

Abstract
Fabry disease is a rare inborn error of the enzyme α-galactosidase (α-Gal) and results in lysosomal substrate accumulation in tissues with a wide range of clinical presentations. The disease has attracted a lot of interest over the last years, in particular since enzyme replacement therapy (ERT) has become widely available in 2001. With rising awareness and rising numbers of (diagnosed) patients, physicians encounter new challenges. Over 900 α-Gal gene mutations are currently known, some with doubtful clinical significance, posing diagnostic and prognostic difficulties for the clinician and a lot of uncertainty for patients. Another challenge are patients who develop neutralising antibodies to ERT, which possibly leads to reduced therapy effectiveness. In this article, we summarise the latest developments in the science community regarding diagnostics and management of this rare lysosomal storage disorder and offer an outlook to future treatments.
Keywords: Fabry disease, biomarkers, antibodies, multidisciplinary working
Introduction
Fabry disease (FD) is of interest to specialties across the spectrum of internal medicine. A range of mutations on the X-chromosome cause a defect in α-galactosidase (α-Gal) enzyme production, resulting in absent or reduced enzyme activity and accumulation of globotriaosylceramide (Gb3) and other sphingolipids in lysosomes in various tissues—mainly cardiac, endothelial, neuronal and renal—resulting in end-organ damage and failure.1
Newborn screening has now been performed in several countries, yielding a prevalence ranging from 1:1368 to 1:8882.2–5 This is in contrast to earlier presumed prevalence of 1:40 000-1:117 0006
Clinical variability of different mutations, variable disease severity and symptom onset make the disease notoriously difficult to diagnose. There is huge clinical variability even within the same family mutation7 8 as well as large intersex variability. Heterozygote females tend to have higher residual enzyme activity as they are mosaic due to variable X-chromosome inactivation.9 Opposed to previous views that women are merely heterozygote carriers with no notable disease activity, we now know that they can display clinical manifestations not dissimilar to males with major organ involvement and associated morbidity/mortality.9
A classification of classic phenotype and late-onset phenotype is widely used as the natural disease process and prognosis differ.10 Typically, classic phenotype patients are null mutant and have no residual enzyme activity, even though due to the method of the enzyme assay they may have reported enzyme activity of 1%–3%. These patients usually display early onset of symptoms—some already in their early childhood— such as neuropathic pain or anhidrosis. They proceed to major organ complications as early as their 20s or 30s. Late-onset patients—typically females or males with non-classical mutations—may present with only one organ involvement, typically in their 50s.10 All organ complications can however occur in both classical and late-onset phenotype (see table 1).
Table 1.
Typical organ involvement in morbus Fabry
| Organ system | Complications |
| Ophtalmological | Cornea verticilllata, tortuous vessels, cataracts58 |
| Dermatological | Angiokeratoma, hypo/anhidrosis, telangiectasia, lymphedema59 |
| Neurological | Neuropathic pain, transient ischaemic attack, stroke, neuropsychiatric complications (depression)60 |
| Cardiac | Conduction abnormalities, left ventricular hypertrophy, sudden cardiac death61 |
| Renal | Proteinuria, reduced glomerular filtration rate62 |
| Gastrointestinal | Diarrhoea, constipation, early satiety, nausea63 |
| Auditory | Hearing loss, tinnitus, vertigo64 |
| Respiratory | Cough, wheezing, airflow limitation65 |
Once suspected, men are diagnosed with FD if enzyme activity is <35% of the mean. The gold standard of diagnosis for females is α-Gal gene sequencing only.11 Genetic analysis is however expensive and only available in specialised laboratories. Moreover, >900 variants of the α-Gal gene have now been identified,12 many of doubtful pathological significance. In these cases, invasive tissue diagnosis investigating the presence of Gb3 deposits is indicated. Therefore, there is a need for more practical and simple diagnostic tools.
Since 2001 widely available enzyme replacement therapy (ERT) with recombinant agalasidase alfa and beta has been a game changer for diagnosing and treating FD13 and has sparked a lot of interest; the rising awareness resulted in high-risk populations being screened (dialysis, renal transplant, left ventricular hypertrophy, cryptogenic stroke)14 and whole families started on treatment.
The current challenges of ERTenzyme replacement therapy are many: first, there is relative lack of efficacy with a lack of visible therapy effect. This results in difficulties with standardised disease activity/therapy monitoring. Antibody formation possibly also contributes to reduced therapy effect.
In this article, we review novel biomarkers for diagnosis and therapy monitoring as well as current practice of treating patients who develop antibodies.
We also discuss novel treatment options with oral therapy and gene therapy, raising the possibility of a cure.
Finally, we highlight the need for systematic multidisciplinary approach to assessment of organ involvement and therapy optimisation.
Biomarkers
Many attempts have been made over the years to find a disease-specific marker that would ideally serve as a rapid screening tool as well as indicator of therapy response.
