

Abstract
Adult-onset leukodystrophies and genetic leukoencephalopathies comprise a diverse group of neurodegenerative disorders of white matter with a wide age of onset and phenotypic spectrum. Patients with white matter abnormalities detected on MRI often present a diagnostic challenge to both general and specialist neurologists. Patients typically present with a progressive syndrome including various combinations of cognitive impairment, movement disorders, ataxia and upper motor neuron signs. There are a number of important and treatable acquired causes for this imaging and clinical presentation. There are also a very large number of genetic causes which due to their relative rarity and sometimes variable and overlapping presentations can be difficult to diagnose. In this review, we provide a structured approach to the diagnosis of inherited disorders of white matter in adults. We describe clinical and radiological clues to aid diagnosis, and we present an overview of both common and rare genetic white matter disorders. We provide advice on testing for acquired causes, on excluding small vessel disease mimics, and detailed advice on metabolic and genetic testing available to the practising neurologist. Common genetic leukoencephalopathies discussed in detail include CSF1R, AARS2, cerebral arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL), and mitochondrial and metabolic disorders.
Keywords: neurogenetics, neuroradiology, dementia, adrenoleukodystrophy, movement disorders
Introduction
Adult patients with extensive white matter hyperintensities on MRI present the neurologist with a complex diagnostic task. There are a wide variety of disorders that can lead to these imaging appearances, including inflammatory, infective and malignant causes, as well as extensive small vessel disease. Often the most difficult patients to diagnose however are those with presumed genetic conditions, including the classical leukodystrophies, where the primary pathology is based in the myelin, or the genetic leukoencephalopathies, where the pathology is mainly neuronal or systemic. Patients may present with a wide spectrum of clinical features, including cognitive and neuropsychiatric changes, movement disorders, spasticity and seizures. The diversity of genetic aetiologies combined with the often-overlapping clinical and radiological phenotypes can make definitive diagnosis challenging.
In 2014 we set out a practical approach to navigate through the maze.1 However, even in the space of 5 years, there has been a substantial increase in the number of implicated genes. Improved phenotype–genotype correlation and increased access to advanced sequencing technologies have therefore changed our diagnostic approach.
This new review provides an opportunity to update and refine our practical approach for the diagnosis of genetic leukoencephalopathies in the era of whole genome sequencing. We would strongly recommend that it be read with the previous paper to avoid any repetition. As before, we emphasise the need to exclude as far as possible any acquired disorder, emphasise facets of the relatively commoner classical leukodystrophies/leukoencephalopathies, then highlight newer emerging diseases. Clinical/MRI features which point towards certain conditions are described. We end with an algorithm that we have found useful and describe our own experience.
For clarity, we are describing an approach where the symptoms have begun after the age of 16 and the MRI shows widespread cranial white matter change on standard (T2, fluid-attenuated inversion recovery (FLAIR)) sequences.
Excluding common acquired leukoencephalopathies
As emphasised previously it is imperative that treatable and acquired causes of white matter disease are ruled out as far as possible, before the patient is referred for investigation of a genetic leukodystrophy. As a minimum, all patients should undergo infective screening, including testing for HIV, syphilis, hepatitis B/C and tuberculosis. Patients with a history of immunosuppression should be tested for progressive multifocal leukoencephalopathy by cerebrospinal fluid (CSF) examination for the presence of JC virus. A high index of suspicion should remain for neoplasia, including primary central nervous system (CNS) lymphoma and gliomatosis cerebri. A careful history will reveal if the patient has been exposed to chemotherapy/radiotherapy (figure 1A) (in particular 5-fluorouracil and methotrexate) and recreational drugs like heroin or methanol, which can all lead to confluent, symmetrical leukoencephalopathy. Treatable inflammatory disorders like systemic lupus erythematosus must be considered (figure 1B,C). Posterior reversible leukoencephalopathy syndrome causes a dramatic leukoencephalopathy often with infarcts and microhaemorrhages (figure 1D), which improves over time. Further guidance on the initial work-up of these patients can be found in round 1 investigations (online supplementary table 1 in: Ahmed et al,1 Journal of Neurology, Neurosurgery, and Psychiatry).
Figure 1.
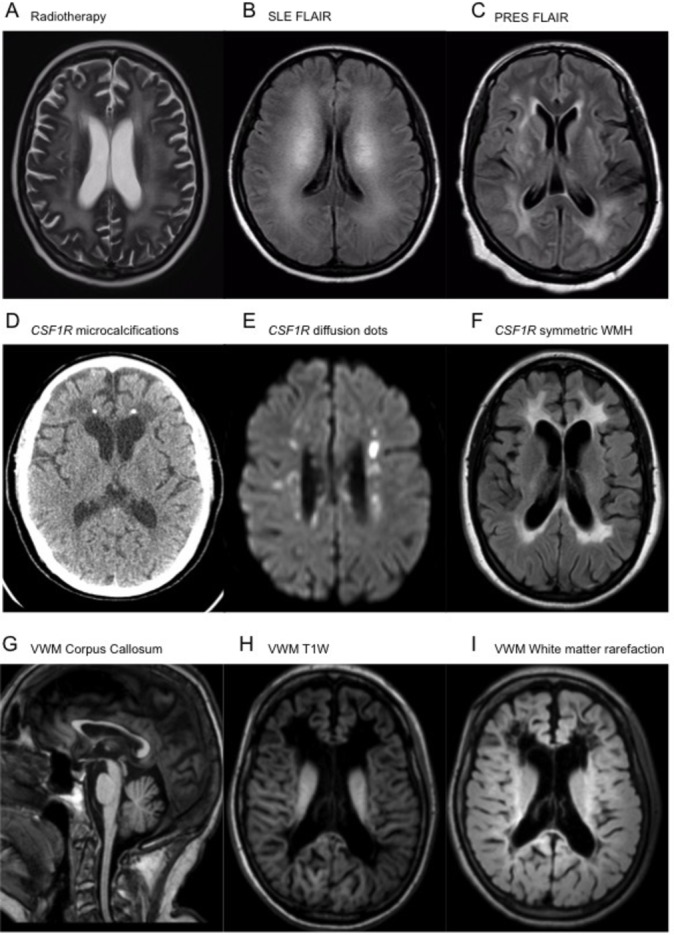
Axial T2-weighted MRI of radiotherapy changes (A), as well as axial FLAIR MRI of cerebral Systemic Lupus Eythematosus (SLE) (B) and PRES (C) demonstrating symmetrical and confluent periventricular and subcortical white matter signal hyperintensity (WMH). Axial non-contrast CT demonstrating subcortical volume loss, punctate calcification and confluent hypodensity adjacent to the frontal horns of the lateral ventricles (D). Axial B1000 DWI and FLAIR MRI acquisitions showing DWI avid punctate lesions (E) as well as confluent periventricular hyperintense signal changes (F) in CSF1R leukodystrophy. MRI sagittal T1W acquisition showing diffuse thinning of the corpus callosum (G), as well as axial T1W (H) and FLAIR (I) sequences demonstrating confluent rarefaction of the subcortical and periventricular white matter adjacent to the frontal horns and ventricular trigones of the lateral ventricles in VWM disease. FLAIR, fluid-attenuated inversion recovery; PRES, posterior reversible leukoencephalopathy syndrome; T1W, T1-weighted; VWM, vanishing white matter.
Features suggestive of an acquired disorder include rapid onset, steroid responsiveness, systemic features, MRI gadolinium enhancement and cervical cord involvement—although of course there are exceptions as discussed below.
Severe small vessel disease or genetic leukoencephalopathy?
A large number of patients referred for investigation will not be diagnosed with a genetic leukodystrophy, but instead will be diagnosed with severe acquired small vessel disease. These patients are more likely to present at an older age, have significant cardiovascular risk factors, and clinically are often asymptomatic or experience slow, indolent decline. They lack a clear family history to suggest a genetic origin. Imaging is likely to show increased T2/FLAIR signal in the periventricular and cerebral white matter, the basal ganglia, pons and cerebellum with chronic lacunes and cerebral microbleeds.2 In contradistinction to many inflammatory disorders such as multiple sclerosis, cervical cord imaging will be normal. In older patients (eg, >60 years), extensive genetic testing is unlikely to reveal a cause.3 However, in patients with this imaging appearance at a younger age, consideration should be given to the genetic forms of small vessel disease (eg, cerebral arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL), COL4A1), which are discussed below.
Clinical and radiological pattern recognition
Many adult-onset leukoencephalopathies present in the same way, with a variable degree of cognitive decline, spasticity, apraxia and ataxia. This presentation is non-specific, and in these cases a non-biased approach using knowledge of the most frequent syndromes and next-generation sequencing technologies is the most efficient route to a diagnosis. However, many of the syndromes described below have particular clues to diagnosis which may be found in a careful history and neurological examination. The mode of inheritance is important and can limit the number of conditions to consider (table 1); however, the most common inheritance pattern in adults is sporadic. This may suggest an autosomal recessive mode of inheritance in a small family, reduced penetrance in an autosomal dominant condition or even a censored family history. The ethnicity of the patient and geographical region of origin are relevant as the frequency of each disorder can vary substantially in different regions.
Table 1.
