 retrotransposition at mammalian telomeres. Nature 446: 208–212. 10.1038/nature05560 [DOI] [PubMed] [Google Scholar]
- Nefedova L, Kim A. 2017. Mechanisms of LTR-retroelement transposition: lessons from Drosophila melanogaster. Viruses 9: E81 10.3390/v9040081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osanai-Futahashi M, Fujiwara H. 2011. Coevolution of telomeric repeats and telomeric repeat-specific non-LTR retrotransposons in insects. Mol Biol Evol 28: 2983–2986. 10.1093/molbev/msr135 [DOI] [PubMed] [Google Scholar]
- Pardue ML, DeBaryshe PG. 2003. Retrotransposons provide an evolutionarily robust non-telomerase mechanism to maintain telomeres. Annu Rev Genet 37: 485–511. 10.1146/annurev.genet.38.072902.093115 [DOI] [PubMed] [Google Scholar]
- Pardue ML, DeBaryshe PG. 2008. Drosophila telomeres: a variation on the telomerase theme. Fly (Austin) 2: 101–110. 10.4161/fly.6393 [DOI] [PubMed] [Google Scholar]
- Pardue ML, DeBaryshe PG. 2011. Retrotransposons that maintain chromosome ends. Proc Natl Acad Sci 108: 20317–20324. 10.1073/pnas.1100278108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Podlevsky JD, Chen JJL. 2016. Evolutionary perspectives of telomerase RNA structure and function. RNA Biol 13: 720–732. 10.1080/15476286.2016.1205768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price MN, Dehal PS, Arkin AP. 2009. FastTree: computing large minimum evolution trees with profiles instead of a distance matrix. Mol Biol Evol 26: 1641–1650. 10.1093/molbev/msp077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rashkova S, Athanasiadis A, Pardue ML. 2003. Intracellular targeting of Gag proteins of the Drosophila telomeric retrotransposons. J Virol 77: 6376–6384. 10.1128/JVI.77.11.6376-6384.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice P, Longden I, Bleasby A. 2000. EMBOSS: the European Molecular Biology Open Software Suite. Trends Genet 16: 276–277. 10.1016/S0168-9525(00)02024-2 [DOI] [PubMed] [Google Scholar]
- Ronquist F, Teslenko M, van der Mark P, Ayres DL, Darling A, Höhna S, Larget B, Liu L, Suchard MA, Huelsenbeck JP. 2012. MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst Biol 61: 539–542. 10.1093/sysbio/sys029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosen M, Edstrom J. 2000. DNA structures common for chironomid telomeres terminating with complex repeats. Insect Mol Biol 9: 341–347. 10.1046/j.1365-2583.2000.00193.x [DOI] [PubMed] [Google Scholar]
- Sakofsky CJ, Malkova A. 2017. Break induced replication in eukaryotes: mechanisms, functions, and consequences. Crit Rev Biochem Mol Biol 52: 395–413. 10.1080/10409238.2017.1314444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slotkin RK, Martienssen R. 2007. Transposable elements and the epigenetic regulation of the genome. Nat Rev Genet 8: 272–285. 10.1038/nrg2072 [DOI] [PubMed] [Google Scholar]
- Smit AFA, Hubley R, Green P. 2015. RepeatMasker Open-4.0. http://www.repeatmasker.org.
- Sobinoff AP, Pickett HA. 2017. Alternative lengthening of telomeres: DNA repair pathways converge. Trends Genet 33: 921–932. 10.1016/j.tig.2017.09.003 [DOI] [PubMed] [Google Scholar]
- Starnes JH, Thornbury DW, Novikova OS, Rehmeyer CJ, Farman ML. 2012. Telomere-targeted retrotransposons in the rice blast fungus Magnaporthe oryzae: agents of telomere instability. Genetics 191: 389–406. 10.1534/genetics.111.137950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van de Lagemaat LN, Landry JR, Mager DL, Medstrand P. 2003. Transposable elements in mammals promote regulatory variation and diversification of genes with specialized functions. Trends Genet 19: 530–536. 10.1016/j.tig.2003.08.004 [DOI] [PubMed] [Google Scholar]
- Villasante A, Abad JP, Planello R, Mendez-Lago M, Celniker SE, de Pablos B. 2007. Drosophila telomeric retrotransposons derived from an ancestral element that was recruited to replace telomerase. Genome Res 17: 1909–1918. 10.1101/gr.6365107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villasante A, de Pablos B, Méndez-Lago M, Abad JP. 2008. Telomere maintenance in Drosophila: rapid transposon evolution at chromosome ends. Cell Cycle 7: 2134–2138. 10.4161/cc.7.14.6275 [DOI] [PubMed] [Google Scholar]
- Watson JD. 1972. Origin of concatemeric T7 DNA. Nat New Biol 239: 197–201. 10.1038/newbio239197a0 [DOI] [PubMed] [Google Scholar]
- Wei KHC, Reddy HM, Rathnam C, Lee J, Lin DAN, Ji SQ, Mason JM, Clark AG, Barbash DA. 2017. A pooled sequencing approach identifies a candidate meiotic driver in Drosophila. Genetics 206: 451–465. 10.1534/genetics.116.197335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werren JH. 2011. Selfish genetic elements, genetic conflict, and evolutionary innovation. Proc Natl Acad Sci 108(Suppl 2): 10863–10870. 10.1073/pnas.1102343108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wicker T, Sabot F, Hua-Van A, Bennetzen JL, Capy P, Chalhoub B, Flavell A, Leroy P, Morgante M, Panaud O, et al. 2007. A unified classification system for eukaryotic transposable elements. Nat Rev Genet 8: 973–982. 10.1038/nrg2165 [DOI] [PubMed] [Google Scholar]
- Xie W, Donohue RC, Birchler JA. 2013. Quantitatively increased somatic transposition of transposable elements in Drosophila strains compromised for RNAi. PLoS One 8: e72163 10.1371/journal.pone.0072163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang HP, Barbash DA. 2008. Abundant and species-specific DINE-1 transposable elements in 12 Drosophila genomes. Genome Biol 9: R39 10.1186/gb-2008-9-2-r39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye C, Hill CM, Wu S, Ruan J, Ma ZS. 2016. DBG2OLC: efficient assembly of large genomes using long erroneous reads of the third generation sequencing technologies. Sci Rep 6: 31900 10.1038/srep31900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zakian VA. 1989. Structure and function of telomeres. Annu Rev Genet 23: 579–604. 10.1146/annurev.ge.23.120189.003051 [DOI] [PubMed] [Google Scholar]
- Zakian VA. 1996. Structure, function, and replication of Saccharomyces cerevisiae telomeres. Annu Rev Genet 30: 141–172. 10.1146/annurev.genet.30.1.141 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.