Gb3
As such Gb3—the end product which accumulates in lysosomes and is routinely used to diagnose the disease on tissue biopsy—heart, kidney15 and most recently skin16—has so far failed to convince as a suitable plasma or urine marker. A study by Young17 performed on hemizygotes, heterozygotes and healthy controls failed to reliably detect increased Gb3 levels in plasma and urine in heterozygotes and hemizygotes with non-classical mutation. Additionally, Schiffmann failed to demonstrate a correlation between plasma or urine Gb3 and clinical response to ERT.18
An interesting approach to detect Gb3 deposits in peripheral blood mononuclear cells (PBMCs)19 represents a low-cost and simple alternative to invasive biopsies. What is more, the Gb3 load in PBMC decreases with long-term ERT and could potentially be used for disease monitoring. It is important to highlight that this method failed to detect Gb3 deposits in non-classical/missense mutations, in particular in four patients with D313Y mutation, which are known to be non-disease-causing mutations.20
LysoGb3
The Gb3 degradation product, globotriaosylsphingosine (lysoGb3), is currently used in disease screening,21 22 but also in the determination of pathogenicity of a mutation for homozygotes as well as heterozygotes. In a prospective evaluation/validation study of 124 patients,23 lysoGb3 levels were correlated to clinical picture, mutation type as well as biochemical and imaging studies and shown to be a reliable predictor of clinically relevant disease. In another study, a raised level of lysoGb3 could be detected even in females with normal α-Gal activity and subsequently those patients were proved to have clinically significant disease.24
Furthermore, lysoGb3 levels have been shown to decrease with ERT, in particular in patients with classical phenotype.25 26 Since these findings, lysoGb3 has been widely accepted as the most accurate marker of disease activity. However, long-term data on clinical outcomes in patients who achieve lysoGb3 reduction are currently not available.
Emerging biomarkers
Several other avenues have been explored but are not yet widely in use due to lack of data. In a pilot study, Cammarata et al identified four micro-RNAs specific for Fabry patients27 regardless of mutation, sex or age. However, two of these micro-RNAs were linked with endothelial dysfunction and the number of patients and controls (30 patients and 30 controls respectively) was too small to derive test sensitivity/specifity.
Further attempts to identify Fabry-specific biomarkers include identification of new Gb3 isoforms using metabolomics28 29 and quantifying abnormal urinary protein excretion using proteome analyses30 31; however, none were able to validate a clinically useful single parameter.
ERT: pitfalls
As indicated above, intravenous ERT has been the gold standard since 2001.
The therapy efficacy monitoring issue has partially been addressed above. Long-term data over the last 17 years are now available to assess clinical response to therapy,32 33 and ERT has been shown to provide the greatest benefit to patients if started early on. On the other hand, during short-term assessments (generally yearly controls), which are relevant to the clinician and patient in standard practice, it can be very difficult to assess progress and make evidence-based decisions on therapy switch or modification, mainly due to slow progression of the disease, coupled with irreversibility of organ damage and poor therapeutic efficacy.
We have already discussed the role of biomarkers measured in plasma and urine and PBMC. A further assessment option is repeated biopsy of affected organs to prove Gb3 deposit reduction,34 which is invasive and impractical. Enzyme activity measurements are equally unsuitable as enzyme activity measured in plasma/leucocytes is sensitive to time after last enzyme infusion.
Therefore, careful clinical assessment, history taking and organ investigations are currently the best tools in the clinician’s box in day-to-day practice, alongside standard adjunctive therapy for specific organ involvement.
The next issue around ERT is antibody formation. The main concern in the first trials were immediate infusion-associated reactions which are IgE mediated. Premedication protocols—usually a combination of single-dose paracetamol or a non-steroidal anti-inflammatory drug, antihistamine and steroid, plus increasing infusion duration to prevent such reactions—have been established successfully and are still used.35
IgG seroconversion has been reported in the first clinical trials in approximately 40% patients; however, it was thought not to influence enzyme efficacy.34
Data from the Fabry registry report seroconversion with neutralising antidrug antibodies (ADA) in 73% males as opposed to 12% females treated with agalsidase beta, which is likely due to complete absence of native enzyme in men with classical mutations. This theory is supported in the fact that men with missense mutations were far less likely to develop antibodies, possibly due to residual native enzyme activity.36
Over the years long-term effect of neutralising antibodies has not been studied in detail, in particular their impact on clinical outcomes.
A study by Benichou et al observed significantly impaired Gb3 clearance in skin biopsies of patients treated with ERT and high antibody titres.37
A correlation between antibody levels and lysoGb3 has been confirmed in a study of 60 Japanese patients.26 Similarly, when analysing in vitro enzyme inhibition in relation to lysoGb3 levels, disease severity scores and subjective symptoms in a group of 168 patients,38 a study confirmed higher lysoGb3 levels as well as worse disease severity scores in patients with serum-mediated enzyme inhibition. There was no difference for the enzymes used (agalsidase alfa or beta) assuming a cross-reactivity and therefore not advocating for a therapy switch in antibody-positive patients. The theory of cross-reactivity is supported by Linhart et al, 39 who proved in vitro inhibition to both agalsidase alfa and beta in ADA-positive patients irrespective of which enzyme they were treated with.