Leukoencephalopathies known to present in adulthood
| Disorder | Inheritance | Investigation | Gene |
| Adult-onset leukodystrophies | |||
| Inherited disorders of metabolism associated with abnormal biochemical tests | |||
| X linked adrenoleukodystrophy | X linked | Very long chain fatty acids | ABCD1 |
| Krabbe disease | AR | WCE (galactocerebrosidase) |
GALC, PSAP |
| Metachromatic leukodystrophy | AR | WCE (arylsulfatase A) Urine sulfatides |
ARSA |
| Cerebrotendinous xanthomatosis | AR | Serum cholestenol, urine bile alcohols | CYP27A1 |
| Methylenetetrahydrofolate reductase deficiency | AR | Plasma amino acid profile | MTHFR |
| Homocystinuria | AR | Plasma amino acid profile | CBS |
| mtDNA mutations | Maternal | mtDNA sequencing Blood/cerebrospinal fluid lactate Muscle biopsy |
Various |
| Disorder | Acronym | Inheritance | Gene |
| Genetic leukoencephalopathies | |||
| Hereditary diffuse leukoencephalopathy with axonal spheroids and pigmented glia | HDLS | AD | CSF1R |
| Cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy | CADASIL | AD | NOTCH3 |
| Cerebral autosomal recessive cerebral arteriopathy with subcortical infarcts and leukoencephalopathy | CARASIL | AR | HTRA1 |
| Cathepsin A-related arteriopathy with strokes and leukoencephalopathy | CARASAL | AD | CTSA |
| Cerebral leukodystrophy with retinal vasculopathy | AD | TREX1 | |
| Small vessel disease with ocular abnormalities | AD | COL4A1 | |
| Alexander disease | AD | GFAP | |
| Adult-onset autosomal dominant leukodystrophy | ADLD | AD | LMNB1 duplication |
| AARS2-related leukodystrophy | AARS2-L | AR | AARS2 |
| Vanishing white matter disease | VWM | AR | EIF2B1-5 |
| Leukoencephalopathy with brainstem and spinal cord involvement and lactate elevation | LBSL | AR | DARS2 |
| Mitochondrial neurogastrointestinal encephalopathy | MNGIE | AR | TYMP |
| Pelizaeus-Merzbacher disease | PMD | X linked dominant | PLP1 |
| Pelizaeus-Merzbacher-like disease | PMLD | AR | GJC2 |
| Adult polyglucosan body disease | APBD | AR | GBE |
| Hypomyelinating leukodystrophy with atrophy of the basal ganglia and cerebellum | H-ABC | AD | TUBB4A |
| Leukoencephalopathy with ataxia | LKPAT | AR | CLCN2 |
| 4H syndrome | AR | POLR3A/POLR3B | |
| Labrune syndrome | AR | SNORD118 | |
| Gordon Holmes syndrome | AR | RNF216 | |
| Polycystic lipomembranous osteodysplasia with sclerosing leukoencephalopathy | PLOSL | AR |
TREM2
TYROBP |
AD, Autosomal Dominant; AR, Autosomal recessive; WCE, White Cell Enzymes.
The most helpful and discriminating symptoms and signs are described in table 2. These include the importance of noting endocrine abnormalities such as premature ovarian failure (POF) (vanishing white matter (VWM) disease, AARS2) or hypogonadotrophic hypogonadism (Gordon Holmes syndrome, POLR3-related disorders). Movement disorders such as prominent parkinsonism or chorea are important, as are the presence of early autonomic (LMNB1) or urinary symptoms (adult polyglucosan body disease). Peripheral demyelinating neuropathy may suggest metachromatic leukodystrophy (MLD), and adrenal failure is highly suggestive of adrenoleukodystrophy (ALD).
Table 2.
Clinical features that can be associated with specific leukodystrophies
| Clinical characteristics of specific leukodystrophies | |
| Premature ovarian failure | VWM. Galactosaemia. AARS2-L. |
| Hypogonadotrophic hypogonadism | Gordon Holmes syndrome. 4H syndrome. |
| Parkinsonism | HDLS. |
| Chorea | Gordon Holmes syndrome. |
| Palatal myoclonus | Alexander disease. |
| Axonal peripheral neuropathy | ALD. LBSL. APBD. CTX. |
| Demyelinating peripheral neuropathy | MLD. |
| Optic atrophy | MLD. Krabbe disease. ALD. PMD mtDNA mutations. |
| Adrenal insufficiency | ALD. |
| Dental abnormalities | 4H syndrome. |
| Tendon xanthomata | CTX. |
| Early urinary frequency | APBD. |
| Early autonomic features | ADLD. |
| Early prominent ataxia | CTX. Gordon Holmes syndrome. LKPAT. |
| Migraine with aura | CADASIL. |
| Cataracts | CTX mtDNA mutations. |
| Occasional rapid progression (<1 year) | ALD. Krabbe disease. |
ADLD, adult-onset, autosomal dominant leukodystrophy; ALD, adrenoleukodystrophy; APBD, adult polyglucosan body disease; CADASIL, cerebral arteriopathy with subcortical infarcts and leukoencephalopathy; CTX, cerebrotendinous xanthomatosis; HDLS, hereditary diffuse leukoencephalopathy with spheroids; LBSL, leukoencephalopathy with brainstem and spinal cord involvement with elevated lactate; LKPAT, leukoencephalopathy with ataxia; MLD, metachromatic leukodystrophy; PMD, Pelizaeus-Merzbacher disease; VWM, vanishing white matter.
Similarly to the clinical presentations, the radiological appearance of genetic leukoencephalopathies can be non-specific, but there are important features to note. Deep white matter diffusion abnormalities have been well described in CSF1R and AARS2, in addition to prominent involvement of the corpus callosum. A patchy leukoencephalopathy with lacunes, microhaemorrhages and anterior temporal lobe involvement is suggestive of a vascular disorder such as CADASIL or cathepsin A-related arteriopathy with strokes and leukoencephalopathy (CARASAL). Hypomyelination is an important sign to note, as there are only a small number of genetic diseases known to cause this radiological appearance. Useful radiological signs can be found in table 3.
Table 3.
Imaging features associated with specific leukodystrophies
| Imaging characteristics of specific leukodystrophies | |
| Frontal predominance | Alexander disease. |
| Basal ganglia abnormalities | H-ABC mtDNA mutations. PLOSL (calcifications). |
| Hypomyelination | PMD/PMLD. H-ABC. 4H syndrome. |
| Anterior temporal lobe signal abnormality | CADASIL. CARASIL. CARASAL. |
| Deep white matter diffusion dots | HDLS. AARS2-L. |
| Corpus callosum involvement | HDLS. AARS2-L. HSP genes (SPG11, SPG15, Fa2H). |
| ’Tadpole brainstem’ (atrophy of the cervical cord and medulla with preserved pons) | Alexander disease. |
| Dentate nucleus signal change/cysts | CTX. |
| Spinal cord abnormalities | Adrenomyeloneuropathy. LBSL. APBD. Alexander disease. |
| Middle cerebellar and cerebral peduncles | LKPAT. |
| Contrast enhancement | ALD. Alexander disease. Krabbe disease. |
| Periventricular microcalcifications (visible on CT) | HDLS. |
| Extensive calcifications and cysts | Labrune syndrome. |
| Bone cysts on X-ray | PLOSL. |
ALD, adrenoleukodystrophy; APBD, adult polyglucosan body disease; CADASIL, cerebral arteriopathy with subcortical infarcts and leukoencephalopathy; CARASAL, cathepsin A-related arteriopathy with strokes and leukoencephalopathy; CARASIL, cerebral autosomal recessive arteriopathy with subcortical infarcts and leukoencephalopathy; CTX, cerebrotendinous xanthomatosis; H-ABC, Hypomyelination with atrophy of the basal ganglia and cerebellum; HDLS, hereditary diffuse leukoencephalopathy with spheroids; HSP, Hereditary Spastic Paraplegia; LBSL, leukoencephalopathy with brainstem and spinal cord involvement with elevated lactate; LKPAT, leukoencephalopathy with ataxia; PLOSL, polycystic lipomembranous osteodysplasia with sclerosing leukoencephalopathy; PMD, Pelizaeus-Merzbacher disease; PMLD, Pelizaeus-Merzbacher-like disorder.
Metabolic leukodystrophies
After excluding common acquired leukoencephalopathies, the first priority should be to identify patients with an inherited metabolic disorder. A small number of tests will detect the diseases that present in adulthood. Recommended testing includes a very long chain fatty acid (VLCFA) profile in men (ALD), specific white cell enzyme activities (Krabbe disease, MLD), raised serum cholestanol/urinary bile alcohols (cerebrotendinous xanthomatosis (CTX)) and plasma amino acids (methylenetetrahydrofolate reductase (MTHFR) deficiency and homocystinuria) (table 1).
X linked ALD
Adult-onset ALD is a rare X linked metabolic disorder of peroxisomal fatty acid beta-oxidation which results in the accumulation of VLCFA in plasma and tissues, including white matter and the adrenal cortex.4
ALD is associated with three main phenotypes, including Addison-only disease, adrenomyeloneuropathy (AMN) and cerebral ALD (CALD). Addison-only presents most commonly by age 8 years, and although it begins without evidence of neurological abnormality some degree of disability (most commonly AMN) usually develops later. Indeed, virtually all patients with ALD who reach adulthood develop AMN, usually between 20 and 30 years of age. AMN is a non-inflammatory distal axonopathy, and is characterised by progressive spastic paraparesis, sensory ataxia, sphincter dysfunction, impotence and pain. CALD usually only affects men and presents with rapidly progressive inflammatory demyelination in the brain, leading to rapid cognitive and neurological decline, dementia, ataxia, seizures and death.