In contrast two other studies40 41 postulated a reduction in plasma lysoGb3 with switch from agalsidase alfa to agalsidase beta in antibody-positive patients, however, not reaching levels as low as antibody-negative patients. This could be explained by the administration of a higher dose of agalsidase beta with full antibody saturation rather than assumption of non-cross-reactivity. This theory was supported by a study of ADA levels immediately after enzyme infusion, where circulating ADA levels were significantly lower following a higher dose enzyme infusion.42
More recent studies have raised the question of immune modulation therapy in response to antibody formation as experienced in other lysosomal storage disorders eligible for ERT.43 In particular, a study investigating the effect of immunosuppression in transplant patients on antibody formation delivered valuable insights44—immunosuppression has been shown to decrease ADAs significantly in therapy naive as well as previously treated patients. Further prospective studies are needed to evaluate clinical impact of lowering ADAs and correlation with biomarkers would be particularly interesting.
To conclude, routine antibody screening is currently not part of clinical practice but may explain why some patients suffer significant disease progression with ERT. Further data are needed to develop strategies for affected patients, be it ERT switch, dose increase, immunomodulation treatment or a switch to a different therapy option.
Novel approaches
Migalastat
A new therapy approach with migalastat was established in 2016. Migalastat is a small-molecule chaperone which facilitates enzyme trafficking to lysosomes in certain mutant enzymes.45 The advantage for patients is oral administration, thus avoiding the need for biweekly enzyme infusions and possible associated adverse reactions. In theory, migalastat has a higher volume of distribution than ERT46 and given consistently may result in better functional enzyme availability. On the downside, migalastat can only be used in suitable mutations—most commonly single missense mutations47, which is the case for an estimated 30% of all Fabry patients.48 A full updated list of amenable mutations is provided by the manufacturer Amicus Therapeutics. A first randomised controlled trial comparing migalastat with ERT has shown comparable effect on renal function, cardiac and composite outcomes,48 first cardiac MRI analyses show a possible positive impact on fibrosis and cardiac hypertrophy.49
To date, one phase II study has examined the combination of ERT with migalastat and showed promising results with a 1.2-fold to 5.1-fold increase of enzyme activity compared with ERT alone.50 Large co-administration studies are needed to confirm these findings and evaluate long-term clinical outcomes.
Lucerastat
Substrate reduction therapy (SRT) uses yet another approach that was tried in other lysosmal storage disorders such as miglustat in Gaucher disease51 and offers the advantages of oral therapy irrespective of genotype. A new drug lucerastat—a direct inhibitor of glucosylceramide synthase—is currently in the later stages of clinical trial development. Glucosylceramide synthase catalyses the first step to glycosphingolipid synthesis, therefore by direct inhibition a lower Gb3 load is achieved.52 Again, a combination of SRT with ERT and/or chaperone therapy may result in more favourable outcomes for Fabry patients.
Gene therapy
Lastly, first gene therapy clinical trials are under way (https://clinicaltrials.gov/ct2/show/NCT03454893?cond=Fabry+Disease&draw=3&rank=21). Animal studies in Fabry knockout mice have succeeded in introducing a working copy of the α-Gal gene via viral vectors (adenovirus or retrovirus), resulting in increased enzymatic activity at 6 months.53 A similar therapeutic approach has been successfully used in human haemophiliacs.54 The prospect of a cure with a single infusion of vector gene is exciting; however, there are currently several limitations of vector use—namely vector neutralising antibodies, which in theory can be overcome by emerging non-viral vectors. Other limitations of course include lack of long-term effects data as well as ethical issues of administering genome-modifying treatment to children.
Multidisciplinary team: FAZiT
Because of the rarity and chronicity of the disease, as well as diagnostic and therapeutic uncertainties, we believe that specialist multidisciplinary assessment is of great benefit to our patients, in particular in the setting of lack of national and international guidelines on assessment and management. The first benefits of multidisciplinary work were reported by the oncologists55 who are historically champions of comprehensive care of their patients. In the rare diseases community, similar attempts have been successful in treating patients with Gaucher disease.56
Our large national centre FAZiT (Fabry Zentrum für interdisziplinäre Therapie – Fabry Centre for Multidisciplinary Therapy), attached to the University Hospital Würzburg, Germany, incorporates the core specialties nephrology, cardiology and neurology, who are involved at each patient visit, with collaboration from other departments - radiology, ear, nose, and throat, ophthalmology, gastroenterology, paediatrics, psychiatry and social services.
For our standard patient journey, see figures 1 and 2.
Figure 1.
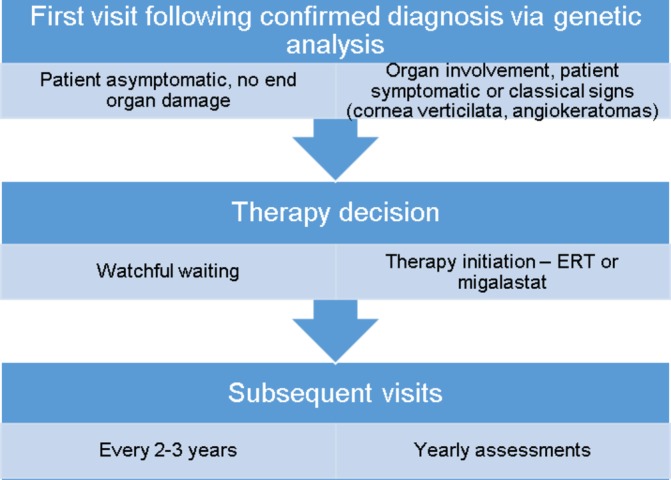
Patient journey in FAZiT. ERT:enzyme replacement therapy.
Figure 2.
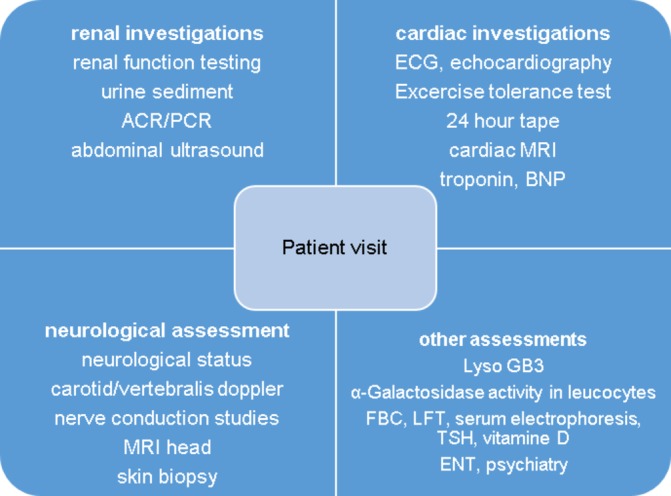
Specialist investigations. ENT, ear, nose and throat; LFT, liver function tests; FBC, full blood count; TSH, thyroid stimulating hormone; BNP, brain natriuretic peptide; MRI, magnetic resonance imaging; ACR, albumin-creatinine-ratio; PCR, protein-creatinine-ratio.
Putting patient care in the centre of our focus, we are actively involved with patient groups and organise yearly educational meetings with talks by experts in their respective fields. This is a great opportunity for patients to share their experience and support each other.
Lastly, we of course contribute to further research of diagnostic tools and therapies and contribute to international consensus on disease management.57
Main messages.
Gold standard diagnosis of Fabry disease is genetic analysis, but distinguishing non-pathogenic variants can still be a challenge.
Novel biomarkers such as lyso Gb3 add to establishing diagnosis and treatment monitoring.
Relative lack of enzyme replacement therapy efficacy, partly due to antibody formation, may be alleviated by combination with new oral therapy approaches.
Multidisciplinary work is key to comprehensive management of Fabry patients.
Current research questions.
Can we develop a reliable diagnostic tool/score to identify non-pathogenic mutations?
Is there a role for immunomodulation in antibody-positive patients on enzyme replacement therapy (ERT)?
Will future randomised controlled trials show that combination of ERT and chaperone/substrate reduction therapy add benefit to clinical outcomes?
Will gene therapy bypass all current difficulties and deliver a cure?
Key references.
Niemann M, Rolfs A, Stork S, et al. Gene mutations vs clinically relevant phenotypes: lyso-Gb3 defines Fabry disease. Circ Cardiovasc Genet 2014;7:8–16
Lenders M, Stypmann J, Duning T, et al. Serum-Mediated Inhibition of Enzyme Replacement Therapy in Fabry Disease. J Am Soc Nephrol 2016;27:256–64.
Hughes DA, Nicholls K, Shankar SP, et al. Oral pharmacological chaperone migalastat compared with enzyme replacement therapy in Fabry disease: 18 month results from the randomised phase III ATTRACT study. J Med Genet 2017;54:288–96.
Guerard N, Oder D, Nordbeck P, et al. Lucerastat, an Iminosugar for Substrate Reduction Therapy: Tolerability, Pharmacodynamics, and Pharmacokinetics in Patients With Fabry Disease on Enzyme Replacement. Clin Pharmacol Ther 2018;103:703–11.
Wanner C, Arad M, Baron R, et al. European expert consensus statement on therapeutic goals in Fabry disease. Mol Genet Metab 2018;124:189–203.
Self-assessment questions.