MRI is always abnormal in neurologically symptomatic men with cerebral disease. Early changes include T2/FLAIR hyperintensities in the parieto-occipital regions and splenium of the corpus callosum. A minority of patients will demonstrate signal abnormalities predominantly in the frontal lobes and genu of the corpus callosum. Peripheral rim enhancement is a typical feature.4 5
Elevated levels of VLCFAs in the blood are suggestive of a peroxisomal disorder, and the diagnosis of ALD can be confirmed by sequencing of the ABCD1 gene. Early diagnosis and family screening are essential to reduce the risk of untreated hypoadrenalism, and to identify early those patients who may benefit from haematopoietic stem cell transplantation (HSCT).6
Krabbe disease
Krabbe disease is a rare autosomal recessive disorder caused by loss of function mutations in GALC, leading to deficiency of galactocerebrosidase, a lysosomal enzyme responsible for the degradation of galactocerebroside to ceramide and galactose. In Krabbe disease, galactocerebroside accumulates in the peripheral and central nervous system producing cerebral atrophy, loss of myelin, gliosis and globoid cells.7
There are three forms of Krabbe disease: infantile, juvenile and adult. The infantile form is the most severe and usually presents between 3 and 6 months of age. After a normal neonatal period, those affected develop a rapidly progressive course involving irritability, hyperaesthesia, visual and hearing loss, severe cognitive and motor deterioration, and seizures. This group rarely survives beyond 2 years. Nerve conduction studies show a demyelinating peripheral neuropathy and CSF analysis may reveal increased total protein concentration.8
Juvenile and adult (late) forms of the disease have a milder, more varied phenotype, a slower rate of progression and a significantly longer lifespan (although we have observed a rapid deterioration over a year in one case). Typical features include spasticity, dementia, ataxia, peripheral neuropathy and visual loss. The T2/FLAIR signal abnormality in Krabbe disease predominantly affects the corticospinal tracts, from the cortex, through the corona radiata internal capsules and cerebral peduncles.9 The optic radiations are frequently involved,9 and intracranial calcifications have been reported, but may only be apparent on CT imaging.10
The diagnosis of Krabbe disease can be made by assay of galactosylceramidase activity via white cell enzyme testing.
Metachromatic leukodystrophy
MLD is an autosomal recessive disorder caused mainly by deficient activity of arylsulfatase A (ARSA). ARSA is responsible for the desulfation of cerebroside sulfate, a major glycolipid of myelin, and decreased ARSA activity leads to the accumulation of cerebroside sulfate in the CNS and peripheral nerves (as well as the kidneys and other visceral organs). The result is central and peripheral demyelination.11
In almost all cases, recessive mutations in the ARSA gene are responsible, although very rarely, MLD can be caused by mutations in the PSAP gene.12 Three major subtypes exist—late infantile (age 6 months to 2 years), juvenile (age 3–16 years) and adult (age >16). Peripheral neuropathy occurs in all forms and gallbladder involvement (hyperplastic polyps) is also common. Late infantile onset is associated with poor prognosis (death typically occurs within 5–6 years) and manifests as regression of motor skills, gait abnormalities, seizures, ataxia, hypotonia, extensor plantars and optic atrophy. Juvenile disease presents similarly but is more heterogeneous. Progression is slower, and these children may survive until early adulthood. Adult onset is usually heralded by dementia, behavioural difficulties, and in a minority with psychosis.13
Nerve conduction studies show marked slowing. Brain MRI reveals symmetric periventricular white matter lesions and cortical atrophy, often with a tigroid or stripe pattern, caused by the appearance of the spared perivascular white matter.14 Diagnosis is established by demonstrating deficient ARSA activity in leucocytes (white cell enzyme testing) or cultured skin fibroblasts. Clinicians should be aware of the pseudodeficiency state, in which ARSA activity levels are low, but do not cause disease. In pseudodeficiency, the activity level is typically 5%–20% of controls.15 In affected individuals with MLD, urine sulfatide excretion is increased, generally tenfold to a hundred-fold greater than controls. Urine sulfatide excretion may therefore be used to differentiate pseudodeficiency from deficiency. However, the assay for urine sulfatide excretion is not currently routinely available in the UK and, as testing for the pseudodeficiency variants can be done rapidly, this is the usual first-line test.
No curative treatment is currently available, but HSCT has slowed disease progression in some patients.16 Other promising novel treatments include gene therapy and enzyme replacement.17
Cerebrotendinous xanthomatosis
CTX is an autosomal recessive disorder of bile acid synthesis caused by mutation of the cytochrome P450 gene CYP27A1, resulting in the production of defective sterol 27-hydroxylase. Consequently, CTX is associated with high levels of cholestanol in plasma and its accumulation in tissue. This gives rise to hallmark clinical manifestations of chronic diarrhoea, bilateral cataracts, tendon xanthomas or tendon thickening, and neurological dysfunction.18
Typical neurological manifestations can include intellectual disability, autism, behavioural and psychiatric problems, dementia, ataxia and epilepsy. MRI studies show cerebral and cerebellar atrophy, extensive white mater lesions of the spinal cord, and bilateral T2/FLAIR hyperintensity of the dentate nuclei and surrounding white matter.
Cholestanol concentrations are increased in plasma, brain, xanthomas and bile. Increased quantities of bile alcohols are useful as a secondary diagnostic test. CSF levels of cholestanol, cholesterol, apolipoprotein B, apolipoprotein A1 and albumin are increased.18
Treatment is with chenodeoxycholic acid (a cholesterol 7α-hydroxylase enzyme inhibitor), which is effective in improving biochemical findings, and in those without advanced disease may improve or stabilise neurological features.19
MTHFR deficiency
Autosomal recessive mutations in the MTHFR gene cause a spectrum of symptoms, with onset ranging from childhood to adulthood. Biochemically, there are elevated plasma homocysteine, homocystinuria and low methionine levels. Patients may develop seizures, cognitive decline, recurrent thrombotic stroke and leukoencephalopathy. The neurological signs may improve with treatment, the mainstay of which is folic acid and betaine supplementation.20
Genetic leukoencephalopathies
Since our previous review, a number of conditions have gained in prominence or have been newly described. They are diagnosed genetically, although as shown in tables 2 and 3 there can be typical diagnostic features to focus the genetic analysis.
CSF1R
Mutations in the CSF1R (colony stimulating factor-1 receptor) gene are known to cause an adult-onset leukodystrophy termed hereditary diffuse leukoencephalopathy with spheroids (HDLS).21 This condition has been shown to be one of the most common causes of adult-onset leukodystrophy, accounting for approximately 10% of cases.22 First described in a large Swedish kindred,23 this is an autosomal dominant disorder in which affected members develop a clinical course characterised by dementia, psychiatric changes and motor decline. The age of onset is variable, even within families, but typically patients develop symptoms in their 40s (range 18–78 years). The penetrance of the disorder is not known, but is likely to be high. Because of incomplete penetrance (and the possibility of de novo mutations), some affected patients will not have a family history and may appear to have a sporadic disorder.
The condition is found worldwide and has a well-described clinical phenotype. Prominent symptoms include parkinsonism,24 which is usually not levodopa-responsive, and upper motor neuron signs such as spasticity and ataxia. A recent large review found the mean disease duration to be 6.8 years from symptom onset to death.25
Pathologically the disease is characterised by the presence of astrogliosis with myelin and axonal loss and frequent axonal spheroids in the cerebral white matter. The spheroids are visible with H&E staining, but also stain positive for neurofilaments, p62, amyloid precursor protein APP and β amyloid. There are typically pigmented microglia present which are autofluorescent and stain positive for CD68. The pathological appearance of this disorder is relatively specific and lends the disease its name. Prior to the identification of the responsible gene, the diagnosis of HDLS could only be confirmed pathologically.
The MRI appearance of HDLS is consistent, with typical features including confluent, largely symmetric T2 hyperintense/T1 hypointense signal abnormality in the frontoparietal and periventricular white matter, which spares the U-fibres, and involves the pyramidal tracts and corpus callosum.26 Punctate areas of restricted diffusion may be present (termed deep white matter diffusion dots) and their appearance may even mimic CNS vasculitis.22 Calcifications may be present, particularly in the periventricular white matter adjacent to the frontal horns (figure 1D–F).
It was shown in 2011 that HDLS is largely caused by mutations in the CSF1R gene.21 27 Almost all mutations are found in the tyrosine kinase domain of the CSF1R protein and the majority of mutations are missense. A small number of splice site, frameshift and indel mutations have also been identified.
AARS2
Mutations in AARS2 (alanyl-transfer (t)RNA synthetase 2) are emerging as a rare cause of leukodystrophy with similar clinical, imaging and radiological phenotype to CSF1R mutations.28 29 This is an autosomal recessive disorder, but frequently appears sporadic, with a younger age of onset than CSF1R (mean age 29 years, range 15–44 years). However the initial symptoms are very similar, with psychiatric changes and cognitive decline, parkinsonism, pyramidal signs, ataxia and seizures. Almost all female cases have experienced POF, although no endocrine or reproductive abnormalities are found in men. MRI features include largely symmetric and confluent T2 hyperintense/T1 hypointense white matter signal change in the frontoparietal white matter, corpus callosum and pyramidal tracts. There are punctate areas of restricted diffusion on diffusion weighted imaging (DWI) often running parallel to the ventricles. A distinguishing feature from CSF1R is that the white matter signal suppresses on FLAIR imaging, indicating a degree of rarefaction.26
The clinical picture over time remains similar to CSF1R, with early cognitive symptoms followed by severe and rapid motor decline. Most patients are fully dependent within 5 years of symptom onset. In one case, pathological features of HDLS were found on brain biopsy, with axon and myelin loss and frequent axonal spheroids containing neurofilament, p62, APP and β amyloid. Pigmented microglia were also seen, and the pathological features were felt to be indistinguishable from HDLS. In another case, no pathological abnormalities were found in a brain biopsy,29 30 and further pathological descriptions are required.
AARS2 encodes a t-RNA synthetase responsible for correctly loading alanine onto tRNA-ala for the translation of mitochondrial proteins. Most commonly patients will have compound heterozygous AARS2 mutations, although homozygous cases have also been reported. Loss of function mutations (frameshift, nonsense, splice site) are the most common, but there are some pathogenic missense mutations, including the recurrent R199C mutation. Mutations in this class of genes (tRNA synthetases) are increasingly implicated in a diverse range of neurological disorders.31 32
VWM disease
VWM disease is an autosomal recessive disorder characterised by progressive neurological impairment and the cystic degeneration of white matter that is visible on MRI. It most commonly presents in childhood, but many adult-onset cases have been reported. The most frequent presenting features include ataxia, spasticity, seizures and cognitive decline. Often patients will experience episodes of severe neurological deterioration after a minor insult, such as a head trauma, an infection or even emotional distress.33 An additional feature in women with VWM is POF; hence, VWM is sometimes referred to as ovarioleukodystrophy.
In one study of adult-onset VWM, the mean age of onset was found to be 31 years, with clinical features including neurological and psychiatric presentations. Frequent MRI features included cerebral and cerebellar atrophy and cavitating leukoencephalopathy with involvement of the corpus callosum.34 The characteristic feature is that the periventricular white matter takes on the same signal intensity as the CSF on T2-weighted and FLAIR imaging (figure 1G–I). VWM is caused by homozygous and compound heterozygous mutations in any of the five genes that encode the subunits of the translation initiation factor EIF2B (EIF2B1–EIF2B5).35
Vascular leukoencephalopathies
A number of different genes come under the umbrella term of vascular leukoencephalopathies, including NOTCH3 (CADASIL), HTRA1 (cerebral autosomal recessive arteriopathy with subcortical infarcts and leukoencephalopathy (CARASIL)), CTSA (CARASAL), COL4A1 and TREX1.
Cerebral arteriopathy with subcortical infarcts and leukoencephalopathy
CADASIL is caused by heterozygous mutations in the NOTCH3 gene. The condition is autosomal dominant and is characterised by recurrent stroke at a young age, cognitive decline, migraine with aura and depression.36 The mean age of first stroke is 46 years and typically the events are subcortical, ischaemic lacunes. Reversible episodes of encephalopathy may also occur. On brain MRI, the first abnormality detected is often white matter signal abnormality in the temporal poles. Over time the burden of white matter lesions increases, with the periventricular, frontoparietal and external capsular white matter most affected.37 There are frequently dilated perivascular spaces and subcortical lacunes. Microhaemorrhages may be detected on gradient echo imaging (figure 2A–C). The diagnosis is supported by the finding of granular osmiophilic material in small arterioles by electron microscopic examination of tissue, usually a skin biopsy. Pathogenic NOTCH3 mutations lead to the gain or loss of a cysteine residue in one of the epidermal growth factor-like repeat (EGFr) domains of the NOTCH3 protein. These EGFr domains normally contain six cysteine residues, and any alteration in this number has been shown to lead to NOTCH3 aggregation. NOTCH3 mutations are not fully penetrant, but it has been shown that cysteine altering mutations affecting the first six EGFr domains are the most penetrant and are associated with the highest burden of white matter disease.38
Figure 2.
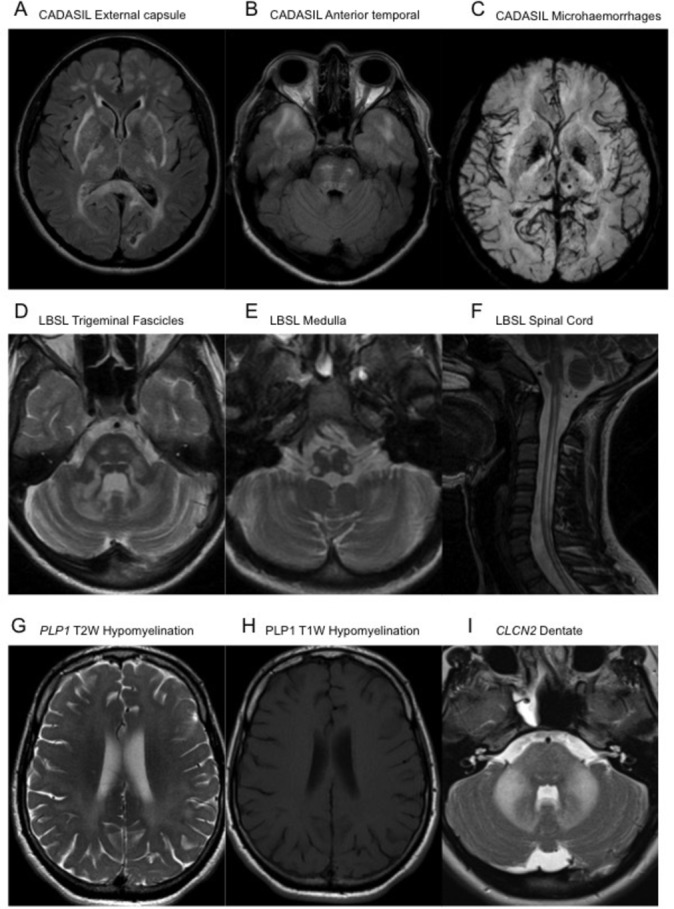
Axial FLAIR MRI sequences demonstrating confluent signal hyperintensity involving the external capsules, posterior limb of the internal capsules, peritrigonal white matter and splenium of the corpus callosum (A), as well as of the subcortical white matter of the temporal poles and central pons (B) in CADASIL. Minimal intensity projection from a susceptibility weighted imaging acquisition MRI demonstrating multiple punctate foci of paramagnetic susceptibility limited within the thalamus, left putamen and subcortical white matter of the left occipital lobe (C), in keeping with multiple microhaemorrhages in CADASIL. Axial T2W MRI sequences demonstrating signal hyperintensity within the trigeminal fascicles and cerebellar white matter (D), as well as within the pyramids, decussation of the medial lemnisci and inferior cerebellar peduncles at the level of the medulla oblongata (E) in LBSL. A sagittal T2W sequence of the upper spinal cord in the same patient demonstrates contiguous longitudinally extensive signal hyperintensity of the dorsal columns and lateral cortical spinal tracts (F). Axial T2W (G) and T1W (H) MRI sequences in a patient with Pelizaeus-Merzbacher disease illustrating the confluent diffuse T2W hyperintense signal within the white matter of the cerebrum, appearing unremarkable on the T1W sequences, suggestive of hypomyelination. Axial T2W sequences demonstrating confluent hyperintense signal change within the pons, middle cerebellar peduncles and dentate nuclei in a patient with a CNCL2 leukodystrophy (I). CADASIL, cerebral arteriopathy with subcortical infarcts and leukoencephalopathy; FLAIR, fluid-attenuated inversion recovery; T1W, T1-weighted; LBSL, leukoencephalopathy with brainstem and spinal cord involvement with elevated lactate; T2W, T2-weighted.
Cerebral autosomal recessive arteriopathy with subcortical infarcts and leukoencephalopathy
This autosomal recessive disorder is caused by biallelic mutations in the HTRA1 gene. CARASIL was first identified and is most common in Japan, but cases in Europe and South America have also been identified.39 40 Clinically it is characterised by dementia, parkinsonism, upper motor neuron signs, and extraneurological features including alopecia and back pain with spondylosis deformans. Acute strokes are common and the typical age of onset is the teenage years to 20s. MRI appearance is similar to CADASIL (above), with spondylosis deformans potentially visible on spinal imaging.41
Recently, it has been proposed that heterozygous HTRA1 mutations can also lead to autosomal dominant severe small vessel disease. In one study these patients were termed ‘manifesting heterozygotes with CARASIL’ and were described with a similar but less severe phenotype to recessive mutations.42 Spondylosis was found in all manifesting heterozygotes. However, another recent study found that the phenotype of heterozygous HTRA1 mutations was different from both CADASIL and CARASIL, and no extraneurological features were observed, although the imaging appearance was similar.43
Cathepsin A-related arteriopathy with strokes and leukoencephalopathy
This very recently described entity due to heterozygous mutations in the CTSA gene is named cathepsin A-related arteriopathy with strokes and leukoencephalopathy (CARASAL). The initial report described two Dutch families, distantly related, in whom members were affected by a severe leukoencephalopathy, accompanied often by ischaemic or haemorrhagic stroke, therapy-resistant hypertension and later cognitive decline.44 The MRI pattern was very similar to CADASIL. Since the initial report, another case has been described from the UK with identical imaging and a similar phenotype, although with additional prominent brainstem features reported.3 45 To date, all patients described have carried the same R325C mutation in CTSA. It is unclear whether this is the only mutation in CTSA which is causal for CARASAL or whether further mutations will be identified.