Please answer true or false
Children—in particular boys—who suffer from Fabry disease can develop neuropathic pain as young as age 3–4.
Due to the x-linked nature of the disease, women are carriers but do not develop significant disease.
Male patients with Fabry disease always develop angiokeratomas.
The main cause of death in classical phenotype patients is end-stage kidney disease.
The most reliable biomarker for disease monitoring is lysoGb3.
Answers.
True.
False.
False.
False.
True.
Acknowledgments
The authors thank Ms Irina Schumacher for her long-standing dedicated work at their centre.
Footnotes
Contributors: CW: conception of work and approval of final version of the manuscript. TC: drafting manuscript and literature search. LS and JG: assistance with graphics and literature search. JM and PN: critical revision for important intellectual content. All authors have reviewed the final version of the manuscript and are accountable for all aspects of the work.
Competing interests: TC has received travel assistance from Genzyme/Sanofi. JM has received travel assistance and speaker honoraria from Genzyme/Sanofi and travel assistance from Amicus Therapeutics and Shire. JG and LS have nothing to declare. PN has received honoraria for lecturing and advisory board participation from Amicus Therapeutics, Genzyme/Sanofi and Shire. CW has received honoraria for lecturing and advisory board participation from Genzyme/Sanofi, Actelion and Protalix and Shire. Research grants were given to the Institution from Genzyme/Sanofi and Shire.
Patient consent for publication: Not required.
Provenance and peer review: Not commissioned; internally peer reviewed.
Data sharing statement: No additional data are available.
References
- 1. Desnick RJ, Astrin KH, Bishop DF. Fabry disease: molecular genetics of the inherited nephropathy. Adv Nephrol Necker Hosp 1989;18:113–27. [PubMed] [Google Scholar]
- 2. Burlina AB, Polo G, Salviati L, et al. Newborn screening for lysosomal storage disorders by tandem mass spectrometry in North East Italy. J Inherit Metab Dis 2018;41:209–19. 10.1007/s10545-017-0098-3 [DOI] [PubMed] [Google Scholar]
- 3. Lin HY, Chong KW, Hsu JH, et al. High incidence of the cardiac variant of Fabry disease revealed by newborn screening in the Taiwan Chinese population. Circ Cardiovasc Genet 2009;2:450–6. 10.1161/CIRCGENETICS.109.862920 [DOI] [PubMed] [Google Scholar]
- 4. Inoue T, Hattori K, Ihara K, et al. Newborn screening for Fabry disease in Japan: prevalence and genotypes of Fabry disease in a pilot study. J Hum Genet 2013;58:548–52. 10.1038/jhg.2013.48 [DOI] [PubMed] [Google Scholar]
- 5. Mechtler TP, Stary S, Metz TF, et al. Neonatal screening for lysosomal storage disorders: feasibility and incidence from a nationwide study in Austria. The Lancet 2012;379:335–41. 10.1016/S0140-6736(11)61266-X [DOI] [PubMed] [Google Scholar]
- 6. Mehta A, Ricci R, Widmer U, et al. Fabry disease defined: baseline clinical manifestations of 366 patients in the Fabry Outcome Survey. Eur J Clin Invest 2004;34:236–42. 10.1111/j.1365-2362.2004.01309.x [DOI] [PubMed] [Google Scholar]
- 7. Beirão I, Cabrita A, Torres M, et al. Biomarkers and imaging findings of anderson–Fabry Disease—what we know now. Diseases 2017;5:15 10.3390/diseases5020015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Verovnik F, Benko D, Vujkovac B, et al. Remarkable variability in renal disease in a large Slovenian family with Fabry disease. Eur J Hum Genet 2004;12:678–81. 10.1038/sj.ejhg.5201184 [DOI] [PubMed] [Google Scholar]
- 9. Deegan PB, Baehner AF, Barba Romero MA, et al. Natural history of Fabry disease in females in the Fabry Outcome Survey. J Med Genet 2006;43:347–52. 10.1136/jmg.2005.036327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Arends M, Wanner C, Hughes D, et al. Characterization of Classical and Nonclassical Fabry Disease: A Multicenter Study. J Am Soc Nephrol 2017;28:1631–41. 10.1681/ASN.2016090964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Zarate YA, Hopkin RJ. Fabry's disease. The Lancet 2008;372:1427–35. 10.1016/S0140-6736(08)61589-5 [DOI] [PubMed] [Google Scholar]