COL4A1 and TREX1
Both COL4A1 and TREX1 are associated with leukoencephalopathies in which ocular or retinal abnormalities may be present. COL4A1 is characterised by diffuse leukoencephalopathy, often with dilated perivascular spaces and microhaemorrhages, and retinal abnormalities including arteriolar tortuosity or retinal haemorrhage.46 It is an autosomal dominant disorder and shows incomplete penetrance. Additional CNS features include intracerebral haemorrhage, calcifications and cerebellar atrophy. Ocular abnormalities are not exclusively confined to the retina in COL4A1 with anterior segment dysgenesis, congenital cataract and nystagmus all reported.47 A second form of leukoencephalopathy with retinal vasculopathy is caused by heterozygous frameshift mutations in the c-terminus of the TREX1 gene.48 In addition to the microvascular disease of the retina which can lead to visual loss, Raynaud’s phenomenon and migraine are both frequently found.49 A phenotype of TREX1 mutations called hereditary endotheliopathy with retinopathy, nephropathy and stroke may present with progressive, contrast-enhancing lesions with surrounding oedema, which may be mistaken for tumours.50 Calcifications are found in more than 50% of patients.51
Mitochondrial DNA mutations
Mitochondrial DNA mutations are a rare cause of adult-onset leukoencephalopathy. Typically patients will present with a multisystem disorder, often including short stature, migraine, sensorineural deafness, cardiac defects and exercise intolerance. There is often involvement of the deep grey structures on imaging, such as the basal ganglia and thalamus, but symmetric and confluent white matter abnormalities can occur.52 Stroke-like episodes or cortical blindness would point to mitochondrial encephalopathy with lactic acidosis and stroke-like episodes (m.3243A>G); myoclonus, seizures and ataxia are suggestive of mitochondrial encephalopathy with ragged red fibres (m.8344A>G). Periventricular signal abnormalities have been reported in Leber’s hereditary optic neuropathy, but only as a minor feature.53 Common mitochondrial point mutations can be readily screened in blood, but if these are negative and a suspicion of mitochondrial disease remains, next-generation sequencing of the complete mitochondrial genome from the muscle may be necessary. Clinicians should be aware that exome sequencing and panel-based sequencing typically target only nuclear genes, that is, not mitochondrial DNA. There are methods to extract mitochondrial DNA sequence from exome data, but this is not commonly done.
Nuclear encoded mitochondrial genes
There are a large number of nuclear encoded genes with established or putative mitochondrial functions (>1000 in MitoCarta).54 This section could never be exhaustive, and some genes, such as AARS2 and DARS2, are discussed elsewhere in detail. Many of these disorders are only known to affect children, but two disorders of mtDNA maintenance can present with prominent leukoencephalopathy in adulthood, namely mitochondrial neurogastrointestinal encephalomyopathy (MNGIE) and POLG mutations. MNGIE is an autosomal recessive disorder caused by mutations in the TYMP gene.55 It usually presents in adulthood with ptosis, chronic progressive external ophthalmoplegia, gastrointestinal dysmotility including pseudo-obstruction, peripheral neuropathy, and diffuse leukoencephalopathy. A similar disorder can be caused by mutations in POLG, although these cases usually have less prominent leukoencephalopathy.56 Involvement of the pulvinar of the thalamus has been reported in POLG mutations.57 Nuclear encoded mitochondrial gene defects can be identified by whole exome or panel sequencing. Variants, either single nucleotide variants or deletions, of mtDNA are usually identified by specific mtDNA sequencing from affected tissue, often muscle.
Leukoencephalopathy with brainstem and spinal cord involvement with elevated lactate
Leukoencephalopathy with brainstem and spinal cord involvement with elevated lactate (LBSL) is an unusual and rare leukoencephalopathy caused by autosomal recessive mutations in the DARS2 gene.58 Usually patients develop symptoms in childhood, but the condition tends to be slowly progressive and evolves over time, so the diagnosis is usually made when the patient is an adult. The most common presentation consists of childhood onset of distal weakness and wasting, with pes cavus, and initially patients may be given a diagnosis of hereditary sensory motor neuropathy. Over time, upper motor neuron signs including spasticity and extensor plantar responses develop. Slowly progressive ataxia is another common feature, and is due to both cerebellar involvement and dorsal column loss. The neuroimaging appearance of LBSL is quite distinct, with a diffuse T2 hyperintense signal abnormality of the cerebral and cerebellar white matter, corpus callosum, medullary pyramids, medial lemniscus, intraparenchymal course of the trigeminal nerves, superior, middle and inferior cerebellar peduncles, and the dorsal columns and lateral corticospinal tracts of the spinal cord. A lactate peak is seen on magnetic resonance spectroscopy, but blood and CSF lactate are not elevated59 (figure 2D–F). DARS2 mutations are usually compound heterozygous, and the most frequent variant is a splice site mutation in intron 2.58 This region is poorly covered by next-generation sequencing approaches, and single-gene testing is recommended if the clinical and radiological picture is suggestive of LBSL.
Alexander disease
Caused by heterozygous mutations in GFAP, Alexander disease has infantile, juvenile and adult onset forms.60 Mutations are often de novo, and young children typically present with seizures, motor regression and macrocephaly. GFAP encodes the glial fibrillar acidic protein, an essential component of astrocytes. Alexander disease is characterised pathologically by the presence of Rosenthal fibres, which are long, filamentous eosinophilic fibres largely composed of GFAP protein.61
In infantile cases, death occurs before 2 years. In juvenile cases, onset is between 4 and 10 years and is typified by ataxia, cognitive decline and bulbar symptoms. Survival can be up to 20–30 years. Adults tend to present with bulbar or pseudobulbar palsy, spasticity, ataxia, cognitive decline and dysautonomia.
The MRI pattern in Alexander disease often falls into two extremes. In most infantile and juvenile cases, there is extensive white matter abnormality with a frontal predominance. In adults, however, the abnormalities are often restricted to the posterior fossa, particularly atrophy of the medulla and cervical spinal cord. Often the lesions are contrast-enhancing, or there may be contrast enhancement of the ventricular lining or periventricular rim.62
LMNB1 duplication
Duplication of LMNB1 causes a form of adult-onset, autosomal dominant leukodystrophy (ADLD) characterised by autonomic dysfunction, spasticity, ataxia and cognitive decline. Symptoms usually develop in the fifth or sixth decade.63 Autonomic symptoms may be prominent and include orthostatic hypotension, urinary incontinence, constipation and erectile dysfunction. MRI features include T2W/FLAIR hyperintensity of the subcortical and deep cerebral white matter, the cerebellar peduncles, the pyramidal tracts and brainstem.64 It is important to note that copy number variants are not reliably identified by next-generation sequencing. Therefore, if ADLD is suspected clinically, specific testing for LMNB1 duplications should be performed.
Hypomyelinating disorders (PLP1, GJC2, TUBB4A, POLR3A, POLR3B, CLCN2, NKX6-2)
Hypomyelination is an extremely useful radiological sign, and in adults can significantly reduce the number of potential genes to test. Hypomyelination should be considered where the T2/FLAIR hyperintensity is diffuse, and often seems to be affecting all of the white matter uniformly. In contrast to demyelinating disorders where the white matter signal is hypointense on T1 imaging, in hypomyelination it may be isointense, or mildly hypointense or hyperintense.65 This sign is easily overlooked if the T1 sequences are not examined (figure 2G–H).
The most common cause of hypomyelinating leukoencephalopathy is Pelizaeus-Merzbacher disease (PMD), caused by mutations in the proteolipid gene PLP1. The most common presentations of PMD include the connatal form, where symptoms are present at the time of birth, and the classical form, where symptoms develop within the first months of life.66 Symptoms begin with nystagmus, hypotonia, ataxia and head tremor. Later motor and language development is delayed. PLP1 duplications are the most common mutation type in children. As PLP1 is an X linked gene, typically male patients are more severely affected than female patients. The connatal and classical forms are not seen in female patients. However, female PLP1 mutation carriers can develop symptoms in adulthood. The phenotype in women often consists of slowly progressive spastic paraplegia and this presentation has been assigned SPG2.67 The most common mutation type in female carriers who manifest symptoms is a nonsense mutation.68 Adult-onset disease in male patients is more aggressive, with prominent head tremor, ataxia, spasticity and cognitive decline. MRI in both male and female patients shows a diffuse hypomyelinating leukodystrophy which is hyperintense on T2/FLAIR and often isointense or mildly hyperintense on T1. The pattern can resemble tiger stripes (tigroid pattern).
Autosomal recessive mutations in GJC2 are a cause of a Pelizaeus-Merzbacher-like disorder.69 The phenotype is similar to that of PLP1 mutations but male and female patients are equally affected. Like PLP1, most patients have onset of symptoms in early life, but there are reports of later onset with slow progression into adulthood, again mostly manifesting by spastic paraplegia, cerebellar signs and a hypomyelinating picture on MRI.70
Autosomal recessive mutations in CLCN2 are a very rare cause of adult-onset leukoencephalopathy, with findings mainly limited to the posterior limb of the internal capsules, the dentate nucleus, the cerebral peduncles and middle cerebellar peduncles (figure 2I). There may be evidence of restricted diffusion in these areas. There may be more extensive involvement of cerebellar white matter, and cerebral white matter may demonstrate features of hypomyelination. In adults, the most common presentation is mild cerebellar ataxia.71
A number of other genes have been associated with hypomyelination, including TUBB4A (with basal ganglia abnormalities),72 POLR3-related disorders (dental abnormalities and hypogonadotrophic hypogonadism)73 and NKX6-2,74 although to date these syndromes are only described with childhood onset.
Very rare genes (RNF216, TREM2, SNORD118)
Gordon Holmes syndrome is a rare autosomal recessive disorder characterised by hypogonadotrophic hypogonadism and ataxia. It is usually accompanied by cognitive decline, diffuse leukoencephalopathy with cerebellar atrophy and chorea. Age of onset is variable and the disorder is caused by mutations in the gene RNF216. The combination of chorea and dementia is reminiscent of an autosomal recessive form of Huntington disease.75
Polycystic lipomembranous osteodysplasia with sclerosing leukoencephalopathy, also known as Nasu-Hakola disease, is a rare syndrome caused by autosomal recessive mutations in TREM2 or TYROBP. The disorder usually presents in adulthood, with bone pain and pathological fractures due to bone cysts. Later, a severe neuropsychiatric syndrome develops due to a progressive leukoencephalopathy. It is most common in Japan and Finland.76 Different TREM2 variants are also significant risk factors for Alzheimer disease.77
Labrune syndrome, also known as leukoencephalopathy with brain calcification and cysts, is a rare autosomal recessive disorder caused by mutations in SNORD118. Labrune syndrome most commonly presents in early life, but there are reports of patients presenting in their 50s. The syndrome consists of progressive dementia, with motor decline and seizures. Large areas of cystic degeneration and calcification occur with a symmetric leukoencephalopathy affecting the periventricular, deep and subcortical white matter.78 SNORD118 is not well covered by next-generation sequencing, and in the right clinical context single-gene sequencing is recommended.
The Queen Square Adult Leukodystrophy Group
The Queen Square Adult Leukodystrophy Group (QSALG) is a multidisciplinary special interest group with input from neuroinflammation, neurogenetics, inherited metabolic disease, cognitive neurology and neuroradiology. Since inception, the group has investigated 116 patients with a wide variety of white matter syndromes, referred from hospitals worldwide. In our experience, a diagnosis can be made in approximately half of patients (54). The most common diagnoses are found in online supplementary figure 1. The most common diagnosis reached is of small vessel disease, followed by CSF1R mutations, mitochondrial diseases, ALD and multiple sclerosis. The remainder of diagnoses were highly heterogeneous, with often only one or at most two cases for each disorder. There were 15 diagnoses that affected only one patient each, and these included CADASIL and CARASIL, PLP1 mutations, Alexander disease and Fabry disease, VWM disease and familial British dementia. This review is representative of the diagnostic approach of the QSALG.
jnnp-2018-319481supp001.png (51.5KB, png)
{kind=link}
Diagnostic algorithm
Figure 3 shows an algorithm that clinicians can use when evaluating adults presenting with a suspected leukoencephalopathy. As described above, initial assessment should focus on the exclusion of common acquired causes and severe small vessel disease (round 1). If these initial tests are negative and the patient is suspected to have a genetic disorder, then the first line of testing should include white cell enzyme activities, a VLCFA profile (in men), plasma cholestanol and bile alcohols and plasma amino acids, to exclude the classical leukodystrophies which can present in adulthood (round 2). This step is important, as enzyme replacement therapy and stem cell transplant are being explored as treatment options for some of these conditions.
Figure 3.

A schematic diagram to illustrate currently available sequencing technologies. Single-gene (Sanger) sequencing is widely available and is performed by amplifying coding regions of the gene of interest by PCR, before sequencing these regions. Introns and promoter regions are not routinely sequenced. Next-generation sequencing-based panel or exome sequencing is becoming more widely available clinically. In this technique, DNA is fragmented, and the exons of multiple genes of interest are selected out for sequencing. Although many genes are sequenced, again only the coding regions are selected. In whole genome sequencing, which is not routinely available to clinicians yet, the DNA is also fragmented, but all the resulting DNA is sequencing, without a selection step. This results in a large amount of data, as the coding regions, intergenic regions, introns and promoters of all human genes are sequenced. Advanced bioinformatics and computing technology are a required part of whole genome sequencing.
If metabolic testing is not revealing, then the clinician should move on to genetic testing in the context of round 3 clinical or imaging patterns. A schematic illustrating the differences between sequencing approaches can be found in figure 4. Next-generation sequencing via diagnostic panels is becoming more widely available through regional genetic testing services, and these can be the most rapid and cost-effective route to diagnosis. It is worth determining which genes are covered by the panel requested, as each panel will differ depending on its design. If there is no access to a diagnostic panel, then consideration should be given to single-gene testing guided by the clinical and radiological phenotypes described above, and found in tables 1 and 2. It should be noted that single-gene testing is a low-yield route to diagnosis, and if a number of genes are sequenced the cost will quickly exceed that of a diagnostic panel. There are a small number of genes where a panel will not be sufficient for diagnosis, usually where copy number variants are a significant concern, and these would include LMNB1 and PLP1. In these cases, clinical suspicion should guide directed testing.
Figure 4.

A recommended algorithm for the evaluation of adults with suspected inherited white matter disorders. CSF, cerebrospinal fluid; mtDNA, mitochondrial DNA; PML, progressive multifocal leukoencephalopathy; VLCFA, very long chain fatty acids; WCE, White cell enzymes.
If a panel-based approach is not successful but a genetic leukoencephalopathy is still considered likely, then consideration should be given to whether the disorder might be due to a mutation in the mitochondrial genome (not targeted by traditional panels). It may be necessary to contact a research group who can offer more extensive genetic testing including whole exome or even whole genome sequencing. In these cases, it is helpful to have obtained DNA from as many affected and unaffected family members as possible.
In our experience of difficult-to-diagnose patients who have already been extensively investigated, a focused exome panel led to a diagnosis in almost 30% of patients.3
Discussion
Leukoencephalopathies are a heterogeneous group of disorders with a wide range of genetic and acquired causes. They are frequently difficult to diagnose, and patients with these disorders typically undergo a large number of expensive, time-consuming and often invasive tests over a long timeframe. This presents a significant burden to patients who usually are deteriorating from a severe and progressive neurological syndrome. The aim of the treating neurologist should be to make a definitive diagnosis, as this allows for better prognostication, more definitive family counselling and prevents further worry about the diagnosis. We hope that we have given a current useful, straightforward logical algorithm that others will find useful. It is clear that careful clinical and radiological assessment, in combination with early use of focused genetic testing, is needed to maximise the probability of making these diagnoses. However, even with this approach, more than half of patients currently do not receive a definitive diagnosis.79 This may be because the disorders are so heterogeneous or because there are still more genes to be described. Advances in genetic technology like whole genome sequencing may solve some cases, but only in combination with a thorough phenotypic approach.
Ultimately, these diagnoses are important because the development of novel therapies for these disorders depends primarily on forming cohorts of patients for clinical trials. HSCT from donors is a potential treatment for ALD, MLD and a number of other disorders. Recently, gene correction has also shown promise. In principle, gene correction (eg, through lentiviral transduction of the patient’s own cells) should be possible in most loss of function genetic disorders, but to show efficacy in rare diseases sufficient numbers of patients must be identified.80
As in 2014, the authors are happy to be contacted to discuss or see adult-onset patients with possible leukodystrophies or genetic leukoencephalopathies.
Footnotes
Contributors: DSL, CW, ARBdP, NJ ciritically reviewed the literature and drafted the manuscript. JAM, AM, RMA, CJM, JDW, JMS, NCF, HH, MEA, ID, EM, JC critically reviewed and edited the manuscript. DSL, JC devised and oversaw the study.
Funding: We would like to gratefully acknowledge our funders, which include the Leonard Wolfson Experimental Neurology Centre, the UK NIHR UCL/UCLH Biomedical Research Centre, the Wellcome Trust and the Medical Research Council.
Competing interests: None declared.
Patient consent: Not required.
Ethics approval: Ethical approval was not required for this study, which did not involve human participants or their data.
Provenance and peer review: Not commissioned; externally peer reviewed.
Data sharing statement: There are no additional unpublished data.