- 12. Ferri L, Malesci D, Fioravanti A, et al. Functional and pharmacological evaluation of novel GLA variants in Fabry disease identifies six (two de novo) causative mutations and two amenable variants to the chaperone DGJ. Clinica Chimica Acta 2018;481:25–33. 10.1016/j.cca.2018.02.021 [DOI] [PubMed] [Google Scholar]
- 13. El Dib R, Gomaa H, Ortiz A, et al. Enzyme replacement therapy for Anderson-Fabry disease: A complementary overview of a Cochrane publication through a linear regression and a pooled analysis of proportions from cohort studies. PLoS One 2017;12:e0173358 10.1371/journal.pone.0173358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Linthorst GE, Bouwman MG, Wijburg FA, et al. Screening for Fabry disease in high-risk populations: a systematic review. J Med Genet 2010;47:217–22. 10.1136/jmg.2009.072116 [DOI] [PubMed] [Google Scholar]
- 15. Schiffmann R, Hughes DA, Linthorst GE, et al. Screening, diagnosis, and management of patients with Fabry disease: conclusions from a "Kidney Disease: Improving Global Outcomes" (KDIGO) Controversies Conference. Kidney Int 2017;91:284–93. 10.1016/j.kint.2016.10.004 [DOI] [PubMed] [Google Scholar]
- 16. Üçeyler N, Schröter N, Kafke W, et al. Skin Globotriaosylceramide 3 Load Is Increased in Men with Advanced Fabry Disease. PLoS One 2016;11:e0166484 10.1371/journal.pone.0166484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Young E, Mills K, Morris P, et al. Is globotriaosylceramide a useful biomarker in Fabry disease? Acta Paediatrica 2005;94:51–4. 10.1080/08035320510028111 [DOI] [PubMed] [Google Scholar]
- 18. Schiffmann R, Ries M, Blankenship D, et al. Changes in plasma and urine globotriaosylceramide levels do not predict Fabry disease progression over 1 year of agalsidase alfa. Genet Med 2013;15:983–9. 10.1038/gim.2013.56 [DOI] [PubMed] [Google Scholar]
- 19. Uceyler N, Bottger J, Henkel L, et al. Detecton of blood Gb3 deposits as a new tool for diagnosis and therapy monitoring in patients with classic Fabry disease. J Intern Med 2018. [DOI] [PubMed] [Google Scholar]
- 20. Oder D, Wanner C, Nordbeck P. The D313Y genotype-Pathogenic mutation or polymorphism? Clin Genet 2018;93:1257 10.1111/cge.13237 [DOI] [PubMed] [Google Scholar]
- 21. Aerts JM, Groener JE, Kuiper S, et al. Elevated globotriaosylsphingosine is a hallmark of Fabry disease. Proc Natl Acad Sci U S A 2008;105:2812–7. 10.1073/pnas.0712309105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Maruyama H, Miyata K, Mikame M, et al. Correction: Effectiveness of plasma lyso-Gb3 as a biomarker for selecting high-risk patients with Fabry disease from multispecialty clinics for genetic analysis. Genet Med [Epub ahead of print: 12 Sep 2018]. 10.1038/s41436-018-0258-3 [DOI] [Google Scholar]
- 23. Niemann M, Rolfs A, Störk S, et al. Gene mutations versus clinically relevant phenotypes: lyso-Gb3 defines Fabry disease. Circ Cardiovasc Genet 2014;7:8–16. 10.1161/CIRCGENETICS.113.000249 [DOI] [PubMed] [Google Scholar]
- 24. Nowak A, Mechtler TP, Desnick RJ, et al. Plasma LysoGb3: A useful biomarker for the diagnosis and treatment of Fabry disease heterozygotes. Mol Genet Metab 2017;120:57–61. 10.1016/j.ymgme.2016.10.006 [DOI] [PubMed] [Google Scholar]
- 25. van Breemen MJ, Rombach SM, Dekker N. Reduction of elevated plasma globotriaosylsphingosine in patients with classic Fabry disease following enzyme replacement therapy. Biochim Biophys Acta 1812;2011:70–6. [DOI] [PubMed] [Google Scholar]
- 26. Sakuraba H, Togawa T, Tsukimura T, et al. Plasma lyso-Gb3: a biomarker for monitoring fabry patients during enzyme replacement therapy. Clin Exp Nephrol 2018;22:843–9. 10.1007/s10157-017-1525-3 [DOI] [PubMed] [Google Scholar]
- 27. Cammarata G, Scalia S, Colomba P, et al. A pilot study of circulating microRNAs as potential biomarkers of Fabry disease. Oncotarget 2018;9:27333–45. 10.18632/oncotarget.25542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Auray-Blais C, Boutin M. Novel gb(3) isoforms detected in urine of fabry disease patients: a metabolomic study. Curr Med Chem 2012;19:3241–52. 10.2174/092986712800784739 [DOI] [PubMed] [Google Scholar]
- 29. Boutin M, Auray-Blais C. Metabolomic discovery of novel urinary galabiosylceramide analogs as Fabry disease biomarkers. J Am Soc Mass Spectrom 2015;26:499–510. 10.1007/s13361-014-1060-3 [DOI] [PubMed] [Google Scholar]