References
- 1. Ahmed RM, Murphy E, Davagnanam I, et al. A practical approach to diagnosing adult onset leukodystrophies. J Neurol Neurosurg Psychiatry 2014;85:770–81. 10.1136/jnnp-2013-305888 [DOI] [PubMed] [Google Scholar]
- 2. Heiss WD, Rosenberg GA, Thiel A, et al. Neuroimaging in vascular cognitive impairment: a state-of-the-art review. BMC Med 2016;14:174 10.1186/s12916-016-0725-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Lynch DS, Rodrigues Brandão de Paiva A, Zhang WJ, et al. Clinical and genetic characterization of leukoencephalopathies in adults. Brain 2017;140:1204–11. 10.1093/brain/awx045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Engelen M, Kemp S, de Visser M, et al. X-linked adrenoleukodystrophy (X-ALD): clinical presentation and guidelines for diagnosis, follow-up and management. Orphanet J Rare Dis 2012;7:51 10.1186/1750-1172-7-51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Loes DJ, Fatemi A, Melhem ER, et al. Analysis of MRI patterns aids prediction of progression in X-linked adrenoleukodystrophy. Neurology 2003;61:369–74. 10.1212/01.WNL.0000079050.91337.83 [DOI] [PubMed] [Google Scholar]
- 6. Eichler F, Duncan C, Musolino PL, et al. Hematopoietic stem-cell gene therapy for cerebral adrenoleukodystrophy. N Engl J Med 2017;377:1630–8. 10.1056/NEJMoa1700554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wenger DA. Krabbe disease. GeneReviews(®), 1993. [Google Scholar]
- 8. Kolodny EH, Raghavan S, Krivit W. Late-onset Krabbe disease (globoid cell leukodystrophy): clinical and biochemical features of 15 cases. Dev Neurosci 1991;13:232–9. 10.1159/000112166 [DOI] [PubMed] [Google Scholar]
- 9. Debs R, Froissart R, Aubourg P, et al. Krabbe disease in adults: phenotypic and genotypic update from a series of 11 cases and a review. J Inherit Metab Dis 2013;36:859–68. 10.1007/s10545-012-9560-4 [DOI] [PubMed] [Google Scholar]
- 10. Livingston JH, Graziano C, Pysden K, et al. Intracranial calcification in early infantile Krabbe disease: nothing new under the sun. Dev Med Child Neurol 2012;54:376–9. 10.1111/j.1469-8749.2011.04167.x [DOI] [PubMed] [Google Scholar]
- 11. Sedel F, Tourbah A, Fontaine B, et al. Leukoencephalopathies associated with inborn errors of metabolism in adults. J Inherit Metab Dis 2008;31:295–307. 10.1007/s10545-008-0778-0 [DOI] [PubMed] [Google Scholar]
- 12. Cesani M, Lorioli L, Grossi S, et al. Mutation update of ARSA and PSAP genes causing metachromatic leukodystrophy. Hum Mutat 2016;37:16–27. 10.1002/humu.22919 [DOI] [PubMed] [Google Scholar]
- 13. Rauschka H, Colsch B, Baumann N, et al. Late-onset metachromatic leukodystrophy: genotype strongly influences phenotype. Neurology 2006;67:859–63. 10.1212/01.wnl.0000234129.97727.4d [DOI] [PubMed] [Google Scholar]
- 14. Cheon JE, Kim IO, Hwang YS, et al. Leukodystrophy in children: a pictorial review of MR imaging features. Radiographics 2002;22:461–76. 10.1148/radiographics.22.3.g02ma01461 [DOI] [PubMed] [Google Scholar]
- 15. Barth ML, Ward C, Harris A, et al. Frequency of arylsulphatase a pseudodeficiency associated mutations in a healthy population. J Med Genet 1994;31:667–71. 10.1136/jmg.31.9.667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. de Hosson LD, van de Warrenburg BP, Preijers FW, et al. Adult metachromatic leukodystrophy treated by allo-SCT and a review of the literature. Bone Marrow Transplant 2011;46:1071–6. 10.1038/bmt.2010.252 [DOI] [PubMed] [Google Scholar]
- 17. Rosenberg JB, Kaminsky SM, Aubourg P, et al. Gene therapy for metachromatic leukodystrophy. J Neurosci Res 2016;94:1169–79. 10.1002/jnr.23792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Nie S, Chen G, Cao X, et al. Cerebrotendinous xanthomatosis: a comprehensive review of pathogenesis, clinical manifestations, diagnosis, and management. Orphanet J Rare Dis 2014;9:179 10.1186/s13023-014-0179-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Pilo-de-la-Fuente B, Jimenez-Escrig A, Lorenzo JR, et al. Cerebrotendinous xanthomatosis in Spain: clinical, prognostic, and genetic survey. Eur J Neurol 2011;18:1203–11. 10.1111/j.1468-1331.2011.03439.x [DOI] [PubMed] [Google Scholar]
- 20. Froese DS, Huemer M, Suormala T, et al. Mutation update and review of severe methylenetetrahydrofolate reductase deficiency. Hum Mutat 2016;37:427–38. 10.1002/humu.22970 [DOI] [PubMed] [Google Scholar]
- 21. Rademakers R, Baker M, Nicholson AM, et al. Mutations in the Colony Stimulating Factor 1 receptor (CSF1R) gene cause hereditary diffuse leukoencephalopathy with spheroids. Nat Genet 2011;44:200–5. 10.1038/ng.1027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lynch DS. Hereditary leukoencephalopathy with axonal spheroids: a spectrum of phenotypes from CNS vasculitis to parkinsonism in an adult onset leukodystrophy series. J Neurol Neurosurg Psychiatry 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Axelsson R, Röyttä M, Sourander P, et al. Hereditary diffuse leucoencephalopathy with spheroids. Acta Psychiatr Scand Suppl 1984;314:1–65. [PubMed] [Google Scholar]
- 24. Sundal C, Fujioka S, Van Gerpen JA, et al. Parkinsonian features in Hereditary Diffuse Leukoencephalopathy with Spheroids (HDLS) and CSF1R mutations. Parkinsonism Relat Disord 2013;19:869–77. 10.1016/j.parkreldis.2013.05.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Konno T, Yoshida K, Mizuno T, et al. Clinical and genetic characterization of adult-onset leukoencephalopathy with axonal spheroids and pigmented glia associated with CSF1R mutation. Eur J Neurol 2017;24:37–45. 10.1111/ene.13125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lakshmanan R, Adams ME, Lynch DS, et al. Redefining the phenotype of ALSP and AARS2 mutation-related leukodystrophy. Neurol Genet 2017;3:e135 10.1212/NXG.0000000000000135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Guerreiro R, Kara E, Le Ber I, et al. Genetic analysis of inherited leukodystrophies: genotype-phenotype correlations in the CSF1R gene. JAMA Neurol 2013;70:875–82. 10.1001/jamaneurol.2013.698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lynch DS, Zhang WJ, Lakshmanan R, et al. Analysis of mutations in AARS2 in a series of CSF1R-negative patients with adult-onset leukoencephalopathy with axonal spheroids and pigmented glia. JAMA Neurol 2016;73:1433–500. 10.1001/jamaneurol.2016.2229 [DOI] [PubMed] [Google Scholar]
- 29. Dallabona C, Diodato D, Kevelam SH, et al. Novel (ovario) leukodystrophy related to AARS2 mutations. Neurology 2014;82:2063–71. 10.1212/WNL.0000000000000497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Szpisjak L, Zsindely N, Engelhardt JI, et al. Novel AARS2 gene mutation producing leukodystrophy: a case report. J Hum Genet 2017;62:329–33. 10.1038/jhg.2016.126 [DOI] [PubMed] [Google Scholar]
- 31. Yao P, Fox PL. Aminoacyl-tRNA synthetases in medicine and disease. EMBO Mol Med 2013;5:332–43. 10.1002/emmm.201100626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Boczonadi V, Jennings MJ, Horvath R. The role of tRNA synthetases in neurological and neuromuscular disorders. FEBS Lett 2018;592:703–17. 10.1002/1873-3468.12962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Vermeulen G, Seidl R, Mercimek-Mahmutoglu S, et al. Fright is a provoking factor in vanishing white matter disease. Ann Neurol 2005;57:560–3. 10.1002/ana.20418 [DOI] [PubMed] [Google Scholar]
- 34. Labauge P, Horzinski L, Ayrignac X, et al. Natural history of adult-onset eIF2B-related disorders: a multi-centric survey of 16 cases. Brain 2009;132(Pt 8):2161–9. 10.1093/brain/awp171 [DOI] [PubMed] [Google Scholar]
- 35. van der Knaap MS, Leegwater PA, Könst AA, et al. Mutations in each of the five subunits of translation initiation factor eIF2B can cause leukoencephalopathy with vanishing white matter. Ann Neurol 2002;51:264–70. 10.1002/ana.10112 [DOI] [PubMed] [Google Scholar]
- 36. Chabriat H, Joutel A, Dichgans M, et al. Cadasil. Lancet Neurol 2009;8:643–53. 10.1016/S1474-4422(09)70127-9 [DOI] [PubMed] [Google Scholar]
- 37. Di Donato I, Bianchi S, De Stefano N, et al. Cerebral Autosomal Dominant Arteriopathy with Subcortical Infarcts and Leukoencephalopathy (CADASIL) as a model of small vessel disease: update on clinical, diagnostic, and management aspects. BMC Med 2017;15:41 10.1186/s12916-017-0778-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Rutten JW, Dauwerse HG, Gravesteijn G, et al. Archetypal NOTCH3 mutations frequent in public exome: implications for CADASIL. Ann Clin Transl Neurol 2016;3:844–53. 10.1002/acn3.344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Bianchi S, Di Palma C, Gallus GN, et al. Two novel HTRA1 mutations in a European CARASIL patient. Neurology 2014;82:898–900. 10.1212/WNL.0000000000000202 [DOI] [PubMed] [Google Scholar]
- 40. Fukutake T, Hirayama K. Familial young-adult-onset arteriosclerotic leukoencephalopathy with alopecia and lumbago without arterial hypertension. Eur Neurol 1995;35:69–79. 10.1159/000117096 [DOI] [PubMed] [Google Scholar]
- 41. Nozaki H, Nishizawa M, Onodera O. Features of cerebral autosomal recessive arteriopathy with subcortical infarcts and leukoencephalopathy. Stroke 2014;45:3447–53. 10.1161/STROKEAHA.114.004236 [DOI] [PubMed] [Google Scholar]
- 42. Nozaki H, Kato T, Nihonmatsu M, et al. Distinct molecular mechanisms of HTRA1 mutants in manifesting heterozygotes with CARASIL. Neurology 2016;86:1964–74. 10.1212/WNL.0000000000002694 [DOI] [PubMed] [Google Scholar]
- 43. Verdura E, Hervé D, Scharrer E, et al. Heterozygous HTRA1 mutations are associated with autosomal dominant cerebral small vessel disease. Brain 2015;138(Pt 8):2347–58. 10.1093/brain/awv155 [DOI] [PubMed] [Google Scholar]
- 44. Bugiani M, Kevelam SH, Bakels HS, et al. Cathepsin A-related arteriopathy with strokes and leukoencephalopathy (CARASAL). Neurology 2016;87:1777–86. 10.1212/WNL.0000000000003251 [DOI] [PubMed] [Google Scholar]
- 45. Hwang YT, Lakshmanan R, Davagnanam I, et al. Brainstem phenotype of cathepsin a-related arteriopathy with strokes and leukoencephalopathy. Neurol Genet 2017;3:e165 10.1212/NXG.0000000000000165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Vahedi K, Massin P, Guichard JP, et al. Hereditary infantile hemiparesis, retinal arteriolar tortuosity, and leukoencephalopathy. Neurology 2003;60:57–63. 10.1212/WNL.60.1.57 [DOI] [PubMed] [Google Scholar]
- 47. Meuwissen ME, Halley DJ, Smit LS, et al. The expanding phenotype of COL4A1 and COL4A2 mutations: clinical data on 13 newly identified families and a review of the literature. Genet Med 2015;17:843–53. 10.1038/gim.2014.210 [DOI] [PubMed] [Google Scholar]
- 48. Richards A, van den Maagdenberg AM, Jen JC, et al. C-terminal truncations in human 3'-5' DNA exonuclease TREX1 cause autosomal dominant retinal vasculopathy with cerebral leukodystrophy. Nat Genet 2007;39:1068–70. 10.1038/ng2082 [DOI] [PubMed] [Google Scholar]
- 49. Ophoff RA, DeYoung J, Service SK, et al. Hereditary vascular retinopathy, cerebroretinal vasculopathy, and hereditary endotheliopathy with retinopathy, nephropathy, and stroke map to a single locus on chromosome 3p21.1-p21.3. Am J Hum Genet 2001;69:447–53. 10.1086/321975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Jen J, Cohen AH, Yue Q, et al. Hereditary endotheliopathy with retinopathy, nephropathy, and stroke (HERNS). Neurology 1997;49:1322–30. 10.1212/WNL.49.5.1322 [DOI] [PubMed] [Google Scholar]
- 51. Stam AH, Kothari PH, Shaikh A, et al. Retinal vasculopathy with cerebral leukoencephalopathy and systemic manifestations. Brain 2016;139:2909–22. 10.1093/brain/aww217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Wong LJ. Mitochondrial syndromes with leukoencephalopathies. Semin Neurol 2012;32:055–61. 10.1055/s-0032-1306387 [DOI] [PubMed] [Google Scholar]
- 53. Küker W, Weir A, Quaghebeur G, et al. White matter changes in Leber's hereditary optic neuropathy: MRI findings. Eur J Neurol 2007;14:591–3. 10.1111/j.1468-1331.2007.01757.x [DOI] [PubMed] [Google Scholar]
- 54. Calvo SE, Clauser KR, Mootha VK. MitoCarta2.0: an updated inventory of mammalian mitochondrial proteins. Nucleic Acids Res 2016;44(D1):D1251–57. 10.1093/nar/gkv1003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Nishino I, Spinazzola A, Hirano M. Thymidine phosphorylase gene mutations in MNGIE, a human mitochondrial disorder. Science 1999;283:689–92. 10.1126/science.283.5402.689 [DOI] [PubMed] [Google Scholar]
- 56. Tang S, Dimberg EL, Milone M, et al. Mitochondrial neurogastrointestinal encephalomyopathy (MNGIE)-like phenotype: an expanded clinical spectrum of POLG1 mutations. J Neurol 2012;259:862–8. 10.1007/s00415-011-6268-6 [DOI] [PubMed] [Google Scholar]
- 57. Tzoulis C, Neckelmann G, Mørk SJ, et al. Localized cerebral energy failure in DNA polymerase gamma-associated encephalopathy syndromes. Brain 2010;133(Pt 5):1428–37. 10.1093/brain/awq067 [DOI] [PubMed] [Google Scholar]
- 58. Scheper GC, van der Klok T, van Andel RJ, et al. Mitochondrial aspartyl-tRNA synthetase deficiency causes leukoencephalopathy with brain stem and spinal cord involvement and lactate elevation. Nat Genet 2007;39:534–9. 10.1038/ng2013 [DOI] [PubMed] [Google Scholar]
- 59. van Berge L, Hamilton EM, Linnankivi T, et al. Leukoencephalopathy with brainstem and spinal cord involvement and lactate elevation: clinical and genetic characterization and target for therapy. Brain 2014;137(Pt 4):1019–29. 10.1093/brain/awu026 [DOI] [PubMed] [Google Scholar]
- 60. Brenner M, Johnson AB, Boespflug-Tanguy O, et al. Mutations in GFAP, encoding glial fibrillary acidic protein, are associated with Alexander disease. Nat Genet 2001;27:117–20. 10.1038/83679 [DOI] [PubMed] [Google Scholar]
- 61. Prust M, Wang J, Morizono H, et al. GFAP mutations, age at onset, and clinical subtypes in Alexander disease. Neurology 2011;77:1287–94. 10.1212/WNL.0b013e3182309f72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Farina L, Pareyson D, Minati L, et al. Can MR imaging diagnose adult-onset Alexander disease? AJNR Am J Neuroradiol 2008;29:1190–6. 10.3174/ajnr.A1060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Padiath QS, Saigoh K, Schiffmann R, et al. Lamin B1 duplications cause autosomal dominant leukodystrophy. Nat Genet 2006;38:1114–23. 10.1038/ng1872 [DOI] [PubMed] [Google Scholar]
- 64. Schuster J, Sundblom J, Thuresson AC, et al. Genomic duplications mediate overexpression of lamin B1 in adult-onset autosomal dominant leukodystrophy (ADLD) with autonomic symptoms. Neurogenetics 2011;12:65–72. 10.1007/s10048-010-0269-y [DOI] [PubMed] [Google Scholar]
- 65. Schiffmann R, van der Knaap MS. Invited article: an MRI-based approach to the diagnosis of white matter disorders. Neurology 2009;72:750–9. 10.1212/01.wnl.0000343049.00540.c8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Hodes ME, Pratt VM, Dlouhy SR. Genetics of Pelizaeus-Merzbacher disease. Dev Neurosci 1993;15:383–94. 10.1159/000111361 [DOI] [PubMed] [Google Scholar]
- 67. Cailloux F, Gauthier-Barichard F, Mimault C, et al. Genotype-phenotype correlation in inherited brain myelination defects due to proteolipid protein gene mutations. Clinical European network on brain dysmyelinating disease. Eur J Hum Genet 2000;8:837–45. 10.1038/sj.ejhg.5200537 [DOI] [PubMed] [Google Scholar]
- 68. Hurst S, Garbern J, Trepanier A, et al. Quantifying the carrier female phenotype in Pelizaeus-Merzbacher disease. Genet Med 2006;8:371–8. 10.1097/01.gim.0000223551.95862.c3 [DOI] [PubMed] [Google Scholar]
- 69. Uhlenberg B, Schuelke M, Rüschendorf F, et al. Mutations in the gene encoding gap junction protein alpha 12 (connexin 46.6) cause Pelizaeus-Merzbacher-like disease. Am J Hum Genet 2004;75:251–60. 10.1086/422763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Orthmann-Murphy JL, Salsano E, Abrams CK, et al. Hereditary spastic paraplegia is a novel phenotype for GJA12/GJC2 mutations. Brain 2009;132(Pt 2):426–38. 10.1093/brain/awn328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Depienne C, Bugiani M, Dupuits C, et al. Brain white matter oedema due to ClC-2 chloride channel deficiency: an observational analytical study. Lancet Neurol 2013;12:659–68. 10.1016/S1474-4422(13)70053-X [DOI] [PubMed] [Google Scholar]
- 72. Pizzino A, Pierson TM, Guo Y, et al. TUBB4A de novo mutations cause isolated hypomyelination. Neurology 2014;83:898–902. 10.1212/WNL.0000000000000754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Wolf NI, Vanderver A, van Spaendonk RM, et al. Clinical spectrum of 4H leukodystrophy caused by POLR3A and POLR3B mutations. Neurology 2014;83:1898–905. 10.1212/WNL.0000000000001002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Chelban V, Patel N, Vandrovcova J, et al. Mutations in NKX6-2 cause progressive spastic ataxia and hypomyelination. Am J Hum Genet 2017;100:969–77. 10.1016/j.ajhg.2017.05.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Santens P, Van Damme T, Steyaert W, et al. RNF216 mutations as a novel cause of autosomal recessive Huntington-like disorder. Neurology 2015;84:1760–6. 10.1212/WNL.0000000000001521 [DOI] [PubMed] [Google Scholar]
- 76. Paloneva J, Manninen T, Christman G, et al. Mutations in two genes encoding different subunits of a receptor signaling complex result in an identical disease phenotype. Am J Hum Genet 2002;71:656–62. 10.1086/342259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Guerreiro R, Wojtas A, Bras J, et al. TREM2 variants in Alzheimer's disease. N Engl J Med 2013;368:117–27. 10.1056/NEJMoa1211851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Jenkinson EM, Rodero MP, Kasher PR, et al. Mutations in SNORD118 cause the cerebral microangiopathy leukoencephalopathy with calcifications and cysts. Nat Genet 2016;48:1185–92. 10.1038/ng.3661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Richards J, Korgenski EK, Taft RJ, et al. Targeted leukodystrophy diagnosis based on charges and yields for testing. Am J Med Genet A 2015;167A:2541–3. 10.1002/ajmg.a.37215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Simonato M, Bennett J, Boulis NM, et al. Progress in gene therapy for neurological disorders. Nat Rev Neurol 2013;9:277–91. 10.1038/nrneurol.2013.56 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
jnnp-2018-319481supp001.png (51.5KB, png)