- 30. Vojtová L, Zima T, Tesař V, et al. Study of urinary proteomes in Anderson-Fabry disease. Ren Fail 2010;32:1202–9. 10.3109/0886022X.2010.516859 [DOI] [PubMed] [Google Scholar]
- 31. Matafora V, Cuccurullo M, Beneduci A, et al. Early markers of Fabry disease revealed by proteomics. Mol Biosyst 2015;11:1543–51. 10.1039/C4MB00707G [DOI] [PubMed] [Google Scholar]
- 32. Hughes DA. Fabry disease: will markers of early disease enable early treatment and better outcomes? Curr Opin Cardiol 2016;31:434–9. 10.1097/HCO.0000000000000308 [DOI] [PubMed] [Google Scholar]
- 33. Oder D, Nordbeck P, Wanner C. Long term treatment with enzyme replacement therapy in patients with Fabry Disease. Nephron 2016;134:30–6. 10.1159/000448968 [DOI] [PubMed] [Google Scholar]
- 34. Schiffmann R, Kopp JB, Austin HA 3rd. Enzyme replacement therapy in Fabry disease: a randomized controlled trial. Jama 2001;285:2743–9. [DOI] [PubMed] [Google Scholar]
- 35. Bodensteiner D, Scott CR, Sims KB, et al. Successful reinstitution of agalsidase beta therapy in Fabry disease patients with previous IgE-antibody or skin-test reactivity to the recombinant enzyme. Genet Med 2008;10:353–8. 10.1097/GIM.0b013e318170f868 [DOI] [PubMed] [Google Scholar]
- 36. Wilcox WR, Linthorst GE, Germain DP, et al. Anti-α-galactosidase A antibody response to agalsidase beta treatment: data from the Fabry Registry. Mol Genet Metab 2012;105:443–9. 10.1016/j.ymgme.2011.12.006 [DOI] [PubMed] [Google Scholar]
- 37. Bénichou B, Goyal S, Sung C, et al. A retrospective analysis of the potential impact of IgG antibodies to agalsidase beta on efficacy during enzyme replacement therapy for Fabry disease. Mol Genet Metab 2009;96:4–12. 10.1016/j.ymgme.2008.10.004 [DOI] [PubMed] [Google Scholar]
- 38. Lenders M, Stypmann J, Duning T, et al. Serum-mediated inhibition of enzyme replacement therapy in Fabry Disease. J Am Soc Nephrol 2016;27:256–64. 10.1681/ASN.2014121226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Linthorst GE, Hollak CE, Donker-Koopman WE, et al. Enzyme therapy for Fabry disease: neutralizing antibodies toward agalsidase alpha and beta. Kidney Int 2004;66:1589–95. 10.1111/j.1523-1755.2004.00924.x [DOI] [PubMed] [Google Scholar]
- 40. Rombach SM, Aerts JM, Poorthuis BJ, et al. Long-term effect of antibodies against infused alpha-galactosidase A in Fabry disease on plasma and urinary (lyso)Gb3 reduction and treatment outcome. PLoS One 2012;7:e47805 10.1371/journal.pone.0047805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Goker-Alpan O, Gambello MJ, Maegawa GH, et al. Reduction of plasma globotriaosylsphingosine levels after switching from agalsidase alfa to agalsidase beta as enzyme replacement therapy for Fabry Disease. JIMD Rep 2016;25:95–106. 10.1007/8904_2015_483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Lenders M, Schmitz B, Brand SM, et al. Characterization of drug-neutralizing antibodies in patients with Fabry disease during infusion. J Allergy Clin Immunol 2018;141:2289–92. 10.1016/j.jaci.2017.12.1001 [DOI] [PubMed] [Google Scholar]
- 43. Bigger BW, Saif M, Linthorst GE. The role of antibodies in enzyme treatments and therapeutic strategies. Best Pract Res Clin Endocrinol Metab 2015;29:183–94. 10.1016/j.beem.2015.01.006 [DOI] [PubMed] [Google Scholar]
- 44. Lenders M, Oder D, Nowak A, et al. Impact of immunosuppressive therapy on therapy-neutralizing antibodies in transplanted patients with Fabry disease. J Intern Med 2017;282:241–53. 10.1111/joim.12647 [DOI] [PubMed] [Google Scholar]
- 45. Germain DP, Hughes DA, Nicholls K, et al. Treatment of Fabry's Disease with the Pharmacologic Chaperone Migalastat. N Engl J Med 2016;375:545–55. 10.1056/NEJMoa1510198 [DOI] [PubMed] [Google Scholar]
- 46. Johnson FK, Mudd PN, Bragat A, et al. Pharmacokinetics and Safety of Migalastat HCl and Effects on Agalsidase Activity in Healthy Volunteers. Clin Pharmacol Drug Dev 2013;2:120–32. 10.1002/cpdd.1 [DOI] [PubMed] [Google Scholar]
- 47. Benjamin ER, Della Valle MC, Wu X, et al. The validation of pharmacogenetics for the identification of Fabry patients to be treated with migalastat. Genet Med 2017;19:430–8. 10.1038/gim.2016.122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Hughes DA, Nicholls K, Shankar SP, et al. Oral pharmacological chaperone migalastat compared with enzyme replacement therapy in Fabry disease: 18-month results from the randomised phase III ATTRACT study. J Med Genet 2017;54:288–96. 10.1136/jmedgenet-2016-104178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Müntze J, Salinger T, Gensler D, et al. Treatment of hypertrophic cardiomyopathy caused by cardiospecific variants of Fabry disease with chaperone therapy. Eur Heart J 2018;39:1861–2. 10.1093/eurheartj/ehy072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Warnock DG, Bichet DG, Holida M, et al. Oral Migalastat HCl leads to greater systemic exposure and tissue levels of active α-galactosidase A in Fabry patients when co-administered with infused agalsidase. PLoS One 2015;10:e0134341 10.1371/journal.pone.0134341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Kuter DJ, Mehta A, Hollak CEM, et al. Miglustat therapy in type 1 Gaucher disease: Clinical and safety outcomes in a multicenter retrospective cohort study. Blood Cells Mol Dis 2013;51:116–24. 10.1016/j.bcmd.2013.04.005 [DOI] [PubMed] [Google Scholar]
- 52. Guérard N, Oder D, Nordbeck P, et al. Lucerastat, an iminosugar for substrate reduction therapy: Tolerability, pharmacodynamics, and pharmacokinetics in patients with Fabry Disease on Enzyme Replacement. Clin Pharmacol Ther 2018;103:703–11. 10.1002/cpt.790 [DOI] [PubMed] [Google Scholar]
- 53. Simonetta I, Tuttolomondo A, Di Chiara T, et al. Genetics and gene therapy of Anderson-Fabry Disease. Curr Gene Ther 2018;18:96–106. 10.2174/1566523218666180404161315 [DOI] [PubMed] [Google Scholar]
- 54. Ohmori T. Advances in gene therapy for hemophilia: basis, current status, and future perspectives. Int J Hematol 2018;2012(Suppl 2). 10.1007/s12185-018-2513-4 [DOI] [PubMed] [Google Scholar]
- 55. Chang JH, Vines E, Bertsch H, et al. The impact of a multidisciplinary breast cancer center on recommendations for patient management: the University of Pennsylvania experience. Cancer 2001;91:1231–7. [DOI] [PubMed] [Google Scholar]
- 56. Torralba-Cabeza Miguel-Ángel, Olivera-González S, Sierra-Monzón J-L. The importance of a multidisciplinary approach in the management of a patient with type I Gaucher Disease. Diseases 2018;6 10.3390/diseases6030069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Wanner C, Arad M, Baron R, et al. European expert consensus statement on therapeutic goals in Fabry disease. Mol Genet Metab 2018;124:189–203. 10.1016/j.ymgme.2018.06.004 [DOI] [PubMed] [Google Scholar]
- 58. Pitz S, Kalkum G, Arash L, et al. Ocular signs correlate well with disease severity and genotype in Fabry disease. PLoS One 2015;10:e0120814 10.1371/journal.pone.0120814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Giuseppe P, Daniele R, Rita BM. Cutaneous complications of Anderson-Fabry disease. Curr Pharm Des 2013;19:6031–6. [DOI] [PubMed] [Google Scholar]
- 60. Tuttolomondo A, Pecoraro R, Simonetta I, et al. Neurological complications of Anderson-Fabry disease. Curr Pharm Des 2013;19:6014–30. 10.2174/13816128113199990387 [DOI] [PubMed] [Google Scholar]
- 61. Morrissey RP, Philip KJ, Schwarz ER. Cardiac abnormalities in Anderson-Fabry disease and Fabry's cardiomyopathy. Cardiovasc J Afr 2011;22:38–44. [PMC free article] [PubMed] [Google Scholar]
- 62. Waldek S, Feriozzi S. Fabry nephropathy: a review - how can we optimize the management of Fabry nephropathy? BMC Nephrol 2014;15:72 10.1186/1471-2369-15-72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Zar-Kessler C, Karaa A, Sims KB, et al. Understanding the gastrointestinal manifestations of Fabry disease: promoting prompt diagnosis. Therap Adv Gastroenterol 2016;9:626–34. 10.1177/1756283X16642936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Köping M, Shehata-Dieler W, Schneider D, et al. Characterization of vertigo and hearing loss in patients with Fabry disease. Orphanet J Rare Dis 2018;13:137 10.1186/s13023-018-0882-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Svensson CK, Feldt-Rasmussen U, Backer V. Fabry disease, respiratory symptoms, and airway limitation – a systematic review. Eur Clin Respir J 2015;2:26721 10.3402/ecrj.v2.26721 [DOI] [PMC free article] [PubMed] [Google Scholar]